

Summary
The objective of this study was to investigate the mechanisms of T helper type 17 (Th17) expansion in lupus nephritis (LN) patients, and to determine whether or not it is associated with impaired function of regulatory T cells (Treg). Major effector subsets of peripheral blood CD4+ T cells were assessed by flow cytometry in 33 LN patients with different activity of the disease and 19 healthy controls. The percentage of circulating Th17 cells was increased in LN (median = 1·2% of CD4+ compared to 0·6% in the control group, P < 0·01), while Treg cells remained unchanged (12·3 versus 12·1% in controls), resulting in a significantly lower Treg/Th17 ratio. Th17 expansion in the patient group was not related to LN activity, renal histology or blood and urine inflammatory biomarkers, but has been associated with a higher cumulative dose of cyclophosphamide. Treg cells in LN displayed mainly effector memory phenotype and expressed higher levels of transforming growth factor (TGF)‐β; however, their suppressant activity in lymphocyte proliferation assay was diminished compared to controls (~fourfold, P < 0·05). Co‐culture of Treg and conventional CD4+ T cells resulted in marked suppression of the Th1 subset in both of the groups studied, but also in a potent expansion of Th17 cells, which in LN was twofold higher, as in controls (P < 0·05). In conclusion, our results demonstrate that Th17 expansion in LN is not increased during disease exacerbation, but is related to chronic immunosuppressive therapy. This immune signature is probably linked to the abnormal function of Treg cells, which were less suppressive in LN patients and even facilitated differentiation of Th17 cells.
Keywords: lupus nephritis, regulatory T cells, systemic lupus erythematosus, Th17 cells
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disorder characterized by aberrant activation of autoreactive T and B cells, tissue deposition of immune complexes and subsequent inflammation with organ damage [1]. Renal involvement, called lupus nephritis (LN), remains the most important predictor of disease severity and mortality in SLE [2]. Recent evidence has suggested that T helper type 17 (Th17)‐mediated responses might be involved in the pathomechanism of immune‐mediated glomerular diseases [3]. Th17 cells constitute a distinct lineage of T helper cells characterized by the expression of transcription factor RAR‐related orphan nuclear receptor (ROR)γt and by the production of proinflammatory cytokines [e.g. interleukin (IL)‐17A and IL‐17F] [4]. Apart from their protective role in infections, Th17 cells have been linked with pathogenesis of various autoimmune disorders [5]. Nevertheless, the role of the Th17‐axis in systemic lupus erythematosus (SLE) has not been resolved, due to large discrepancies in both experimental models and human studies. Animal models of LN have documented involvement of the Th17 response in autoimmune renal damage [6]; however, if repeated on an IL‐17A‐deficent background they did not confirm the crucial role of Th17 cells in nephritis [7]. Similarly, several reports have demonstrated increased systemic levels of IL‐17A [8, 9, 10, 11, 12] and a higher fraction of circulating Th17 cells in human SLE [13, 14, 15]. However, there were also studies which did not show significant over‐expansion of Th17 cells [16, 17, 18, 19]. The role of the IL‐17/Th17 axis in LN is not well understood, as there are contradictory results concerning the contribution of Th17 cells in active renal lesions [19, 20]. Recently, we compared urine mRNA expression in LN, but did not show a clear up‐regulation of signature genes of the Th17 axis in patients with renal flare [21].
As the development of Th17 cells shares many similarities with regulatory T cells (Treg) and is reciprocally regulated, recent studies point to the imbalance between these two cell subsets as a major immune deregulation occurring in SLE [17, 18]. Treg lymphocytes constitute a separate lineage of CD4+ T cells which maintain immunotolerance by a direct suppressant effect on conventional effector T cells or by secretion of immunoregulatory cytokines [22]. It has been suggested that a Treg/Th17 imbalance could lead to a putative escape of autoreactive Th17 cells from Treg control, and thus facilitate autoimmunization [23]. Little is known, however, about how such imbalance develops and whether expansion of Th17 cells is an intrinsic trait of LN or if it develops gradually during the course of the disease.
The aim of our study was to evaluate a fraction of Th17 and Treg cells in the peripheral blood of LN patients, in search of a potential link between the Th17 immune axis and clinical manifestation of SLE and nephritis activity. Additionally, we investigated the in‐vitro cross‐talk between Th17 and Treg cells in LN patients in order to determine whether the potentially enhanced systemic Th17 response in LN results from quantitative or qualitative deficiency in the Treg subset.
Materials and methods
Characteristics of the patients
We enrolled 33 SLE patients who fulfilled the American College of Rheumatology criteria [24] and had clinically overt renal symptoms (clinical characteristics summarized in Table 1). In 29 subjects (88%) LN was confirmed by renal biopsy and staged according to International Society of Nephrology (ISN)/Renal Pathology Society (RPS) criteria [25]. In total, we analysed 16 patients with active LN [active urine sediment, proteinuria > 1 g/24 h or > twofold increase, SLE Disease Activity Index (SLEDAI) > 6 and 17 with inactive disease (stable proteinuria < 1 g/24 h, SLEDAI < 6]. In inactive LN the disease was quiescent for > 6 months. All patients were not treated with cyclophosphamide (CTX) or mycophenolate mofetil (MMF) in the preceding 6 months. Nineteen healthy individuals served as a control group. The study was approved by the Ethics Committee of the Jagiellonian University and informed written consent was obtained from all participants.
Table 1.
Characteristics of the subjects studied
| Active LN (n = 16) | Inactive LN (n = 17) | Healthy controls (n = 19) | |
|---|---|---|---|
| Age (years) | 37 (34–47) | 35 (29–47) | 34 (27–41) |
| Sex (female, %) | 12 (75%) | 14 (82%) | 15 (79%) |
| Age at diagnosis (years) | 27.5 (23–32) | 23 (22–33) | n.a. |
| SLE duration (years) | 11 (5·5–16·5) | 8 (4–16) | n.a. |
| SLEDAI (score) | 21 (14–23)## | 2 (0–4) | n.a. |
| First renal exac./relapse (n) | 7/9 | n.a. | n.a. |
| Previous renal exac. (n) range1 | 3 (1–6) | 1 (1–5) | n.a. |
| LN class II/III/IV/V (unknown)2 | 0/2/10/2(2) | 2/1/11/1(2) | n.a. |
| Proteinuria (g/day) | 3.1 (1.9–5.2)## | 0.11 (0.06–0.32) | n.a. |
| Creatinine (serum, μmol/l) | 72 (66–99) | 65 (60–82) | 71 (65–79) |
| GFR (ml/min/1·73m2)3 | 88.4 (71–97) | 96 (80–115) | 94 (85–99) |
| C3c (g/l) | 0·560 (0·48–0·780)**## | 1·04 (0·83–1·19) | 1·15 (0·96–1·41) |
| C4 (g/l) | 0·070 (0·04–0·1)**## | 0·165 (0·130–0·19)* | 0·205 (0·164–0·308) |
| CRP (mg/l) | 17·1 (9·1–25·8)**## | 1·8 (0·65–8·1)* | 0·74 (0·32–1·1) |
| ANA (titre), range | 10 240 (1280–20 480)# | 5120 (160–20 480) | None |
| Anti‐dsDNA (titre), range | 640 ( < 10–10 240)## | 20 ( < 10–320) | None |
| Current systemic GCS (mg/day)4 | 10 (5–16)# | 4 (0–4) | None |
| Current AZA therapy (%)5 | 3 (19%) | 2 (12%) | None |
| Previous CTX treatment (yes, %) | 7 (44%)## | 15 (88%) | None |
| Cumulative CTX dose (g) | 0 (0–9·8)# | 9 (5·8–25) | n.a. |
LN = lupus nephritis; SLE = systemic lupus erythematosus; SLEDAI = SLE Disease Activity Index; GFR = glomerular filtration rate; C3c = complement component C3c; C4 = complement component C4; CRP = C‐reactive protein; ANA = anti‐nuclear antibodies; GCS = glucocorticoids; AZA = azathioprine; CTX = cyclophosphamide. 1 Including current in case of active LN group; 2 according to International Society of Nephrology (ISN)/Renal Pathology Society (RPS) [25]; 3 according to the Modification of Diet in Renal Disease Study (MDRD) formula; 4 adjusted to methylprednisolone; 5 at the study entry, and during the preceding 6 months, patients were not treated with other IS drugs [e.g. CTX or mycophenolate mofetil (MMF)]. *P < 0·05 and **P < 0·01 in comparison to controls; # P < 0·05 and ## P < 0·01 in comparison to inactive LN.
