

Abstract
EGFRC797S mutation inducing resistance against third generation EGFR inhibitor drugs is an emerging “unmet clinical need” for nonsmall cell lung cancer patients. The pyrimidopyrimidinone derivative JND3229 was identified as a new highly potent EGFRC797S inhibitor with single digit nM potency. It also exhibited good in vitro and in vivo monodrug anticancer efficacy in a xenograft mouse model of BaF3/EGFR19D/T790M/C797S cells. A high-resolution X-ray crystallographic structure was also determined to elucidate the interactions between JND3229 and EGFRT790M/C797S. Our study provides an important structural and chemical basis for future development of new generation EGFRC797S inhibitors as anticancer drugs.
Keywords: EGFRC797S, Clinical resistance, Fourth-generation inhibitors, Monodrug efficacy
The epidermal growth factor receptor (EGFR, ErbB1, HER1) is one of the most validated molecular targets for anticancer drug discovery.1 Three generations of EGFR inhibitors, e.g., gefitinib,2 erlotinib,3 afatinib,4 and osimertinib (AZD9291),5 have been approved by US FDA and achieved significantly clinical benefit in nonsmall cell lung cancer (NSCLC) patients. Particularly, the wild-type sparing third generation EGFR threonine790 to methionine790 mutant (T790M) inhibitor osimertinib demonstrates ∼75% overall response rate (ORR) in EGFRT790M mutation-positive NSCLC patients and was approved as the first-line treatment for metastatic NSCLC patients in 2018,6,7 representing one of the most advanced progresses in human cancer therapy.
However, a tertiary Cys797 to Ser797 (C797S) point mutation, disturbing covalent bond formation with the irreversible third generation EGFR inhibitors, becomes a leading mechanism of clinically acquired resistance in ∼40% of patients treated with osimertinib.8−10 An allosteric inhibitor, EAI045 (1, Figure 1),11 has been discovered to exhibit low nM IC50 values against EGFRT790M and EGFRC797S mutants. However, the combination with an EGFR antibody cetuximab was required for EAI045 to demonstrate in vivo therapeutic efficacy because of asymmetric dimerization of the receptor.11 (2-Hydroxy)phenyl-4-substituted quinazoline derivatives (2a and 2b),12 trisubstituted imidazole (3),13−15 and a pan kinase inhibitor Gö6976 (4)16 were also discovered to display strong inhibition against EGFRC797S kinase, but neither cell-based activity nor in vivo efficacy of these molecules was disclosed. Encouragingly, brigatinib (5),17 an FDA-approved ALK kinase inhibitor, was recently reported to strongly suppress the kinase activity of EGFRC797S and inhibit the proliferation of NSCLC cancer cells harboring the EGFRC797S mutation with IC50 values in nM ranges. Moreover, brigatinib also exhibited significant antitumor efficacy in a xenograft mouse model of PC9 and MGH121-res2 NSCLC cancer cells with EGFRC797S mutations by combinations with EGFR antibody cetuximab or pantitumumab.17 Nevertheless, it is still highly valuable to discover novel EGFRC797S inhibitors with monodrug efficacy for overcoming the required resistance against third generation EGFR inhibitors.18−20 Herein, we report the identification of JND3229 (6, Figure 1) as a new reversible EGFRC797S mutant inhibitor demonstrating both in vitro and in vivo monodrug efficacy against the proliferation of EGFRC797S mutated cancer cells.
Figure 1.

Chemical structures of the representative EGFRC797S inhibitors 1–6.
For new EGFRC797S inhibitors to be discovered, a random screening was initially conducted by utilizing our in-house kinase inhibitor library containing ∼3000 compounds.21−26 The effort helped us to identify JND3229 (Figure 1), a pyrimidopyrimidinone derivative, as a new EGFRC797S inhibitor. The molecule was readily synthesized by using a protocol outlined in Scheme 1. Briefly, a commercially available ethyl 4-chloro-2-(methylthio)-pyrimidine-5-carboxylate (7) was reacted with 1-Boc-1,4-cyclohexanediamine (8) followed by reduction, oxidation, and Borch reductive amination and cyclization to yield key pyrimidopyrimidinone 14. Oxidation of 14 with 3-chloroperbenzoic acid (m-CPBA) yielded the sulfone 15, which was consequently subjected to nucleophilic deprotection and acylation reaction to give the title compound JND3229.
