

Abstract
The surge in meals high in calories has prompted an epidemic of metabolic disorders around the world such that the elevated incidence of obese and diabetic individuals is alarming. New research indicates that metabolic disorders pose a risk for neurological and psychiatric conditions including stroke, Alzheimer’s disease, Huntington’s disease, and depression, all of which have a metabolic component. These relationships are rooted to a dysfunctional interaction between molecular processes that regulate energy metabolism and synaptic plasticity. The strong adaptive force of dietary factors on shaping the brain during evolution can be manipulated to trans form the interaction between cell bioenergetics and epigenome with the aptitude to promote long-lasting brain healthiness. A thorough understanding of the association between the broad action of nutrients and brain fitness requires high level data processing empowered with the capacity to integrate information from a multitude of molecular entities and pathways. Nutritional systems biology is emerging as a viable approach to elucidate the multiple molecular layers involved in information processing in cells, tissues, and organ systems in response to diet. Information about the wide range of cellular and molecular interactions elicited by foods on the brain and cognitive plasticity is crucial for the design of public health initiatives for curtailing the epidemic of metabolic and brain disorders.
Keywords: Neurotrophin, Bioenergetic, Metabolism, Brain disorders, Diet, Lifestyle, Systems biology, Systems nutrigenomics
1. Introduction
The advent of mechanization has dramatically changed peoples lifestyle in the last few decades. In particular, the surge in meals rich in caloric contents an adoption of a sedentary life style has triggered an epidemic of metabolic disorders (Haffner and Taegtmeyer, 2003; Lutsey et al., 2008; Trushnikova et al., 2014) with subsequent detriment of brain function. There is immense necessity to understand the action of diet on gene regulatory mechanisms, which are endowed with the capacity to direct all molecular and physiological processes governing brain function. The process of adjustment to environmental challenges has played an imperative role for species survival, and we are starting to understand the engagement of cellular, molecular, and ultimately gene regulatory events. In particular, it is becoming evident that diet influences the genome, and that epigenetic alterations enable the organism to elaborate complex multiorgan responses and behaviors that may extend throughout lifetime and across generations. The human genome comprises an entire collection of genes that are encoded and controlled by more than 3 billion DNA base pairs, which contain all of the information needed to maintain an organ system. Such information is subject to the effects of environmental modulations which determine the balance between health and dis ease.
We are starting to learn that adjustments in cell metabolism are the driving force for genomic adaptations and epigenomic alterations, and that reprograming of the epigenome is a fundamental mechanism by which the effects of diet are saved in our genes (Albuquerque et al., 2015). Energy use and conservation are essential needs of all living organisms. The brain has tremendous dependency for energy, particularly in humans where the brain contributes to about 20% of resting metabolism while only accounting for 2% of the total body mass. Maintenance of caloric homeostasis has been a powerful driving force in evolution such that alterations in cell metabolism have a strong impact on gene regulatory mechanisms that determine cellular features and ultimately, the phenotypic identity of an individual throughout its life-course. Herein we discuss how bioenergetic operations driven by environmental challenges can reprogram genes resulting in alterations of cellular and phenotypic identity underlying brain function. The broad impact of dietary nutrients can engage the entire cascade of gene regulatory mechanisms including epigenomic (modifications of DNA elements) and transcrip-tomic (abundance and alternative splicing), and their functional protein and metabolite products. In addition, thousands of endogenous microbes (microbiome) that live in multiple tissues and organ systems are surfacing as important modulators of the action of ingested foods on body and brain (Org et al., 2015; Parks et al., 2013). Growing evidence indicates that alterations in these regulatory mechanisms are core as pects of wide-ranging pathogenesis and also amenable to the manipulation of scientifically-based lifestyle management (Gomez-Pinilla, 2008; Meng et al., 2016). Here, we explore how diet affects the interrelationship between cell metabolism and genetic/epigenetic regulation with the potential to impact cellular processes sub-serving cognitive function.
Given their broad spectrum of action, foods can act on a multitude of molecular interfaces that control cellular homeostasis. A thorough understanding of the molecular-level associations between nutrients and complex brain functions requires high-level integrative analysis. Recently, nutritional systems biology harnessing advances of diverse omics technologies has emerged with the merit to help assess the consequences of nutritional intake at several levels of molecular information processing at the cell and organismal levels. These associations range from the DNA elements composing the genome, modifications in the genome (epigenome), protein products of genes (proteome), metabolite products of metabolic pathways (metabolome), to the microbiome (Zhao et al., 2015). These genome-wide approaches have the power to acquire multi-dimensional evidence to achieve a systems-level comprehension of the regulatory mechanisms governing the effects of foods on the brain. Information about these regulatory mechanisms can be used to guide public health programs to improve mental health and quality of life across lifespan, with particular relevance to the growing metabolic disease epidemic. Here we review the increasing evidence bonding metabolic dysregulation to brain disorders, the role of diet in this connection, and the mechanistic insights gained through both conventional and emerging systems biology approaches.
2. The threat of the new epidemic of metabolic disorders
2.1. Obesity and type-2 diabetes
The adoption of dietary habits abundant in calories and the lack of exercise in modern societies are perceived as main contributors to the mounting contemporary incidence of metabolic disorders. The metabolic syndrome (MetS), defined as a cluster of metabolic dysfunctions (triglycerides, abdominal fat, and blood pressure, and low HDL and insulin sensitivity) that predispose individuals to cardiometabolic disorders including obesity, diabetes, and cardiovascular disease, is attaining epidemic levels around the world. Type 2 diabetes (T2D) has significantly increased in recent decades in both the adult and child populations—a change in incidence rate that is documented to be escalating in association with the adoption of a sedentary lifestyle and excessive caloric-intake (Flegal et al., 1998; Mokdad et al., 2001; Ogden et al., 2002; Pinhas-Hamiel et al., 1996). Moreover, T2D ranks amongst the fastest growing health problems worldwide, and the number of diabetic individuals is predicted to increase to 350 million worldwide by the year 2020. It is appraised that the prevalence of obesity around the world has increased 41% between the years 1980 and 2013 (Ng et al., 2014). According to data from the National Health and Nutrition Examination Survey (Ogden et al, 2012) obesity in USA reached up to 36% in adult men and women and 17% in children between years 2009–2010 −0. Currently, more than 2 billion people worldwide are overweight or obese (González-Muniesa et al., 2017), and projections predict that the density of obese individuals will keep increasing for the next three decades up to 60% in adult men, 40% in adult women, and 25% in children (CDC Report 2014, http://www.cdc.gov/obesity/data/adult.html). Obesity is also linked to an increased risk of T2D, cardiovascular disease, cancer, and mortality (Hedley et al., 2004). Clearly, MetS, obesity and associated metabolic diseases present a worldwide clinical and public health burden that needs to be urgently addressed.
2.2. Metabolic perturbations increase risk of neurological and psychiatric disorders
The threat posed by metabolic disorders on the brain, especially MetS, is particularly frightening on the face that most brain disorders have some malfunction in the capacity of brain cells to metabolize energy (Chan, 2005; Cheng et al., 2012; Du et al., 2013). The growing prevalence of metabolic disorders appears to have grave consequences for the maintenance of brain metabolic homeostasis and cognitive function. Both obesity and T2D are significant risk factors for neurodegenerative diseases such as Alzheimer s disease (AD) and dementia, which constitutively involve hippocampal atrophy. Diabetic individuals show impairments in multiple aspects of cognitive function. Moreover, a high body mass index (BMI) in otherwise healthy adults has been associated with executive dysfunction. Insulin resistance associated with T2D is implicated in the pathogenesis of AD (Jiang et al., 2007) and may affect as high as 80% of the AD population (Janson et al., 2004). In fact, it appears that the cognitive dysfunction observed in many AD patients could be associated with impaired insulin signaling or sensitivity—a condition that has gained the recognition of being a type 3 diabetes. Animal models of diabetes further corroborate that cognitive impairment is a central characteristic of metabolic disorders, which entails a dysfunction in hippocampal synaptic plasticity (Stranahan et al., 2008). Similar to findings from human studies, obese mice exhibit memory impairments on multiple cognitive domains (Farr et al., 2008). At the molecular level, it has been shown that the brain-derived neurotrophic factor (BDNF) system, which is central to the regulation of both energy homeostasis and neurocognitive processes, is compromised in disorders of both energy metabolism such as obesity and insulin resistance (Kernie et al., 2000; Lyons et al., 1999) and cognitive function such as dementia, AD, Huntington s Disease, Parkinson s disease, and schizophrenia (Caraci et al., 2018; Zuccato and Cattaneo, 2009).
In addition to cognitive disorders, several lines of evidence have implicated abnormal metabolism in the pathophysiology of psychiatric disorders. An increased incidence rate of T2D, impaired glucose tolerance, and insulin resistance occur in the psychiatric population (Dixon et al., 2000; Lilliker, 1980; Mukherjee et al., 1996; Pasinetti et al., 2007; Rasgon et al., 2010; Regenold et al., 2002). In diabetics, the commonness of depression is 2 3 folds higher (Anderson et al., 2001; Gavard et al., 1993), and there is high risk of developing diabetes during the 13-year period following diagnosis of major depression (Eaton et al., 1996). Both lines of evidence highlight the co-occurrence of metabolic diseases and depression. A similar panorama exists for other psychiatric illnesses, including manic depression. For example, a 3-fold increase in the incidence of diabetes has been reported in a study of 203 manic-depressive subjects (Lilliker, 1980). The association between metabolism and long-term vulnerability to neurological disorders has received indirect support by several epidemiological studies. For example, people who were either conceived or in utero during the Dutch Famine of 1944–45 or the Chinese Famine of 1959–1961 had a two-fold greater risk of schizophrenia as adults (Brown and Susser, 2008; Susser and St Clair, 2013; Xu et al., 2009).
2.3. Diet as an escalator of MetS and brain dysfunction (Figs. 1 and 2)
Fig. 1.
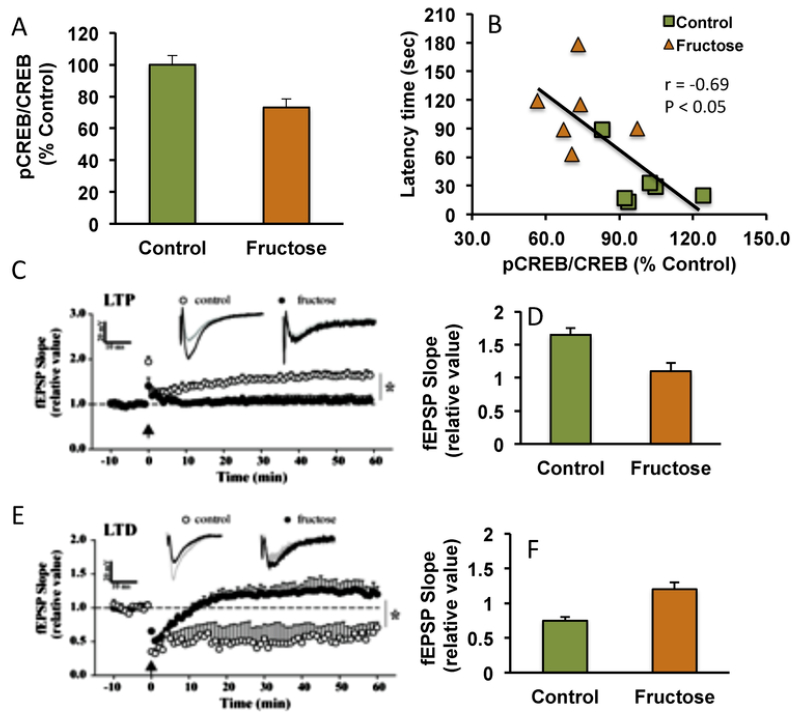
Influence of metabolic perturbation on brain plasticity and behavior. High fructose consumption for a period longer than six weeks promotes metabolic syndrome in rodents. (A) The effects of fructose on the brain are evidenced by reductions in the activation of molecular systems important for synaptic plasticity such as cAMP-response element binding (pCREB/CREB) in the hippocampus; for reviews see (Agrawal and Gomez-Pinilla, 2012). CREB belongs to a family of transcription factors that play a major role in synaptic plasticity and learning and memory (Benito and Barco, 2010). (B) Fructose treatment also results in memory deficit in the Barnes maze as evidenced by a decrease in latency time to find the escape hole, in proportion to an increase in CREB activation. (C, D) Fructose exposure also reduces hippocampal excitability in the form of long-term potentiation (LTP), and (E, F) synaptic strength or long-term depression (LTD); for reviews see (Cisternas et al., 2015). This information highlight the deleterious effects of metabolic perturbations on the substrates for synaptic plasticity and cognitive function in the brain.
Fig. 2.
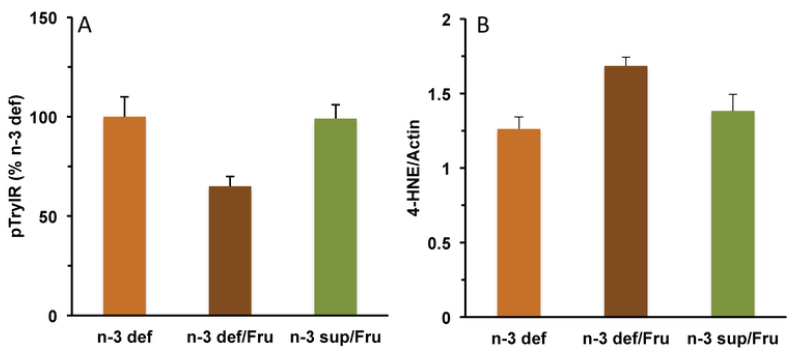
Interactive actions of fructose and omega-3 fatty acids consumption on the management of metabolic dysfunction in the hippocampus. (A) Although deficiency of omega-3 fatty acids (n-3 def) does not alter insulin receptor signaling, concurrent fructose administration reduces insulin signaling (n-3 def/Fru). N-3 supplementation provides protection against the effects of fructose (n-3 sup/Fru); see for reviews (Agrawal and Gomez-Pinilla, 2012). The results highlight the importance of dietary omega-3 fatty acids for maintaining proper insulin signaling in brain, particularly under the metabolic challenges posed by metabolic dysfunction carried by fructose consumption. (B) Deficiency in omega-3 fatty acids also makes the brain vulnerable to lipid peroxidation carried by fructose exposure. N-3 supplementation appears to protect against the lipid peroxidation driven by fructose. A failure in cell metabolism can result in lipid peroxidation of the plasma membrane by engaging membrane bound the omega-3 fatty acid DHA, and the omega-6 arachidonic acid (AA) resulting in the generation of 4-HNE and 4HHE, respectively (see Fig. 3). Insulin receptor activation examined by immunoprecipitation of phosphotyrosine (pTyr) followed by immunoblotting with insulin receptor.