Isolation of peripheral blood lymphocytes and cell cultures
Peripheral blood mononuclear cells (PBMC) were isolated from heparinized blood using Histopaque gradient centrifugation (Sigma‐Aldrich, St Louis, MO, USA). For detection of intracellular cytokines, PBMC were stimulated for 5 h with phorbol myristate acetate (PMA) (50 ng/ml) and ionomycin (0·7 μg/ml) in the presence of brefeldin A (10 μg/ml, all reagents from Sigma‐Aldrich) in RPMI‐1640 supplemented with 10% fetal calf serum (Pan Biotech, Aidenbach, Germany). Cells were stained for effector cytokines and analysed by flow cytometry (FC).
For the in‐vitro suppression assay, immunomagnetically separated (Miltenyi Biotec, Bergisch‐Gladbach, Germany) CD4+CD25– (Tconv, conventional) and CD4+CD25+ (Treg) cells were cultured (at ratios of 2 : 1, 1 : 1 and 1 : 2, final 0·2 × 106/well) in aCD3‐coated (BioLegend, San Diego, CA, USA) 96‐well plates in X‐VIVO‐15 medium (Lonza, Basel, Switzerland) with 2·5% human AB‐serum (Pan Biotech) and aCD28 (1 μg/ml, BioLegend). Viable cells [7‐aminoactinomycin D (7‐AAD), BD Biosciences, San Jose, CA, USA] were counted at baseline and after 5 days by FC. To reproduce Th17‐like differentiation, Tconv were stimulated with aCD3/aCD28 and cultured without cytokines (control) or in the presence of cytokine mixture (all reagents from R&D Systems, Minneapolis, MN, USA): IL‐1β (final 10 ng/ml), IL‐6 (10 ng/ml), IL‐23 (50 ng/ml) and transforming growth factor (TGF)‐β1 (1 ng/ml), anti‐interferon (IFN)‐γ and anti‐IL‐4 (6 μg/ml each). In a parallel set of wells purified Treg cells were added at different ratios. Lymphocytes were restimulated on day 5 with PMA/ionomycin (as described) and analysed by FC. To analyse latent TGF‐β expression by Treg, PBMC were stimulated for 24 h with aCD3/aCD28 and stained for FC.
Flow cytometry
Aliquots of blood were stained with combinations of the following antibodies (all from BD Biosciences, if not specified): CD45‐V450, CD3‐fluorescein isothiocyanate (FITC), CD4‐peridinin chlorophyll‐cyanin (PerCP‐Cy)5·5, CD8‐phycoerythrin (PE)‐Cy7, CD8‐allophycocyanin (APC)‐Cy7 (BioLegend), CD16/CD56‐PE, CD19‐APC, CD45RA‐PE, CD45RO‐FITC, CD25‐PE, CD127‐PE‐Cy7 (BioLegend), CXCR3‐PE‐Cy7, CCR4‐AlexaFluor‐647, CCR6‐V450, CCR7‐AlexaFluor‐647 and CCR10‐PE (R&D Systems). Treg cells were identified as CD4+CD25highCD127low, which was confirmed by co‐expression of forkhead box protein 3 (FoxP3) transcription factor. Absolute cell numbers were calculated based on white blood cell (WBC) counts and cell differential measured by automated haematology analyser. To detect intracellular cytokines, lymphocytes were labelled with CD4‐PerCP‐Cy5.5 and CD8‐APC‐Cy7 (BioLegend), stained for viability (FVS450, BD Biosciences), fixed/permeabilized (Cytofix/Cytoperm Kit, BD Biosciences), and stained for cytokines: IL‐4‐AlexaFluor467 (BioLegend), IFN‐γ‐FITC (BioLegend), IL‐22‐PE‐Cy7 (eBioScience, San Diego, CA, USA) and IL‐17A‐PE (eBioScience). To detect a latent‐TGF‐β/glycoprotein A repetitions predominant (GARP) complex, activated PBMC were surface‐stained with CD4‐PerCP‐Cy5·5, CD127‐FITC, CD25‐APC‐Cy7 (BioLegend), GARP‐eFluor660 (eBioScience) and latency‐associated peptide (LAP)‐PE‐Cy7 (eBioScience) and viable (FVS450 exclusion) Treg were identified by intracellular staining with FoxP3‐PE (eBioScience). Samples were analysed by multi‐colour FC [FACS Canto II, BD Biosciences] and FACS Diva Software (BD Biosciences).
Serum and urine biomarkers
Levels of serum and urine cytokines were measured by Luminex (Magpix; Luminex Corp., Austin, TX, USA) using predesigned multiplex panels (eBioscience). Urinary sediment mRNA expression was assessed by real‐time polymerase chain reaction (PCR) (43 genes; Applied Biosystems, Foster City, CA, USA) on a 7900HT real‐time PCR platform (Applied Biosystems). Detailed methods were presented in our previous study, which focused on molecular biomarkers of LN [21].
Statistical methods
Statistical analysis was performed with Statistica version 13.1 (Tibco Software Inc., Palo Alto, CA, USA) and GraphPad Prism version 7.0 (GraphPad Software, Inc., La Jolla, CA, USA). Data are presented as medians and 0·25–0·75 quartile range, if not stated otherwise. Differences between groups were determined by a Mann–Whitney U‐ or Fisher’s exact test. In in‐vitro experiments assessing the influence of Treg or cytokines, statistical significance was determined by analysis of variance (anova) and repeated‐measures two‐way anova or Kruskal–Wallis test, if justified. Spearman’s rank sum test was used to analyse correlations; a P‐value < 0·05 was considered significant.
Results
Increased fraction of Th17 cells in LN
All LN patients were characterized by a considerable lymphopenia affecting all lymphocyte subsets (Supporting information, Table S1). We observed large variability in the fraction of cytokine‐producing CD4+ T cells both in LN patients and controls (Fig. 1a,b). However, only a percentage of Th17 cells (i.e. producing IL‐17A but not IFN‐γ) was increased significantly in both active and inactive LN (median 1·2%) compared to control (median 0·6%, Fig. 1c, Supporting information, Table S2). Nevertheless, a marked increase in Th17 cells (i.e. > 90th percentile as determined in the control group) was seen only in four active and three inactive LN patients. These patients did not differ in demographic and clinical characteristics, except for a ~twofold higher cumulative CTX dose compared to patients with a normal percentage of Th17 cells (Supporting information, Table S2). As expected, absolute counts of cytokine‐producing CD4+ T cells were decreased in LN patients due to SLE‐related lymphopenia (Fig. 1d, Supporting information, Table S3). The proliferation potential of Th17 cells was investigated using short‐term cultures of Tconv cells in control conditions or in the presence of cytokines promoting type 17 differentiation (Fig. 1e). Cytokine treatment resulted in a significant increase in the fraction of Th17 cells in LN patients (Fig. 1f,g). Interestingly, the effect of cytokines (i.e. fold increase in the number of Th17 cells compared to control conditions) was exactly the same in LN patients and controls (Fig. 1h). These data were confirmed by FC analysis of the chemokine receptor signature of CD4+ memory T cells (Supporting information, Fig. S1a), which showed an increase in the percentage of the Th17‐like subset (i.e. CXCR3–CCR4+CCR6+) in LN patients (median 8·4%) compared to controls (5·9%, P < 0·05). There was a marked correlation between corresponding CD4+ subsets (e.g. Th17 cells identified either by intracellular cytokines or chemokine receptors) and a similar decline in cell numbers in different study groups (Supporting information, Fig. S1b,c).
Figure 1.
