

Abstract
Several aryl hydrocarbon receptor (AhR)-active pharmaceuticals were screened as inhibitors of pancreatic cancer cell invasion and identified two compounds, omeprazole, that inhibited invasion. Inhibition of highly invasive Panc1 cell invasion by omeprazole involves an AhR-dependent non-genomic pathway, and omeprazole-mediated inhibition of Panc1 cell invasion was dependent on Jun-N-terminal kinase (JNK) and mitogen-activated kinase kinase 7 (MKK7). The failure of omeprazole to induce nuclear translocation of the AhR was not due to overexpression of cytosolic AhR partner proteins Hsp90 or XAP2, and results of DNA sequencing show that the AhR expressed in Panc1 cells was not mutated. Results of RNAseq studies indicate that omeprazole induced an AhR-dependent downregulation of several pro-invasion factors including activated leukocyte cell adhesion molecule (ALCAM), long chain fatty acid CoA-synthase (CSL4), stathmin 3 (STMN3) and neuropillin 2 (NRP2), and the specific functions of these genes are currently being investigated.
Keywords: omeprazole, pancreatic cancer, invasion, inhibition, JNK
Graphical Abstract
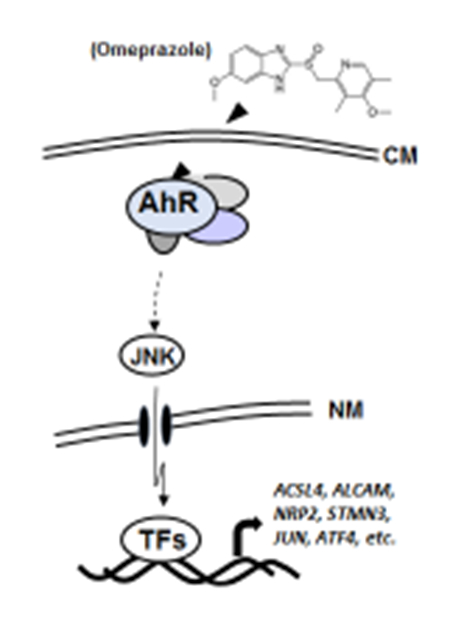
1. INTRODUCTION
The AhR is a ligand-activated transcription factor that was initially identified as the intracellular protein that bound the environmental toxicant 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and related compounds, and subsequent studies showed that the AhR binds structurally and functionally diverse ligands including health-promoting phytochemicals and pharmaceuticals [1–4]. The classical mechanism of action associated with ligand-dependent activation of the AhR involves heterodimer formation with the AhR nuclear translocator (Arnt) and binding to cis-acting xenobiotic response elements on target gene promoters [1, 2]. The nuclear AhR interacts with other partners and other response elements, and there are also reports of non-genomic (or extranuclear) AhR-dependent pathways [5–8] and the AhR plays an essential role in normal physiology and pathophysiology [9–12].
Studies in this laboratory have been investigating AhR-active pharmaceuticals and re-purposing them as AhR-dependent anticancer agents in breast and pancreatic cancer [13–15]. The advantage in identifying previously approved pharmaceuticals (e.g. omeprazole, a proton pump inhibitor) for applications in cancer chemotherapy is their relatively rapid approval. After screening several AhR-active pharmaceuticals in highly aggressive Panc1 cells, we identified omeprazole as an inhibitor of Panc1 cell migration/invasion and this response was AhR-dependent and involved a non-genomic pathway [15]. In this paper, our mechanistic studies show that AhR-dependent inhibition of Panc1 cell invasion by omeprazole involves activation of JNK and multiple downstream genes.
2. MATERIALS AND METHODS
Cell lines, antibodies, and reagents.