Dietary factors high in caloric contents such as sugars and high saturated fat are considered important contributors to the epidemics of MetS (Gerrits and Tsalikian, 1993; Zivkovic et al., 2007). Rodents exposed to a fructose diet display physiological signs of MetS such as increased hepatic lipid and triglyceride level (Agrawal and Gomez-Pinilla, 2012; Kelley et al., 2004), and peripheral insulin resistance (Agrawal and Gomez-Pinilla, 2012; Dekker et al., 2010). Consumption of fructose has recently been recognized as a critical instigator of the current epidemic of metabolic disorders in humans (Dhingra et al., 2007; Ishimoto et al., 2013; Lanaspa et al., 2013; Lyssiotis and Cantley, 2013; Stanhope et al., 2009), and its effects on compromising neuronal function are becoming alarming (Farooqui et al., 2011; Newcomer, 2007; Raji et al., 2010; Zhang et al., 2009). We have adopted fructose in the drinking water as a rodent model to promote metabolic disturbances such as peripheral and central insulin resistance, lipid dysregulation, and disrupted insulin signaling (Figs. 2 and 3) (Agrawal and Gomez-Pinilla, 2012). Our research has revealed the harmful impact of fructose on the brain (Agrawal and Gomez-Pinilla, 2012; Cisternas et al., 2015; Gomez-Pinilla, 2008) such that the animals on fructose show impaired memory and learning skills, as well as reductions in proteins re lated to brain plasticity and mitochondrial bioenergetics (Figs. 1–3) (Agrawal and Gomez-Pinilla, 2012). In addition, the capacity of the hippocampus to sustain synaptic plasticity in the forms of long-term potentiation (LTP) and long-term depression (LTD) was seriously compromised (Cisternas et al., 2015). Fructose exposure also reduced the number of contact zones and the size of postsynaptic densities (PSDs) in the hippocampus, which changes were concomitant to a decrease in hippocampal neurogenesis (Cisternas et al., 2015). The results of these studies challenge the current view that the effects of diet-induced metabolic abnormalities are restricted to the periphery. As discussed below, diet may also use epigenetic mechanisms to promote long-term alterations in the brain (Tyagi et al., 2015).
Fig. 3.
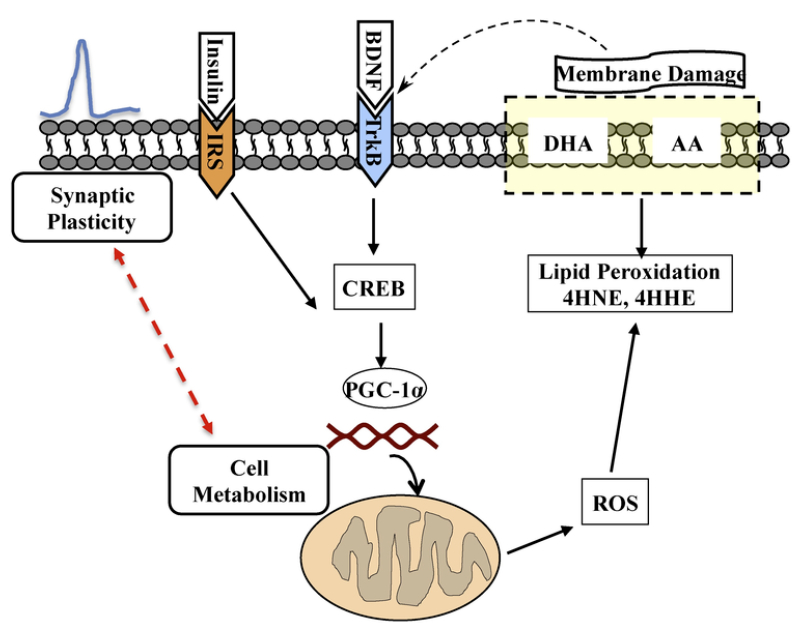
The diagram illustrates the working association between cell metabolism and synaptic plasticity, which is a target of dietary interventions. Most signaling receptors are embedded in the plasma membrane such that the functionality of the membrane is crucial for regulation of neuronal excitability and communication. In this hypothetical model, activation of the BDNF receptor (TrkB) or insulin receptor (IRS) signal a molecular cascade that regulates synaptic plasticity and cell metabolism. Insulin activates regulators of mitochondrial biogenesis, such as the peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1α), a member of a family of transcription co-activators (Ventura-Clapier et al., 2008), using downstream systems overlaping with those of BDNF. A failure in cell metabolism involving the mitochondria can result in overproduction of reactive oxygen species (ROS), lipid peroxidation of the plasma membrane by engaging membrane bound omega-3 fatty acid docosahexaenoic (DHA), and the omega-6 arachidonic acid (AA) resulting in the generation of 4-HNE and 4HHE, respectively. Peroxidation of membrane produces alterations in the function of key membrane proteins including glucose transporter, glutamate transporter, and sodium potassium ATPases (Lauderback et al., 2001); (Mark et al., 1997). The DHA is an important component of the membrane and crucial to maintain membrane function. Both BDNF/trkB (Chen et al., 2009) and insulin receptor (Lee et al., 2005) pathways convergence on signaling pathways regulating synaptic activity and learning and memory (Alonso et al., 2002). Through TrkB receptor, BDNF leads to the activation of CREB, which is a potent activator of PGC-1α (Herzig et al., 2001). Therefore, the effects of fructose on CREB activation in rats (Figs. 1 and 2) emphasize the interactive actions of metabolic and plasticity signals. As discussed in the text, metabolic dysfunction involving mitochondrial abnormalities is getting recognition as a common feature in the pathogenesis of neurological disorders.
In addition to high fructose diet, high fat diet (HFD) composed by high contents of sugars and saturated fats is another established promotor of MetS and contributes significantly to harm brain function and plasticity (Buettner et al., 2007; Freeman et al., 2014; Zivkovic et al., 2007), disrupting signaling through insulin receptors which are abundant in brain areas associated with cognitive function such as the hippocampus (Agrawal et al., 2009). The effects of the HFD has also been allied to reduced cognitive capacity which is accompanied by impaired synaptic plasticity and elevated inflammatory parameters in the brain (Hao et al., 2016; Hsu et al., 1989; Molteni et al., 2002).
In turn, poor consumption of dietary omega-3-fatty acids poses a risk to disturbances in insulin signaling that are common in diabetes (Agrawal and Gomez-Pinilla, 2012). The necessary action of dietary omega-3 fatty acids is also observed in studies showing that their dietary deficiency during brain formation exacerbates the outcome of metabolic disorders in the adult brain (Bhatia et al., 2011). The omega-3 fatty acid docosahexaenoic (DHA), an important supporter of metabolic homeostasis, has been demonstrated to be instrumental to counteract the action of fructose (Agrawal and Gomez-Pinilla, 2012). We discuss below how diet can reprogram metabolic pathways in the brain and can determine lifelong predisposition to metabolic disorders.
2.4. Cell metabolic dysfunction disrupts brain plasticity (Fig. 3)
Energy efficiency and accumulation of high-quality structural materials provided by diet have been primordial elements for successful biological adaptation (Gomez-Pinilla, 2008), and new studies show that these properties are operational for the maintenance of proper brain function. Alterations in bioenergetics mechanisms likely contribute to the cellular and molecular abnormalities that characterize various metabolic and neurodegenerative diseases. The functional significance of a disturbance in cell energy metabolism becomes evident when we examine it in the context of neurocognitive deficits common to ageing and neurodegenerative diseases. It has been reported that both senile dementia of the Alzheimer’s type (Schubert, 2005) and T2D (Wren and Garner, 2005) are characterized by a decrease in cerebral glucose metabolism and associated with disturbances in acetyl-CoA synthesis and oxidative phosphorylation (Meier-Ruge et al., 1994). Multiple studies demonstrate that metabolic derangements, including oxidative stress and glucose dysregulation, accelerate brain ageing (Nunomura et al., 2006; Yaffe et al., 2004). Interestingly, a perturbed energy status related to a dysregulation in cellular energy transfer mechanisms (creatine synthesis and/or uptake) is implicated in the pathoetiology of AD (Nunomura et al., 2006). A mitochondrial deficit constitutes one of the main features in all major age-related neurodegenerative diseases and during normal ageing (see review (Reeve et al., 2008)). Notably, the incidence of T2D is increased in association with ageing (Kelley, 2002; Mootha et al., 2003; Patti et al., 2003). Moreover, the levels of BDNF, a key neurotrophin involved in both cell metabolism and neuroplasticity, decrease progressively during the ageing process (Cotman, 2005; Yamanaka et al., 2007), especially being prominent in hippocampal pyramidal and dentate granule cells. Conspicuously, the age-related BDNF decline in the hippocampus occurs in concert with neurocognitive impairments in both animals and humans (Cotman, 2005; Kramer et al., 1999; Laurin et al., 2001). Exemplative findings are encountered in human AD studies, in which AD brains exhibit decreased hippocampal BDNF levels (Phillips et al., 1991) and impairments in energy metabolism (Hoyer, 2004a, b) such as altered cytochrome c oxidase activity (Valla et al., 2001). Overexpression of BDNF and CREB has been shown to attenuate cognitive deficits in rodent models of AD (Caccamo et al., 2010; Nagahara et al., 2013).
Given the strong dependency of brain function and plasticity on proper regulation of cellular energy, they are highly amenable to fluctuations in the energy status of the cell occurring during development, in response to environmental challenges, and in disease states (Gomez-Pinilla and Tyagi, 2013; Mattson, 2012). Across many species, the energy demands of the central nervous system are directly related to the density of neuronal connectivity irrespective of brain mass (Herculano-Houzel, 2011). Cell energy metabolism is an important driving force of gene regulation and epigenetic mechanisms. As discussed above, this relationship is conspicuously revealed in conditions in which energy homeostasis is compromised in the pathoetiology of numerous metabolic and neurocognitive disorders including T2D, dementia, depression, addiction, and neurodegenerative diseases. Recent evidence demonstrates that diet have a powerful capacity to foster biological adaptability to support cognitive aptitude by engaging epigenetic mechanisms (Gomez-Pinilla and Tyagi, 2013).
3. Epigenetics bridging diet and long-term brain plasticity (Fig. 4)
Fig. 4.
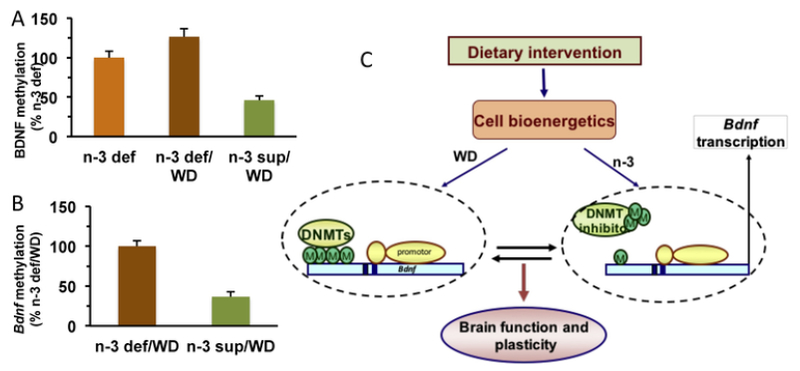
Dietary exposure is operational for building an “epigenetic memory” influencing long-term brain function and plasticity. Methylation of DNA occurs in about 80% of all CpG in mammals (about 80% of CpG are methylated). Methylation impacts transcription of genes by impeding binding of proteins or attracting binding of methyl-CpG-binding proteins such that reduction of methylation stimulate transcription of Bdnf gene. (A) Prior exposure to a diet rich in omega-3 fatty acids protects against the effects of switching to a western diet (WD) high in calories. Animals that had been exposed to a diet rich in omega 3 fatty acids (n-3 sup) show reduced methylation of the Bdnf promoter IV after switching to a WD, relative to animals that had been exposed to a diet deficient in omega-3 fatty acids (n-3 def/WD). The methylation was blocked using the methylation inhibitor decitabine (DEC). See for reviews (Tyagi et al., 2015). (C) Potential mechanism by which cell metabolism influenced by diet can serve to regulate methylation (M) of the Bdnf gene based on (Tyagi et al., 2015). Dietary interventions affect cell energy regulators such as NAD/NADH, SIRT1, and PGC-1α, which in turn act on DNA methyltransferase (DNMT) to regulate Bdnf methylation and transcription. The pre-exposure to dietary n-3 fatty acid (n-3 diet) may act as a determining factor for the outcome of WD exposure. Omega-3 fatty acids and WD can have opposite actions on DNMTs by promoting hypomethylation and hypermethylation of the Bdnf exon IV, respectively.
3.1. Interaction between diet and epigenetics
Despite the association between diet and brain health supported by epidemiological and clinical evidence, we are just starting to understand mechanisms involved. Increasing evidence from the clinical, epidemiological, and experimental literature supports the idea that diet can promote stable modifications in the brain that can last for a long time up to the point to affect future generations. New research indicates that the powerful action of diet on cell metabolism is operational for reprograming gene regulatory mechanisms underpinning brain function and plasticity, and disease susceptibiljty. As discussed below, cumulative evidence over the past decade shows the potential of select food components to alter the functional program of genes using epigenetic modifications. Epigenetics denotes changes in the genome that can alter gene express ion without involving alterations in DNA sequence (e.g., C to T nucleotide change on the DNA) as in the case of genetic variations. Exciting new Information has revealed clues how the interaction between cell metabolism and epigenetic is significant for control of patho-physiological processes, which are amenable to dietary regulation.