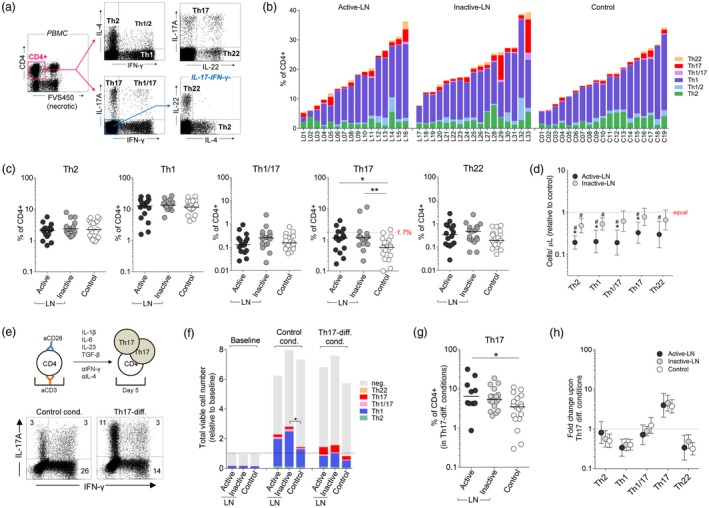
Cytokine production by in‐vitro activated CD4+ T cells. (a) Peripheral blood mononuclear cells (PBMC) were stimulated with phorbol myristate acetate (PMA)/ionomycin and intracellulary stained for T effector signature cytokines. Representative flow cytometry dot‐plots and gating strategy used to identify cytokine producing CD4+ T cells. (b) Graph showing cumulative percentages of major functional subsets of CD4+ T cells in individual lupus nephritis (LN) patients and controls. (c) The percentage of CD4+ T cells with certain functional phenotypes: T helper type 2 (Th2), Th1, Th1/17, Th17 (dashed line = 90th percentile of the control group) and Th22 (Th1/2 cells not shown). Horizontal bars represent medians. *P < 0·05, **P < 0·01. (d) Absolute numbers of CD4+ T cell subtypes (peripheral blood). Data presented as fold difference in comparison to median value in the healthy controls group. #P < 0·05 in comparison to the control group; *P < 0·05 in comparison to inactive LN. (e) Th17‐expansion assay. Magnetically separated CD4+CD25– T cells (i.e. conventional CD4+ T cells) were stimulated in vitro with aCD3/aCD28 and cultured for 5 days in medium alone or in the presence of cytokines inducing Th17‐differentiation and blocking immunoglobulin (Ig)G (as indicated). Representative flow cytometry dot plots showing distribution of Th17 [interferon (IFN)‐γ IL‐17A+], Th1 (IFN‐γ+ IL‐17A‒) and Th1/17 (both positive) cells upon culture in Th17‐prone conditions [both graphs show 50 000 events (viable CD4+ gate)]. (f) The cumulative numbers (mean values) of cytokine positive CD4+ T cells (neg. = cytokine‐negative) in different culture conditions (data presented as relative to baseline, i.e. value 2 means twofold increase in the cell count after 5 days of culture). *P < 0·05 significant difference in the number of cytokine‐positive cells (all combined). (g) Percentage of Th17 cells after in‐vitro expansion in cytokine milieu [as in (e)]. *P < 0·05. (h) Comparable increase in the number of Th17 cells during in‐vitro cell culture of CD4+ Tconv cells from LN patients and controls. Data presented as mean change (95% confidence interval) in comparison to control conditions. There was a significant (P < 0·01) increase in the number of Th17 cells and a decrease of Th1, Th2 (except active LN) and Th22 cells in cytokine conditions (statistics symbols not shown for clarity).
No linkage between peripheral blood Th17 and LN activity and renal histology status
To investigate the association between the systemic Th17 response and LN clinics we stratified patients based on the median percentage of circulating Th17 cells. Th17high (≥ 1·2% of CD4+) and Th17low ( < 1·2%) patients did not differ in clinical characteristics (Supporting information, Table S4), except for a trend towards lower proteinuria (P = 0·052) and higher cumulative CTX dose (P = 0·056) in the case of the Th17high endotype. Similarly, a significant correlation (r = 0.59, P < 0·01) was found between the percentage of Th17‐like cells and cumulative CTX dose (Supporting information, Fig. S1d–f). There was no association between percentage of circulating Th17 cells and other clinical characteristics of LN (Supporting information, Fig. S2) or renal histology (Supporting information, Table S5, Fig. S3). There was only a weak positive correlation between percentage of circulating Th17 cells and Th17‐axis gene expression (e.g. IL17A, CCL20) in the urine sediment (Supporting information, Fig. S4A). Additionally, serum IL‐17A was detected only in two active LN patients; both had an increased percentage of circulating Th17 cells (data not shown). Nevertheless, the most reliable cytokine [e.g. urine chemokine C‐X‐C motif ligand 10 (CXCL10) and chemokine C‐C motif ligand 2 (CCL2)] and mRNA (TBX21, CCL2, CD3G) biomarkers of active LN [21] were not elevated in patients with the Th17high endotype (Supporting information, Fig. S4b,c). These results indicate that an increased systemic Th17 response in LN only weakly translates into the type 17 signature in the urine, and is not linked with more active disease or certain patterns of renal pathology.
Increased fraction of effector memory Treg cells in LN
In the next part of the study we analysed whether or not expansion of Th17 subset in LN is associated with functional abnormality of Treg cells. As shown in Fig. 2a, we did not detect any decline in the percentage of Treg cells (CD4+CD25highCD127low) in LN compared to controls. A fraction of CD25highCD127low cells expressing the FoxP3 transcription factor was also not decreased in active LN (median 6·9% of CD4+) compared to inactive disease (6·5%) and normal donors (5·4%). Interestingly, we observed a switch towards a more mature phenotype of Treg cells in LN, as the percentage of Treg with effector memory (EM) phenotype (Fig. 2b,c) was increased both in active (median 47%) and inactive LN (36%) compared to control (28%, P < 0·01 both). As expected, there was a marked decline (~fourfold) in the absolute count of peripheral blood Treg cells in all LN subjects compared to control (Fig. 2d).
Figure 2.
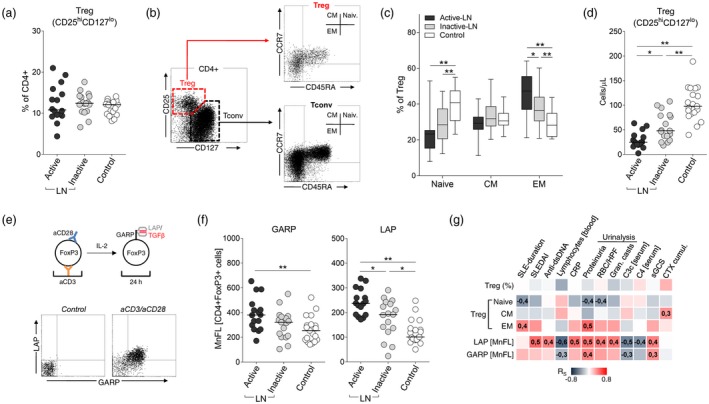
Regulatory T cell (Treg) phenotype and function. (a) No difference in the percentage of peripheral blood CD25hiCD127lo CD4+ cells (Treg) between lupus nephritis (LN) patients and control subjects. (b) Flow cytometry (FC) gating strategy to identify memory (EM = effector memory, CM = central memory) subsets of Treg and conventional T cells (Tconv). (c) Decreased fraction of naive and increase in EM Treg cells (gated on CD4+CD25hiCD127lo) in LN patients. *P < 0·05, **P < 0·01. (d) Treg cell counts (peripheral blood) in LN patients and control subjects. (e) Transforming growth factor (TGF)‐β expression by in‐vitro activated Treg cells. Peripheral blood mononuclear cells (PBMC) were activated (aCD3/aCD28) for 24 h in the presence of interleukin (IL)‐2, stained for surface latency‐associated peptide (LAP) and glycoprotein A repetitions predominant (GARP) and intracellular forkhead box protein 3 (FoxP3), and analysed by flow cytometry (FC). Dot‐plots show expression of LAP and GARP in CD4+FoxP3+ cells in different culture conditions. (f) Higher surface expression of GARP and LAP by in‐vitro stimulated lymphocytes from active LN patients. Data presented as mean fluorescence intensity (MFI) in the FoxP3+CD4+ gate. *P < 0·05, **P < 0·01. (g) Matrix of correlation coefficients (R S – Spearman) in cross‐comparison of Treg phenotype [naive, CM and effector memory (EM) (% of Treg)], and expression of LAP/GARP, with major clinical and laboratory measures of LN. Data from active and inactive LN were combined. Numerical values indicate significant (P < 0·05) correlations.
