

Abstract
Background
Altered methylation patterns are driving forces in colorectal carcinogenesis. The serrated adenocarcinoma (SAC) and sporadic colorectal carcinoma showing histological and molecular features of microsatellite instability (hmMSI-H) are two endpoints of the so-called serrated pathological route sharing some characteristics but displaying a totally different immune response and clinical outcome. However, there are no studies comparing the methylome of these two subtypes of colorectal carcinomas. The methylation status of 450,000 CpG sites using the Infinium Human Methylation 450 BeadChip array was investigated in 48 colorectal specimens, including 39 SACs and 9 matched hmMSI-H.
Results
Microarray data comparing SAC and hmMSI-H showed an enrichment in functions related to morphogenesis, neurogenesis, cytoskeleton, metabolism, vesicle transport and immune response and also significant differential methylation of 1540 genes, including CD14 and HLA-DOA which were more methylated in hmMSI-H than in SAC and subsequently validated at the CpG, mRNA and protein level using pyrosequencing, quantitative polymerase chain reaction (qPCR) and immunohistochemistry.
Conclusions
These results demonstrate particular epigenetic regulation patterns in SAC which may help to define key molecules responsible for the characteristic weak immune response of SAC and identify potential targets for treating SAC, which lacks molecular targeted therapy.
Electronic supplementary material
The online version of this article (10.1186/s13148-018-0571-3) contains supplementary material, which is available to authorized users.
Keywords: Colorectal cancer, Methylome, Serrated, Immune response, Microsatellite instability, Epigenetics, Colon carcinogenesis, CD14, HLA-DOA
Background
Serrated polyp pathway is considered as an alternative pathological sequence to the so-called adenoma-carcinoma sequence which is typically characterized by chromosomal instability and by ending up in the development of conventional carcinoma (CC) [1]. Less is known about the CRCs developed mainly in proximal colon through the serrated pathway, although high-level of microsatellite instability (MSI-H), BRAF mutation and CpG island methylation phenotype (CIMP) seems to be the driven forces in this carcinogenic process. The CRC showing histological and molecular features of MSI-H (hmMSI-H) [2, 3] is considered as one endpoint of the serrated route as well as serrated adenocarcinoma (SAC) which has a typical serrated morphology and remnants of serrated polyps (SP) adjacent to the invasive tumour [4, 5]. SAC has been recognized in the latest WHO classification of tumours of the digestive system as a new subtype of colorectal cancer (CRC) [6], accounting for 7.5 to 8.7% of all CRCs [5, 7] approximately one third of serrated pathway CRCs [8] and most are microsatellite stable (MSS) and can be either BRAF or KRAS mutated [9–11]. Criteria for SAC histologic diagnosis have been proposed [7] and recently validated in a series of 85 cases, and it has been shown to have a worse prognosis than conventional carcinoma (CC) [5]. Accordingly, SAC displays a higher frequency of adverse histological features at the invasive front including high-grade tumour budding and cytoplasmic pseudofragments, infiltrating growth pattern and weak peritumoural lymphocyte response [12]. Besides, mRNA microarray studies have demonstrated that SAC has a different expression profile compared to CC [13, 14]. Despite these features, there are no studies assessing the differences in the molecular signatures of SAC and the typical hmMSI-H nor specific markers nor the clinical differences between these two CRC subtypes.
Based on an initial histological evaluation, we aimed in this work to investigate the following issues:
-To discern which are the differentially methylated functions between SAC and hmMSI-H and if these could explain the histologic characteristics of these two entities
-To identify and validate at different levels those differentially methylated genes so they could be used as diagnostic markers or potential therapeutic targets
Results
The clinico-pathological features of the patients have been previously reported [5, 10] and are shown in Table 1. SACs were diagnosed on the basis of criteria proposed by Mäkinen et al. [7] and hmMSI-H according to prior established criteria [2] (Fig. 1). No significant differences were observed for confounding variables between SAC and hmMSI-H in the training and validation series (Table 1).
Table 1.
Clinico-pathological features of the study cases
| Training set | DNA validation set (pyrosequencing) | RNA validation set (qPCR) | Protein validation set (IHC) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SAC | hmMSI-H | SAC | hmMSI-H | SAC | hmMSI-H | SAC | hmMSI-H | |||||
| n = 39 (%) | n = 9 (%) | p value | n = 16 (%) | n = 9 (%) | p value | n = 19 (%) | n = 22 (%) | p value | n = 26 (%) | n = 21 (%) | p value | |
| Gender | ||||||||||||
| Female | 20 (51.3) | 7 (77.8) | 8 (50) | 7 (77.8) | 15 (78.9) | 14 (63.6) | 19 (73.1) | 12 (57.1) | ||||
| Male | 19 (48.7) | 2 (22.2) | 0.284 | 8 (50) | 2 (22.2) | 0.174 | 4 (21.1) | 8 (36.4) | 0.234 | 7 (26.9) | 9 (42.9) | 0.201 |
| Age [SD] | 71.6 [10.0] | 66.4 [15.22] | 0.094 | 72.0 [9.5] | 70.3 [8.4] | 0.267 | 69.0 [8.43] | 69.75 [13.9] | 0.839 | 69.9 [6.8] | 70 [10.7] | 0.969 |
| Location | ||||||||||||
| Proximal | 26 (66.7) | 8 (100) | 9 (56.3) | 8 (88.9) | 10 (52.6) | 15 (68.2) | 14 (53.8) | 13 (61.9) | ||||
| Distal/rectum | 13 (33.3) | 1 (0) | 0.182 | 7 (43.8) | 1 (11.1) | 0.107 | 9 (47.4) | 7 (31.8) | 0.309 | 12 (46.2) | 8 (38.1) | 0.399 |
| Dukes’ stage | ||||||||||||
| A | 4 (10.3) | 1 (11.1) | 2 (12.5) | 1 (11.1) | 3 (15.8) | 6 (27.3) | 4 (15.4) | 4 (19.0) | ||||
| B | 13 (33.3) | 5 (55.6) | 7 (43.8) | 5 (55.6) | 7 (36.8) | 9 (40.9) | 9 (34.6) | 10 (47.6) | ||||
| C | 17 (43.6) | 3 (33.3) | 0.846 | 7 (43.8) | 3 (33.3) | 0.846 | 9 (47.4) | 7 (31.8) | 0.522 | 13 (50.0) | 7 (33.3) | 0.523 |
| WHO grade | ||||||||||||
| High | 5 (12.8) | 0 (0) | 1 (6.3) | 0 (0) | 1 (5.3) | 0 (0) | 0 (0) | 0 (0) | ||||
| Low | 34 (87.2) | 9 (100) | 0.596 | 15 (93.8) | 9 (100) | 0.64 | 18 (94.7) | 22 (100) | 0.463 | 26 (100) | 21 (100) | NA |
| Type | ||||||||||||
| Non-mucinous | 34 (87.1) | 6 (66.7) | 14 (87.5) | 6 (66.7) | 17 (89.5) | 19 (86.4) | 22 (84.6) | 18 (85.7) | ||||
| Mucinous | 5 (12.8) | 3 (33.3) | 0.321 | 2 (12.5) | 3 (33.3) | 0.23 | 2 (10.5) | 3 (13.6) | 0.572 | 4 (15.4) | 3 (14.3) | 0.623 |
SAC serrated adenocarcinoma, hmMSI-H colorectal carcinoma showing histological and molecular features of microsatellite instability, qPCR quantitative PCR, IHC immunohistochemistry, SD standard deviation
Fig. 1.