3.2. Mechanisms of epigenetics
Unlike genetic variations that are relatively static, epigenetic alterations are dynamic and potentially reversible such that nutritional supplementation or pharmaceutical agents may be developed to counteract undesirable epigenomic profiles. DNA methylation and various histone modifications such as methylation, acetylation, phosphorylation, ribosylation, and ubiquitination are widely studied epigenetics modifications, although microRNAs and long noncoding RNAs (lncRNAs) are also considered epigenetic regulators (Cech and Steitz, 2014; Rivera and Ren, 2013). The idea behind is that accessibility of DNA to the transcriptional machinery is crucial to regulate gene expression and DNA transcription. Heterochromatin regions that are tightly wound-up tend to be expressionally repressed, whereas euchromatin regions are open and conducive to gene transcription. Therefore, agents that alter the structure of chromatin can change the transition between silent to transcriptionally permissive chromatin leading to changes in gene expression (Fischle et al., 2003; Sng et al., 2006).
Nutrients and dietary factors can promote epigenetic functional changes by acting at various levels of chromatin regulation such as a source of methyl groups or as co-enzymes involved in methyl transfer for one-carbon metabolism (Kim et al., 2009). It has also been shown that select bioactive food components directly affect enzymes involved in the catalysis of DNA methylation and histone modifications (Choi 2010). Interestingly, the vitamin B9 or folate influences the epigenome, and in combination with vitamin B12, acts as a cofactor in generating the major source of methyl groups in the brain, namely S-adenosylme-thionine (SAMe). Folate is heavily involved in cell metabolism by interacting with the mitochondria such that folate dietary deprivation results in mitochondrial dysfunction (Ye et al., 2010). Though folate is a methyl donor in one-carbon metabolism (Lucock, 2000), high levels of folate during pregnancy cause both hyper and hypomethylation of offspring DNA in a gene-specific manner (Barua et al., 2014). It has been shown that doses of folate around those prescribed (4 mg/day) to women with a history of neural tube defect pregnancy, can alter the methylation status of candidate autism genes in the DNA of the offspring (Barua et al., 2014). It has also been shown that high folate supplementation during pregnancy promotes a reversible methylation of the promotor for the anorexigenic peptide pro-opiodmelanocortin (POMC) in the hypothalamus of the offspring (Cho et al., 2013), reducing POMC expression, and ultimately causing hyperphagia and obesity in the offspring. The impact of foods on epigenetic mechanisms is additionally relevant on the face that epigenetic alterations are potentially reversible opening to the possibility to employ diet to offset unhealthy epigenetic marks. In these terms, epigenetic regulation is perceived as a fundamental tool used by evolution to ensure species adaptation to challenging environmental conditions. The same property is under the scope of several pharmaceutical strategies to combat several diseases.
3.3. Epigenetic regulation of Bdnf transcription, and implications for brain disorders
DNA methylation is altered globally with age and neurological conditions (Teschendorff, 2013; Teschendorff et al., 2013). For example, methylation of histone 3 lysine 9 (H3K9), which has a repressive function (Lachner et al., 2001), has been found elevated in timing with cognitive deficits in an animal model of AD, causing reduced Bdnf expression (Walker et al., 2013). Several studies have reported the influences of foods on epigenetic regulation including DNA methylation and his tone modifications, and are reviewed by Choi et al (Choi et al., 2013). In particular, it has been reported that Bdnf methylation in the human frontal cortex changes proportionally to age (Keleshian et al., 2013). In primary neuronal cultures, both ageing and a rodent model of AD like pathology reduced histone acetylation in hippocampal/cortical neurons. In the blood, DNA methylation on the CpG island of the promotor region for exon I, but not IV, was significantly associated with major depression (Fuchikami et al., 2011). The clustering of disorders related to impaired glucose metabolism, i.e., T2D, and psychiatric, i.e., dementia and depression are prominently associated with lowered levels of BDNF (Krabbe et al., 2007). Lowered levels of serum BDNF has been described in patients with major depression (Karege et al., 2002), and the protective action of BDNF against the pathogenesis of depressive disorders has received abundant experimental support (Filus and Rybakowski, 2005). Psychiatric disorders such as depression, bipolar, and in many instances, severe cognitive impairments have been coupled with epigenetic regulation of the BDNF gene (Ikegame et al., 2013; Kang et al., 2013). Though these studies were done in humans and therefore may only reflect peripheral changes in BDNF regulation, data suggest that methylation status of the BDNF promoter may be consistent across brain and peripheral tissues, reflecting the possibility that methylation of peripheral Banff gene may serve as a biomarker for Bdnf methylation status in the brain (Ikegame et al., 2013). Although low levels of BDNF and elevated methylation of the BDNF promoter are not correlated with the severity of symptoms (Birkenhäger et al., 2012; Molendijk et al., 2011), epigenetic changes may be related to the interaction between the disorder and antidepressant treatment (Carlberg et al., 2014; Lopez et al., 2013). The frontal cortex of patients with bipolar disorder and AD have been found hypermethylated in the CpG islands of the BDNF promotor region (Rao et al., 2012). Reduced levels of BDNF have been found in the hippocampus, prefrontal cortex (Dwivedi, 2009; Karege et al., 2005; Pandey et al., 2008) and plasma (Kim et al., 2007) of suicidal patient. One study found hypermethylation of the BDNF promotor region in Wernickes area in the brain of patients who had committed suicide (Keller et al., 2010).
3.4. Diet and epigenetic alterations (Fig. 4)
It has been reported in rodents (Tyagi et al., 2015) that the type of food consumed in early life can imprint the genome by promoting epigenetic alterations in the Bdnf gene. These studies showed that prior exposure to a diet with adequate levels of omega-3 fatty acids reduced the cognitive decay originated from subsequent exposure to a Western diet (WD) high in saturated fat and sugar. Abundant evidence indicates that dietary exposure to the omega 3 fatty acid docosahexaenoic or DHA promotes resilience against several types of injuries and neuropsychiatric disorders. Interestingly, Tyagi et al (Tyagi et al., 2015) also reported that exposure either to WD or omega-3 fatty acids exerted opposite actions on the methylation of the Bdnf promotor exon IV. The metabolic sensor NAD/NADH and SIRT1 metabolic regulator appeared to act as an intermediate step between the diet and Bdnf methylation. The use of a DNA methytransferase inhibitor counteracted the Bdnf hypermethylation and cognitive decay, and blocked the WD-reducing effects on the mitochondrial transcription regulator PGC-1α suggesting that the PGC-1α action is downstream to the Bdnf methylation. The results of this study support the possibility that the effects of the omega-3 fatty acid diet are saved in the epigenome as an epigenetic memory by interplaying with metabolic signals. This epigenetic memory could serve as an underlying mechanism for protecting the brain during periods of hardship. Exemplifying an association between cell metabolism and epigenetics, it has been shown that caloric intake regulates cell metabolic factors involved in chromatin modifications such as ATP, acetyl-CoA, s-adenosyl-L-methionine, and NADH (Ramsey et al., 2007; Wallace and Fan, 2010). In addition, glucose availability affects ATP- dependent histone acetylation (Wellen et al., 2009).
Another front for the action of foods on epigenetics is the control and temporal preservation of signals related to the hedonic reward system. Chronic administration of a WD has been shown to impact sensitivity to rewarding behaviors by increasing DNA methylation of the opioid receptor (Vucetic et al., 2011b). The decision to eat or not to eat is susceptible to epigenetic regulation of the POMC promotor region, which is affected by folate supplementation during pregnancy (Cho et al., 2013). Too much folate during pregnancy may lead to overconsumption of salient foods, which can cause epigenetic silencing of the μ-opioid receptor (Vucetic et al., 2011b), which has been suggested to increase susceptibility to over-indulging in rewarding behaviors such as consuming highly palatable foods (Vucetic et al., 2011a). Together, the results of these studies support the idea that this type of epigenetic alterations may result in overconsumption of foods and obesity, and increased vulnerability to cognitive disorders.
3.5. Transgenerational inheritance
An increasing number of epidemiological studies suggest the possibility that the epigenomic consequences of dietary habits can be inheritable with the intrinsic capacity to influence vulnerability to metabolic disorders such as diabetes in the posterity (reviewed in Ref. Wang et al., 2011). This capacity may extend to cognitive disorders, for example, both over and under nutrition during pregnancy increases susceptibility of the offspring to neurodegenerative diseases (Chouliaras et al., 2010). As discussed below, the propensity to metabolic disorders and obesity becomes even more alarming for public health in the context of transference of information across generations.
It is increasingly recognized that epigenetic variability can last for considerable time. Although still debatable, the concept of epigenetics brings in heritable changes in gene function that are not attributable to alterations in DNA sequence, that entails the transmission of epigenetic marks (e.g. the methylation pattern of certain genes) from parent to daughter. There is captivating evidence in the epidemiological literature that an environmentally induced phenotype can be inherited (Ramsey et al., 2007). Edvinsson and colleagues examined the records of the 1890, 1905, and 1920 cohorts from Overkalix, an isolated community in Northern Sweden, resulting in the intriguing discovery that longevity (Bygren et al., 2001) and diabetic mortality (Kaati et al., 2002) are linked to ancestral food supply. Prominently, it was found that diabetes mortality in subsequent generations increased if the paternal grandfather was exposed to a surplus of food during his mid-childhood (the slow growth period occurring before the prepubertal growth peak) (Kaati et al., 2002). In a subsequent study, they found that the paternal grandfathers food supply during mid-childhood was linked to mortality risk in the grandsons, but not in the granddaughters. These results in humans support the existence of a sex-specific, male-line transgenerational inheritance system that records and utilizes nutritionally related information from previous generations, or rather, a system that enables the memory of an environmental event to be passed on to subsequent generations (Kaati et al., 2007). Genomic imprinting embraces the principle of transgenerational epigenetic inheritance in which an epigenetic signature in one-generation influences gene expression in subsequent generations (Ferguson-Smith and Surani, 2001). Additionally, complex diseases such as T2D, do not seem to have a gene that predetermines a person to develop the disease—only genes that influence predisposition to the disease (Edwards and Myers, 2007), which are likely subject to epigenetic modification. Therefore, Conrads Waddington’s original definition is highly relevant when we consider the ability of the environment to impact the genome through experience-dependent epigenetic mechanisms.
A growing line of evidence in rodents has provided experimental support to the possibility that epigenetic traits can be transmitted from one generation to others. Maternal-pup licking behavior, which seems to be coded in experience-dependent epigenetic modifications, can be transmitted to her offspring and persist over many generations (Weaver et al., 2004). The quality of maternal care may “program” the genome through epigenetic mechanisms to modulate the stress response in the offspring (Weaver et al., 2004). Through epigenetic modifications, acquired traits such as maternal and paternal conditions (Champagne, 2016; Peter et al., 2016) (Ng et al., 2010), hormonal (Stolzenberg and Champagne, 2016) may be transmitted to offspring. In support of a link between cell metabolism and epigenetics, it has been shown that chronic consumption of a caloric diet can program pancreatic ß-cell dysfunction, impaired insulin secretion and glucose tolerance in the female rat offspring (Ng et al., 2010).
4. Challenges posed by the multiple effects of diet on brain can be met by the new science of nutritional system biology (Fig. 5)
Fig. 5.
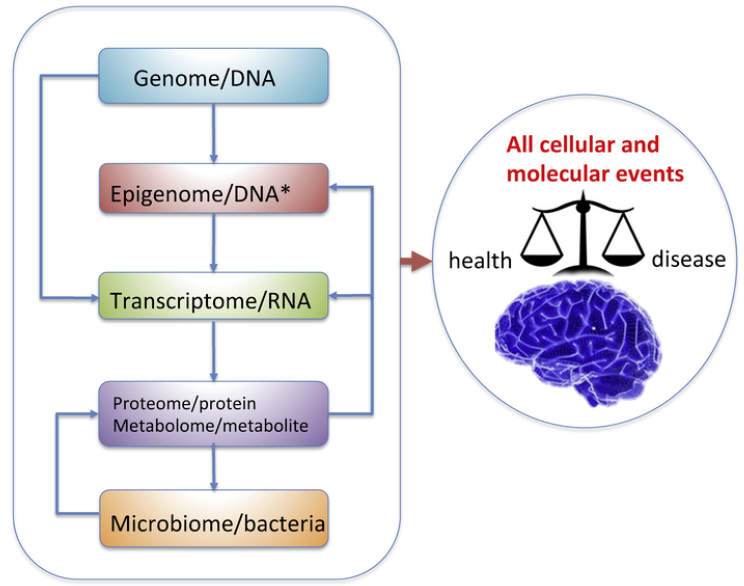
Nutritional system biology provides a thorough representation of the multitude of molecular interactions affected by diet to ultimately regulate brain homeostasis and disease. The heritable genome has the program to direct all cellular and molecular events, and determines the expression of genes in the transcriptome of individual cells, which in turn instruct the proteome to synthesize functional protein products that are critical for cell to cell communication and processing of higher order information. Lifestyle factors such as diet can interact with the genome and modify the epigenome to affect the transcriptome. Byproducts of the gut microbiota can modulate the host immune system as well as enzymes that assist in metabolism to influence metabolite diversity and quantity. Metabolites, in turn, play important roles in gene expression regulation by contributing to modifications of the epigenome that subsequently change the transcriptional regulatory machinery. These complex cascades and interactions are critical elements for consideration in multidimensional data integration that can provide meaningful information about brain function and pathogenesis.