Surprisingly, in‐vitro activated Treg cells from LN patients showed very high expression of both latent TGF‐β and its coupling receptor GARP (Fig. 2e,f). This was due probably to a higher abundance of EM cells within the Treg fraction, as we have shown a positive correlation between percentage of EM Treg cells and relative expression of latent TGF‐β (r = 0·56, P < 0·01). Additionally, we showed associations between expression of latent TGF‐β and several clinical and laboratory parameters of the disease (Fig. 2g): for example, C3c levels (r = –0·49), SLEDAI score (r = 0·55) or urine protein excretion rate (r = 0·45).
Treg/Th17 imbalance in LN is not related to laboratory and clinical disease parameters
Comparing the numbers of Treg and conventional T helper subsets we found that only the ratio of Treg to Th17 cells was decreased significantly in LN (Supporting information, Table S6). When stratified based on the median value of the Treg/Th17 ratio, LN patients with lower Treg/Th17 values (i.e. imbalance promoting Th17 response) did not differ in clinical parameters (not shown) and renal biopsy status (Supporting information, Fig. S5). Levels of serum and urine biomarkers (e.g. urine mRNA expression) were also comparable in patients with high and low Treg/Th17 ratios ( Supporting information, Fig. S6, transcriptome data not shown).
Treg cells are less suppressive and facilitate proliferation of Th17 lymphocytes in LN patients
To investigate potential defects in Treg suppression we first analysed the capacity of Treg cells to modulate proliferation of conventional CD4+ T cells. As shown in Fig. 3a, the suppressant activity of Treg was significantly lower in LN patients (~10% decreased proliferation in Tconv: Treg 1 : 1 ratio) compared to healthy subjects (~35% decrease, P < 0·01), with no difference between active and inactive LN (not shown). To further verify which Tconv fraction is predominantly inhibited, we analysed cytokine production by proliferating cells after restimulation with PMA/ionomycin (Fig. 3b). Co‐culture of Tconv and Treg cells resulted in a marked increase in the fraction of Th17 cells, which was more pronounced in LN patients compared to the control group (Fig. 3c). In control subjects, Treg cells inhibited mainly the proliferation of CD4+ T cells not expressing effector cytokines (i.e. naive), Th1 cells and, to a lesser extent, Th2 cells, while in LN patients we showed predominantly inhibition of Th1 cells, with a somewhat opposite effect on Th2 cells (Fig. 3d). In both study groups Treg cells markedly facilitated the expansion of Th17 cells (three‐ to eightfold increase), but this effect was ~twofold stronger in LN patients compared to controls (Fig. 3d). A similar but smaller effect of Treg was seen in cells cultured in the presence of cytokines facilitating Th17 expansion (not shown). Together, these data suggest that expansion of Th17 cells in LN could be related in part to the skewed memory phenotype of Treg cells or their plasticity. This was supported by a positive correlation of the baseline percentage of EM Treg with the fraction of Th17 cells in Tconv plus Treg co‐cultures (Fig. 3e).
Figure 3.
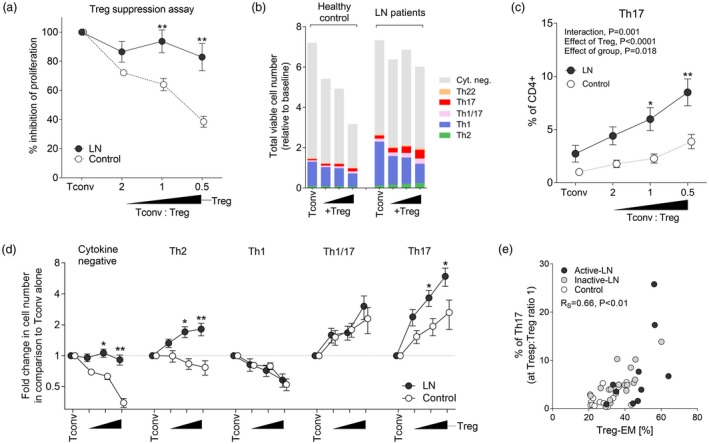
Regulatory T cell (Treg) suppression in lupus nephritis (LN). (a) Suppression assay. Activated (aCD3/aCD28) CD4+CD25– (Tconv) and CD4+CD25+ (Treg) cells were cultured for 5 days at different ratios (triangle). Results are presented as % inhibition of proliferation (compared to Tconv alone). LN data were combined, as there was no difference between active and inactive patients. **P < 0·01 compared to control subjects. (b) Influence of Treg on CD4+ T cell proliferation and cytokine production. Stacked graph summarizes change in numbers of cytokine‐producing Tconv cells (neg. = cytokine negative) at day 5 compared to baseline (day 0). Experimental setup as in (a). (c) Percentage of T helper type 17 (Th17) cells in CD4+ T cell cultures (day 5) in LN patients (data combined) and control. Two‐way analysis of variance (anova) statistics and post‐hoc tests: *P < 0·05, **P < 0·01 in comparison to the control group. (d) Fold change in the number of cytokine producing CD4+ T cell subsets at different Tconv : Treg ratios [as in (a)]. Data were standardized to experimental control (Tconv alone). *P < 0·05, **P < 0·01 compared to control group (anova and post‐hoc test). (e) Correlation between the percentage of effector memory (EM) Treg cells and the fraction of Th17 cells in the cell culture at a 1 : 1 Tconv:Treg ratio.
Discussion
In this study, we analysed functional subsets of peripheral blood T helper cells in a well‐defined cohort of SLE patients with renal disease. LN patients showed an increased fraction of circulating Th17 cells and lower Treg/Th17 ratio compared to normal donors, regardless of disease activity and leading clinical symptoms. We also demonstrated that expansion of Th17 cells in LN might be related to the abnormal function of Treg cells.
In our study, the LN endotype with a higher systemic percentage of Th17 cells was not related to more active disease, a specific pattern in renal histology or blood and urine inflammatory biomarkers. Our results are in accordance with previous studies which demonstrated up‐regulation of the Th17‐axis in SLE compared to control, but failed to find an association between Th17 responses and disease activity [9, 10, 11]. Moreover, in the available literature there was usually no association between increased levels of IL‐17A or a higher fraction of Th17 cells and the presence of nephritis in SLE patients [8, 9, 10, 11, 14] with only few studies demonstrating clear expansion of Th17 in more severe proliferative LN [26, 27]. Inconsistencies between various studies may be attributed to different strategies in detection of the Th17 subset, e.g. cytokine expression versus surface phenotype. Here, we used both methodological approaches, and the results were convergent. Altogether, our study indicates that Th17 expansion in LN is a general feature of SLE, but not a characteristic trait of active lupus nephritis.
The contribution of Th17 cells in the pathogenesis of LN is still not fully understood. In previous reports, IL‐17‐producing lymphocytes were detected easily in the kidneys of LN patients and correlated with some urinalysis markers of kidney involvement [20, 28]. The kidney expression of IL‐17A cells was usually increased in patients characterized by severe disease or proliferative nephropathy [12, 15]. Nevertheless, Th17 cells did not constitute the predominant effector T cell fraction in LN lesions, as they were largely outnumbered by Th1 cells both in renal tissues and urine samples [29, 30]. This was confirmed by gene expression studies, which demonstrated a predominant type‐1 immune pattern both in renal biopsies [31] and in the urine sediment of active LN patients [32]. Our recent results corroborated these findings, showing higher mRNA expression of Th1 signature genes (e.g. TBX21) in the urine samples from active LN, with no evident up‐regulation of the Th17 axis [21]. In the current study we re‐analysed urine biomarker data in search for a potential linkage between systemic Th17 response and renal inflammation. It emerged that the percentage of circulating Th17 cells correlated only weakly with urinary expression of Th17‐related genes, with no linkage between systemic Th17 cells and recognized urinary biomarkers of active LN. Additionally, recent studies showed a somewhat inverse correlation between urine biomarkers of the Th17‐axis and LN activity [19, 33, 34]. Taken together, these data indicate that even though the Th17 signature can be detected in renal lesions of LN patients, there is no clear evidence of the essential contribution of Th17 cells in LN exacerbations. It is interesting in this respect that recent blood transcriptome profiling in a large cohort of paediatric SLE patients showed predominant type I IFN and plasmablast signatures in the early stage of the disease, with gradual progression towards a full‐blown inflammatory pattern, marked by neutrophil signature, during the development of nephritis [35]. This observation, together with earlier data on the Th17 to Th1 shift during experimental glomerulonephritis [3], suggests that different immune mechanisms are operational at different stages of LN, with a predominance of Th1‐mediated responses in advanced disease.