Panc1 human pancreatic cancer cell lines were obtained from the American Type Culture Collection (Manassas, VA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) nutrient mixture supplemented with 0.22% sodium bicarbonate, 0.011% sodium pyruvate, 10% fetal bovine serum (FBS) at 37°C in the presence of 5% CO2. pcDNA3-Flag-MKK4, pcDNA3-Flag-MKK7-JNK1 WT, and pcDNA3-Flag-MKK7-JNK1 APF plasmid clones were purchased from Addgene (Cambridge, MA). CYP1A1, AHR, and antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and SAPK/JNK pathway, AKT pathway, MAPK family, and GAPDH antibody were purchased from Cell Signaling Biotechnology (Danvers, MA). All compounds used in this study and anti-Flag M2 antibody were purchased from Sigma-Aldrich (St. Louis, MO).
Quantitative real-time PCR.
cDNA was prepared from the total RNA of cells using High capacity RNA-to-cDNA Kit (Applied Biosystems, Foster City, CA). Each PCR was carried out in triplicate using iTaq universal SYBR Green mix (Bio-Rad) and Bio-Rad CFX384 (C1000) real-time PCR System. Values for each gene were normalized to expression levels of TATA-binding protein (TBP). The sequences of the primers used for real-time PCR are listed in Supplemental Table 1.
Western blot analysis.
Cells (3 × 105) were plated in six-well plates in DMEM media containing 2.5% FBS for 16 hr, treated with different concentrations of the compounds, and cellular lysates were analyzed by western blot analysis as previously described [13–15].
Invasion assay.
For invasion assay of Panc1 cells, the BD-Matrigel Invasion Chamber (24-transwell with 8 μm pore size polycarbonate membrane) was used in a process of modified Boyden chamber assay as previously described [13–15].
Transfection of siRNA.
Cells (1.5 × 105 cells/well) were plated in 6-well plates in DMEM media supplemented with 10% FBS. After 16 hr, the cells were transfected with 100 nM of each siRNA duplex for 6 hr using Lipofectamine 2000 reagent (Invitrogen) following the manufacturer’s protocol.
RNA-seq analysis.
Total RNA was extracted from cells using Trizol reagent (Invitrogen) and RNA mini prep kit (Qiagen) according to the manufacturer’s protocol and contaminated chromosomal DNA was removed by treatment of DNase I (Qiagen). Sequencing and analysis were performed by Genomics and Bioinformatics Service center (www.txgen.tamu.edu). FASTQ sequences were mapped to human genome hg19 (GRCh37) downloaded from UCSC genome browser website and aligned using TopHat2 [16]. Transcript structure and abundance were estimated using Cufflinks [17]. The differential expression analysis was performed using DESeq2 (version 1.10.1) [18] and R (version 3.4.0). Briefly, count matrices were fit for a generalized linear model per gene following a negative binomial distribution. Dispersion estimates for each gene within groups were shrunk using an empirical Bayes approach. Log fold-changes were compared between disease groups using the Wald test. Pairwise correlation was calculated with Pearson’s method in R. Hierarchical clustering analyses performed using DESeq2 rlog-normalized RNA-seq data.
Tumor sphere formation assay.
Cells were plated in 12-well ultralow attachment plates (Corning) at a density of 3,000 cells/ml in serum-free DMEM:F12 (Gibco) supplemented with 20 ng/ml recombinant human EGF (PeproTech), 100 ng/ml recombinant human FGF-basic (Gibco) and 1:50 B27 supplement (Gibco), and incubated at 37°C in the presence of 5% CO2 for 7 days.
Apoptosis assay.
Cells were treated with 0.1% DMSO, 200 μM omeprazole in 2.5% FBS-DMEM for 24 hr and stained using the Alexa Fluor® 488 Annexin V/Dead Cell Apoptosis Kit according to the manufacturer’s protocol (Molecular Probes). Annexin V and PI were analyzed by flow cytometry. Annexin V-positive cells were counted as Apoptosis cells in total 500,000 cells per each sample.
Statistics.
All of the experiments were repeated a minimum of three times. The data are expressed as the means (SD). Statistical significance was analyzed using either Unpaired-Student’s t-test (two-tailed) or analysis of variance (ANOVA) test. A P value of less than 0.05 was considered statistically significant.