4.1. Challenges: the need for multiple events consideration and big data processing
As discussed above, a growing body of evidence denotes that diet influences brain function and plasticity by engaging epigenetic regulation of genes that can extend the effects of diet for considerable time up to the point to determine vulnerability to neurological disorders. However, the greater part of previous studies focus on individual candidate genes, pathways, and molecular events, offering limited mechanistic insights. A thorough understanding of the association between nutrients and disease requires the use of approaches to integrate information derived from a multitude of molecular entities and pathways involved. Recently, nutritional systems biology has emerged as a powerful approach to address challenges in nutritional research using advanced omics technologies (de Graaf et al., 2009; Kaput and Morine, 2012; Panagiotou and Nielsen, 2009; van Ommen et al., 2008; Zhao et al., 2015). In principle, nutritional systems biology purposes to assess the interactions and influences of nutritional intake at multiple molecular layers involved in information processing in cells, tissues, and organ systems. The molecular level information involves the genome (DNA sequence), transcriptome (gene expression, particularly mRNAs) epigenome (DNA and chromatin modifications), proteome (protein products of coding genes), metabolome (metabolic products), and the gut microbiome interacting with the host.
The various levels of molecular entities carry intrinsic relationships. According to the central dogma, all instructions for protein synthesis are encoded in the DNA (genome) which is transcribed into mRNA (transcriptome), and subsequently translated into proteins (proteome). However, recent studies have revealed the critical role of epigenetic mechanisms (epigenome) as an additional dimension in gene regulation through DNA and histone modifications, chromatin remodeling, or noncoding RNAs in the absence of alterations of the DNA coding information (Cech and Steitz, 2014; Rivera and Ren, 2013). Coding genes in the transcriptome, in turn, produce a wide range of proteins (pro-teome) which are the basic functional units of cellular structures as well as metabolic, biochemical and signaling pathways. Proteins involved in metabolic reactions are responsible for producing various metabolites (metabolome). Many proteins, such as DNA/RNA binding proteins, transcription factors, splicing factors, DNA methyltransferases, and histone deacetylases, are also essential regulators of transcriptional and translational processes. In addition to these molecular entities belong to the host, gut microbiome residing in host digestive tract has recently been recognized as our second genome. Gut microbiome has been found to modulate host metabolism and influence the metabolome, which can in turn modify the epigenome to regulate gene expression.
Genome-wide approaches enabling the capturing of multi-dimensional signals involved with the action of diet can help derive a systems-level landscape of the molecular underpinnings of the broad effects of diet on the brain. We define such diet or nutrition related omics studies “nutri-omics”. However, most of current dietary studies involving omics have targeted peripheral tissues such as liver, adipose, pancreas, and muscle, and very few studies have investigated the effects of foods on the brain (Zhao et al., 2015). In the sections below, we introduce the concepts of individual nutri-omics and highlight the corresponding recent application examples that utilize these concepts to study the dietary effect on the brain.
4.2. Nutrigenetics
Individuals can respond to nutrients or diets differently depending on the genetic background. Nutrigenetics investigates how nutrients or diets interact with genetic variations in individual genomes to impose personalized dietary response (Ioannidis et al., 2011). The term genetic variation is generally used to describe DNA sequence differences between individuals in a population. DNA sequence variations include single nucleotide polymorphisms (SNPs), insertions, deletions, copy number variations (CNVs), and inversions, with the most common be-DNA variations can be correlated with dietary responses across individuals to identify which genetic variants are associated with differential response to a diet across the population. For instance, individuals with the ApoE-ε4 genetic polymorphism, an established genetic risk for Alzheimer’s disease, show little protection against AD when consuming fish (Plourde et al., 2009) based on decreased half-life and enhanced β- oxidation of ingested DHA (Chouinard-Watkins et al., 2013).
Of note, when genome-wide analysis is conducted, that is, examining the interactions between millions of genetic variants with a particular diet or nutrient, the term “nutrigenomics” is more appropriate because strictly speaking, “genomics’ is used to define studies of DNA sequences at global scale. However, the term “nutrigenomics’ has been traditionally reserved to describe studies of the nutritional effect on gene expression at messenger RNA level (see next section). In addition, the majority of the genetic studies of nutritional response are candidate gene based, rather than genome-wide, and replication of findings has been difficult (Ioannidis et al., 2011). For these reasons, we retain the use of “nutrigenetics’ in the current review as traditionally referred to. Future global nutrigenetic studies examining the relationship between the larger numbers of genetic variations with dietary response are warranted to offer a more comprehensive understanding regarding how gene by diet interactions modulate the balance between health and disease. The information on which genetic variants determine beneficial or deleterious dietary responses in terms of disease risks from such studies are critical for designing personalized nutritional remedies to prevent or reverse diseases.
4.3. Nutritranscriptomics (Fig. 6)
Fig. 6.
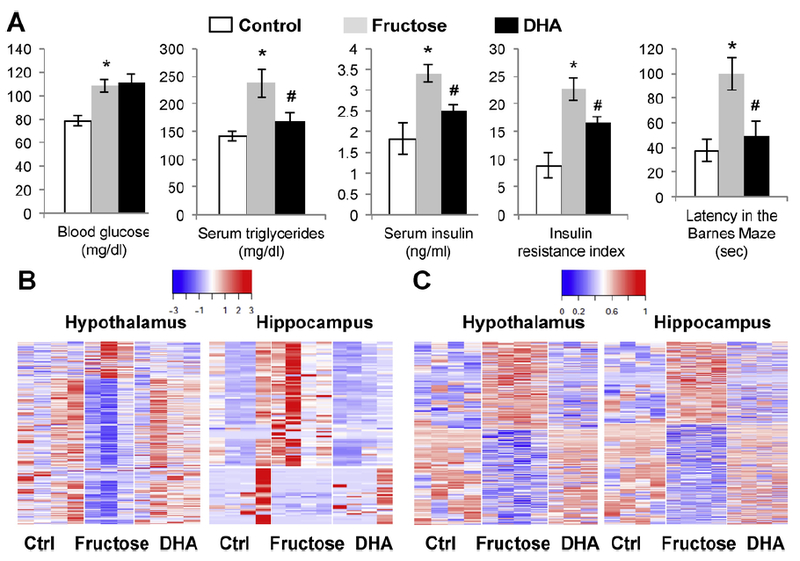
Changes in metabolic and behavior phenotypes, transcriptome, DNA methylome, and biological pathways in response to fructose treatment and supplementation with the omega- 3 fatty acid DHA, according to (Meng et al., 2016). (A) Metabolic and behavior phenotypes. From left to right: blood glucose, serum triglycerides, serum insulin, insulin resistance index, latency time in the Barnes Maze test. Fructose group was compared with the control group, and the DHA + fructose group (DHA) was compared to the fructose only group. *p < 0.01 and #p < 0.05 by 2-sided Student’s t-test. Error bars in the plots are standard errors (n = 8/group. (B) Heatmap of gene expression changes in hypothalamus and hippocampus. Blue to red colors indicate low to high expression values. (C) Heatmap of DNA methylation changes in hypothalamus and hippocampus. Blue to red colors indicate low to high methylation levels.
Diets or nutrients can also alter the transcriptional activities of genes, that is, the convertion of DNA to messenger RNAs (mRNAs), by modulating the expression levels of genes or alterative splicing, which can then further induce physiological or pathophysiological effects. Transcriptomics studies that measure >20,000 expressed genes or transcripts in response to nutritional or dietary challenges are the most abundant type of omics studies in nutritional systems biology (Capozzi and Bordoni, 2013). Such studies have been termed “nutrigenomics” in the past. Here we adopt the term “nutritranscriptomics’ to more accurately describe the molecular entities under investigation being mRNA transcription, rather than DNA sequences as examined in a genetic or genomic study. In a nutritranscriptomic study, the transcriptional activities of individual genes and transcripts can be measured with either microarray technologies or more recently RNA sequencing approaches, and then correlated with dietary information to identify all genes whose activities are influenced by a diet.
One of the first demonstrations about the contribution of nutritran-scriptomics to nutritional neuroscience is highlighted by a recent study showing the influence of fructose and DHA on gene regulatory mechanisms in the brain (Fig. 6) (Meng et al., 2016). Fructose consumption has become a major risk factor for a broad spectrum of complex metabolic disorders such as obesity, diabetes, and cardiovascular diseases, and its impact on the pathology of neurological and psychiatric disorders is starting to be recognized. The application of nutritranscrip tomics revealed that fructose and DHA each affects hundreds of genes by altering expression levels or modulating alternativesplicing and elicit opposite actions on gene regulatory mechanisms in the hypothalamus and hippocampus brain regions in rodents. The transcriptional alterations carried by fructose were shown to converge with genes posing genetic risks of metabolic and neuropsychiatric disorders uncovered by human genome-wide association studies. More interestingly, fructose and DHA elicit opposite actions on gene regulatory mechanisms, posing the potential to engage DHA supplementation to counteract the deleterious effects of fructose on the brain.
4.4. Nutriepigenomics (Fig. 6)
As discussed previously, there has been tremendous progress in the last few years about the mechanisms through which environmental exposures are “remembered”, and how they may be accountable for dysfunctions observed in various brain disorders (Nestler, 2009; Nestler et al., 2016; Tshala-Katumbay et al., 2015). The epigenome is dynamic and responsive to environmental stimuli, such that it is becoming known as a main intermediate mechanism by which dietary factors can affect long-term gene expression. As discussed earlier, various epigenetic mechanisms including DNA methylation, histone modification, chromatin remodeling, and noncoding RNAs are all critical transcriptional regulators (Holoch and Moazed, 2015). Recent technological progresses have enabled global epigenetic measurements using array- or sequencing-based methodologies (Morozova and Marra, 2008; Pritchard et al., 2012; Telese et al., 2013; Zhao et al., 2015). The individual epigenomic signals can be associated with dietary treatment information to detect which epigenetic events are perturbed by a particular diet.
Our recent nutriepigenomics study strongly implicated extensive epigenetic reprogramming, far beyond what was explored before based on individual genes such as Bdnf to be involved in the dietary effects on the brain (Meng et al., 2016). Based on DNA methylome profiling of millions of methylation sites via next generation sequencing, fructose diet was found to perturb thousands of DNA methylation sites in both the hypothalamus and hippocampus of rodent brain, which was normalized by the omega-3 fatty acid DHA supplementation. Many of the differentially methylated DNA sites by fructose and DHA co-localized with a subset of genes whose expression was affected by fructose, indicating the cis-regulatory role of DNA methylation in gene transcription. In addition, transregulatory mechanism of DNA methylation was supported by the co-localization of differential methylation sites with a number of transcriptional regulators such as transcription factors and splicing factors and microRNAs (e.g., rno-miR-421, rno-miR-143) whose predicted target genes were amongst the fructose-affected genes. As an increasing body of evidence indicates that neurological and psychiatric disorders have an epigenetic component, these results support that diet has the capacity to regulate the susceptibility to disease by modulating the global epigenetic regulatory machinery, rather than a limited number of epigenetic sites.
4.5. Nutriproteomics and nutrimetabolomics
The functional products of transcriptomic and epigenomic alterations in response to nutrients are reflected in proteins and metabolites. The past few year have seen major progresses in high throughput proteomics and metabolomics technologies (Breker and Schuldiner, 2014; Gibbons et al., 2015; Sauer and Luge, 2015), greatly expanding the informative molecular dimensions when monitoring the effects of nutrition. Now it is possible to measure the abundance and post-trans lational modifications of hundreds of proteins as well as hundreds or thousands of metabolites to capture key pathways affected by nutritional or dietary modulation (Gibbons et al., 2015).
Recent applications of nutriproteomics in neuroscience have revealed that proteins involved in mitochondrial complex, tricarboxylic acid cycle, heat shock, fatty acid utilization are altered in diabetic sensory neurons (Akude et al., 2011), and are associated mitochondrial activity in mouse neurons (Chowdhury et al., 2011). Whole brain proteomics analysis of rats receiving a HFD revealed proteins involved in mitochondria function, oxidative phosphorylation, calcium homeostasis hypoxia inducible factor (HIF) signaling, glycolysis/gluconeogenesis, and cytoskeleton to be affected by HFD (Smine et al., 2017). Interestingly, supplementation with a grape seed and skin extract (GSSE) in the HFD animals reversed the changes in many proteins involved in ox idative phosphorylation, calcium signaling, HIF signaling and glycolysis/gluconeogenesis pathways, supporting a protective role of GSSE in counteracting the HFD effects (Smine et al., 2017). Other processes affected by GSSE included FOXO signaling pathway, chemokine immunity signaling and platelet activation signals. In terms of the nu trimetabolomics, existing applications are largely limited to the peripheral such as plasma, urine, and liver tissues (Llorach et al., 2012; Vasilopoulou et al., 2016; Zhao et al., 2015), and future brain-specific profiling are warranted to increase comprehension of the effects of diet on brain metabolism.
4.6. Nutrimicrobiomics
The intestinal microbiome has profound effects on whole body physiology by regulating metabolism of fatty acids and vitamins and by contributing to host immune homeostasis. As such, it has the capacity to serve as an essential mediator of the effects of nutrients on the pathophysiology of various organ systems. An increasing number of studies indicate that the microbiome acts as a pivotal station for the interaction between gut, immune system, and brain (Fung et al., 2017). It is starting to be understood that the action of the microbiome is important for the regulation of important aspects of brain development, adult plasticity, and resistance to disease via modulation of the immune systems (Sandhu et al., 2017). As the immune system is an important regulator of brain function including inflammation, response to damage, and behavior, the fact that gut microbes influence peripheral immune cells and whole immune response has provided the mechanistic link between the action of microbiome and several aspects of brain function. It is now understood that the microbiota affects the maturation and function of brain cells such as microglia and astrocytes and their sub-products actions on inflammation, neuronal function and signaling (Fung et al., 2017). Dietary composition, and caloric intake appear to strongly and swiftly regulate microbial composition and function, making the microbiota-gut-brain communication a crucial element to understand the action of nutrition and diet on brain function and resistance to diseases.