Histological morphology of colorectal cancer subtypes (H-E staining). a Serrated adenocarcinoma (SAC) showing serrated glands with large, rounded nuclei with prominent nucleoli and ample eosinophilic cytoplasm. Original magnification × 20. b Colorectal carcinoma showing histological and molecular features of microsatellite instability (hmMSI-H). The image represents a microglandular monomorphic pattern with a small area of necrosis. Original magnification × 15
Differentially methylated functions
Bioinformatic analysis revealed a considerable number of GO biological processes (BP) differentially methylated in SAC vs. hmMSI-H: 40 GO terms obtained using ClusterProfiler while 76 terms where found using FatiGo (Additional file 1: Table S1). In general, differentially methylated genes were enriched for GO-BP terms related with biosynthesis (GO:0034654, GO:0031326, GO:0044271, GO:0018130); nitrogen and nucleic acid metabolism (GO:0090304, GO:0060255, GO:0051173, GO:0006807); RNA activity and transcription regulation (GO:0006355, GO:0032774, GO:2001141); protein secretion (GO:0051051, GO:0051047), neurogenesis (GO:0051960, GO:0030182, GO:0022008, GO:0030900), morphogenesis (GO:0048598, GO:0048562, GO:0009887, GO:0016331), sensory perception (GO:0007608, GO:0050911, GO:0050906); cytoskeleton and cell movement (GO:0048870, GO:0016477, GO:0051674) and immune response (GO:0002250).
The scatterplot obtained using REVIGO shows the GO biological processes which are differentially methylated between SAC and hmMSI-H after the redundancy reduction (Fig. 2). GO cellular component and molecular function categories are shown as Additional file 2: Figure S1.
Fig. 2.

Terms of Gene Ontology biological processes enriched in the comparison between SAC and hmMSI-H for their methyloma profile. The scatterplot shows the biological processes which are globally differentially methylated between SAC and hmMSI-H after GO term redundancy reduction. The graph is represented in a two-dimensional space derived by applying multidimensional scaling to a matrix of the GO terms’ semantic similarities [35]. Terms associated with higher methylation in hmMSI-H are shown in red squares. Colour intensity indicates level of significance (log10 p value). Circle diameter indicates number of genes for each biological process
Differentially methylated genes
The analysis of the methylome microarray data identified 1540 differentially methylated genes, 266 of which were more methylated in SAC than in hmMSI-H (Additional file 3: Table S2). No significant methylated genes were observed when comparing normal mucosa from SAC and normal hmMSI-H mucosa or when comparing Spanish and Finnish serrated tumour cases. Table 2 shows the list of the 42 most differentially methylated genes as obtained from the array analysis. Functions associated with these genes are shown in Additional file 4. Based on the extent of differential methylation grade, the importance of the biological functions, the design of suitable primers and the availability of antibodies, we decided to validate HLADOA and CD14 at the DNA, mRNA and protein level. CD14 is a surface antigen, preferentially expressed on monocytes/macrophages that cooperate with other proteins to mediate the innate immune response to bacterial lipopolysaccharide. Alternative splicing results in multiple transcript variants (NCBI RefSeqs NM_000591.3, NM_001040021.2, NM_001174104.1, NM_001174105.1) encoding the same protein. HLA-DOA, in turn, is a member of the HLA class II which forms a heterodimer with HLA-DOB. This heterodimer, HLA-DO, is found in lysosomes in B cells and regulates HLA-DM-mediated peptide loading on MHC class II molecules.
Table 2.
List of the 42 most differentially methylated genes (37 more methylated in hmMSI-H and 5 in SAC) as obtained from the array analysis
| Gene symbol | >methyl. in | adj p val. | Gene name | Chr |
|---|---|---|---|---|
| PDCD2L | hmMSI-H | 0.00101 | Programmed cell death 2-like | 19 |
| FGFR2 | SAC | 0.00147 | Fibroblast growth factor receptor 2 | 10 |
| CTSL2 | hmMSI-H | 0.00159 | Cathepsin L2 | 9 |
| TLR4 | hmMSI-H | 0.00159 | Toll-like receptor 4 | 9 |
| ANKRD10 | hmMSI-H | 0.00350 | Ankyrin repeat domain 10 | 13 |
| COPZ1 | hmMSI-H | 0.00420 | Coatomer protein complex, subunit zeta 1 | 12 |
| C17orf108 | SAC | 0.00420 | Chromosome 17 open reading frame 108 | 17 |
| CEP68 | hmMSI-H | 0.00580 | Centrosomal protein 68 kDa | 2 |
| ZNF652 | hmMSI-H | 0.00580 | Zinc finger protein 652 | 17 |
| PCYOX1L | hmMSI-H | 0.00583 | Prenylcysteine oxidase 1 like | 5 |
| GALNT11 | hmMSI-H | 0.00583 | UDP-NADG:polypeptide N-acetylgalactosaminyltransferase 11 | 7 |
| MICAL2 | hmMSI-H | 0.00583 | Microtubule-associated monoxygenase, calponin and LIM domain 2 | 11 |
| OSCP1 | hmMSI-H | 0.00583 | Organic solute carrier partner 1 | 1 |
| ACAD10 | hmMSI-H | 0.00583 | Acyl-CoA dehydrogenase family, member 10 | 12 |
| MELK | hmMSI-H | 0.00583 | Maternal embryonic leucine zipper kinase | 9 |
| HLA-DOA | hmMSI-H | 0.00583 | Major histocompatibility complex, class II, DO alpha | 6 |
| KRT20 | hmMSI-H | 0.00583 | Keratin 20 | 17 |
| TMEM45B | hmMSI-H | 0.00583 | Transmembrane protein 45B | 11 |
| LOC285401 | SAC | 0.00641 | Uncharacterized LOC285401 | 3 |
| PPIL1 | hmMSI-H | 0.00650 | Peptidylprolyl isomerase (cyclophilin)-like 1 | 6 |
| CA13 | hmMSI-H | 0.00709 | Carbonic anhydrase XIII | 8 |
| BCL7C | hmMSI-H | 0.00709 | B cell CLL/lymphoma 7C | 16 |
| CD14 | hmMSI-H | 0.00850 | CD14 molecule | 5 |