3. RESULTS
Non-genomic activities of the AhR are usually associated with activation of kinase activities [8, 19, 20]. TCDD but not omeprazole decreased phospho-Akt (p-Akt) 2 to 6 hr after treatment (Figs. 1A and 1B). However, omeprazole but not TCDD inhibited Panc1 cell invasion and only omeprazole induced a rapid (0.5 hr) activation of JNK (p-JNK) which also increased slightly after 6 and 12 hr, whereas minimal effects were observed for changes in phosphorylation of p38, ERK and Akt (data not shown and Suppl. Fig. 1). We then investigated the time-dependent effects of omeprazole in wild-type Panc1 cells or cells in which the AhR was silenced (siAhR) by RNA interference (RNAi) (Fig. 1C). The results clearly showed that omeprazole-mediated induction of p-JNK after 0.25 and 0.5 hr were attenuated after AhR knockdown, and similar but not as striking results were observed for phosphorylation of cJun (p-cJun). Interestingly, omeprazole-induced JNK did not modulate Annexin V staining (marker of apoptosis) in Panc1 cells (Suppl. Fig. 2) and this contrasts to the effects of melatonin which induced apoptosis via suppression of NFκB in MiaPaca2 cells [21].
Figure 1.
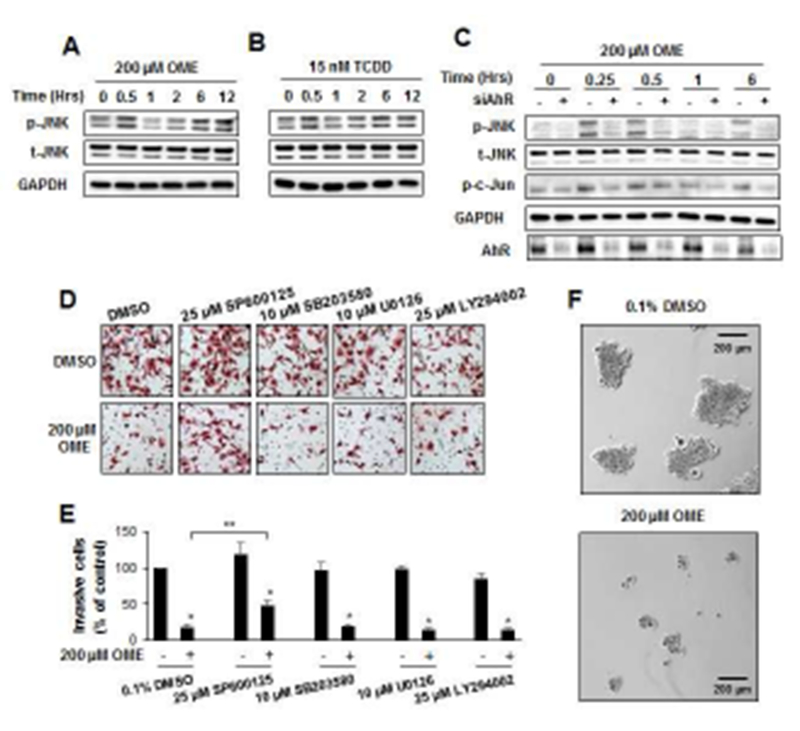
Activation of kinases and their role in Panc1 cell invasion. Panc1 cells were treated with 200 μM omeprazole (A) and 15 nM TCDD (B) for up to 12 hr, and whole cell lysates were analyzed by western blots as outlined in the Materials and Methods. Role of AhR in mediating effects of omeprazole (C). Cells were transfected with a non-specific oligonucleotide (−) or siAhR (+), treated with 200 μM omeprazole for up to 6 hr, and whole cell lysates were analyzed by western blots. Effects of kinase inhibitors on invasion. Panc1 cells were treated with 200 μM omeprazole in the presence or absence of several kinase inhibitors, and effects on cell invasion were determined in a Boyden chamber assay (D) and quantitated (E). (F) Panc1 cell spheroids were treated with DMSO or omeprazole, and spheroid growth was determined as outlined in the Materials and Methods. Results are expressed as means ± SD for at least 3 replicates per treatment group, and significant (p<0.05) inhibition of invasion (*) and reversal of these effects by kinase inhibitors are indicated (**).