Scheme 1. Synthesis Protocol for JND3229 (6).

Reagents and conditions: (a) K2CO3, DMF, 80 °C, 85%; (b) LiAlH4, THF, −40 to 0 °C, 50%; (c) MnO2, DCM, rt, 92%; (d) AcOH, NaBH4, PhMe, 110 °C to rt, 80%; (e) triphosgene, Et3N, DCM, 0 °C to rt, 94%; (f) m-CPBA, DCM, rt, 88%; (g) F3CCOOH, 2-BuOH, 110 °C; (h) F3CCOOH, DCM, rt, 50% (for two steps); (i) propanoic acid, HATU, DIPEA, DCM, rt, 90%.
JND3229 exhibited strong inhibition against the kinase activity of EGFRL858R/T790M/C797S with an IC50 value of 5.8 nM under the conditions of an enzyme-linked immunosorbent assay (ELISA) (Figure 2A). Additionally, the compound also potently suppressed EGFRL858R/T790M and EGFRWT with IC50 values of 30.5 and 6.8 nM, respectively (Figure 2A). Similar to the previous observation, osimertinib exhibited ∼70-fold less potency against the EGFRC797S mutant compared with its strong inhibition against EGFRL858R/T790M (Figure 2B). The pan-kinase inhibitor staurosporine (Figure 2C), EGFRC797S inhibitor 2b (Figure 2D), and brigatinib (Figure 2E), used as positive drugs, also displayed strong inhibition against EGFRL858R/T790M/C797S kinase.
Figure 2.

JND3229 potently inhibits the kinase activities of EGFRL858R/T790M and EGFRL858R/T790M/C797S. The kinase inhibitory efficacy of JND3229 (A), AZD9291 (B), staurosporine (C), inhibitor 2b (D), and brigatinib (E) against EGFRL858R/T790M (blue mark), EGFRL858R/T790M/C797S (red mark), and EGFRWT (purple mark) were tested by ELISA assay, and the IC50 values were calculated based on three independent repeats.
The strong kinase inhibition of JND3229 was further validated by investigating its potential suppression on activity of EGFR signals in BaF3 cells stably transfected with EGFRL858R/T790M/C797S and EGFR19D/T790M/C797S (Figure 3). It was shown that JND3229 potently inhibited the phosphorylation of EGFRL858R/T790M/C797S and EGFR19D/T790M/C797S in a dose-dependent manner. Consistent with their kinase activity, staurosporine also demonstrated strong inhibition of EGFRC797S activation in BaF3 cells, but the effect of osimertinib was minor.
Figure 3.

JND3229 potently inhibits the phosphorylation of EGFRL858R/T790M/C797S and EGFR19D/T790M/C797S in engineering BaF3 Cells. BaF3 cells that overexpressed EGFRL858R/T790M/C797S (A) or EGFR19D/T790M/C797S (B) were treated with the indicated concentrations of JND3229, AZD9291, and staurosporine for 2 h and stimulated by EGF for 15 min. Cell lysates were harvested for Western blot analysis for EGFR phosphorylation.
The antiproliferative activities of JND3229 were also investigated against a panel of cells with different EGFR status. It was shown that JND3229 potently inhibited the proliferation of BaF3 cells harboring the EGFRL858R/T790M/C797S and EGFR19D/T790M/C797S mutations with IC50 values of 0.51 and 0.32 μM, respectively (Table 1), which are comparable to those of brigatinib. It also potently suppressed the growth of NCI-H1975 NSCLC cells with EGFRT790M mutation with an IC50 value of 0.31 μM. Although osimertinib exhibited strong antiproliferative effect on NCI-H1975 cells with an IC50 value of 0.13 μM, its inhibition against BaF3 cells harboring the EGFRC797S mutation was significantly less potent (IC50 > 4 μM). Consistent with its nonselective inhibition against EGFRWT, JND3229 also obviously suppressed the proliferation of A431 cancer cells overexpressing EGFRWT with an IC50 value of 0.27 μM.