| PLA2G4C | hmMSI-H | 0.00905 | Phospholipase A2, group IVC (cytosolic, calcium-independent) | 19 |
| LY6G6D | hmMSI-H | 0.00908 | Lymphocyte antigen 6 complex, locus G6D | 6 |
| TCF7L2 | hmMSI-H | 0.00954 | Transcription factor 7-like 2 (T-cell specific, HMG-box) | 10 |
| RP11-165H20.1 | hmMSI-H | 0.00954 | CHIA-like pseudogene | 1 |
| CHD6 | hmMSI-H | 0.00954 | Chromodomain helicase DNA-binding protein 6 | 20 |
| PARN | hmMSI-H | 0.00954 | Poly(A)-specific ribonuclease | 16 |
| KCNK15 | hmMSI-H | 0.00954 | Potassium channel, subfamily K, member 15 | 20 |
| TMEM209 | hmMSI-H | 0.01015 | Transmembrane protein 209 | 7 |
| GOLM1 | hmMSI-H | 0.01016 | Golgi membrane protein 1 | 9 |
| ARRDC1 | hmMSI-H | 0.01016 | Arrestin domain containing 1 | 9 |
| TRIM21 | hmMSI-H | 0.01016 | Tripartite motif containing 21 | 11 |
| PHAX | hmMSI-H | 0.01016 | Phosphorylated adaptor for RNA export | 5 |
| NMT2 | hmMSI-H | 0.01016 | N-myristoyltransferase 2 | 10 |
| ATP12A | hmMSI-H | 0.01016 | ATPase, H+/K+ transporting, nongastric, alpha polypeptide | 13 |
| ARHGAP30 | SAC | 0.01016 | Rho GTPase-activating protein 30 | 1 |
| WWP1 | hmMSI-H | 0.01055 | WW domain-containing E3 ubiquitin protein ligase 1 | 8 |
| GNAI3 | hmMSI-H | 0.01055 | Guanine nucleotide binding (G protein), α inhibit polypeptide 3 | 1 |
| CD48 | SAC | 0.01055 | CD48 molecule | 1 |
| EXPH5 | hmMSI-H | 0.01070 | Exophilin 5 | 11 |
Genes chosen for validation by pyrosequencing, qPCR, and IHC are written in bold letters
SAC serrated adenocarcinoma, hmMSI-H colorectal carcinoma showing molecular and histological features of microsatellite instability
Validation of methylated sites by pyrosequencing
The distribution of CpG islands in HLA-DOA and CD14 enabled the design of primers to quantify the level of CpG methylation in these genes by pyrosequencing upon bisulfite-treated DNA. As shown in Table 3, the methylation percentage in CD14 was generally lower than that in HLA-DOA CpG sites. Intriguingly, tumoural specimens from SAC, but not hmMSI-H cases, showed lower methylation level of HLA-DOA than normal adjacent samples. In contrast, CpG2 site from CD14 displayed higher methylation in tumoural than in normal specimens from hmMSI-H patients, whereas no such significant difference was observed in SAC cases. Consistent with the microarray results, the percentage of CpG methylation in HLA-DOA and CD14 was higher in hmMSI-H than in SAC at all CpG sites studied except for CpG3 in HLA-DOA (Table 3).
Table 3.
Pyrosequencing for the relative quantitation of CpG methylation in CD14 and HLA-DOA genes
| Gene | CD14 | HLA-DOA | ||||
|---|---|---|---|---|---|---|
| CpG site | CpG1 | CpG2 | CpG1 | CpG2 | CpG3 | |
| SAC-T (n = 16) | Mean | 6.89 | 13.56 | 33.83 | 34.95 | 89.78 |
| SD | 5.53 | 8.06 | 15.47 | 14.91 | 8.04 | |
| hMSI-H-T (n = 9) | Mean | 13.64 | 22.38 | 50.94 | 48.12 | 94.61 |
| SD | 6.15 | 8.9 | 14.81 | 13.62 | 2.36 | |
| SAC-T vs. hmMSI-H | p value | 0.010 | 0.020 | 0.013 | 0.039 | 0.14 |
| SAC-N (n = 8) | Mean | 7.97 | 13.86 | 60.02 | 58.27 | 100 |
| SD | 3.19 | 2.19 | 2.01 | 2.94 | 0 | |
| MSI-N (n = 6) | Mean | 8.25 | 13.72 | 53.13 | 53.26 | 98.69 |
| SD | 2.36 | 2.37 | 6.73 | 8.92 | 2.75 | |
| SAC-T vs. SAC-N | p value | 0.616 | 0.911 | 0.0001 | 0.0003 | 0.006 |
| hmMSI-H-T vs. hmMSI-H-N | p value | 0.064 | 0.038 | 0.742 | 0.432 | 0.002 |
| SAC-N vs hmMSI-N | p value | 0.860 | 0.911 | 0.017 | 0.159 | 0.271 |
SAC-T tumoural tissue from serrated adenocarcinoma, hmMSI-H-T tumoural tissue from histological and molecular high-level microsatellite unstable colorectal carcinoma, SAC-N normal tissue adjacent to SAC, hmMSI-H-N normal tissue adjacent to hmMSI-H
Validation by qPCR
With the aim of finding out whether higher CpG methylation in HLA-DOA and CD14 in hmMSI-H correlates with a decreased expression of these genes, an analysis of the expression of HLA-DOA and CD14 mRNA by quantitative PCR was performed. HLA-DOA expression did not show differences between tumoural and normal mucosa (median 0.00438 vs. 0.00467; p = 0.531), whereas CD14 expression was lower in tumoural than in normal mucosa, although not reaching statistical significance (median 0.0128 vs. 0.0599; p = 0.098). There were no differences when comparing tumoural and normal mucosa for each CRC subtype (Table 4). The qPCR results validated the microarray and pyrosequencing results as CRC cases with serrated histology showed higher expression of CD14 (median 0.133 vs. 0.004; p = 0.004) and HLA-DOA (median 0.099 vs. 0.006; p = 0.047) than hmMSI-H cases, respectively (Fig. 3/Table 4).
Table 4.
Quantitative analysis of the CD14 and HLA-DOA mRNA expression in tumour and adjacent normal mucosa
| Gene | Sample type | N | Mean ± SD | Median | IR |
|---|---|---|---|---|---|
| CD14 | SAC-T | 19 | 0.166 ± 0.16 | 0.133 | 0.509 |
| hmMSI-H-T | 22 | 0.066 ± 0.16 | 0.004 | 0.718 | |
| p value | 0.004 | ||||
| SAC-N | 7 | 0.191 ± 0.17 | 0.198 | 0.386 | |
| hmMSI-H-N | 7 | 0.129 ± 0.24 | 0.037 | 0.667 | |
| p value | 0.383 | ||||
| HLA-DOA | SAC-T | 19 | 0.0617 ± 0.11 | 0.099 | 0.375 |
| hmMSI-H-T | 22 | 0.021 ± 0.03 | 0.006 | 0.138 | |
| p value | 0.047 | ||||
| SAC-N | 7 | 0.135 ± 0.01 | 0.0098 | 0.034 | |
| hmMSI-H-N | 7 | 0.07 ± 0.008 | 0.037 | 0.021 | |
| p value | 0.620 |
SAC-T tumoural tissue from serrated adenocarcinoma, hmMSI-H-T tumoural tissue from histological and molecular high-level microsatellite unstable colorectal carcinoma, SAC-N normal tissue adjacent to SAC, hmMSI-H-N normal tissue adjacent to hmMSI-H
Fig. 3.