We also investigated the effects of several kinase inhibitors on omeprazole-induced Panc1 cell invasion (Figs. 1D and 1E). The JNK inhibitor SP600125 significantly reversed omeprazole-induced invasion, whereas p38, MAPK and Akt inhibitors SB203580, U0126 and LY294002, respectively, did not significantly attenuate invasion induced by these AhR-active pharmaceuticals. We also observed that omeprazole which activated JNK decreased Panc1 tumor sphere formation (Fig. 1F) and this contrasts to a report showing that a JNK inhibitor decreased cancer stem cells [22]. MKK4 and MKK7 are two upstream activators of JNK that have non-redundant functions in vivo [23], and we used wild-type (active) MKK7-JNK-WT chimera and mutant (inactive) MKK7-JNK-APF (Fig. 2A) [24] to determine their effects on kinase activation and Panc1 cell invasion. Only MKK7-JNK-WT activated p-JNK and p-cJun (Fig. 2B) and in invasion assays using these same constructs (Figs. 2C and 2D), only MKK7-JNK-WT inhibited Panc1 cell invasion. In contrast, overexpression of MKK4 did not activate JNK (data not shown). These results further confirm a ligand-activated AhR-JNK connection and that JNK activation is directly involved in the invasion of Panc1 cells.
Figure 2.
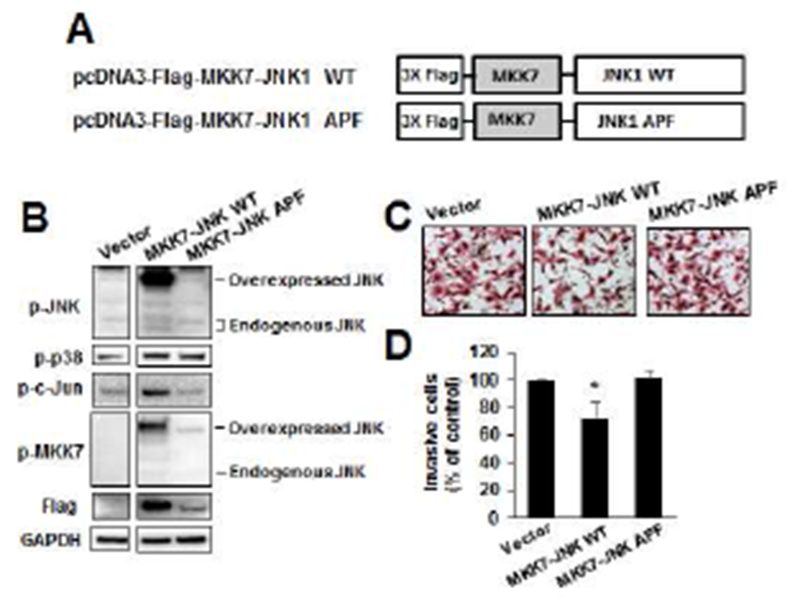
Direct effects of kinases on Panc1 cell invasion. A series of MKK4/7 flag-tagged constructs (A) were transfected into Panc1 cells, and their effects on kinase expression (B) were determined using whole cell lysates. Panc1 cells were transfected with the kinases and their effects on cell invasion were determined in Boyden chamber assays (C) and quantitated (D). Results are expressed as means ± SD for at least 3 replicated determinations for each treatment group, and significant (p<0.05) inhibition is indicated (*).