As with the cases of other omics, microbiome sequencing technologies including 16S rDNA sequencing and metagenomics are enabling high throughput studies of dietary effects on gut microbiota as well as metabolic and brain functions. In particular, high caloric diets, high fiber diets, and food additives have shown profound impact on the gut microbiota. For instance, protein and fat were found to promote Bac-teroides, Alistipes and Parabacteroides, while long-term carbohydrate consumption favors Prevotella, Paraprevotella and Catenibacterium in human subjects (Wu et al., 2011). The specific microbiota altered by diets, in turn, can influence food digestion and nutrient absorption and metabolism, such that obesity-associated bacteria promote energy harvest and inflammation (Hartstra et al., 2015). Butyrate-producing bacteria which produce small chain fatty acids such as butyrate appear to have protective effects on metabolic and brain disorders. Butyrate can enhance mitochondrial activity, prevent metabolic endotoxemia, activate intestinal gluconeogenesis, activate G protein coupled receptors, interact with histone deacetylases to regulate gene expression, and promote microbiome homeostasis (Bourassa et al., 2016; Hartstra et al., 2015). The ability of diets to influence gut microbiota composition and metabolites can be harnessed to explore potential therapeutic avenues. Indeed, gut bacteria and metabolite modulations through oral administration of butyrate or fecal transplantation can reverse metabolic perturbations seen in metabolic syndrome (Chassaing et al., 2015; Gao et al., 2009; Suez et al., 2014; Vrieze et al., 2012) and enhance brain plasticity in neurological disease models ranging from depression and autism to neurodegenerative diseases and cognitive impairment (Bourassa et al., 2016; Stilling et al., 2016). High fiber diet, which promotes butyrate-producing bacteria, has been proposed as a potential remedy to promote mental health (Bourassa et al., 2016).
4.7. Application of nutritional systems biology to clinical neuroscience (Fig. 7)
Fig. 7.
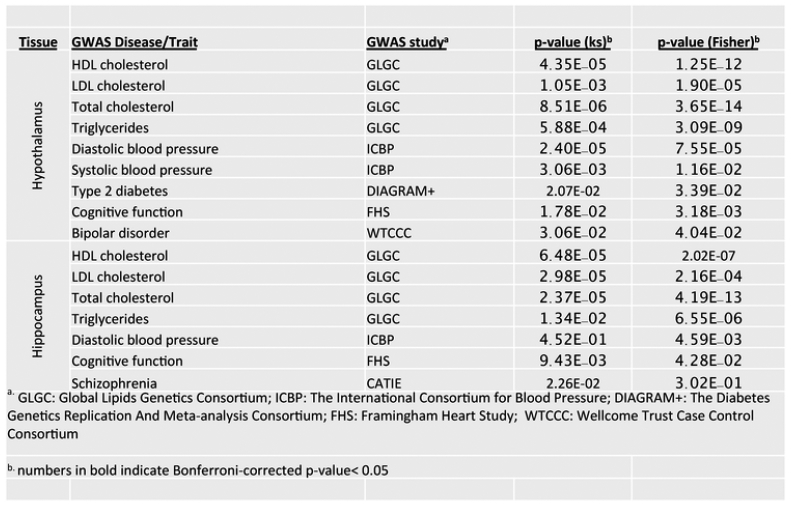
Relevance of fructose signatures and key driver genes (KDs) to human diseases, according to (Meng et al., 2016). To assess the relevance of nutrigenomic signals from the rodent model to human pathophysiology, the fructose signature genes as well as the KDs were compared to human GWAS of metabolic disorders and brain related diseases or traits. Numerous overlapping genes were found between fructose signature genes and top GWAS hits for a broad range of brain disorders (e.g., Alzheimer’s disease, attention deficient hyperactive disorder, depression, addiction, Parkinsons disease) and MetDs-related diseases or traits (e.g., blood pressure, cardiovascular disease, lipids.
It is becoming an alarming concern that metabolic disorders can compromise brain function and precipitate to neurological disorders. Most of the existing approaches to understanding the impact of metabolism on the brain have been focused on the action of a few genes and proteins or examining individual types of molecular information in individual tissues one at a time, which hardly explain the whole dimension of the pathology. Our group has recently applied the nutritional system biology concepts described above to combine dietary treatment, genome-wide transcriptome, and DNA methylome analyses to extract information about crucial gene regulatory mechanisms (expression level, alternative splicing, epigenetic regulation) underlying the action of nutrients on multiple brain regions, setting the start of the systems nutrigenomics field in neuroscience (Meng et al., 2016).
Information about the intersection between rodent modeling with human genome-wide association studies (GWAS) of brain disorders is useful to infer translatability to human pathophysiology. For example, the system biology approach allows to compare signature genes activated by fructose as well as the key driver genes (KDs) identified from hypothalamus and hippocampus to large-scale human GWAS of metabolic disorders and brain related diseases or traits (Meng et al., 2016). Numerous overlapping genes between fructose signature genes and top GWAS hits were found for a broad range of brain disorders (e.g., Alzheimer’s disease, attention deficient hyperactive disorder, depression, addiction, Parkinsons disease) and metabolic-related diseases or traits (e.g., blood pressure, cardiovascular disease, lipids, obesity, and T2D). We found that human orthologs of the fructose signature genes from the hypothalamus and the hippocampus are highly enriched for genetic polymorphisms associated with many of these diseases or traits (Fig. 7). As the human GWAS genes are based on genetic evidence and imply causal information, these results support that the hypothalamus and hippocampus genes perturbed by metabolic fructose treatment in the rodent model likely play causal roles in both metabolic and brain disorders in humans. As human GWAS studies identify causal genes for a variety of human diseases, the convergence between human disease causal genes and those affected by nutrition or diet has raised the exciting possibility that the effects of foods and metabolic alterations can be saved in the organism in the form of genomic and epigenomic modifications that can predict disease susceptibility. Of course, comprehensive characterization of additional layers of molecular information such as genetic variations, proteins, metabolites, and gut microbiome are needed to further complement the existing studies.
5. Consequences and considerations
5.1. Changes in environmental conditions: the need for lifestyle intervention
Globally, the prevalence of obesity in adults is now estimated to be about 36%, which is an 8% increase from 1980 (Trushnikova et al., 2014). Metabolic disorders such as T2D rank amongst the fastest growing health problems worldwide (Padwal, 2014; Verma and Hussain, 2017). In recent years, metabolic disorders have demonstrated a tendency to manifest earlier and earlier in life and interestingly, at a higher rate in women (Attig et al., 2013). Particularly disconcerting is the rise of T2D in the child/adolescent population, even in children as young as 8 years of age (Libman and Arslanian, 1999). These findings have critical implications in light of the growing body of evidence demonstrating the establishment of the epigenome can be affected during critical developmental periods (Attig et al., 2013). In particular, studies have shown that the quality of a mother s nutrition during her childhood years can influence her childrens risk for developing cardiovascular disease, T2D, and hypertension in adulthood (Attig et al., 2010, 2013; Dyer and Rosenfeld, 2011). Moreover, the nutritional condition of the grandmother during her pregnancy can influence the mothers nutrition in utero, which in turn can influence the grandchild’s birth weight (Heijmans et al., 2008). Particularly, epigenetic modifications during the perinatal period may have a powerful ability to induce persistent changes in behavioral adaptations related to energy metabolism and the availability of dietary methyl donors (Attig et al., 2013; Dominguez-Salas et al., 2014; Tyagi et al., 2015; Waterland, 2014). Increasing evidence for epigenetic effects at the level of the cell, organism, and family lineage, warrant considering the implications and consequences of the metabolic epidemic, i.e., that the increased rate of metabolic diseases in women may have detrimental and long-lasting consequences in their offspring and possibly function to perpetuate the prevalence of metabolic syndromes world-wide.
Human habits are deeply rooted to culture and society such that the increase in automation in the contemporary society is strongly linked to deviations in traditional dietary habits in conjunction with a paucity of the practice of physical activity. In particular, conventional dietary routines based on the feasting of fish, lean meat, vegetables, fruits, and whole grains have become gradually superseded by processed foods including refined grains and sugars, fried meals, and fatty meats. The increased pace of life secondary to industrialization has resulted in the rise of fast food restaurants and consumption of “junk food/’ Excessive energy intake conjoint with a sedentary lifestyle poses a risk for developing late-life cognitive impairment, including AD (Janson et al., 2004; Malek-Ahmadi et al., 2013; Stranahan et al., 2008). Moreover, studies in humans show a strong association between western diet and the risk of mood disorders such as depression and anxiety (Jacka et al., 2010).
5.2. Advances in technologies: the need for nutritional systems biology
There is an urgent need for nutritional systems biology to delineate the effects of dietary manipulations at the various levels of information processing from gene transcripts, proteins, and metabolites, to the microbiome and epigenomics (responsible for mediating environmental responses). The remarkable technological advances we are experiencing today offer the ideal incubator for such systems level mechanistic studies to better understand the actions of diet and nutrition on diverse molecules and pathways in individual cells and tissues in a dynamic manner. When coupled with similar systems biology studies which delineate key disease causal molecular events and pathways, as implemented in the iPOP project (Li-Pook-Than and Snyder, 2013), it is possible to predict the consequences of various lifestyle and dietary conditions for early diagnosis and disease prevention. An added advantage is that the system biology information processing is based on molecular mechanistic information rather than traditional symptom-based approaches. Emerging evidence from gene-lifestyle interaction studies of brain disorders supports that life style modifications can counteract the biological effects of genetic predisposition (Temelkova-Kurktschiev and Stefanov, 2012). Nutritional systems biology promises comprehensive molecular signature maps of dietary components posing neurological and psychiatric risks as well as those with the potential to normalize disease risks.
6. Concluding remarks
The rise in consumption of high caloric foods has triggered an epidemic of metabolic disorders (Haffner and Taegtmeyer, 2003; Lutsey et al., 2008) such that the number of obese, diabetic and pre-diabetic persons in the U.S. is now estimated at over 40% of the population (Cowie et al., 2009). Epidemiological studies have attributed a majority of obesity, T2D, and MetS cases to major lifestyle factors such as unhealthy diet and physical inactivity and subsequent toll in brain function (Gomez-Pinilla, 2008). T2D is more prevalent in populations consuming the so-called western or conservative diet, which is high in carbohydrates (from refined grains and sugar), red meat, and saturated fat. On the other hand, dietary interventions involving increased polyunsaturated fat and fiber reduce T2D risk (Agrawal and Gomez- Pinilla, 2012; Gomez-Pinilla and Tyagi, 2013; Lim et al., 2005; Meng et al., 2016). It is becoming understood that diabetes and other metabolic disorders such as obesity increase the vulnerability to neurological disorders (Gomez-Pinilla, 2008; Newcomer, 2007). These associations are not surprising given that brain operation is heavily dependent on accuracy of mechanisms that manage supply of functional energy to cells. Metabolic syndrome is a risk factor for several neurological disorders such as brain trauma, stroke, Alzheimer s disease, and Huntington s disease, all of which have a metabolic component (Farooqui et al., 2011; Freiherr et al., 2013; Malek-Ahmadi et al., 2013).
Accumulating evidence points out that the remarkable capacity of the brain for cognitive plasticity is heavily rooted in a working connection between the gene regulation and cell metabolism. The advent of nutritional system biology offers the unique opportunity to integrate a multitude of molecular variables to have a thorough view of the broad impact of diet on the brain. The results from system nutrigenomic research is starting to reveal the power of diet and cell metabolism to modify the program of genes with subsequent implications for brain pathogenesis. Lifestyle factors that intrinsically alter cell energy metabolism, such as exercise and diet, are particularly influential on the epigenome. Diet and exercise engage epigenetic mechanisms to promote stable modifications of the broader realm of molecular systems that regulate synaptic plasticity and behavior. Epigenetic information which is potentially heritable has many implications for evolution and the etiology of diseases, including coping behaviors and psychological disturbances. In harmony with their strong adaptive force, dietary factors and exercise are surfacing as critical promotors of biological adjustments that can tip the balance between health and disease. Procurement of system level information concern-ing the action of diet on brain is insightful for the design of public health initiatives to prevent or redirect the course of the epidemic of metabolic and brain disorders.
Acknowledgements
We thank Zhe Ying for assistance with the illustrations. FGP is supported by National Institute of Health grants R01NS050465, R21N-S103088, and R01DK104363. XY is supported by National Institute of Health grant R01DK104363, R21NS103088, American Heart Association Scientist Development Grant 13SDG17290032, Leducq Foundation, Hellman Fellows Award.