qPCR results of the mRNA expression of CD14 and HLA-DOA genes in SAC and CC tumoural tissue as well as in adjacent non-tumoural specimens. Asterisk indicates statistical significance.*p > 0.05; ***p > 0.005
Validation by immunohistochemistry
In order to investigate whether differential methylated status of HLA-DOA and CD14 in SAC compared to hmMSI-H could have an effect on protein expression within the tissue cells, immunohistochemistry for both HLA-DOA and CD14 was performed. Figure 4 shows the staining pattern for CD14 being cytoplasmic in stromal cells, not only in myeloid cells from monocyte/macrophague linage but also in some endothelial, (lower left image) whereas for HLA-DOA mantle zone cells from a lymph node were positive (lower right image). In SAC cases, a positive membranous staining for CD14 was observed in some stromal cells and lymphocytes with no expression in neoplastic glands. As shown in Fig. 4, the expression of CD14 was also absent in neoplastic glands from hmMSI-H cases and only few stromal cells stained positive and no staining was observed in lymphocytes. In tumoural cases, an intense cytoplasmic and luminal secretion staining for HLA-DOA was observed in SAC neoplastic glands whereas weak cytoplasmic expression considered as negative was observed in hmMSI-H tumour cells. In both cases, stromal cells were negative (Fig. 4).
Fig. 4.
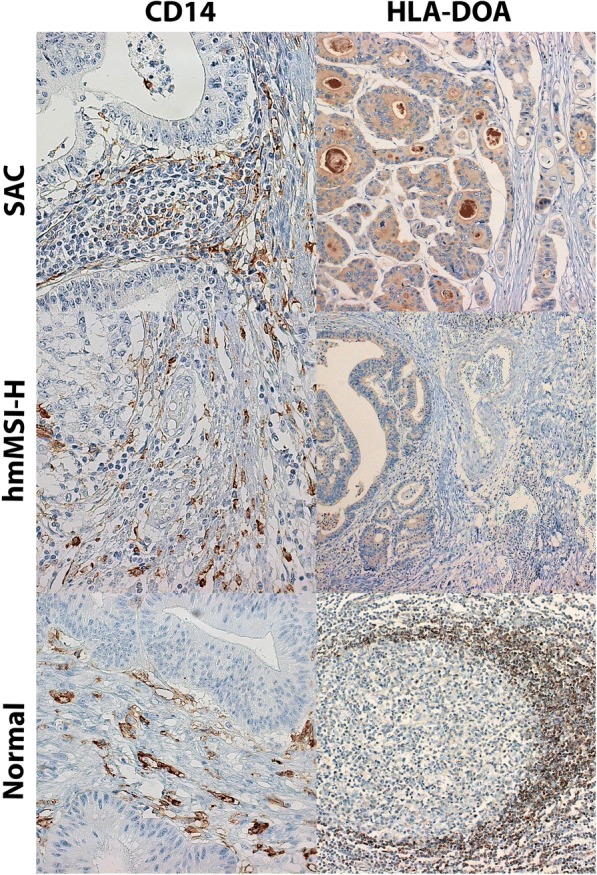
Immunohistochemical expression of CD14 and HLA-DOA in serrated adenocarcinoma (SAC) and CRC showing histological and molecular features of microsatellite instability (hmMSI-H) and in normal tissue (intestinal lamina propria and lymph node, respectively). Original × 40 magnification in all except in HLA-DOA in hmMSI-H (× 20)
Surprisingly, negative or weak expression of CD14 was statistically more frequent in SAC than in hmMSI-H (81.8% vs. 42.9%; p = 0.008) (Fig. 4; Table 5). As expected, moderate/strong HLA-DOA staining was higher in SAC than in hmMSI-H (30.8% vs 0%; p = 0.005), thus confirming array, pyrosequencing and qPCR results.
Table 5.
Immunohistochemical expression of CD14 and HLA-DOA proteins
| Protein | Sample type | n | Distribution (A–C) Intensity (1–3) score |
Two-tier score | p value (Fisher) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| A1 | A3 | B2 | B3 | C3 | Negative/weak (A1-B2) | Moderate/strong (B3-C3) | ||||
| CD14 | SAC | 26 | 11 | 10 | 0 | 3 | 2 | 21 (81.8) | 5 (19.2) | |
| hmMSI-H | 21 | 3 | 6 | 0 | 5 | 7 | 9 (42.9) | 12 (57.1) | 0.008 | |
| HLA-DOA | SAC | 26 | 9 | 8 | 1 | 8 | 0 | 18 (69.2) | 8 (30.8) | |
| hmMSI-H | 21 | 17 | 4 | 0 | 0 | 0 | 21 (100) | 0 | 0.005 | |
Immunohistochemical markers in the tumours were evaluated by considering the distribution area score (A < one third, B = between one and two thirds, C = two-thirds-whole area) and the staining intensity (1 = none or weak staining, 2 = moderate, 3 = strong)
SAC serrated adenocarcinoma, hmMSI-H-T colorectal carcinoma with histological and molecular high-level microsatellite instability
Association of CD14/HLA-DOA methylation with KRAS/BRAF/MSI-H status
In order to ascertain the relationship between the methylation of these genes with common biomarkers used in the clinical management of CRC, we performed an association study using the cases of DNA validation set. The results in Table 6 show that CD14 methylation is significantly associated with wild-type KRAS, V600E mutant BRAF and microsatellite instability. HLA-DOA methylation did not correlate with these markers except for a tendency with microsatellite instability (p = 0.069).
Table 6.