Omeprazole-induced activation of JNK which is important for Panc1 cell invasion (Figs. 1–2) involves the cytosolic AhR [15], and we therefore investigated possible factors that block nuclear accumulation of the AhR in highly invasive pancreatic cancer cells. We hypothesized that the AhR in Panc1 cells may be mutated in the region containing a nuclear localization signal or in other domains of the AhR; however, we sequenced all 11 exons and did not detect any mutation (Fig. 3A) and this complemented previous studies showing that overexpression of wild-type AhR did not result in nuclear uptake [15]. We then used two well-characterized assays that denote a functional nuclear AhR complex, namely induction of CYP1A1 and CYP1B1 (Figs. 3B and 3C) coupled with knockdown of various factors that could influence omeprazole-dependent induction of these genes. Omeprazole decreased basal CYP1A1 and CYP1B1 mRNA levels and effects of TCDD an inducer of these genes was minimal (<2-fold) in control cells (transfected with siCtl – a non-specific oligonucleotide) and previous studies showed that omeprazole-mediated effects on CYP1A1/CYP1B1 were AhR-independent [15]. Knockdown of Arnt (siArnt) decreased mRNA levels and the silencing of XAP2 (siXAP2) and Hsp90 (siHsp90) which bind to the cytosolic AhR did not affect induction of CYP1A1 or CYP1B1 by omeprazole. We also examined the effects of three additional genes that interact with the AhR, namely mitochondrial ribosomal protein 40 (MRPL40), eukaryotic translation elongation factor 1α1 (Eef1A1), and activated leukocyte cell adhesion molecule CD166 (ALCAM) [25]. Knockdown of these three factors also did not enhance CYP1A1/CYP1B1 induction, and currently we are investigating other factors expressed in Panc1 cells that inhibit nuclear translocation of the AhR. The efficiencies of gene knockdown by these oligonucleotides is summarized in Supplemental Figure 3.
Figure 3.
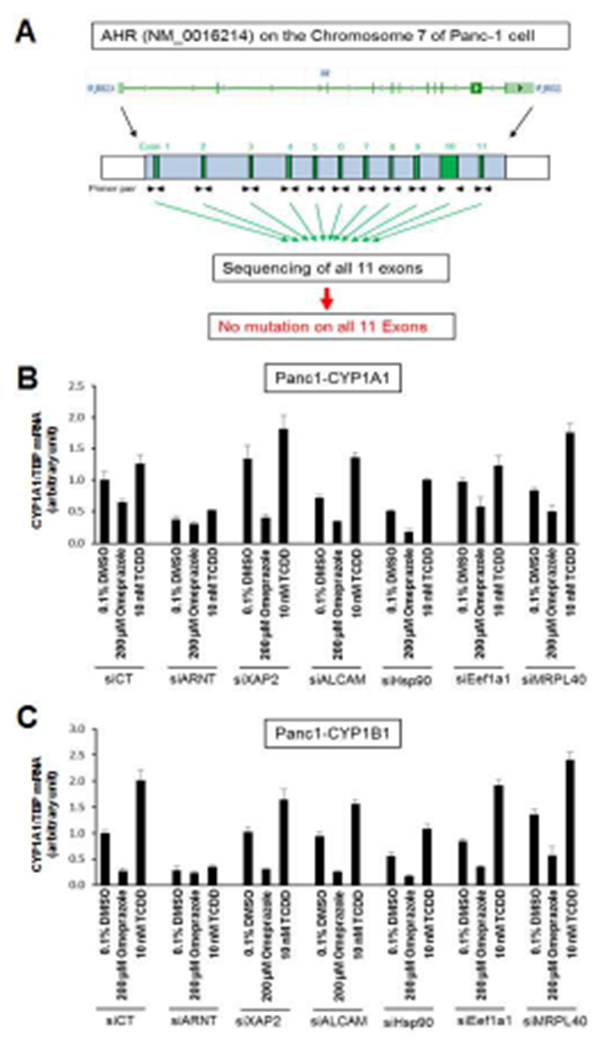
Factors affecting Ah-responsiveness. (A) Diagramatic illustration of the AhR gene and its 11 exons. Panc1 cells were transfected with a non-specific control oligonucleotide/DMSO and other small inhibitory RNAs, and restoration of nuclear AhR functions was determined by measuring changes in CYP1A1 (B) or CYP1B1 (C) expression by real time PCR. Results are expressed as means ± SD for at least 3 replicates determinations for each treatment group.