Table 1. Antiproliferative Activities of JND3229 against Cells with Different Mutant EGFR.
| antiproliferation
IC50 (μM) |
||||
|---|---|---|---|---|
| cells | EGFR status | JND3229 | AZD9291 | brigatinib |
| BaF3 | L858R/T790M/C797S | 0.51 ± 0.08 | 5.15 ± 1.57 | 0.42 ± 0.08 |
| 19D/T790M/C797S | 0.32 ± 0.11 | 4.61 ± 2.34 | 0.26 ± 0.02 | |
| NCI-H1975 | L858R/T790M | 0.31 ± 0.01 | 0.13 ± 0.04 | 1.09 ± 0.24 |
| A431 | WT | 0.27 ± 0.18 | 1.24 ± 0.37 | |
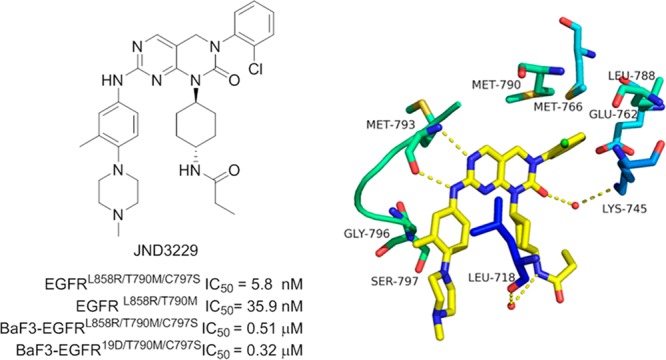
A 2.65 Å resolution X-ray crystallographic structure of JND3229-EGFRT790M/C797S complex was also determined to elucidate detailed interactions between the inhibitor and the kinase (Figure 4 and Table S1). It was shown that JND3229 was accommodated in the ATP binding site of the C797S-mutated EGFR with a reversible “U-shaped” configuration. The pyrido[2,3-d]pyrimidine-7-one core formed a bidentate hydrogen bond interaction with the “hinge” residue Met793 of the protein. The 2-chlorophenyl group was directed toward the hydrophobic back pocket composed by Lys745, Glu762, Leu788, Met766, and Met790, and the carbonyl of pyrido[2,3-d]pyrimidine-7-one formed a hydrogen bond with the nitrogen of Lys745 mediated by a water molecule. The propionamide group was located in a solvent-exposing region, and the NH moiety formed a hydrogen bond with Leu718 mediated by another water molecule. The left-hand methyl-substituted phenyl group interacted with Gly796 by van der Waals, and the hydrophilic methyl piperazine moiety was extended directly to the solvent.
Figure 4.
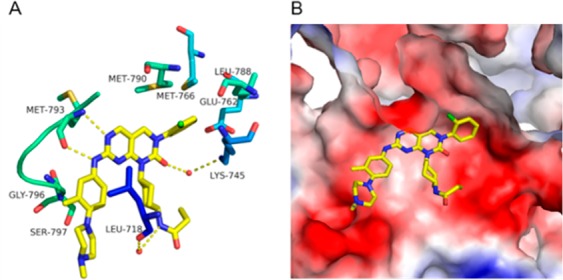
X-ray crystal structure of JND3229 with EGFRL858R/T790M/C797S (PDB ID: 5ZTO). (A) EGFR kinase shown in green and blue stick and ribbon representation. JND3229 is shown in yellow stick structure. Hydrogen bonds are indicated by yellow hatched lines to key amino acids. Water is represented as red dots. (B) X-ray crystal structure with the interaction surface.
The in vivo anticancer efficacy of JND3229 was also examined using a xenograft mouse model. BALB/c mice bearing established BaF3-EGFR19D/T790M/C797S mouse xenograft tumors were treated with JND3229 twice daily at a dose of 10 mg/kg by intraperitoneal injection or by vehicle control for 10 days. EAI045 (60 mg/kg, once daily by oral gavage) combination with cetuximab (1 mg/kg, once every other day by intraperitoneal injection) were used as positive control. The data showed that administration of JND3229 caused an obvious suppression of tumor growth with a tumor growth inhibition (TGI) value of 42.2% (Figure 5A), which was more potent than that of EAI045/cetuximab combination (TGI = 22.3%, Figure 5B). In addition, JND3229 was well tolerated, and there is no obvious body weight loss or other obvious toxic sign in the treated animals. Further immunohistochemistry analysis demonstrated that JND3229 treatment significantly decreased the level of phosphorylated EGFR (p-EGFR) in the tumor tissues, confirming in vivo target inhibition of the compound (Figure 5C).
Figure 5.