Association of CD14 and HLA-DOA methylation (measured as percentage of methylated cytosine) with common colorectal carcinoma biomarkers (KRAS, BRAF, MSI)
| Biomarker* | CD14 | HLA-DOA | |||||
|---|---|---|---|---|---|---|---|
| Median | IR | p value | Median | IR | p value | ||
| KRAS | Mutated | 9.20 | 5.9 | 35.97 | 38.8 | ||
| Wild-type | 15.355 | 12 | 0.022 | 40.37 | 33.7 | 0.290 | |
| BRAF | Mutated | 16.68 | 21.7 | 39.63 | 35.5 | ||
| Wild-type | 12.845 | 6.4 | 0.035 | 37.53 | 36.2 | 0.82 | |
| MSI | Unstable | 16.7 | 19.1 | 40.95 | 30.2 | ||
| Stable | 11.32 | 6.1 | 0.001 | 31.9 | 39.6 | 0.069 | |
*Codon 12 and 13 mutations in KRAS and V600E mutation in BRAF were considered. CpG site 2 for CD14 and CpG site 2 for HLA-DOA were considered. Similar results were obtained for site 1 (data not shown)
IR interquartile range, MSI microsatellite instability
Discussion
A two-arm model has been proposed to explain the progression of the serrated pathway, the aberrant crypt foci-hyperplastic type being the earliest lesion which may develop into hyperplastic polyps and sessile serrated adenomas, or into traditional serrated adenomas, both of which may progress to CRC having serrated histology such as SAC and to CRC with histological and molecular features of microsatellite instability (hmMSI-H). Despite that these two CRC subtypes seem to share a common pathological route, the clinical manifestation and prognosis is surprisingly different. On the one hand, hmMSI is associated with an abundant immune response and good prognosis [15]. On the other hand, SAC has a worse outcome that conventional CRC as it displays a high frequency of histological adverse prognostic factors, the weak immune response being one of them, along with high tumour budding and infiltrative growth pattern. It is therefore important to study which molecular features distinguish SAC and hmMSI-H, not only for understanding the serrated polyp pathway but also for providing diagnostic markers and molecular targets for histology-based specific treatments. In this study, which is the first comparing the methylome of these two entities, we have observed important differences in the functions associated with methylated genes from each tumour type. First, most of these genes were more methylated in hmMSI-H than in SAC, thus suggesting that, in general, MSI-H development might more associated with aberrant hypermethylated, and thus silencing, genes than SAC. Given that differentially methylated genes were more methylated in SAC than in CC [16], SAC would show an intermediate hypermethylation status between hmMSI-H and CC. Functions differentially methylated are mostly related to metabolism, morphogenesis, neurogenesis, cytoskeleton and immune response. These findings are consistent with previous works analysing the expression profile of SAC highlighting the over-representation of these biological activities when compared to CC [13, 14]. Evasion of immune response has been proposed as an emerging hallmark of cancer, and in fact, weak peritumoural lymphocytic infiltration (PLI) is considered as one histological adverse prognostic factor [17, 18]. All this evidence is also consistent with the higher frequency of tumour infiltrating and peritumoural lymphocytes and “Crohn-like” inflammatory response found in hmMSI-H and the weak PLI and worse prognosis which characterize SAC [12]. This finding raises the question of which molecular factors determine these differences between these two CRCs. One possible explanation comes from the fact that hmMSI-H CRC, due to the dysfunction of the DNA mismatch repair machinery, generates a high number of neoantigens that stimulate the immune system whose effector cells are attracted to the tumour invasive front [19]. Given the allegedly common origin of hmMSI-H and SAC and some molecular similarities, one challenging therapeutic approach for SAC might be to make this CRC subtype recognizable for the immune system. In order to know which steps in the immune response could be affected, a list of significantly methylated genes has been obtained in this work most of them belonging to immune response.
From those significantly methylated genes, CD14 and HLADOA were chosen to be validated at the DNA, mRNA and protein level. CD14 is protein involved expressed mainly by monocyte/macrophage cell although some non-myeloid lineage cells such epithelial can also express it [20]. Apart from its role in response against sepsis, recent reports have found that CD14+ circulating monocytes and CD14+ tumour infiltrating macrophages are more frequently found in patients with CRC compared to healthy controls, these findings being associated with a higher plasma concentration of the immunosuppressive cytokine, IL10 [21]. These CD14+ monocytes are a subset of myeloid-derived suppressor cells (MDSC) which are key mediators in the negative regulation of immune responses [22]. Therefore, the observed methylation and CD14 gene silencing could have an immunostimulatory effect in MSI-H CRCs in contrast to SAC where CD14 gene is less methylated and, subsequently, more expressed. Despite that our study validates CD14 microarray result at the DNA and mRNA level, it was not able to confirm a lower expression of the CD14 protein in hmMSI-H. Possible reasons for that finding could be the expression of specific CD14 transcripts by myeloid cells or non-myeloid with other functions than immune tolerance or the fact that CD14 is also expressed by non-myeloid cells [20], thus making complicated its histological evaluation. Supporting the conflicting nature of this issue, some other studies have reported that CD14+ macrophages at the invasive front correlates with a more favourable prognosis in CRC patients with metastasis [23]. The expression of molecules expressed by immune cells such as CD14 will be dependent on the overall presence of these cells in the tumour microenvironment. One of our aims was to characterize molecules or immune cell types that are responsible for the difference observed between SAC and hmMSI-H. The higher CD14 methylation and the decreased gene expression observed in hmMSI-H stress the point that, despite hmMSI-H having a remarkable immune response, some kind of difference in the CD14 function may exist between SAC and hmMSI-H.
Human leukocyte antigen (HLA) family members are antigen-presenting molecules which are expressed by virtually all cells in human body, including cancer cells, although the downregulation or aberrant expression of these molecules are used by the tumour to avoid immune response [24]. As regards to HLA-DOA, we validated the higher methylation and lower mRNA and protein expression in hmMSI-H compared to SAC. Very little is known about that protein as there are no studies linking it to colorectal carcinoma. Archer et al., by analysing the methylation status of 1505 CpG sites in hepatocellular carcinoma (HCC) tissue and comparing it with paired pre-neoplastic non-tumourous specimens from the same patients, observed that HLA-DOA was hypomethylated in tumoural tissue [25]. This finding supports a role of HLA-DOA, not only in HCC but in CRC tumour development. In agreement, our results in CRC also demonstrated a lower methylation in CRC compared to adjacent normal specimens, regardless the histological type of CRC. This observation did not reach statistical significance at the mRNA expression level, thus suggesting additional factors involved in the regulation of HLA-DOA expression. Very few articles correlated HLA-DOA with immune response features. Wang et al. reported that hypomethylation of CpGs in 6p21.3, where HLA-DOA is located, was associated with increased CD8 T cell tumour infiltration in serous ovarian cancer [26] whereas Ningappa et al. and Sindhi et al. demonstrated that HLA-DOA inhibits B cell presentation of antigen and consequently these authors proposed a potentially novel antirejection drug target [27, 28]. Our results are in line with these latter works; as transcription of major histocompatibility genes is silenced by DNA methylation [29] of upstream promoters, it would not be surprising that higher HLA-DOA methylation in hmMSI-H might have an effect on enhancing antigen presentation by B cells and generate a more prominent immune response.
Conclusions
Our study identifies key functions and genes that might be important for understanding two CRC histological subtypes sharing a common pathological route but developing a dissimilar immune response and having a different clinical outcome. However, subsequent studies are needed to further characterize the interplay between the immune microenvironment and the tumour CRC cells in the serrated pathway.
Material and methods
Patients and tumour samples
SACs were diagnosed on the basis of criteria proposed by Mäkinen et al. (epithelial serrations, clear or eosinophilic cytoplasm, abundant cytoplasm, vesicular nuclei, absence of or less than 10% necrosis of the total surface area, mucin production and cell balls and papillary rods in mucinous areas of a tumour) [7]. hmMSI-H were diagnosed according to prior established criteria (mucinous, signet-ring cell, and medullary carcinoma, tumour infiltrating and peritumoural lymphocytes, “Crohn-like” inflammatory response, poor differentiation, tumour heterogeneity and “pushing” tumour border) (Fig. 1) [2]. Frozen samples of 21 and 18 SACs were retrieved from Santa Lucia General University Hospital (HGUSL), Cartagena, Spain, and Oulu University Hospital, Oulu, Finland, respectively. Additionally, nine matched hmMSI-H from HGUSL were included for the methylome microarray study. Validation by methylated sequences was performed on 16 Spanish SAC cases and nine hmMSI-H from the microarray subset. Validation by qPCR was performed upon frozen specimens of 12 SAC and nine hmMSI-H, and in addition, adjacent normal mucosa was also analysed from eight SACs and six hmMSI-H. Paraffin blocks of 26 SAC and 21 matched hmMSI-H, included in previous works, [10, 14] were used for immunohistochemistry (IHC) validation. The assessment of the MSI-H condition was confirmed at the molecular level as described previously by our group [10], and none of the hmMSI-H showed serrated morphology. The cases from DNA validation set were used to assess the correlation between gene methylation and KRAS, BRAF and MSI status. The study was approved by the Hospital Ethics Committee and was carried out in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Written informed consent was obtained from all the patients.