Abbreviations:
- BDNF
brain-derivedneurotrophicfactor
- DHA
docosahexaenoic
- HFD
high fat diet
- HIF
hypoxia inducible factor
- MetS
metabolic syndrome
- POMC
pro-opiodmelanocortin
- T2D
type 2 diabetes
References
- Li-Pook-Than’ J., Snyder M, 2013. iPOP goes the world: integrated personalized Omics profiling and the road toward improved health care. Chem. Biol 20, 660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal R, Gomez-Pinilla F, 2012. ‘Metabolic syndrome in the brain: deficiency in omega-3 fatty acid exacerbates dysfunctions in insulin receptor signalling and cogni tion. J. Physiol. (Paris) 590, 2485–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal R, Tyagi E, Shukla R, Nath C, 2009. A study of brain insulin receptors, AChE activity and oxidative stress in rat model of ICV STZ induced dementia. Neuropharmacology 56, 779–787. [DOI] [PubMed] [Google Scholar]
- Akude E, Zherebitskaya E, Chowdhury SK, Smith DR, Dobrowsky RT, Ferny-hough P, 2011. Diminished superoxide generation is associated with respiratory chain dysfunction and changes in the mitochondrial proteome of sensory neurons from diabetic rats. Diabetes 60, 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albuquerque D, Stice E, Rodríguez-López R, Manco L, Nóbrega C, 2015. Current review of genetics of human obesity: from molecular mechanisms to an evolutionary perspective. Mol. Genet. Genomics 290, 1191–1221. [DOI] [PubMed] [Google Scholar]
- Alonso M, Vianna MR, Izquierdo I, Medina JH, 2002. Signaling mechanisms mediating BDNF modulation of memory formation in vivo in the hippocampus. Cell. Mol. Neurobiol 22, 663–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RJ, Freedland KE, Clouse RE, Lustman PJ, 2001. The prevalence of co-morbid depression in adults with diabetes: a meta-analysis. Diabetes Care 24, 1069–1078. [DOI] [PubMed] [Google Scholar]
- Attig L, Gabory A, Junien C, 2010. Early nutrition and epigenetic programming: chasing shadows. Curr. Opin. Clin. Nutr. Metab. Care 13, 284–293. [DOI] [PubMed] [Google Scholar]
- Attig L, Vigé A, Gabory A, Karimi M, Beauger A, Gross MS, Athias A, Gallou-Ka-bani C, Gambert P, Ekstrom TJ, Jais JP, Junien C, 2013. Dietary alleviation of maternal obesity and diabetes: increased resistance to diet-induced obesity transcrip tional and epigenetic signatures. PLoS One 8, e66816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barua S, Kuizon S, Junaid MA, 2014. Folic acid supplementation in pregnancy and implications in health and disease. J. Biomed. Sci 21, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito E, Barco A, 2010. CREB’s control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci 33, 230–240. [DOI] [PubMed] [Google Scholar]
- Bhatia HS, Agrawal R, Sharma S, Huo YX, Ying Z, Gomez-Pinilla F, 2011. Omega-3 fatty acid deficiency during brain maturation reduces neuronal and behavioral plasticity in adulthood. PLoS One 6, e28451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenhäger TK, Geldermans S, Van den Broek WW, van Beveren N, Fekkes D, 2012. Serum brain-derived neurotrophic factor level in relation to illness severity and episode duration in patients with major depression. J. Psychiatr. Res 46, 285–289. [DOI] [PubMed] [Google Scholar]
- Bourassa MW, Alim I, Bultman SJ, Ratan RR, 2016. Butyrate, neuroepigenetics and the gut microbiome: can a high fiber diet improve brain health?. Neurosci. Lett 625, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breker M, Schuldiner M, 2014. The emergence of proteome-wide technologies: systematic analysis of proteins comes of age. Nat. Rev. Mol. Cell Biol 15, 453–464. [DOI] [PubMed] [Google Scholar]
- Brown AS, Susser ES, 2008. Prenatal nutritional deficiency and risk of adult schizophrenia. Schizophr. Bull 34, 1054–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner R, Scholmerich J, Bollheimer LC, 2007. High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity (Silver Spring) 15, 798–808. [DOI] [PubMed] [Google Scholar]
- Bygren LO, Kaati G, Edvinsson S, 2001. Longevity determined by paternal ancestors’ nutrition during their slow growth period. Acta Biotheor. 49, 53–59. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S, 2010. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 107, 22687–22692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capozzi F, Bordoni A, 2013. Foodomics: a new comprehensive approach to food and nutrition. Genes Nutr 8, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraci F, Copani A, Nicoletti F, Drago F, 2018. Depression and Alzheimer’s disease: neurobiological links and common pharmacological targets. Eur. J. Pharmacol 626, 64–71. [DOI] [PubMed] [Google Scholar]
- Carlberg L, Scheibelreiter J, Hassler MR, Schloegelhofer M, Schmoeger M, Ludwig B, Kasper S, Aschauer H, Egger G, Schosser A, 2014. Brain-derived neu-rotrophic factor (BDNF)-epigenetic regulation in unipolar and bipolar affective disor der. J. Affect. Disord 168, 399–406. [DOI] [PubMed] [Google Scholar]
- Cech TR, Steitz JA, 2014. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157, 77–94. [DOI] [PubMed] [Google Scholar]
- Champagne FA, 2016. Epigenetic legacy of parental experiences: dynamic and interactive pathways to inheritance. Dev. Psychopathol 28, 1219–1228. [DOI] [PubMed] [Google Scholar]
- Chan PH, 2005. Mitochondrial dysfunction and oxidative stress as determinants of cell death/survival in stroke. Ann. N. Y. Acad. Sci 1042, 203–209. [DOI] [PubMed] [Google Scholar]
- Chassaing B, Koren O, Goodrich JK, Poole AC, Srinivasan S, Ley RE, Gewirtz AT, 2015. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 519, 92–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TJ, Wang DC, Chen SS, 2009. Amyloid-beta interrupts the PI3K-Akt-mTOR signaling pathway that could be involved in brain-derived neurotrophic factor-in duced Arc expression in rat cortical neurons. J. Neurosci. Res 87, 2297–2307. [DOI] [PubMed] [Google Scholar]
- Cheng G, Kong RH, Zhang LM, Zhang JN, 2012. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol 167, 699–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho CE, Sánchez-Hernández D, Reza-López SA, Huot PS, Kim YI, Anderson GH, 2013. High folate gestational and post-weaning diets alter hypothalamic feed ing pathways by DNA methylation in Wistar rat offspring. Epigenetics 8, 710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SW, Friso S, 2010. Epigenetics: a new bridge between nutrition and health. Adv. Nutr 1, 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SW, Claycombe KJ, Martinez JA, Friso S, Schalinske KL, 2013. Nutritional epigenomics: a portal to disease prevention. Adv. Nutr 4, 530–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouinard-Watkins R, Rioux-Perreault C, Fortier M, Tremblay-Mercier J, Zhang Y, Lawrence P, Vohl MC, Perron P, Lorrain D, Brenna JT, Cunnane SC, Plourde M, 2013. Disturbance in uniformly 13C-labelled DHA metabolism in elderly human subjects carrying the apoE epsilon4 allele. Br. J. Nutr 110, 1751–1759. [DOI] [PubMed] [Google Scholar]
- Chouliaras L, Rutten BP, Kenis G, Peerbooms O, Visser PJ, Verhey F, van Os J, Steinbusch HW, van den Hove DL, 2010. Epigenetic regulation in the pathophysi ology of Alzheimer s disease. Prog. Neurobiol 90, 498–510. [DOI] [PubMed] [Google Scholar]
- Chowdhury SK, Dobrowsky RT, Fernyhough P, 2011. Nutrient excess and altered mitochondrial proteome and function contribute to neurodegeneration in diabetes. Mitochondrion 11, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisternas P, Salazar P, Serrano FG, Montecinos-Oliva C, Arredondo SB, Varela-Nallar L, Barja S, Vio CP, Gomez-Pinilla F, Inestrosa NC, 2015. Fructose con sumption reduces hippocampal synaptic plasticity underlying cognitive performance. Biochim. Biophys. Acta 1852, 2379–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman CW, 2005. The role of neurotrophins in brain aging: a perspective in honor of Regino Perez-Polo. Neurochem. Res 30, 877–881. [DOI] [PubMed] [Google Scholar]
- Cowie CC, Rust KF, Ford ES, Eberhardt MS, Byrd-Holt DD, Li C, Williams DE, Gregg EW, Bainbridge KE, Saydah SH, Geiss LS, 2009. Full accounting of dia betes and pre-diabetes in the U.S. population in 1988–1994 and 2005–2006. Diabetes Care 32, 287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf AA, Freidig AP, De Roos B, Jamshidi N, Heinemann M, Rullmann JA, Hall KD, Adiels M, van Ommen B, 2009. Nutritional systems biology modeling: from molecular mechanisms to physiology. PLoS Comput. Biol 5, e1000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker MJ, Su Q, Baker C, Rutledge AC, Adeli K, 2010. Fructose: a highly li- pogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab 299, E685–694. [DOI] [PubMed] [Google Scholar]
- Dhingra R, Sullivan L, Jacques PF, Wang TJ, Fox CS, Meigs JB, DAgostino RB, Gaziano JM, Vasan RS, 2007. Soft drink consumption and risk of developing cardiometabolic risk factors and the metabolic syndrome in middle-aged adults in the community. Circulation 116, 480–488. [DOI] [PubMed] [Google Scholar]
- Dixon L, Weiden P, Delahanty J, Goldberg R, Postrado L, Lucksted A, Lehman A, 2000. Prevalence and correlates of diabetes in national schizophrenia samples. Schiz ophr. Bull 26, 903–912. [DOI] [PubMed] [Google Scholar]
- Dominguez-Salas P, Moore SE, Baker MS, Bergen AW, Cox SE, Dyer RA, Ful ford AJ, Guan Y, Laritsky E, Silver MJ, Swan GE, Zeisel SH, Innis SM, Waterland RA, Prentice AM, Hennig BJ, 2014. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat. Commun 5, 3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Ma M, Zhao Q, Fang L, Chang J, Wang Y, Fei R, Song X, 2013. Mitochondrial bioenergetic deficits in the hippocampi of rats with chronic ischemia-induced vascular dementia. Neuroscience 231, 345–352. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, 2009. Brain-derived neurotrophic factor: role in depression and suicide. Neuropsychiatr. Dis. Treat 5, 433–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer JS, Rosenfeld CR, 2011. Metabolic imprinting by prenatal, perinatal, and postnatal overnutrition: a review. Semin. Reprod. Med 29, 266–276. [DOI] [PubMed] [Google Scholar]
- Eaton WW, Armenian H, Gallo J, Pratt L, Ford DE, 1996. Depression and risk for onset of type II diabetes. A prospective population-based study. Diabetes Care 19, 1097–1102. [DOI] [PubMed] [Google Scholar]
- Edwards TM, Myers JP, 2007. Environmental exposures and gene regulation in dis ease etiology. Environ. Health Perspect 115, 1264–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqui AA, Farooqui T, Panza F, Frisardi V, 2011. Metabolic syndrome as a risk factor for neurological disorders. Cell. Mol. Life Sci 10.1007/s00018-011-0840-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr SA, Yamada KA, Butterfield DA, Abdul HM, Xu L, Miller NE, Banks WA, Morley JE, 2008. Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology 149, 2628–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson-Smith AC, Surani MA, 2001. Imprinting and the epigenetic asymmetry between parental genomes. Science 293, 1086–1089. [DOI] [PubMed] [Google Scholar]
- Filus JF, Rybakowski J, 2005. [Neurotrophic factors and their role in the pathogenesis of affective disorders]. Psychiatr. Pol 39, 883–897. [PubMed] [Google Scholar]
- Fischle W, Wang Y, Allis CD, 2003. Histone and chromatin cross-talk. Curr. Opin. Cell Biol 15, 172–183. [DOI] [PubMed] [Google Scholar]
- Flegal KM, Carroll MD, Kuczmarski RJ, Johnson CL, 1998. Overweight and obe sity in the United States: prevalence and trends, 1960–1994. Int. J. Obes. Relat. Metab. Disord 22, 39–47. [DOI] [PubMed] [Google Scholar]
- Freeman LR, Haley-Zitlin V, Rosenberger DS, Granholm AC, 2014. Damaging effects of a high-fat diet to the brain and cognition: a review of proposed mechanisms. Nutr. Neurosci 17, 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiherr J, Hallschmid M, Frey WH, Brünner YF, Chapman CD, Hölscher C, Craft S, De Felice FG, Benedict C, 2013. Intranasal insulin as a treatment for alzheimer s disease: a review of basic research and clinical evidence. CNS Drugs [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchikami M, Morinobu S, Segawa M, Okamoto Y, Yamawaki S, Ozaki N, Inoue T, Kusumi I, Koyama T, Tsuchiyama K, Terao T, 2011. DNA methylation profiles of the brain-derived neurotrophic factor (BDNF) gene as a potent diagnostic biomarker in major depression. PLoS One 6, e23881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung TC, Olson CA, Hsiao EY, 2017. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci 20, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, Cefalu WT, Ye J, 2009. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58, 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavard JA, Lustman PJ, Clouse RE, 1993. Prevalence of depression in adults with diabetes. An epidemiological evaluation. Diabetes Care 16, 1167–1178. [DOI] [PubMed] [Google Scholar]
- Gerrits PM, Tsalikian E, 1993. Diabetes and fructose metabolism. Am. J. Clin. Nutr 58, 796S–799S. [DOI] [PubMed] [Google Scholar]
- Gibbons H, O’Gorman A, Brennan L, 2015. Metabolomics as a tool in nutritional research. Curr. Opin. Lipidol 26, 30–34. [DOI] [PubMed] [Google Scholar]
- Gomez-Pinilla F, 2008. Brain foods: the effects of nutrients on brain function. Nat. Rev. Neurosci 9, 568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Tyagi E, 2013. Diet and cognition: interplay between cell metabolism and neuronal plasticity. Curr. Opin. Clin. Nutr. Metab. Care 16, 726–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Muniesa P, Mártinez-GonzáleZ’ MA, Hu FB, Després JP, Matsuzawa Y, Loos RJF, Moreno LA, Bray GA, Martinez JA, 2017. Obesity. Nat. Rev. Dis. Primers 3, 17034. [DOI] [PubMed] [Google Scholar]