JND3229 shows in vivo antitumor efficacy in the EGFR19D/T790M/C797S mouse xenograft model. Mice bearing BaF3-EGFR19D/T790M/C797S tumor xenografts were treated with JND3229 (10 mg/kg in 0.5% HPMC, ip, Bid) or treated with EAI045 (60 mg/kg in 10% NMP/90% PEG300, po, qd) combined with cetuximab (1 mg/kg in 0.9% w/v NaCl/water, ip, qod). Relative tumor volume (RTV) (A) and body weight (B) of EGFR19D/T790M/C797S mouse xenograft model were measured (n = 6) every 2–3 days. The tumor growth inhibition was measured at the final day of the treatment for the drug-treated group versus the control (*P < 0.05 vs control, Student’s t test). (C) JND3229 suppresses the expression of p-EGFR in the EGFR19D/T790M/C797S xenograft model. Tumor tissue sections were stained by hematoxylin and eosin (H&E) (blue) and p-EGFR antibody (brown), and the relative integrated optical density (IOD) of p-EGFR labeling is presented by quantitative analysis (*P < 0.05 vs control, Student’s t test). IOD, integrated optical density; all values represent mean ± SD or mean ± SEM.
In summary, JND3229 was identified as a new EGFRC797S inhibitor. The compound potently inhibited EGFRC797S mutated kinase with an IC50 value of 5.8 nM and strongly suppressed the proliferation of BaF3 cells harboring the EGFRL858R/T790M/C797S and EGFR19D/T790M/C797S mutations with IC50 values of 0.51 and 0.32 μM, respectively. Moreover, the compound also demonstrated in vivo monodrug anticancer efficacy in a xenograft mouse model of BaF3 cells with EGFR19D/T790M/C797S mutation. A high-resolution X-ray crystallographic structure was also determined to elucidate the interactions between JND3229 and EGFRT790M/C797S protein. Although the relatively low target selectivity (Table S2) may raise some concern about the potential off-target toxicity of this molecule, our study might provide a useful structural and chemical basis for further development of the fourth generation EGFRC797S inhibitors to overcome the acquired resistance against osimertinib.
Acknowledgments
The authors appreciate the financial support from Guangdong Natural Science Funds (2015A030306042, 2105A030312014), National Natural Science Foundation of China (21572230, 81425021, 21702075, 81673285, and 31270769), Guangdong Nanyue-Baijie Award, Guangzhou City Key Laboratory of Precision Chemical Drug Development (201805010007), Institutes for Drug Discovery and Development of Chinese Academy of Science (CASIMM0120185006), and Jinan University.
Glossary
Abbreviations
- EGFR
epidermal growth factor receptor
- NSCLC
nonsmall cell lung cancer
- ORR
overall response rate
- C797S
Cys797 to Ser797
- m-CPBA
3-chloroperbenzoic acid
- ELISA
enzyme-linked immunosorbent assay
- TGI
tumor growth inhibition
- RTV
relative tumor volume
- IOD
integrated optical density.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00373.
Chemistry, biological assay, and X-ray crystallography data (PDF)
Author Contributions
# X.L., T.Z., and S.-J.Z. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Lynch T. J.; Bell D. W.; Sordella R.; Gurubhagavatula S.; Okimoto R. A.; Brannigan B. W.; Harris P. L.; Haserlat S. M.; Supko J. G.; Haluska F. G.; Louis D. N.; Christiani D. C.; Settleman J.; Haber D. A. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Paez J. G.; Janne P. A.; Lee J. C.; Tracy S.; Greulich H.; Gabriel S.; Herman P.; Kaye F. J.; Lindeman N.; Boggon T. J.; Naoki K.; Sasaki H.; Fujii Y.; Eck M. J.; Sellers W. R.; Johnson B. E.; Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Dowell J.; Minna J. D.; Kirkpatrick P. Erlotinib hydrochloride. Nat. Rev. Drug Discovery 2005, 4, 13–14. 10.1038/nrd1612. [DOI] [PubMed] [Google Scholar]
- Dungo R. T.; Keating G. M. Afatinib: first global approval. Drugs 2013, 73, 1503–1515. 10.1007/s40265-013-0111-6. [DOI] [PubMed] [Google Scholar]
- Kim E. S. Olmutinib: first global approval. Drugs 2016, 76, 1153–1157. 10.1007/s40265-016-0606-z. [DOI] [PubMed] [Google Scholar]
- Soria J. C.; Ohe Y.; Vansteenkiste J.; Reungwetwattana T.; Chewaskulyong B.; Lee K. H.; Dechaphunkul A.; Imamura F.; Nogami N.; Kurata T.; Okamoto I.; Zhou C.; Cho B. C.; Cheng Y.; Cho E. K.; Voon P. J.; Planchard D.; Su W. C.; Gray J. E.; Lee S. M.; Hodge R.; Marotti M.; Rukazenkov Y.; Ramalingam S. S. Osimetinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 113–125. 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- FDA Center for Drug Evaluation and Research . www.accessdata.fda.gov/drugsatfda_docs/label/2018/208065s008lbl.pdf (accessed April 19, 2018).