DNA extraction
A volume of approximately 10 mm3 was extracted from each frozen tissue using the disposable sterile biopsy punch. DNA was extracted following the manufacturer’s instructions (Qiagen, Hilden, Germany). Briefly, tissue was disrupted and homogenized in ATL buffer using a Tissueruptor (Qiagen), incubated with proteinase K and the homogenate was subjected to automatic DNA extraction using the Qiacube equipment and the QiaAmp DNA Mini Kit (cat no.:51306), both provided by Qiagen.
Bisulfite treatment and DNA methylation assay
HumanMethylation450K BeadChip (Illumina, Inc., San Diego, CA), using Infinium HD Methylation assay for genome-wide DNA methylation screening, was employed. In brief, genomic DNA (1000 ng) from each sample was bisulfite converted with the EZ DNA Methylation Kit (Zymo Research, Orange, CA) according to the manufacturer’s recommendations. Bisulfite-treated DNA was isothermally amplified at 37 °C (20–24 h), and the DNA product was fragmented by an endpoint enzymatic process, then precipitated, resuspended, applied to an Infinium Human Methylation450K BeadChip (Illumina, San Diego, CA, USA), and hybridized at 48 °C (16–24 h). The fluorescently stained chip was imaged by the Illumina i-SCAN, and Illumina’s Genome Studio program (Methylation Module) was used to analyse BeadArray data to assign site-specific DNA methylation β-values to each CpG site. The data set supporting the results of this article are available in the GEO repository, GSE68060 in https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68060.
Preprocessing of methylation data
Processing of raw data was done using R packages. Probes with a low detection p value (p < 0.01) in more than 95% of the samples and those measuring SNPs or mapping in X or Y chromosomes were removed and normalization followed a three-step procedure. Firstly, a colour bias adjustment was applied using the methylumi R-package [30]. Then, wateRmelon [31] R-package was used to perform between-sample normalization by equalization of type I and type II backgrounds followed by separated quantile normalization of methylated and unmethylated intensities. Finally, A BMIQ [32] intra-sample normalization procedure, included in the wateRmelon R-package, was applied to correct the bias of type II probe values.
Differential methylation functional profiling
The analysis of differentially methylated genes was performed using limma [33] R-package. Data were fitted to a linear model, and differential methylated genes were identified by using the empirical Bayes method included in the package. If the comparison was done between paired samples, a moderated paired t test was applied. A FDR-corrected p value of 0.05 was used as the threshold to select differentially methylated genes. Functional profiling of the differentially methylated genes was performed using ClusterProfiler and the FatiScan method included in the Babelomics [33, 34] web suite. For functional annotation, the Biological Process Database from Gene Ontology (GO) (www.geneontology.org) was used. Differentially methylated GO biological process was represented as scatterplot using REVIGO online package [35].
Pyrosequencing
DNA methylation percentages of five different CpG island sites included in the microarray (two in CD14, three in HLA-DOA) were analysed and quantified by pyrosequencing. Bisulfite-converted DNA was previously amplified by PCR using Hot-Start GoTaq polymerase (Promega, Madison, WI) under the following conditions: 1 ul of DNA, 4 ul of 5X polymerase buffer, 0.2 mM dNTPs, 0.6 mM MgCl2, 0.3 μM of either biotin-labelled forward or reverse primers and 0.05 U/μl Hot-start Go Taq Flexi polymerase (Promega). PCR protocol was performed as follows: initial denaturation at 94 °C for 2 min, 35 cycles of 94 °C 10 s, 64 °C (CD14) or 60 °C (HLA-DOA) 10 s and 72 °C 50 s and a final extension step of 72 °C 7 min. Details of amplicon and primer sequences are provided in Additional file 5: Table S3. PCR products were verified using the QIAxcel DNA high-resolution electrophoresis system. Pyrosequencing of methylated sites was performed using the PyroMark Q24 (Qiagen) according to the manufacturer’s protocol. The methylation level was assessed using the PyroMark Q24 2.0.6 Software (Qiagen) by which the methylation percentage (mC/mC+C) for each CpG was calculated. The results are presented as the percentage (mean ± SD) of the different CpG sites studied for each of the CpG sites analysed whose sequences and relative positions are also shown as Additional file 5: Table S3.
Quantitative PCR for assessing mRNA expression
RNAs from 20 SACs and 22 hmMSI-H, including those from the training set, were extracted with the miRNeasy kit (ref: 217004, Qiagen) and used for validation by qPCR. The retrotranscriptase reaction was performed from a total of 1 μg of DNAseI-treated RNA using the DyNAmo cDNA synthesis Kit (ref: F470L) provided by Thermo Scientific (Rockford, IL). Five microlitres of 1:5 diluted cDNA was added to the qPCR reaction containing 12.5 μl 2X QuantiTect SYBR Green PCR Kit (ref:204145, Qiagen) and 300 nM of each primer in a total volume of 25 μl. qPCR was performed on a 7500F real-time PCR system by Applied Biosystems (Foster City, CA, USA) according to the instruction manual and following the standard protocol: 50 °C 2 min, 95 °C 10 min, 40 cycles of 95 °C 15 s, 60 °C 1 min and a melt curve stage consisting of 95 °C 15 s, 60 °C min, 95 °C 30 s and 60 °C 30 s. Primers were designed using primer3 software and sequences, and amplicon sizes are shown in Additional file 5: Table S3. The relative quantitation was done by the 2-ΔCt method using β-actin as the housekeeping gene.
Immunohistochemistry
The validation subset consisted of 26 SAC and 21 hmMSI-H cases matched for gender, age and location, and a representative area of each tumour was selected by one of us (JGS). Whole 2.5-μm sections were stained with CD14 and HLADOA rabbit antibodies. Details on equipment, antigen retrieval conditions (buffer, pH, temperature, time) and incubation (temperature, time) for both antibodies are as follows: Bechmark Ultra Ventana, (CC1, basic, 95 °C, 56 min) and (overnight, room temperature). Antibody purveyor and type, code (clone) and antibody dilution were as follows: for CD14: Cell Marque, monoclonal, 760-4523 (EPR3653), 1:5, and for HLA-DOA: Sigma Aldrich, polyclonal, HPA045038, 1:200. Endogenous peroxidase activity was blocked using 0.5% H2O2 for 5 min. For visualization of the antigen, the sections were immersed in 3,3′-diaminobenzidine (DAB) and counterstained with Harris’ haematoxylin for 5 min. Following manufacturers´ recommendations sinusoidal histiocytes and mantle zone from a lymph node were used as positive controls for CD14 and HLA-DOA, respectively.