- Haffner S, Taegtmeyer H, 2003. Epidemic obesity and the metabolic syndrome. Circulation 108, 1541–1545. [DOI] [PubMed] [Google Scholar]
- Hao S, Dey A, Yu X, Stranahan AM, 2016. Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav. Immun 51, 230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartstra AV, Bouter KEC, Backhed F, Nieuwdorp M, 2015. Insights into the role of the microbiome in obesity and type 2 diabetes. Diabetes Care 38, 159–165. [DOI] [PubMed] [Google Scholar]
- Hedley AA, Ogden CL, Johnson CL, Carroll MD, Curtin LR, Flegal KM, 2004. Prevalence of overweight and obesity among US children, adolescents, and adults, 1999–2002. JAMA 291, 2847–2850. [DOI] [PubMed] [Google Scholar]
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH, 2008. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. U. S. A 105, 17046–17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herculano-Houzel S, 2011. Brains matter, bodies maybe not: the case for examining neuron numbers irrespective of body size. Ann. N. Y. Acad. Sci 1225, 191–199. [DOI] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M, 2001. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 413, 179–183. [DOI] [PubMed] [Google Scholar]
- Holoch D, Moazed D, 2015. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet 16, 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer S, 2004. Causes and consequences of disturbances of cerebral glucose metabolism in sporadic Alzheimer disease: therapeutic implications. Adv. Exp. Med. Biol 541, 135–152. [DOI] [PubMed] [Google Scholar]
- Hoyer S, 2004. Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur. J. Pharmacol 490, 115–125. [DOI] [PubMed] [Google Scholar]
- Hsu CY, Liu TH, Xu J, Hogan EL, Chao J, Sun G, Tai HH, Beckman JS, Freeman BA, 1989. Arachidonic acid and its metabolites in cerebral ischemia. Ann. N. Y. Acad. Sci 559, 282–295. [DOI] [PubMed] [Google Scholar]
- Ikegame T, Bundo M, Murata Y, Kasai K, Kato T, Iwamoto K, 2013. DNA methylation of the BDNF gene and its relevance to psychiatric disorders. J. Hum. Genet 58, 434–438. [DOI] [PubMed] [Google Scholar]
- Ioannidis JPA, Tarone R, McLaughlin JK, 2011. The false-positive to false-negative ratio in epidemiologic studies. Epidemiology 22, 450–456. [DOI] [PubMed] [Google Scholar]
- Ishimoto T, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, Orlicky DJ, Cicerchi C, McMahan RH, Abdelmalek MF, Rosen HR, Jackman MR, MacLean PS, Diggle CP, Asipu A, Inaba S, Kosugi T, Sato W, Maruyama S, Sánchez-Lozada LG, Sautin YY, Hill JO, Bonthron DT, Johnson RJ, 2013. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructoki nase. Hepatology 58, 1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacka FN, Pasco JA, Mykletun A, Williams LJ, Hodge AM, OReilly SL, Nicholson GC, Kotowicz MA, Berk M, 2010. Association of western and traditional diets with depression and anxiety in women. Am. J. Psychiatry [DOI] [PubMed] [Google Scholar]
- Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC, 2004. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53, 474–481. [DOI] [PubMed] [Google Scholar]
- Jiang J, Wang W, Sun YJ, Hu M, Li F, Zhu DY, 2007. Neuroprotective effect of curcumin on focal cerebral ischemic rats by preventing blood-brain barrier damage. Eur. J. Pharmacol 561, 54–62. [DOI] [PubMed] [Google Scholar]
- Kaati G, Bygren LO, Edvinsson S, 2002. Cardiovascular and diabetes mortality determined by nutrition during parents and grandparents slow growth period. Eur. J. Hum. Genet 10, 682–688. [DOI] [PubMed] [Google Scholar]
- Kaati G, Bygren LO, Pembrey M, Sjöström’ M, 2007. Transgenerational response to nutrition, early life circumstances and longevity. Eur. J. Hum. Genet 15, 784–790. [DOI] [PubMed] [Google Scholar]
- Kang HJ, Kim JM, Lee JY, Kim SY, Bae KY, Kim SW, Shin IS, Kim HR, Shin MG, Yoon JS, 2013. BDNF promoter methylation and suicidal behavior in depressive patients. J. Affect. Disord 151, 679–685. [DOI] [PubMed] [Google Scholar]
- Kaput J, Morine M, 2012. Discovery-based nutritional systems biology: developing N- of-1 nutrigenomic research. Int. J. Vitam. Nutr. Res 82, 333–341. [DOI] [PubMed] [Google Scholar]
- Karege F, Perret G, Bondolfí G, Schwald M, Bertschy G, Aubry JM, 2002. Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res 109, 143–148. [DOI] [PubMed] [Google Scholar]
- Karege F, Bondolfí G, Gervasoni N, Schwald M, Aubry JM, Bertschy G, 2005. Low brain-derived neurotrophic factor (BDNF) levels in serum of depressed patients probably results from lowered platelet BDNF release unrelated to platelet reactivity. Biol. Psychiatry 57, 1068–1072. [DOI] [PubMed] [Google Scholar]
- Keleshian VL, Modi HR, Rapoport SI, Rao JS, 2013. Aging is associated with altered inflammatory, arachidonic acid cascade, and synaptic markers, influenced by epigenetic modifications, in the human frontal cortex. J. Neurochem 125, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Keller S, Sarchiapone M, Zarrilli F, Videtic A, Ferraro A, Carli V, Sacchetti S, Lembo F, Angiolillo A, Jovanovic N, Pisanti F, Tomaiuolo R, Monticelli A, Balazic J, Roy A, Marusic A, Cocozza S, Fusco A, Bruni CB, Castaldo G, Chiariotti L, 2010. Increased BDNF promoter methylation in the Wernicke area of suicide subjects. Arch. Gen. Psychiatry 67, 258–267. [DOI] [PubMed] [Google Scholar]
- Kelley DE, 2002. Action for health in diabetes: the look AHEAD clinical trial. Curr. Diab. Rep 2, 207–209. [DOI] [PubMed] [Google Scholar]
- Kelley GL, Allan G, Azhar S, 2004. High dietary fructose induces a hepatic stress re sponse resulting in cholesterol and lipid dysregulation. Endocrinology 145, 548–555. [DOI] [PubMed] [Google Scholar]
- Kernie SG, Liebl DJ, Parada LF, 2000. BDNF regulates eating behavior and locomo tor activity in mice. EMBO J 19, 1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Lee HP, Won SD, Park EY, Lee HY, Lee BH, Lee SW, Yoon D, Han C, Kim DJ, Choi SH, 2007. Low plasma BDNF is associated with suicidal behav ior in major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 31, 78–85. [DOI] [PubMed] [Google Scholar]
- Kim KC, Friso S, Choi SW, 2009. DNA methylation, an epigenetic mechanism con necting folate to healthy embryonic development and aging. J. Nutr. Biochem 20, 917–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krabbe KS, Nielsen AR, Krogh-Madsen R, Plomgaard P, Rasmussen P, Erikstrup C, Fischer CP, Lindegaard B, Petersen AM, Taudorf S, Secher NH, Pilegaard H, Bruunsgaard H, Pedersen BK, 2007. Brain-derived neurotrophic factor (BDNF) and type 2 diabetes. Diabetologia 50, 431–438. [DOI] [PubMed] [Google Scholar]
- Kramer AF, Hahn S, Cohen NJ, Banich MT, McAuley E, Harrison CR, Chason J, Vakil E, Bardell L, Boileau RA, Colcombe A, 1999. Ageing, fitness and neu- rocognitive function. Nature 400, 418–419. [DOI] [PubMed] [Google Scholar]
- Lachner M, O Carroll D, Rea S, Mechtler K, Jenuwein T, 2001. Methylation of his tone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410, 116–120. [DOI] [PubMed] [Google Scholar]
- Lanaspa MA, Ishimoto T, Li N, Cicerchi C, Orlicky DJ, Ruzycki P, Ruzicky P, Ri vard C, Inaba S, Roncal-Jimenez CA, Bales ES, Diggle CP, Asipu A, Petrash JM, Kosugi T, Maruyama S, Sanchez-Lozada LG, McManaman JL, Bonthron DT, Sautin YY, Johnson RJ, 2013. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Com mun 4, 2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA, 2001. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of Abeta1–42. J. Neurochem 78, 413–416. [DOI] [PubMed] [Google Scholar]
- Laurin D, Verreault R, Lindsay J, MacPherson K, Rockwood K, 2001. Physical activity and risk of cognitive impairment and dementia in elderly persons. Arch. Neu rol 58, 498–504. [DOI] [PubMed] [Google Scholar]
- Lee CC, Huang CC, Wu MY, Hsu KS, 2005. Insulin stimulates postsynaptic density- 95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of ra pamycin signaling pathway. J. Biol. Chem 280, 18543–18550. [DOI] [PubMed] [Google Scholar]
- Libman I, Arslanian SA, 1999. Type II diabetes mellitus: no longer just adults. Pediatr. Ann 28, 589–593. [DOI] [PubMed] [Google Scholar]
- Lilliker SL, 1980. Prevalence of diabetes in a manic-depressive population. Compr. Psy chiatry 21, 270–275. [DOI] [PubMed] [Google Scholar]
- Lim GP, Calon F, Morihara T, Yang F, Teter B, Ubeda O, Salem N Jr, Frautschy SA, Cole GM, 2005. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J. Neurosci 25, 3032–3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorach R, Garcia-Aloy M, Tulipani S, Vazquez-Fresno R, Andres-Lacueva C, 2012. Nutrimetabolomic strategies to develop new biomarkers of intake and health effects. J. Agric. Food Chem 60, 8797–8808. [DOI] [PubMed] [Google Scholar]
- Lopez JP, Mamdani F, Labonte B, Beaulieu MM, Yang JP, Berlim MT, Ernst C, Turecki G, 2013. Epigenetic regulation of BDNF expression according to antidepres sant response. Mol. Psychiatry 18, 398–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucock M, 2000. Folic acid: nutritional biochemistry, molecular biology, and role in dis ease processes. Mol. Genet. Metab 71, 121–138. [DOI] [PubMed] [Google Scholar]
- Lutsey PL, Steffen LM, Stevens J, 2008. Dietary intake and the development of the metabolic syndrome: the Atherosclerosis Risk in Communities study. Circulation 117, 754–761. [DOI] [PubMed] [Google Scholar]
- Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L, 1999. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc. Natl. Acad. Sci. U. S. A 96, 15239–15244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyssiotis CA, Cantley LC, 2013. Metabolic syndrome: F stands for fructose and fat. Na ture 502, 181–182. [DOI] [PubMed] [Google Scholar]
- Malek-Ahmadi M, Beach T, Obradov A, Sue L, Belden C, Davis K, Walker DG, Lue L, Adem A, Sabbagh MN,. Type 2 diabetes is associated with increased alzheimer’s disease ncarriersCurr. Alzheimer Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP, 1997. A role for 4- hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J. Neurochem 68, 255–264. [DOI] [PubMed] [Google Scholar]
- Mattson MP, 2012. Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metab 16, 706–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier-Ruge W, Bertoni-Freddari C, Iwangoff P, 1994. Changes in brain glucose metabolism as a key to the pathogenesis of Alzheimer’s disease. Gerontology 40, 246–252. [DOI] [PubMed] [Google Scholar]
- Meng Q, Ying Z, Noble E, Zhao Y, Agrawal R, Mikhail A, Zhuang Y, Tyagi E, Zhang Q, Lee JH, Morselli M, Orozco L, Guo W, Kilts TM, Zhu J, Zhang B, Pellegrini M, Xiao X, Young MF, Gomez-Pinilla F, Yang X, 2016. Systems nu- trigenomics reveals brain gene networks linking metabolic and brain disorders. EBio Medicine 7, 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP, 2001. The continuing epidemics of obesity and diabetes in the United States. JAMA 286, 1195–1200. [DOI] [PubMed] [Google Scholar]
- Molendijk ML, Bus BA, Spinhoven P, Penninx BW, Kenis G, Prickaerts J, Voshaar RC, Elzinga BM, 2011. Serum levels of brain-derived neurotrophic factor in major depressive disorder: state-trait issues, clinical features and pharmacological treatment. Mol. Psychiatry 16, 1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molteni R, Barnard RJ, Ying Z, Roberts CK, Gomez-Pinilla F, 2002. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience 112, 803–814. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC, 2003. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Morozova O, Marra MA, 2008. Applications of next-generation sequencing technologies in functional genomics. Genomics 92, 255–264. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Decina P, Bocola V, Saraceni F, Scapicchio PL, 1996. Diabetes melli- tus in schizophrenic patients. Compr. Psychiatry 37, 68–73. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Mateling M, Kovacs I, Wang L, Eggert S, Rockenstein E, Koo EH, Masliah E, Tuszynski MH, 2013. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J. Neurosci 33, 15596–15602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, 2009. Epigenetic mechanisms in psychiatry. Biol. Psychiatry 65, 189–190. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Peña CJ, Kundakovic M, Mitchell A, Akbarian S, 2016. Epigenetic basis of mental illness. Neuroscientist 22, 447–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomer JW, 2007. Metabolic syndrome and mental illness. Am. J. Manage. Care 13, S170–177. [PubMed] [Google Scholar]
- Ng SF, Lin RC, Laybutt DR, Barres R, Owens JA, Morris MJ, 2010. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature 467, 963–966. [DOI] [PubMed] [Google Scholar]
- Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, Abraham JP, Abu-Rmeileh NM, Achoki T, AlBuhairan FS, Alemu ZA, Alfonso R, Ali MK, Ali R, Guzman NA, Ammar W, Anwari P, Banerjee A, Barquera S, Basu S, Bennett DA, Bhutta Z, Blore J, Cabral N, Nonato IC, Chang JC, Chowdhury R, Courville KJ, Criqui MH, Cundiff DK, Dabhadkar KC, Dandona L, Davis A, Dayama A, Dharmaratne SD, Ding EL, Durrani AM, Esteghamati A, Farzadfar F, Fay DF, Feigin VL, Flaxman A, Forouzanfar MH, Goto A, Green MA, Gupta R, Hafezi-Nejad N, Hankey GJ, Harewood HC, Havmoeller R, Hay S, Hernandez L, Husseini A, Idrisov BT, Ikeda N, Islami F, Jahangir E, Jassal SK, Jee SH, Jeffreys M, Jonas JB, Kabagambe EK, Khalifa SE, Kengne AP, Khader YS, Khang YH, Kim D, Kimokoti RW, Kinge JM, Kokubo Y, Kosen S, Kwan G, Lai T, Lein-salu M, Li Y, Liang X, Liu S, Logroscino G, Lotufo PA, Lu Y, Ma J, Mainoo NK, Mensah GA, Merriman TR, Mokdad AH, Moschandreas J, Naghavi M, Naheed A, Nand D, Narayan KM, Nelson EL, Neuhouser ML, Nisar MI, Ohkubo T, Oti SO, Pedroza A, Prabhakaran D, Roy N, Sampson U, Seo H, Sepanlou SG, Shibuya K, Shiri R, Shiue I, Singh GM, Singh JA, Skirbekk V, Stapelberg NJ, Sturua L, Sykes BL, Tobias M, Tran BX, Trasande L, Toyoshima H, van de Vijver S, Vasankari TJ, Veerman JL, Velasquez-Melen-dez G, Vlassov VV, Vollset SE, Vos T, Wang C, Wang X, Weiderpass E, Werdecker A, Wright JL, Yang YC, Yatsuya H, Yoon J, Yoon SJ, Zhao Y, Zhou M, Zhu S, Lopez AD, Murray CJ, Gakidou E, 2014. Global, regional, and national prevalence of overweight and obesity in children and adults during 19802013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384, 766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA, 2006. Involvement of oxidative stress in Alzheimer disease. J. Neuropathol. Exp. Neurol 65, 631–641. [DOI] [PubMed] [Google Scholar]