- Piotrowska Z.; Niederst M. J.; Karlovich C. A.; Wakelee H. A.; Neal J. W.; Mino-Kenudson M.; Fulton L.; Hata A. N.; Lockerman E. L.; Kalsy A.; Digumarthy S.; Muzikansky A.; Raponi M.; Garcia A. R.; Mulvey H. E.; Parks M. K.; DiCecca R. H.; Dias-Santagata D.; Iafrate A. J.; Shaw A. T.; Allen A. R.; Engelman J. A.; Sequist L. V. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discovery 2015, 5, 713–722. 10.1158/2159-8290.CD-15-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thress K. S.; Paweletz C. P.; Felip E.; Cho B. C.; Stetson D.; Dougherty B.; Lai Z.; Markovets A.; Vivancos A.; Kuang Y.; Ercan D.; Matthews S. E.; Cantarini M.; Barrett J. C.; Janne P. A.; Oxnard G. R. Acquired EGFR C797S mutation mediated resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederst M. J.; Hu H.; Mulvey H. E.; Lockerman E. L.; Garcia A. R.; Piotrowska Z.; Sequist L. V.; Engelman J. A. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 2015, 21, 3924–3933. 10.1158/1078-0432.CCR-15-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y.; Yun C. H.; Park E.; Ercan D.; Manuia M.; Juarez J.; Xu C.; Rhee K.; Chen T.; Zhang H.; Palakurthi S.; Jang J.; Lelais G.; DiDonato M.; Bursulaya B.; Michellys P. Y.; Epple R.; Marsilje T. H.; McNeill M.; Lu W.; Harris J.; Bender S.; Wong K. K.; Janne P. A.; Eck M. J. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.; Jung H. Y.; Mah S.; Hong S. Discovery of EGF receptor inhibitors that are selective for the d746–750/T790M/C797S mutant through structure-based de novo design. Angew. Chem., Int. Ed. 2017, 56, 7634–7638. 10.1002/anie.201703389. [DOI] [PubMed] [Google Scholar]
- Gunther M.; Juchum M.; Kelter G.; Fiebig H.; Laufer S. Lung cancer: EGFR inhibitors with low nanomolar activity against a therapy-resistant L858R/T790M/C797S mutant. Angew. Chem., Int. Ed. 2016, 55, 10890–10894. 10.1002/anie.201603736. [DOI] [PubMed] [Google Scholar]
- Juchum M.; Gunther M.; Doring E.; Sievers-Engler A.; Lammerhofer M.; Laufer S. Trisubstituted imidazoles with a rigidized hinge binding motif act as single digit nM inhibitors of clinically relevant EGFR L858R/T790M and L858R/T790M/C797S mutants: an example of target hopping. J. Med. Chem. 2017, 60, 4636–4656. 10.1021/acs.jmedchem.7b00178. [DOI] [PubMed] [Google Scholar]
- Gunther M.; Lategahn J.; Juchum M.; Doring E.; Keul M.; Engel J.; Tumbrink H. L.; Rauh D.; Laufer S. Trisubstituted pyridinylimidazoles as potent inhibitors of the clinically resistant L858R/T790M/C797S EGFR mutant: targeting of both hydrophobic regions and the phosphate binding site. J. Med. Chem. 2017, 60, 5613–5637. 10.1021/acs.jmedchem.7b00316. [DOI] [PubMed] [Google Scholar]
- Kong L. L.; Ma R.; Yao M. Y.; Yan X. E.; Zhu S. J.; Zhao P.; Yun C. H. Structural pharmacological studies on EGFR T790M/C797S. Biochem. Biophys. Res. Commun. 2017, 488, 266–272. 10.1016/j.bbrc.2017.04.138. [DOI] [PubMed] [Google Scholar]