The driving force of Th17 cell expansion in only some SLE patients remains unclear. This may be due to repeated stimulation with autoantigens and chronic inflammatory conditions (e.g. increased levels of IL‐1β and IL‐6), which favours Th17 differentiation. Here it is of interest that when we created in‐vitro conditions promoting Th17 differentiation it resulted in marked expansion of Th17 cells in LN. Nevertheless, we used an entire fraction of Tconv cells (both naive and memory cells) for this experiment, therefore the rate of Th17 expansion in vitro probably reflected baseline abundance of this memory subset. As a consequence, the fold increase in the number of Th17 cells in cytokine conditions was exactly the same in LN patients and healthy controls. Alternatively, Th17 expansion in SLE may also be a result of a skewed balance between Th17 and Treg cells. Both subsets require TGF‐β to induce the expression of their master transcription controllers, RORγt and FoxP3, but their final lineage commitment seems to be regulated reciprocally [36]. Similarly, IL‐2, which is required for the expansion of Treg cells, inhibits the differentiation of Th17 cells [37]. Additionally, recent studies have provided evidence for considerable phenotype plasticity of both Th17 and Treg subsets [38]. For example, under pathogenic inflammatory conditions Treg are able to lose FoxP3 expression and transdifferentiate into Th17 cells [39].
Whereas it is generally accepted that Treg cells play a crucial role in maintaining self‐tolerance and preventing autoimmunity, the data on their deficiency or potential dysfunction in SLE are largely confounding. Similarly to our previous study in SLE [18], and many other reports [16, 17, 19, 40], we did not observe any decline in the percentage of Treg cells in LN. In fact, in some patients the fraction of Treg cells was even expanded, as has been described previously [41, 42]. In this study, SLE‐related lymphopenia affected both conventional CD4+ effector T cells and Treg cells, but the absolute numbers of Treg cells were reduced markedly in LN, which resulted in a significantly lower Treg/Th17 ratio. Earlier studies showed similar data, pointing to the imbalance between the two subsets as a phenomenon linked with active disease [17, 27]. Nevertheless, in our study the ratio of Treg to Th17 cells was not related to disease exacerbation, and did not differ in patients stratified based on clinical data or renal biopsy status.
Another interesting observation of our study is that Treg cells from active LN patients showed a marked transition towards the EM phenotype compared to inactive disease and control subjects. Effector Treg cells exert their regulatory actions by direct receptor contact with target cells and partly by secretion of immunosuppressive cytokines, TGF‐β being the most important one [22]. In the current study we showed higher expression of the TGF‐β complex by in‐vitro‐activated FoxP3+ T cells from LN patients. This phenomenon was related to the Treg EM phenotype, and was more pronounced in the active disease. A similar feature of Treg cells was described by Henriques et al. [16], who showed higher mRNA expression of TGF‐β in sorted Treg cells from SLE patients. Together, these data suggest that circulating Treg cells in LN, although limited in number, are still capable of production of high amounts of TGF‐β, and they should have correct suppressant functions. Surprisingly, the Treg suppression assay revealed significantly lower inhibition of T cell proliferation in LN patients compared to healthy subjects, which is in line with earlier reports showing impaired Treg functions in SLE subjects [43]. At first sight these observations are counterintuitive; however, they suggest potential differences in sensitivity of effector T cells subsets to the inhibitory effects of Treg.
To verify this hypothesis, we next determined whether Th17 cells in LN are actually prone to Treg‐mediated suppression. We cultured conventional CD4+ T cells in the presence of increasing numbers of Treg cells, and next determined the proliferation potential of various functional (i.e. producing effector cytokines) T helper subsets. This showed that Treg cells efficiently inhibited the proliferation of Th1 cells both in LN patients and controls. In contrast, Th17 subset expanded significantly in the presence of Treg cells, and this effect was more pronounced in LN. As we adjusted the number of CD4+ T cells in the culture this effect cannot be attributed to SLE‐related lymphopenia, but is probably a characteristic feature of Treg–Th17 cross‐talk in SLE. Additionally, due to considerable phenotypical heterogeneity and plasticity of Treg cells, some in‐vitro expanded cells with the Th17 phenotype were probably derived from the Treg fraction. Unfortunately, we did not track individual cells, so it was not possible to confirm to what extent facilitated in‐vitro expansion of IL‐17A‐expressing cells was due to enhanced proliferation of bona fide Th17 cells, or facilitated transdifferentiation of Treg cells into the IL‐17A‐producing ‘ex‐Treg’ subset. Regardless of which of these mechanisms dominated, these data indicated that SLE‐related expansion of Th17 cells and imbalance in Treg/Th17 may result in part from the facilitated differentiation of cells with Th17‐like effector functions. Interestingly, recent studies confirmed that in certain conditions Treg actually promoted Th17 cells and enhanced their proinflammatory functions [44]. For example, during infections Treg cells not only induced expansion of Th17 cells, but also converted themselves into IL‐17A‐producing lymphocytes which, together, facilitated subsequent clearance of various pathogens [45, 46].
We did not detect any significant linkage between the frequency of circulating Th17 cells (or Treg/Th17 cell imbalance) and disease activity status. Additionally, we showed large heterogeneity in fraction of functional T helper cell subsets, with only a small group of LN patients who showed a truly high expansion of Th17. Such heterogeneity is due probably to a natural variance in LN, or may arise from enrolment of patients at different disease stages or treatment modes. Interestingly, we noted a positive correlation between Th17 percentage and cumulative CTX dose. As in our study patients were not treated with CTX in the recent past (< 6 months), this result suggests that previous courses of induction immunosuppressive therapy might have a far‐reaching effect on the differentiation of Th17 and Treg cells and their respective balance. This is in line with a recent report in LN, which demonstrated a significant increase in the frequency of Th17 but no change in ratio to Treg cells after 6 months of standard induction immunosuppressive therapy with CTX or MMF [19]. Additionally, studies in vasculitis syndromes revealed that initial expansion of Th17 cells did not normalize long after completion of induction therapy, despite achieving complete remission [47, 48]. These effects could be attributed to the different susceptibility of Th17 and Treg cells to CTX. For example, low‐dose CTX was able to promote expansion of Th17 cells both in vitro and during treatment of cancer patients [49]. Conversely, Treg cells were found to be extremely sensitive to CTX‐mediated apoptosis [50]. These data suggest that changes in the rate of Th17 expansion, and subsequent Treg to Th17 imbalance in LN, may depend upon certain therapeutic regimens or on the time that has elapsed since withdrawal from treatment. Moreover, once achieved, imbalance in memory T cell subsets (i.e. after repeated exacerbations and treatment courses) becomes a stable feature of SLE that persists in sustained remission.
In summary, our results indicate that pronounced Th17 expansion and related decline in the Treg to Th17 ratio affects only a subgroup of LN patients, and is not associated with specific clinical pattern or disease activity. Th17 expansion in LN may be related to the altered function of Treg cells, which displayed a less suppressive phenotype in vitro and facilitated differentiation of Th17 cells. Our data do not support a concept of a crucial role of Th17‐axis in development of SLE nephropathy, as there was no linkage with renal histology status or LN activity. Th17‐axis is probably involved in the pathogenesis of SLE, but it does not play a major role in renal exacerbation of the disease. We have found a relationship between the Th17 subset and intensity of immunosuppressive therapy in the past. However, the exact mechanisms underlying this observation require detailed examination. Further prospective studies are needed to determine whether Th17 expansion in some LN patients is associated with worse long‐term prognosis, and if novel therapies targeting Th17 axis would bring clinical benefits in this subset of patients.