These markers were evaluated by considering a staining intensity in both the centre of the tumour and the invasive front (1 = none or weak staining, 2 = moderate, 3 = strong) and a staining area score (A < one third, B = between one and two thirds, C > two thirds) in a given area. For statistical analysis, both intensity and distribution were considered.
Statistical analysis of validation data
For the analysis of quantification of methylated DNA sequences, the data correspond to a split-plot design with one between-subject factor defining six independent groups of cases (SAC, CC, hmMSI-H; tumoural and non-tumoural) and one within-subject factor (CpG sites) defining nine repeated measures for every case. Accordingly, we performed two ANOVA SPF-p-q. The first compared the means of the tumoural vs. non-tumoural groups in each of the nine different CpG sites and the second the means of the six different groups in these sites. For checking the relationship between methylation percentage and binary variables, the t test for independent samples and the Mann-Whitney’s U test were used. Statistical significance in the immunohistochemistry study was assessed using Pearson χ2 or Fisher’s exact test when indicated. Descriptive statistics were computed for real-time PCR. Statistical analysis was performed using the SPSS (Version 22, Chicago, IL) package.
Additional files
Table S1. Gene Ontology biological processes (BP) terms differentially enriched in the comparison SAC vs. hmMSI-H; 40 obtained using ClusterProfiler and 76 using FatiGo. (XLSX 25 kb)
Figure S1. Terms of Gene Ontology Cellular Component and Molecular Functions enriched in the comparison between SAC and hmMSI-H for their methyloma profile. The scatterplot shows the terms which are globally differentially methylated between SAC and hmMSI-H after GO term redundancy reduction. The graph is represented in a two dimensional space derived by applying multidimensional scaling to a matrix of the GO terms’ semantic similarities [35]. (TIF 162 kb)
Table S2. List of the 1540 differentially methylated genes between hmMSI-H and SAC. (XLSX 149 kb)
Molecular functions of the 42 most differentially methylated genes. Source https://www.ncbi.nlm.nih.gov/gene. (DOCX 23 kb)
Table S3. Primer sequences, amplicon sizes and location of CpGs evaluated in the study. (DOC 48 kb)
Acknowledgements
We are grateful to the Madrid division of Spanish Genotyping National Center (Cegen) in the National Oncology Research Center (CNIO), Madrid, Spain, and to Fundación para la Formación e Investigación Sanitarias from Healthcare Council of Murcia Region, Spain.
Funding
This work was supported by a grant from Instituto de Salud Carlos III, Ministerio de Sanidad, Spain, and FEDER funds (refs: PI12-1232 and PI15-0626).
Availability of data and materials
The data set supporting the results of this article are available in the GEO repository, GSE68060 in https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68060.
Abbreviations
- CC
Conventional carcinoma
- CpG
Cytosine-phospho-guanine
- CRC
Colorectal carcinoma
- HLA
Human leukocyte antigen
- hmMSI-H
Colorectal carcinoma showing typical molecular and histological features of MSI-H
- IHC
Immunohistochemistry
- MSI-H
High-grade microsatellite instability
- MSS
Microsatellite stability
- qPCR
Quantitative polymerase chain reaction
- SAC
Serrated adenocarcinoma
- SD
Standard deviation
- WHO
World Health Organization
Authors’ contributions
JGS, MCT, DTM and PCZ participated in data collection and analysis (pyrosequencing. qPCR and IHC). FHL and AC carried out the bioinformatic analysis of array data. AT and MJM contributed in series characterization and data collection. PCZ designed the study and wrote the draft of the manuscript, and AT, MJM and AC revised the final version. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was approved by the Hospital Ethics Committee (HGUSL) and was carried out in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Written informed consent was obtained from all the patients.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer. 2003;3:695–701. doi: 10.1038/nrc1165. [DOI] [PubMed] [Google Scholar]
- 2.Bellizzi AM, Frankel WL. Colorectal cancer due to deficiency in DNA mismatch repair function: a review. Adv Anat Pathol. 2009;16:405–417. doi: 10.1097/PAP.0b013e3181bb6bdc. [DOI] [PubMed] [Google Scholar]
- 3.Bettington M, Walker N, Clouston A, Brown I, Leggett B, Whitehall V. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology. 2013;62:367–386. doi: 10.1111/his.12055. [DOI] [PubMed] [Google Scholar]
- 4.Mäkinen MJ. Colorectal serrated adenocarcinoma. Histopathology. 2007;50:131–150. doi: 10.1111/j.1365-2559.2006.02548.x. [DOI] [PubMed] [Google Scholar]
- 5.García-Solano J, Pérez-Guillermo M, Conesa-Zamora P, et al. Clinicopathologic study of 85 colorectal serrated adenocarcinomas: further insights into the full recognition of a new subset of colorectal carcinoma. Hum Pathol. 2010;41:1359–1368. doi: 10.1016/j.humpath.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Hamilton SR, Bosman FT, Boffetta P, et al. Carcinoma of the colon and rectum. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, et al., editors. WHO classification of tumours of the digestive system. Lyon: IARC; 2010. pp. 134–146.7. [Google Scholar]
- 7.Tuppurainen K, Mäkinen JM, Junttila O, Liakka A, Kyllönen AP, Tuominen H, et al. Morphology and microsatellite instability in sporadic serrated and non-serrated colorectal cancer. J Pathol. 2005;207:285–294. doi: 10.1002/path.1850. [DOI] [PubMed] [Google Scholar]
- 8.O'Brien MJ, Yang S, Mack C, et al. Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am J Surg Pathol. 2006;30:1491–1501. doi: 10.1097/01.pas.0000213313.36306.85. [DOI] [PubMed] [Google Scholar]
- 9.Stefanius K, Ylitalo L, Tuomisto A, et al. Frequent mutations of KRAS in addition to BRAF in colorectal serrated adenocarcinoma. Histopathology. 2011;58:679–692. doi: 10.1111/j.1365-2559.2011.03821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.García-Solano J, Conesa-Zamora P, Carbonell P, et al. Colorectal serrated adenocarcinoma shows a different profile of oncogene mutations, MSI status and DNA repair protein expression compared to conventional and sporadic MSI-H carcinomas. Int J Cancer. 2012;131:1790–1799. doi: 10.1002/ijc.27454. [DOI] [PubMed] [Google Scholar]
- 11.García-Solano J, Conesa-Zamora P, Carbonell P, et al. Microsatellite pathologic score does not efficiently identify high microsatellite instability in colorectal serrated adenocarcinoma. Hum Pathol. 2013;44:759–765. doi: 10.1016/j.humpath.2012.07.020. [DOI] [PubMed] [Google Scholar]