- Uchibori K.; Inase N.; Araki M.; Kamada M.; Sato S.; Okuno Y.; Fujita N.; Katayama R. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat. Commun. 2017, 8, 14768. 10.1038/ncomms14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.; Yu L.; Zhang Z.; Ren X.; Smaill J. B.; Ding K. Targeting EGFRL858R/T790M and EGFRL858R/T790M/C797S resistance mutations in NSCLC: Current developments in medicinal chemistry. Med. Res. Rev. 2018, 38, 1550–1581. 10.1002/med.21488. [DOI] [PubMed] [Google Scholar]
- Chen L.; Fu W.; Zheng L.; Liu Z.; Liang G. Recent progress of small-molecule epidermal growth factor receptor (EGFR) inhibitors against C797S resistance in non-small-cell lung Cancer. J. Med. Chem. 2018, 61, 4290–4300. 10.1021/acs.jmedchem.7b01310. [DOI] [PubMed] [Google Scholar]
- Grabe T.; Lategahn J.; Rauh D. C797S Resistance: The Undruggable EGFR Mutation in Non-Small Cell Lung Cancer?. ACS Med. Chem. Lett. 2018, 9, 779–782. 10.1021/acsmedchemlett.8b00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S.; Xu T.; Zhang L.; Zhang Z.; Luo J.; Liu Y.; Lu X.; Tu Z.; Ren X.; Ding K. Design, synthesis, and biological evaluation of 2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidinyl derivatives as new irreversible epidermal growth factor receptor inhibitors with improved pharmacokinetic properties. J. Med. Chem. 2013, 56, 8803–8813. 10.1021/jm4012388. [DOI] [PubMed] [Google Scholar]
- Tan L.; Zhang Z.; Gao D.; Luo J.; Tu Z. C.; Li Z.; Peng L.; Ren X.; Ding K. 4-Oxo-1,4-dihydroquinoline-3-carboxamide derivatives as new Axl kinase inhibitors. J. Med. Chem. 2016, 59, 6807–6825. 10.1021/acs.jmedchem.6b00608. [DOI] [PubMed] [Google Scholar]
- Xun Q.; Zhang Z.; Luo J.; Tong L.; Huang M.; Wang Z.; Zou J.; Liu Y.; Xu Y.; Xie H.; Tu Z. C.; Lu X.; Ding K. Design, synthesis, and structure-activity relationship study of 2-Oxo-3,4-dihydropyrimido[4,5-d]pyrimidines as new colony stimulating factor 1 receptor (CSF1R) kinase inhibitors. J. Med. Chem. 2018, 61, 2353–2371. 10.1021/acs.jmedchem.7b01612. [DOI] [PubMed] [Google Scholar]
- Chang S.; Zhang L.; Xu S.; Luo J.; Lu X.; Zhang Z.; Xu T.; Liu Y.; Tu Z.; Xu Y.; Ren X.; Geng M.; Ding J.; Pei D.; Ding K. Design, synthesis, and biological evaluation of novel conformationally constrained inhibitors targeting epidermal growth factor receptor threonine790 → methionine790 mutant. J. Med. Chem. 2012, 55, 2711–2723. 10.1021/jm201591k. [DOI] [PubMed] [Google Scholar]
- Xu T.; Zhang L.; Xu S.; Yang C. Y.; Luo J.; Ding F.; Lu X.; Liu Y.; Tu Z.; Li S.; Pei D.; Cai Q.; Li H.; Ren X.; Wang S.; Ding K. Pyrimido[4,5-d]pyrimidin-4(1H)-one derivatives as selective inhibitors of EGFR threonine790 to methionine790 (T790M) mutants. Angew. Chem., Int. Ed. 2013, 52, 8387–8390. 10.1002/anie.201302313. [DOI] [PubMed] [Google Scholar]
- Yu L.; Huang M.; Xu T.; Tong L.; Yan X. E.; Zhang Z.; Xu Y.; Yun C.; Xie H.; Ding K.; Lu X. A structure-guided optimization of pyrido[2,3-d]pyrimidin-7-ones as selective inhibitors of EGFRL858R/T790M mutant with improved pharmacokinetic properties. Eur. J. Med. Chem. 2017, 126, 1107–1117. 10.1016/j.ejmech.2016.12.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.