Author contributions
B. J., J. K. and J. M. designed the study; J. K. and S. B. recruited patients and performed clinical data analysis; B. J., J. K. and H. P. performed the experiments; B. J., J. K. and M. S. analysed the data; B. J., S. B. and J. M. wrote the paper.
Disclosures
The authors declare they have no conflicts of interest.
Supporting information
Fig. S1. Subsets of CD4+ memory T‐cells in LN. (a) Major memory CD4+ T‐cell subsets were identified by the surface expression of chemokine receptors (flow cytometry, additional staining for CD45RA not shown for clarity). (b) Cells were classified as: Th1‐like (CXCR3+ CCR6‐), Th1/17‐like (CXCR3+ CCR6+), Th2‐like (CXCR3‐ CCR4+ CCR6‐ CCR10‐), Th17‐like (CXCR3‐ CCR4+ CCR6+ CCR10‐), and Th22‐like (CXCR3‐ CCR4+ CCR6+ CCR22+). Data presented as medians (quartiles). *P < 0·05. (c) Correlation between percentage of type‐17 T‐cells identified based on intracellular cytokine detection, or chemokine receptor staining. (d) Decline in the number of memory CD4+ T‐cells (peripheral blood) in LN patients. Data presented as fold difference (mean and 95%CI) in comparison to median value in the healthy controls group. #P < 0·05 in comparison to control group. *P < 0·05 in comparison to inactive LN. (e) Matrix of correlation coefficients (RS – Spearman) in cross‐comparison of CD4+ T‐cell subsets distinguished by chemokine receptors (% of CD4+), with major clinical and laboratory measures of LN. Active and inactive LN data were combined. Numeric values mark significant (P < 0.05) correlations. (f) Correlation between percentage of Th17‐like cells (% of CD4+) and cumulative dose of cyclophosphamide (CTX, g). (g) Percentage of Th17‐like cells in LN patients (active and inactive LN combined) stratified based on cumulative CTX dose. Horizontal lines represent median values. **P < 0·01, *P < 0·05.
Fig. S2. Matrix of correlation coefficients (RS – Spearman) in cross‐comparison of cytokine expressing CD4+ T‐cell subsets with major clinical and laboratory measures of LN. Active and inactive LN data were combined. Numeric values mark significant (P < 0·05) correlations.
Fig. S3. No linkage between fraction of systemic Th17 cells and renal histology status. (a) Percentage of circulating Th17 cells (IL‐17A+ IFNγ‐) in LN patients stratified based on disease activity status and renal histology class (ISN/RPS [Ref. 25 in the main body of manuscript]). (b) Th17 cell fraction in LN patients with different activity and chronicity of glomerular lesions (L – low, M – moderate, H – high). (c) Histology patterns in LN patients with contrasting numbers of Th17 cells. LN patients were stratified as Th17‐low and Th17‐high, based on the median percentage of circulating Th17 cells (< or > 1.2% of CD4+). There was no statistical difference in the frequency of certain lesions between the two designated groups. (d) Th17 percentages in patients with crescents and leukocyte infiltration in glomeruli as opposed to those without a lesion. On each graph horizontal bars represent median values. Only patients with III and IV ISN/RPS classes were shown in (b), (c) and (d).
Fig. S4. Correlation between systemic Th17 response and markers of renal inflammation. (a) Matrix of correlation coefficients (RS – Spearman) in cross‐correlation of major CD4+ T‐cell subsets (% of CD4+) and Treg/Th17 ratio with serum and urine biomarkers. Only most important cytokines linked with active LN were included in the analysis (based on our previous study [Ref. 21 in the main body of the manuscript]). Lower part shows a list of gene transcripts significantly upregulated (red) or downregulated (blue) in urine sediment from active LN patients. Just for comparison we also show genes (black) linked with Th17 response (e.g. RORC stands for RORγc transcription factor). Active and inactive LN data were combined. There was no significant correlation in majority of cross‐comparisons except for weak (RS~0.4) linkage between systemic Th17 (and Treg/Th17 ratio) and urine gene expression of CCL20, CXCL8 and IL17A. Numeric values mark significant (P<0.05) correlations. (b) Volcano plot showing differential expression of genes (panel of 43 immune genes as listed in Ref. 21) in the urine sediment cells from patients with Th17‐high (i.e. Th17 >median value 1.2% of CD4+) as compared to Th17‐low (<1.2%) endotype, separately for active and inactive disease. The transcriptome profiles did not differ except for moderately increased expression of TGFB1 (red dot) in inactive‐LN patients with Th17‐high endotype. (c) Relative mRNA expression (expressed as relative to GAPDH x103) of TBX21, CCL2, CD3G, CXCL8, IL17A, and TGFB1 in LN patients with Th17‐high and –low endotypes. TBX21 and CCL2 mRNA were previously found to be the most reliable biomarkers of active LN. Horizontal lines represent medians. *P < 0·05.
Fig. S5. No linkage between ratio of systemic Treg to Th17 cells and renal histology status. (a) Treg/Th17 ratio in LN patients stratified based on disease activity status and renal histology class (ISN/RPS). (b) Treg/Th17 ratios in LN patients with different activity and chronicity of glomerular lesions (L – low, M – moderate, H – high). (c) Histology patterns in LN patients with contrasting values of Treg/Th17 ratio (low <10, high >10). There was no difference in the frequency of lesions between the two groups. (d) Treg/Th17 ratios in patients with or without crescents and leukocyte infiltration in glomeruli.
Fig. S6. No difference in serum (a) and urine (b) levels of cytokines in LN patients with low or high systemic Treg/Th17 ratios. Horizontal lines represent median values. (c) Matrix of correlation coefficients (RS – Spearman) in cross‐comparison of major T‐helper cell subset ratios with major clinical and laboratory measures of LN. Active and inactive LN data were combined. Most important biomarkers of active LN are marked red (i.e. urine CXCL10 and CCL2). Numeric values mark significant (P < 0·05) correlations. There was no linkage between Treg/Th17 ratio and LN clinics.
Table S1. Basic subsets of peripheral blood T‐cells in LN patients (also stratified into active and inactive groups) and control.
Table S2. Clinical characteristics of active and inactive LN patients with markedly elevated Th17 cells (i.e. >1.7% of CD4+) in peripheral blood.
Table S3. Functional subsets of peripheral blood T‐cells in LN patients and control group.
Table S4. Clinical characteristics of active and inactive LN patients stratified based on the percentage of Th17 cells in peripheral blood.
Table S5. No linkage between percentage of circulating Th17 cells and renal biopsy ststus.
Table S6. Ratio of main subsets of cytokine producing CD4+ T‐cells and Treg cells.
Acknowledgements
This work was supported by the National Science Center, Poland, grant number NN402‐627940.