- 12.García-Solano J, Conesa-Zamora P, Trujillo-Santos J, Mäkinen MJ, Pérez-Guillermo M. Tumour budding and other prognostic pathological features at invasive margins in serrated colorectal adenocarcinoma: a comparative study with conventional carcinoma. Histopathology. 2011;59:1046–1056. doi: 10.1111/j.1365-2559.2011.04043.x. [DOI] [PubMed] [Google Scholar]
- 13.Laiho P, Kokko A, Vanharanta S, et al. Serrated carcinomas form a subclass of colorectal cancer with distinct molecular basis. Oncogene. 2007;26:312–320. doi: 10.1038/sj.onc.1209778. [DOI] [PubMed] [Google Scholar]
- 14.Conesa-Zamora P, García-Solano J, García-García F, et al. Expression profiling shows differential molecular pathways and provides potential new diagnostic biomarkers for colorectal serrated adenocarcinoma. Int J Cancer. 2013;132:297–307. doi: 10.1002/ijc.27674. [DOI] [PubMed] [Google Scholar]
- 15.Kang S, Na Y, Joung SY, Lee SI, Oh SC, Min BW. The significance of microsatellite instability in colorectal cancer after controlling for clinicopathological factors. Medicine (Baltimore) 2018;97:e0019. doi: 10.1097/MD.0000000000010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conesa-Zamora P, García-Solano J, Turpin Mdel C, et al. Methylome profiling reveals functions and genes which are differentially methylated in serrated compared to conventional colorectal carcinoma. Clin Epigenetics. 2015;7:101. doi: 10.1186/s13148-015-0128-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jass JR, Love SB, Northover JM. A new prognostic classification of rectal cancer. Lancet. 1987;1:1303–1306. doi: 10.1016/S0140-6736(87)90552-6. [DOI] [PubMed] [Google Scholar]
- 18.Roxburgh CS, Salmond JM, Horgan PG, Oien KA, McMillan DC. Tumour inflammatory infiltrate predicts survival following curative resection for node-negative colorectal cancer. Eur J Cancer. 2009;45:2138–2145. doi: 10.1016/j.ejca.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 19.Maby P, Tougeron D, Hamieh M, et al. Correlation between density of CD8+ T-cell infiltrate in microsatellite unstable colorectal cancers and frameshift mutations: a rationale for personalized immunotherapy. Cancer Res. 2015;75:3446–3455. doi: 10.1158/0008-5472.CAN-14-3051. [DOI] [PubMed] [Google Scholar]
- 20.Jersmann HP. Time to abandon dogma: CD14 is expressed by non-myeloid lineage cells. Immunol Cell Biol. 2005;83:462–467. doi: 10.1111/j.1440-1711.2005.01370.x. [DOI] [PubMed] [Google Scholar]
- 21.Li C, Luo X, Lin Y, et al. A higher frequency of CD14+ CD169+ monocytes/macrophages in patients with colorectal cancer. PLoS One. 2015;10:e0141817. doi: 10.1371/journal.pone.0141817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toor SM, Syed Khaja AS, El Salhat H, et al. Increased levels of circulating and tumor-infiltrating granulocytic myeloid cells in colorectal cancer patients. Front Immunol. 2016;7:560. doi: 10.3389/fimmu.2016.00560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kinouchi M, Miura K, Mizoi T, et al. Infiltration of CD14-positive macrophages at the invasive front indicates a favorable prognosis in colorectal cancer patients with lymph node metastasis. Hepato-Gastroenterology. 2011;58:352–358. [PubMed] [Google Scholar]
- 24.Leone P, De Re V, Vacca A, Dammacco F, Racanelli V. Cancer treatment and the KIR-HLA system: an overview. Clin Exp Med. 2017;17:419–429. doi: 10.1007/s10238-017-0455-4. [DOI] [PubMed] [Google Scholar]
- 25.Archer KJ, Mas VR, Maluf DG, Fisher RA. High-throughput assessment of CpG site methylation for distinguishing between HCV-cirrhosis and HCV-associated hepatocellular carcinoma. Mol Gen Genomics. 2010;283:341–349. doi: 10.1007/s00438-010-0522-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang C, Cicek MS, Charbonneau B, et al. Tumor hypomethylation at 6p21.3 associates with longer time to recurrence of high-grade serous epithelial ovarian cancer. Cancer Res. 2014;74:3084–3091. doi: 10.1158/0008-5472.CAN-13-3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ningappa M, Ashokkumar C, Higgs BW, et al. Enhanced B cell alloantigen presentation and its epigenetic dysregulation in liver transplant rejection. Am J Transplant. 2016;16:497–508. doi: 10.1111/ajt.13509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sindhi R, Higgs BW, Weeks DE, et al. Genetic variants in major histocompatibility complex-linked genes associate with pediatric liver transplant rejection. Gastroenterology. 2008;135(3):830–839. doi: 10.1053/j.gastro.2008.05.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Majumder P, Boss JM. CTCF controls expression and chromatin architecture of the human major histocompatibility complex class II locus. Mol Cell Biol. 2010;30:4211–4223. doi: 10.1128/MCB.00327-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis S, Du P, Bilke S, Triche T, Bootwalla M. methylumi: Handle Illumina methylation data. 2014. R package version 2.10.0. Available at https://bioconductor.org/packages/release/bioc/html/methylumi.html
- 31.Pidsley Ruth, Y Wong Chloe C, Volta Manuela, Lunnon Katie, Mill Jonathan, Schalkwyk Leonard C. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14(1):293. doi: 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teschendorff AE, Marabita F, Lechner M, et al. A beta-mixture quantile normalisation method for correcting probe design bias in Illumina Infinium 450k DNA methylation data. Bioinformatics. 2013;29:189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Al-Shahrour F, Carbonell J, Minguez P, et al. Babelomics: advanced functional profiling of transcriptomics, proteomics and genomics experiments. Nucleic Acids Res. 2008;36:W341–W346. doi: 10.1093/nar/gkn318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ashburner M, Ball CA, Blake JA, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Gene Ontology biological processes (BP) terms differentially enriched in the comparison SAC vs. hmMSI-H; 40 obtained using ClusterProfiler and 76 using FatiGo. (XLSX 25 kb)
Figure S1. Terms of Gene Ontology Cellular Component and Molecular Functions enriched in the comparison between SAC and hmMSI-H for their methyloma profile. The scatterplot shows the terms which are globally differentially methylated between SAC and hmMSI-H after GO term redundancy reduction. The graph is represented in a two dimensional space derived by applying multidimensional scaling to a matrix of the GO terms’ semantic similarities [35]. (TIF 162 kb)
Table S2. List of the 1540 differentially methylated genes between hmMSI-H and SAC. (XLSX 149 kb)
Molecular functions of the 42 most differentially methylated genes. Source https://www.ncbi.nlm.nih.gov/gene. (DOCX 23 kb)
Table S3. Primer sequences, amplicon sizes and location of CpGs evaluated in the study. (DOC 48 kb)
Data Availability Statement
The data set supporting the results of this article are available in the GEO repository, GSE68060 in https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68060.