We also used RNAseq analysis to investigate AhR-regulated genes that were altered after knockdown of the AhR (siAhR) and also omeprazole-induced/repressed genes that are altered after AhR depletion by siRNA. We identified 343, 163 and 183 AhR/omeprazole (AhR-dependent)-regulated genes associated with total, metastasis and JNK cascade genes, and a heat map diagram for metastasis genes is illustrated in Figure 4A. Four of these genes, long chain fatty acyl-CoA synthetase-4 (ACSL4), ALCAM, stathmin-3 (STMN3), and neuropilin 2 (NRP2), were decreased by treatment with omeprazole and this response was attenuated after AhR knockdown (Fig. 4B). Four and a half LIM domains 2 (FHL2) and zyxin (ZYX), were induced by omeprazole, and the induced response was significantly inhibited by knockdown of the AhR. Knockdown efficiencies are summarized in Supplemental Figure 3. There is evidence that ACSL4, ALCAM, STMN3 and NRP2 are associated with migration or other pro-oncogenic activities [26–29] and downregulation of these genes by omeprazole is consistent with the anti-invasion effects observed for omeprazole. ZYX and FLH2 exhibit both tumor suppressive and tumor promoter activities [30–33], although the latter gene plays a role in pancreatic cancer cell radio-resistance [32]. Current studies are evaluating the relative contributions and mechanisms of induction of these and other omeprazole-regulated genes in pancreatic cancer.
Figure 4.
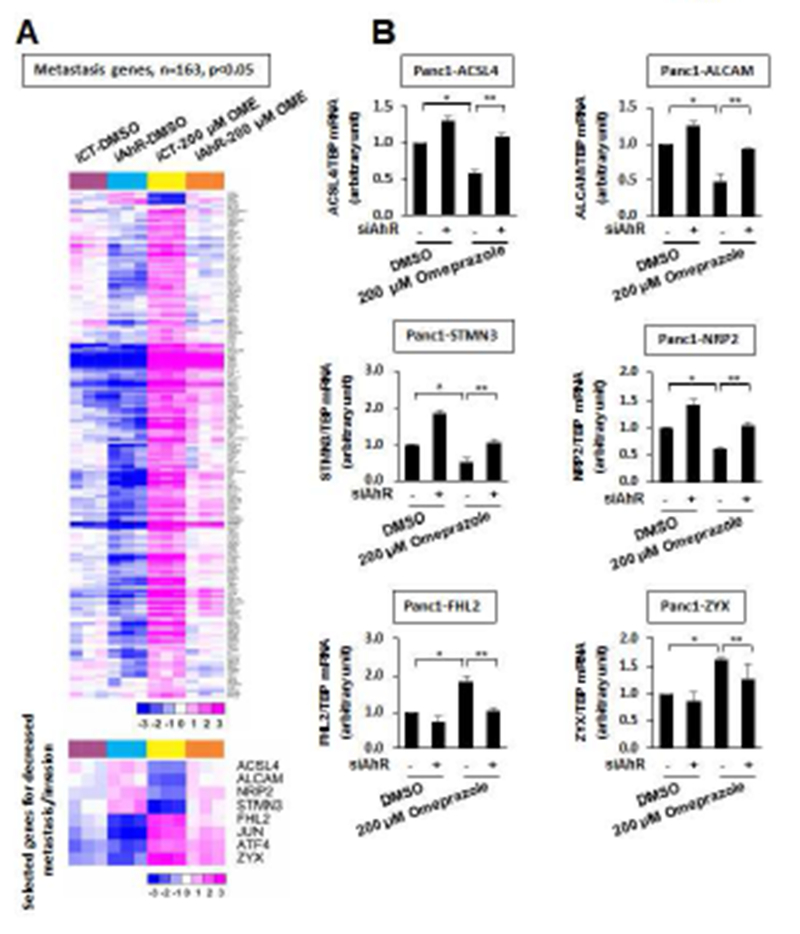
RNAseq analysis of “metastasis genes”. (A) Map illustrating changes in expression of metastasis genes in various treatment groups. (B) Real time PCR analysis of selected genes. Panc1 cells were treated with DMSO or omeprazole in the presence or absence of siAhR (AhR knockdown), and changes in gene expression were determined by real time PCR as outlined in the Materials and Methods. Results are expressed as means ± SD for at least 3 replicate determinations in each treatment group. Significant (p<0.05) effects by omeprazole (*) and reversal of the effects by AhR knockdown (**) are indicated.