References
- 1. Kaul A, Gordon C, Crow MK et al. Systemic lupus erythematosus. Nat Rev Dis Primers 2016;2:16039. [DOI] [PubMed] [Google Scholar]
- 2. Borchers AT, Leibushor N, Naguwa SM, Cheema GS, Shoenfeld Y, Gershwin ME. Lupus nephritis: a critical review. Autoimmun Rev 2012;12:174–94. [DOI] [PubMed] [Google Scholar]
- 3. Krebs CF, Schmidt T, Riedel JH, Panzer U. T helper type 17 cells in immune‐mediated glomerular disease. Nat Rev Nephrol 2017;13:647–59. [DOI] [PubMed] [Google Scholar]
- 4. Hemdan NY, Birkenmeier G, Wichmann G, Abu El‐Saad AM, Krieger T, Conrad K, Sack U. Interleukin‐17‐producing T helper cells in autoimmunity. Autoimmun Rev 2010;9:785–92. [DOI] [PubMed] [Google Scholar]
- 5. Burkett PR, Meyer zu Horste G, Kuchroo VK. Pouring fuel on the fire: Th17 cells, the environment, and autoimmunity. J Clin Invest 2015;125:2211–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang Z, Kyttaris VC, Tsokos GC. The role of IL‐23/IL‐17 axis in lupus nephritis. J Immunol 2009;183:3160–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schmidt T, Paust HJ, Krebs CF et al Function of the Th17/interleukin‐17A immune response in murine lupus nephritis. Arthritis Rheumatol 2015;67:475–87. [DOI] [PubMed] [Google Scholar]
- 8. Wong CK, Lit LC, Tam LS, Li EK, Wong PT, Lam CW. Hyperproduction of IL‐23 and IL‐17 in patients with systemic lupus erythematosus: implications for Th17‐mediated inflammation in auto‐immunity. Clin Immunol 2008;127:385–93. [DOI] [PubMed] [Google Scholar]
- 9. Zhao XF, Pan HF, Yuan H et al Increased serum interleukin 17 in patients with systemic lupus erythematosus. Mol Biol Rep 2010;37:81–5. [DOI] [PubMed] [Google Scholar]
- 10. Vincent FB, Northcott M, Hoi A, Mackay F, Morand EF. Clinical associations of serum interleukin‐17 in systemic lupus erythematosus. Arthritis Res Ther 2013;15:R97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mok MY, Wu HJ, Lo Y, Lau CS. The relation of interleukin 17 (IL‐17) and IL‐23 to Th1/Th2 cytokines and disease activity in systemic lupus erythematosus. J Rheumatol 2010;37:2046–52. [DOI] [PubMed] [Google Scholar]
- 12. Zickert A, Amoudruz P, Sundström Y, Rönnelid J, Malmström V, Gunnarsson I. IL‐17 and IL‐23 in lupus nephritis – association to histopathology and response to treatment. BMC Immunol 2015;16:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang J, Chu Y, Yang X et al Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum 2009;60:1472–83. [DOI] [PubMed] [Google Scholar]
- 14. Shah K, Lee WW, Lee SH et al. Dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Res Ther 2010;12:R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen DY, Chen YM, Wen MC, Hsieh TY, Hung WT, Lan JL. The potential role of Th17 cells and Th17‐related cytokines in the pathogenesis of lupus nephritis. Lupus 2012;21:1385–96. [DOI] [PubMed] [Google Scholar]
- 16. Henriques A, Inês L, Couto M et al Frequency and functional activity of Th17, Tc17 and other T‐cell subsets in systemic lupus erythematosus. Cell Immunol 2010;264:97‐103. [DOI] [PubMed] [Google Scholar]
- 17. Ma J, Yu J, Tao X, Cai L, Wang J, Zheng SG. The imbalance between regulatory and IL‐17‐secreting CD4+ T cells in lupus patients. Clin Rheumatol 2010;29:1251–8. [DOI] [PubMed] [Google Scholar]
- 18. Kleczynska W, Jakiela B, Plutecka H, Milewski M, Sanak M, Musial J. Imbalance between Th17 and regulatory T‐cells in systemic lupus erythematosus. Folia Histochem Cytobiol 2011;49:646–53. [DOI] [PubMed] [Google Scholar]
- 19. Mesquita D Jr, Kirsztajn GM, Franco MF et al CD4(+) T helper cells and regulatory T cells in active lupus nephritis: an imbalance towards a predominant Th1 response? Clin Exp Immunol 2018;191:50–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Y, Ito S, Chino Y et al Laser microdissection‐based analysis of cytokine balance in the kidneys of patients with lupus nephritis. Clin Exp Immunol 2010;159:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jakiela B, Kosałka J, Plutecka H et al. Urinary cytokines and mRNA expression as biomarkers of disease activity in lupus nephritis. Lupus 2018;27:1259–70. [DOI] [PubMed] [Google Scholar]
- 22. Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol 2010;10:849–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuhn A, Beissert S, Krammer PH. CD4(+)CD25 (+) regulatory T cells in human lupus erythematosus. Arch Dermatol Res 2009;301:71–81. [DOI] [PubMed] [Google Scholar]
- 24. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997;40:1725. [DOI] [PubMed] [Google Scholar]
- 25. Weening JJ, D’Agati VD, Schwartz MM et al The classification of glomerulonephritis in systemic lupus erythematosus revisited. J Am Soc Nephrol 2004;15:241–50. [DOI] [PubMed] [Google Scholar]
- 26. Shenoy S, Chaurasia S, Edavalath S et al. Effect of induction therapy on circulating T‐helper 17 and T‐regulatory cells in active proliferative lupus nephritis. Int J Rheum Dis 2018. doi: 10.1111/1756-185X.13272. [DOI] [PubMed] [Google Scholar]
- 27. Xing Q, Wang B, Su H, Cui J, Li J. Elevated Th17 cells are accompanied by FoxP3+ Treg cells decrease in patients with lupus nephritis. Rheumatol Int 2011;32:949–58. [DOI] [PubMed] [Google Scholar]
- 28. Crispín JC, Oukka M, Bayliss G et al Expanded double negative T cells in patients with systemic lupus erythematosus produce IL‐17 and infiltrate the kidneys. J Immunol 2008;181:8761–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Enghard P, Humrich JY, Rudolph B et al CXCR29+CD4+ T cells are enriched in inflamed kidneys and urine and provide a new biomarker for acute nephritis flares in systemic lupus erythematosus patients. Arthritis Rheum 2009;60:199–206. [DOI] [PubMed] [Google Scholar]
- 30. Klocke J, Kopetschke K, Grießbach AS et al Mapping urinary chemokines in human lupus nephritis: Potentially redundant pathways recruit CD4(+) and CD8(+) T cells and macrophages. Eur J Immunol 2017;47:180–92. [DOI] [PubMed] [Google Scholar]
- 31. Chan RW, Lai FM, Li EK et al. Intrarenal cytokine gene expression in lupus nephritis. Ann Rheum Dis 2007;66:886–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chan RW, Lai FM, Li EK, Tam LS, Chow KM, Li PK, Szeto CC. Expression of T‐bet, a type 1 T‐helper cell transcription factor, in the urinary sediment of lupus patients predicts disease flare. Rheumatology (Oxf) 2007;46:44–8. [DOI] [PubMed] [Google Scholar]
- 33. Kwan BC, Tam LS, Lai KB et al The gene expression of type 17 T‐helper cell‐related cytokines in the urinary sediment of patients with systemic lupus erythematosus. Rheumatology (Oxf) 2009;48:1491–7. [DOI] [PubMed] [Google Scholar]
- 34. Szeto CC, Tam LS, Kwan BC et al Monitoring of urinary messenger RNA levels for the prediction of flare In systemic lupus erythematosus. Clin Chim Acta 2012;413:448–55. [DOI] [PubMed] [Google Scholar]
- 35. Banchereau R, Hong S, Cantarel B et al Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell 2016;165:551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou L, Lopes JE, Chong MM et al TGF‐beta‐induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 2008;453:236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zheng SG, Wang J, Horwitz DA. Cutting edge: Foxp3+CD4+CD25+ regulatory T cells induced by IL‐2 and TGF‐beta are resistant to Th17 conversion by IL‐6. J Immunol 2008;180:7112–6. [DOI] [PubMed] [Google Scholar]
- 38. Stadhouders R, Lubberts E, Hendriks RW. A cellular and molecular view of Thelper 17 cell plasticity in autoimmunity. J Autoimmun 2018;87:1–15. [DOI] [PubMed] [Google Scholar]
- 39. Komatsu N, Okamoto K, Sawa S et al Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med 2014;20:62–8. [DOI] [PubMed] [Google Scholar]
- 40. Alvarado‐Sánchez B, Hernández‐Castro B, Portales‐Pérez D et al. Regulatory T cells in patients with systemic lupus erythematosus. J Autoimmun 2006;27:110–8. [DOI] [PubMed] [Google Scholar]
- 41. Yan B, Ye S, Chen G, Kuang M, Shen N, Chen S. Dysfunctional CD4+, CD25+ regulatory T cells in untreated active systemic lupus erythematosus secondary to interferon‐alpha‐producing antigen‐presenting cells. Arthritis Rheum 2008;58:801–12. [DOI] [PubMed] [Google Scholar]