- Ogden CL, Flegal KM, Carroll MD, Johnson CL, 2002. Prevalence and trends in overweight among US children and adolescents, 1999–2000. JAMA 288, 1728–1732. [DOI] [PubMed] [Google Scholar]
- Org E, Parks BW, Joo JW, Emert B, Schwartzman W, Kang EY, Mehrabian M, Pan C, Knight R, Gunsalus R, Drake TA, Eskin E, Lusis AJ, 2015. Genetic and environmental control of host-gut microbiota interactions. Genome Res 25, 1558–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padwal RS, 2014. Obesity, diabetes, and the metabolic syndrome: the global scourge. Can. J. Cardiol 30, 467–472. [DOI] [PubMed] [Google Scholar]
- Panagiotou G, Nielsen J, 2009. Nutritional systems biology: definitions and approaches. Annu. Rev. Nutr 29, 329–339. [DOI] [PubMed] [Google Scholar]
- Pandey GN, Rizavi HS, Dwivedi Y, Pavuluri MN, 2008. Brain-derived neurotrophic factor gene expression in pediatric bipolar disorder: effects of treatment and clinical response. J. Am. Acad. Child Adolesc. Psychiatry 47, 1077–1085. [DOI] [PubMed] [Google Scholar]
- Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, Pan C, Civelek M, Rau CD, Bennett BJ, Mehrabian M, Ursell LK, He A, Castellani LW, Zinker B, Kirby M, Drake TA, Drevon CA, Knight R, Gargalovic P, Kirchgessner T, Eskin E, Lusis AJ, 2013. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab 17, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinetti GM, Zhao Z, Qin W, Ho L, Shrishailam Y, Macgrogan D, Ressmann W, Humala N, Liu X, Romero C, Stetka B, Chen L, Ksiezak-Reding H, Wang J, 2007. Caloric intake and Alzheimer’s disease. Experimental approaches and therapeutic implications. Interdiscip. Top. Gerontol 35, 159–175. [DOI] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Ko-hane I, Costello M, Saccone R, Landaker EJ, Goldfíne AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ, 2003. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. U. S. A 100, 8466–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter CJ, Fischer LK, Kundakovic M, Garg P, Jakovcevski M, Dincer A, Amaral AC, Ginns EI, Galdzicka M, Bryce CP, Ratner C, Waber DP, Mokler D, Med ford G, Champagne FA, Rosene DL, McGaughy JA, Sharp AJ, Galler JR, Akbarian S, 2016. DNA methylation signatures of early childhood malnutrition as sociated with impairments in attention and cognition. Biol. Psychiatry 80, 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW, 1991. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer s disease. J. Neurosci 7, 695–702. [DOI] [PubMed] [Google Scholar]
- Pinhas-Hamiel O, Dolan LM, Daniels SR, Standiford D, Khoury PR, Zeitler P, 1996. Increased incidence of non-insulin-dependent diabetes mellitus among adolescents.J. Pediatr. (Rio J) 128, 608–615. [DOI] [PubMed] [Google Scholar]
- Plourde M, Vohl MC, Vandal M, Couture P, Lemieux S, Cunnane SC, 2009. Plasma n-3 fatty acid response to an n-3 fatty acid supplement is modulated by apoE epsilon4 but not by the common PPAR-alpha L162V polymorphism in men. Br. J. Nutr 102, 1121–1124. [DOI] [PubMed] [Google Scholar]
- Pritchard CC, Cheng HH, Tewari M, 2012. MicroRNA profiling: approaches and considerations. Nat. Rev. Genet 13, 358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raji CA, Ho AJ, Parikshak NN, Becker JT, Lopez OL, Kuller LH, Hua X, Leow AD, Toga AW, Thompson PM, 2010. Brain structure and obesity. Hum. Brain Mapp 31, 353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey KM, Marcheva B, Kohsaka A, Bass J, 2007. The clockwork of metabolism. Annu. Rev. Nutr 27, 219–240. [DOI] [PubMed] [Google Scholar]
- Rao JS, Keleshian VL, Klein S, Rapoport SI, 2012. Epigenetic modifications in frontal cortex from Alzheimer s disease and bipolar disorder patients. Transl. Psychi atry 2, e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasgon NL, Kenna HA, Reynolds-May MF, Stemmle PG, Vemuri M, Marsh W, Wang P, Ketter TA, 2010. Metabolic dysfunction in women with bipolar disorder: the potential influence of family history of type 2 diabetes mellitus. Bipolar Disord 12, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve AK, Krishnan KJ, Turnbull DM, 2008. Age related mitochondrial degenera tive disorders in humans. Biotechnol. J 3, 750–756. [DOI] [PubMed] [Google Scholar]
- Regenold WT, Thapar RK, Marano C, Gavirneni S, Kondapavuluru PV, 2002. Increased prevalence of type 2 diabetes mellitus among psychiatric inpatients with bipolar I affective and schizoaffective disorders independent of psychotropic drug use. J. Affect. Disord 70, 19–26. [DOI] [PubMed] [Google Scholar]
- Rivera CM, Ren B, 2013. Mapping human epigenomes. Cell 155, 39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandhu KV, Sherwin E, Schellekens H, Stanton C, Dinan TG, Cryan JF, 2017. Feeding the microbiota-gut-brain axis: diet, microbiome, and neuropsychiatry. Transl. Res 179, 223–244. [DOI] [PubMed] [Google Scholar]
- Sauer S, Luge T, 2015. Nutriproteomics: facts, concepts, and perspectives. Proteomics 15, 997–1013. [DOI] [PubMed] [Google Scholar]
- Schubert D, 2005. Glucose metabolism and Alzheimer s disease. Ageing Res. Rev 4, 240–257. [DOI] [PubMed] [Google Scholar]
- Smine S, Obry A, Kadri S, Hardouin J, Freret M, Amri M, Jouenne T, Limam F, Cosette P, Aouani E, 2017. Brain proteomic modifications associated to protective effect of grape extract in a murine model of obesity. Biochim. Biophys. Acta 1865, 578–588. [DOI] [PubMed] [Google Scholar]
- Sng JC, Taniura H, Yoneda Y, 2006. Histone modifications in kainate-induced status epilepticus. Eur. J. Neurosci 23, 1269–1282. [DOI] [PubMed] [Google Scholar]
- Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, Hatcher B, Cox CL, Dyachenko A, Zhang W, McGahan JP, Seibert A, Krauss RM, Chiu S, Schaefer EJ, Ai M, Otokozawa S, Nakajima K, Nakano T, Bey-sen C, Hellerstein MK, Berglund L, Havel PJ, 2009. Consuming fructose-sweet ened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Invest 119, 1322–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stilling RM, van de Wouw M, Clarke G, Stanton C, Dinan TG, Cryan JF, 2016. The neuropharmacology of butyrate: The bread and butter of the microbiota-gut-brain axis?. Neurochem. Int 99, 110–132. [DOI] [PubMed] [Google Scholar]
- Stolzenberg DS, Champagne FA, 2016. Hormonal and non-hormonal bases of maternal behavior: the role of experience and epigenetic mechanisms. Horm. Behav 77, 204–210. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM, Matt son MP, 2008. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus 18, 1085–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suez J, Korem T, Zeevi D, Zilberman-Schapira G, Thaiss CA, Maza O, Israeli D, Zmora N, Gilad S, Weinberger A, Kuperman Y, Harmelin A, Kolodkin-Gal I, Shapiro H, Halpern Z, Segal E, Elinav E, 2014. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 514, 181–186. [DOI] [PubMed] [Google Scholar]
- Susser E, St Clair D, 2013. Prenatal famine and adult mental illness: interpreting concordant and discordant results from the Dutch and Chinese Famines. Soc. Sci. Med 97, 325–330. [DOI] [PubMed] [Google Scholar]
- Telese F, Gamliel A, Skowronska-Krawczyk D, Garcia-Bassets I, Rosenfeld MG, 2013. “Seq-ing” insights into the epigenetics of neuronal gene regulation. Neuron 77, 606–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temelkova-Kurktschiev T, Stefanov T, 2012. Lifestyle and genetics in obesity and type 2 diabetes. Exp. Clin. Endocrinol. Diabetes 120, 1–6. [DOI] [PubMed] [Google Scholar]
- Teschendorff AE, 2013. Epigenetic aging: insights from network biology. Aging (Albany NY) 5, 719–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, West J, Beck S, 2013. Age-associated epigenetic drift: implications, and a case of epigenetic thrift?. Hum. Mol. Genet 22, R7–R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushnikova TN, Medvedeva EL, Baidina TV, Danilova MA, 2014. Brain-derived and ciliary neurotrophic factors in patients with multiple sclerosis. Zh. Nevrol. Psikhiatr. Im. S. S 114, 33–36. [PubMed] [Google Scholar]
- Tshala-Katumbay D, Mwanza JC, Rohlman DS, Maestre G, Oriá RB, 2015. A global perspective on the influence of environmental exposures on the nervous system. Nature 527, S187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi E, Zhuang Y, Agrawal R, Ying Z, Gomez-Pinilla F, 2015. Interactive actions of Bdnf methylation and cell metabolism for building neural resilience under the in fluence of diet. Neurobiol. Dis 73, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valla J, Berndt JD, Gonzalez-Lima F, 2001. Energy hypometabolism in posterior cin- gulate cortex of Alzheimer’s patients: superficial laminar cytochrome oxidase associated with disease duration. J. Neurosci 21, 4923–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ommen B, Cavallieri D, Roche HM, Klein UI, Daniel H, 2008. The challenges for molecular nutrition research 4: the nutritional systems biology level. Genes Nutr 3, 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilopoulou CG, Margarity M, Klapa MI, 2016. Metabolomic analysis in brain research: opportunities and challenges. Front. Physiol 7, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura-Clapier R, Garnier A, Veksler V, 2008. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc. Res 79, 208–217. [DOI] [PubMed] [Google Scholar]
- Verma S, Hussain ME, 2017. Obesity and diabetes: an update. Diabetes Metab. Syndr 11, 73–79. [DOI] [PubMed] [Google Scholar]
- Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, Derrien M, Druesne A, Van Hylckama Vlieg JE, Bloks VW, Groen AK, Heilig HG, Zoetendal EG, Stroes ES, de Vos WM, Hoekstra JB, Nieuwdorp M, 2012. Transfer of intesti nal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143, 913–916, e917. [DOI] [PubMed] [Google Scholar]
- Vucetic Z, Kimmel J, Reyes TM, 2011. Chronic high-fat diet drives postnatal epigenetic regulation of μ-opioid receptor in the brain. Neuropsychopharmacology 36, 1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucetic Z, Kimmel J, Reyes TM, 2011. Chronic high-fat diet drives postnatal epigenetic regulation of μ-opioid receptor in the brain. Neuropsychopharmacology 36, 1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MP, LaFerla FM, Oddo SS, Brewer GJ, 2013. Reversible epigenetic histone modifications and Bdnf expression in neurons with aging and from a mouse model of Alzheimer’s disease. Age (Dordr) 35, 519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Fan W, 2010. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 10, 12–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wu Z, Li D, Li N, Dindot S, Satterfield MC, Bazer FW, Wu G,. Nutrition, epigenetics, and metabolic syndrome. Antioxid. Redox Signal [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterland RA, 2014. Epigenetic mechanisms affecting regulation of energy balance: many questions, few answers. Annu. Rev. Nutr 34, 337–355. [DOI] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dy-mov S, Szyf M, Meaney MJ, 2004. Epigenetic programming by maternal behavior. Nat. Neurosci 7, 847–854. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB, 2009. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wren JD, Garner HR, 2005. Data-mining analysis suggests an epigenetic pathogenesis for type 2 diabetes. J. Biomed. Biotechnol 2005, 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, Sinha R, Gilroy E, Gupta K, Baldassano R, Nessel L, Li H, Bushman FD, Lewis JD, 2011. Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Woodroffe A, Rodriguez-Murillo L, Roos JL, van Rensburg EJ, Abecasis GR, Gogos JA, Karayiorgou M, 2009. Elucidating the genetic architecture of fa milial schizophrenia using rare copy number variant and linkage scans. Proc. Natl. Acad. Sci. U. S. A 106, 16746–16751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe K, Kanaya A, Lindquist K, Simonsick EM, Harris T, Shorr RI, Tylavsky FA, Newman AB, 2004. The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA 292, 2237–2242. [DOI] [PubMed] [Google Scholar]
- Yamanaka M, Tsuchida A, Nakagawa T, Nonomura T, Ono-Kishino M, Sugaru E, Noguchi H, Taiji M, 2007. Brain-derived neurotrophic factor enhances glucose utilization in peripheral tissues of diabetic mice. Diabetes Obes. Metab 9, 59–64. [DOI] [PubMed] [Google Scholar]
- Ye YL, Chan YT, Liu HC, Lu HT, Huang RF, 2010. Depleted folate pool and dysfunctional mitochondria associated with defective mitochondrial folate proteins sen sitize Chinese ovary cell mutants to tert-butylhydroperoxide-induced oxidative stress and apoptosis. J. Nutr. Biochem 21, 793–800. [DOI] [PubMed] [Google Scholar]
- Zhang L, Bruce-Keller AJ, Dasuri K, Nguyen AT, Liu Y, Keller JN, 2009. Diet-induced metabolic disturbances as modulators of brain homeostasis. Biochim. Biophys. Acta 1792, 417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Barrere-Cain RE, Yang X, 2015. Nutritional systems biology of type 2 diabetes. Genes Nutr 10, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivkovic AM, German JB, Sanyal AJ, 2007. Comparative review of diets for the metabolic syndrome: implications for nonalcoholic fatty liver disease. Am. J. Clin. Nutr 86, 285–300. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E, 2009. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol 5, 311–322. [DOI] [PubMed] [Google Scholar]