4. DISCUSSION
The AhR plays a role in maintaining cellular homeostasis and various diseases and like other receptors, selective AhR modulators (SAhRMs) or ligands can be developed for applications as potential chemotherapeutic agents [2–4, 12]. For example, the AhR pathway plays a negative role in glioblastoma and there is evidence that kynurenine, a tryptophan metabolite, decreases immune surveillance and induces cancer cell growth via AhR-dependent pathways which can be inhibited by AhR antagonists [34, 35]. Research in this laboratory has focused on re-purposing AhR-active pharmaceuticals for clinical applications in cancer treatment since the drug approval process would be significantly shortened [13–15].
In previous studies, we showed that omeprazole inhibited estrogen receptor-negative breast cancer cell invasion through downregulation of the pro-oncogenic factor CXCR4 via the nuclear AhR complex [13]. The surprising observation regarding the inhibition of Panc1 cell invasion was that this response was dependent on the cytosolic AhR and independent of Arnt [15] and this non-genomic pathway was observed only in highly invasive quasimesenchymal pancreatic ductal adenoma (QM-PDA) cells (such as Panc1 cells) but not in less invasive cell lines (e.g. BxPC3, L3.6pL). Previous studies show that the cytosolic AhR acts, in part, by modulating kinases [8, 19, 20], suggesting that ligand-induced effects in Panc1 cells may be due to activation of one or more kinases and our results showed that activation of JNK was observed after treatment with omeprazole for 0.25 to 0.5 hr (Fig. 1). These data, coupled with the loss of drug-induced JNK activation after knockdown of the AhR and inhibition of drug-induced Panc1 cell invasion by the JNK inhibitor SP600125 demonstrate the role of JNK in mediating the AhR-dependent inhibition of invasion by omeprazole. In contrast, omeprazole did not activate JNK in BxPC3 pancreatic cancer cells (data not shown), a cell line that we previously showed formed a nuclear AhR complex [15].
Nuclear translocation of the AhR is dependent on multiple factors including the ligand and AhR structure, the ability to dissociate from AhR binding proteins such as XAP2 and Hsp90, and to form a heterodimer with Arnt [36–41]. We sequenced the 11 exons of the AhR from Panc1 cells and did not observe any mutations (Fig. 3A) and previously showed that Arnt could undergo hypoxia-induced nuclear uptake in Panc1 cells [15], suggesting that Arnt was not defective. Previous studies showed that overexpression of Hsp90 inhibited Ah-responsiveness and XAP2 played an important role in cytosolic to nuclear shuttling of the AhR and also Ah-responsiveness [36–41]. Therefore, we knocked down Arnt, Hsp90, XAP2 and other genes known to interact with the AhR [25] and observed that Ah-responsiveness (i.e. induction of CYP1A1/CYP1B1) was not restored (Fig. 3), and we are currently examining the roles of other factors expressed in Panc1 cells that affect intracellular shuttling of the AhR.