- 42. Pan X, Yuan X, Zheng Y et al Increased CD45RA+ FoxP3(low) regulatory T cells with impaired suppressive function in patients with systemic lupus erythematosus. PLOS ONE 2012;7:e34662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vargas‐Rojas MI, Crispín JC, Richaud‐Patin Y, Alcocer‐Varela J. Quantitative and qualitative normal regulatory T cells are not capable of inducing suppression in SLE patients due to T‐cell resistance. Lupus 2008;17:289–94. [DOI] [PubMed] [Google Scholar]
- 44. Chen X, Oppenheim JJ. Th17 cells and Tregs: unlikely allies. J Leukoc Biol 2014;95:723–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pandiyan P, Conti HR, Zheng L, et al CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse Candida albicans Th17 cell infection model. Immunity 2011;34:422–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moore‐Connors JM, Fraser R, Halperin SA, Wang J. CD4⁺CD25⁺Foxp3⁺ regulatory T cells promote Th17 responses and genital tract inflammation upon intracellular Chlamydia muridarum infection. J Immunol 2013;191:3430–9. [DOI] [PubMed] [Google Scholar]
- 47. Wilde B, Thewissen M, Damoiseaux J et al. Th17 expansion in granulomatosis with polyangiitis (Wegener’s): the role of disease activity, immune regulation and therapy. Arthritis Res Ther 2012;14:R227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Szczeklik W, Jakieła B, Wawrzycka‐Adamczyk K et al Skewing toward Treg and Th2 responses is a characteristic feature of sustained remission in ANCA‐positive granulomatosis with polyangiitis. Eur J Immunol 2017;47:724–33. [DOI] [PubMed] [Google Scholar]
- 49. Viaud S, Flament C, Zoubir M et al Cyclophosphamide induces differentiation of Th17 cells in cancer patients. Cancer Res 2011;71:661–5. [DOI] [PubMed] [Google Scholar]
- 50. Heylmann D, Bauer M, Becker H, van Gool S, Bacher N, Steinbrink K, Kaina B. Human CD4+CD25+ regulatory T cells are sensitive to low dose cyclophosphamide: implications for the immune response. PLOS ONE 2013;8:e83384. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Subsets of CD4+ memory T‐cells in LN. (a) Major memory CD4+ T‐cell subsets were identified by the surface expression of chemokine receptors (flow cytometry, additional staining for CD45RA not shown for clarity). (b) Cells were classified as: Th1‐like (CXCR3+ CCR6‐), Th1/17‐like (CXCR3+ CCR6+), Th2‐like (CXCR3‐ CCR4+ CCR6‐ CCR10‐), Th17‐like (CXCR3‐ CCR4+ CCR6+ CCR10‐), and Th22‐like (CXCR3‐ CCR4+ CCR6+ CCR22+). Data presented as medians (quartiles). *P < 0·05. (c) Correlation between percentage of type‐17 T‐cells identified based on intracellular cytokine detection, or chemokine receptor staining. (d) Decline in the number of memory CD4+ T‐cells (peripheral blood) in LN patients. Data presented as fold difference (mean and 95%CI) in comparison to median value in the healthy controls group. #P < 0·05 in comparison to control group. *P < 0·05 in comparison to inactive LN. (e) Matrix of correlation coefficients (RS – Spearman) in cross‐comparison of CD4+ T‐cell subsets distinguished by chemokine receptors (% of CD4+), with major clinical and laboratory measures of LN. Active and inactive LN data were combined. Numeric values mark significant (P < 0.05) correlations. (f) Correlation between percentage of Th17‐like cells (% of CD4+) and cumulative dose of cyclophosphamide (CTX, g). (g) Percentage of Th17‐like cells in LN patients (active and inactive LN combined) stratified based on cumulative CTX dose. Horizontal lines represent median values. **P < 0·01, *P < 0·05.
Fig. S2. Matrix of correlation coefficients (RS – Spearman) in cross‐comparison of cytokine expressing CD4+ T‐cell subsets with major clinical and laboratory measures of LN. Active and inactive LN data were combined. Numeric values mark significant (P < 0·05) correlations.
Fig. S3. No linkage between fraction of systemic Th17 cells and renal histology status. (a) Percentage of circulating Th17 cells (IL‐17A+ IFNγ‐) in LN patients stratified based on disease activity status and renal histology class (ISN/RPS [Ref. 25 in the main body of manuscript]). (b) Th17 cell fraction in LN patients with different activity and chronicity of glomerular lesions (L – low, M – moderate, H – high). (c) Histology patterns in LN patients with contrasting numbers of Th17 cells. LN patients were stratified as Th17‐low and Th17‐high, based on the median percentage of circulating Th17 cells (< or > 1.2% of CD4+). There was no statistical difference in the frequency of certain lesions between the two designated groups. (d) Th17 percentages in patients with crescents and leukocyte infiltration in glomeruli as opposed to those without a lesion. On each graph horizontal bars represent median values. Only patients with III and IV ISN/RPS classes were shown in (b), (c) and (d).
Fig. S4. Correlation between systemic Th17 response and markers of renal inflammation. (a) Matrix of correlation coefficients (RS – Spearman) in cross‐correlation of major CD4+ T‐cell subsets (% of CD4+) and Treg/Th17 ratio with serum and urine biomarkers. Only most important cytokines linked with active LN were included in the analysis (based on our previous study [Ref. 21 in the main body of the manuscript]). Lower part shows a list of gene transcripts significantly upregulated (red) or downregulated (blue) in urine sediment from active LN patients. Just for comparison we also show genes (black) linked with Th17 response (e.g. RORC stands for RORγc transcription factor). Active and inactive LN data were combined. There was no significant correlation in majority of cross‐comparisons except for weak (RS~0.4) linkage between systemic Th17 (and Treg/Th17 ratio) and urine gene expression of CCL20, CXCL8 and IL17A. Numeric values mark significant (P<0.05) correlations. (b) Volcano plot showing differential expression of genes (panel of 43 immune genes as listed in Ref. 21) in the urine sediment cells from patients with Th17‐high (i.e. Th17 >median value 1.2% of CD4+) as compared to Th17‐low (<1.2%) endotype, separately for active and inactive disease. The transcriptome profiles did not differ except for moderately increased expression of TGFB1 (red dot) in inactive‐LN patients with Th17‐high endotype. (c) Relative mRNA expression (expressed as relative to GAPDH x103) of TBX21, CCL2, CD3G, CXCL8, IL17A, and TGFB1 in LN patients with Th17‐high and –low endotypes. TBX21 and CCL2 mRNA were previously found to be the most reliable biomarkers of active LN. Horizontal lines represent medians. *P < 0·05.
Fig. S5. No linkage between ratio of systemic Treg to Th17 cells and renal histology status. (a) Treg/Th17 ratio in LN patients stratified based on disease activity status and renal histology class (ISN/RPS). (b) Treg/Th17 ratios in LN patients with different activity and chronicity of glomerular lesions (L – low, M – moderate, H – high). (c) Histology patterns in LN patients with contrasting values of Treg/Th17 ratio (low <10, high >10). There was no difference in the frequency of lesions between the two groups. (d) Treg/Th17 ratios in patients with or without crescents and leukocyte infiltration in glomeruli.
Fig. S6. No difference in serum (a) and urine (b) levels of cytokines in LN patients with low or high systemic Treg/Th17 ratios. Horizontal lines represent median values. (c) Matrix of correlation coefficients (RS – Spearman) in cross‐comparison of major T‐helper cell subset ratios with major clinical and laboratory measures of LN. Active and inactive LN data were combined. Most important biomarkers of active LN are marked red (i.e. urine CXCL10 and CCL2). Numeric values mark significant (P < 0·05) correlations. There was no linkage between Treg/Th17 ratio and LN clinics.
Table S1. Basic subsets of peripheral blood T‐cells in LN patients (also stratified into active and inactive groups) and control.
Table S2. Clinical characteristics of active and inactive LN patients with markedly elevated Th17 cells (i.e. >1.7% of CD4+) in peripheral blood.
Table S3. Functional subsets of peripheral blood T‐cells in LN patients and control group.
Table S4. Clinical characteristics of active and inactive LN patients stratified based on the percentage of Th17 cells in peripheral blood.
Table S5. No linkage between percentage of circulating Th17 cells and renal biopsy ststus.
Table S6. Ratio of main subsets of cytokine producing CD4+ T‐cells and Treg cells.