In summary, this study shows that omeprazole-induced inhibition of Panc1 cell invasion is due to AhR-dependent induction of JNK which in turn modulates expression of several genes and this includes inhibition of pro-oncogenic factors (Fig. 4). Although this mechanism is associated with highly invasion QM-PDA cancer types [15], the overall contributions of non-genomic vs. genomic AhR-regulated pathways in pancreatic cancer are unknown, even though this receptor is overexpressed in human pancreatic tumors [15]. Our current studies are focused on addressing these problems and this include identification of specific genes that block AhR nuclear uptake since these genes are key mechanistic links and may be druggable. There is evidence from some epidemiological studies that long term use of proton pump inhibitors increases the risk of several cancers including pancreatic cancer [42, 43]. However, a recent review [44] also points out that proton pump inhibitors used as part of a therapeutic regimen can be beneficial for several cancers and this is consistent with studies showing the anticancer activity of omeprazole in pancreatic and other cancers [13–15, 44, 45].
Supplementary Material
Highlights.
Omeprazole inhibition of Panc1 cell invasion is JNK- and AhR-dependent.
The non-genomic pathway is not due to AhR mutations.
Omeprazole-AhR-JNK modulates expression of invasion genes.
Acknowledgements:
The assistance of Jennifer Goldsby and Yating Cheng with bioinformatics is gratefully acknowledged.
Funding Sources: The financial assistance of Texas A&M AgriLife Research, the Sid Kyle Chair Endowment, and the National Institutes of Health (P30-ES023512 and R21-ES025839) is gratefully acknowledged.
Abbreviations:
- AhR
aryl hydrocarbon receptor
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- JNK
c-jun N-terminal kinase
- PAH
polynuclear aromatic hydrocarbons
- QM-PDA
quasimensenchymal pancreatic ductal adenocarcinoma
- SAhRMs
selective aryl hydrocarbon receptor modulators
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: None
LITERATURE CITED
- [1].Gu YZ, Hogenesch JB, Bradfield CA, The PAS superfamily: sensors of environmental and developmental signals, Annu. Rev. Pharmacol. Toxicol, 40 (2000) 519–561. [DOI] [PubMed] [Google Scholar]
- [2].Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B, Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor, Toxicol. Sci, 124 (2011) 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Safe S, Chadalapaka G, Jutooru I, AHR-reactive compounds in the human diet, in: Pohjanvirta R (Ed.) The Ah Receptor in Biology and Toxicology, John Wiler & Sons, Hoboken, NJ, 2012, pp. 331–342. [Google Scholar]
- [4].Hu W, Sorrentino C, Denison MS, Kolaja K, Fielden MR, Induction of cyp1a1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro, Mol. Pharmacol, 71 (2007) 1475–1486. [DOI] [PubMed] [Google Scholar]
- [5].Vogel CF, Matsumura F, A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-kappaB family, Biochem. Pharmacol, 77 (2009) 734–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Singh NP, Nagarkatti M, Nagarkatti PS, Role of dioxin response element and nuclear factor-kappaB motifs in 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated regulation of Fas and Fas ligand expression, Mol. Pharmacol, 71 (2007) 145–157. [DOI] [PubMed] [Google Scholar]
- [7].Huang G, Elferink CJ, A novel nonconsensus xenobiotic response element capable of mediating aryl hydrocarbon receptor-dependent gene expression, Mol. Pharmacol, 81 (2012) 338–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Blankenship A, Matsumura F, 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) causes an Ah receptor-dependent and ARNT-independent increase in membrane levels and activity of p60(Src), Environ. Toxicol. Pharmacol, 3 (1997) 211–220. [DOI] [PubMed] [Google Scholar]
- [9].Stevens EA, Mezrich JD, Bradfield CA, The aryl hydrocarbon receptor: a perspective on potential roles in the immune system, Immunology, 127 (2009) 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Marshall NB, Kerkvliet NI, Dioxin and immune regulation: emerging role of aryl hydrocarbon receptor in the generation of regulatory T cells, Ann. N. Y. Acad. Sci, 1183 (2010) 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Busbee PB, Rouse M, Nagarkatti M, Nagarkatti PS, Use of natural AhR ligands as potential therapeutic modalities against inflammatory disorders, Nutr. Rev., 71 (2013) 353–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Murray IA, Patterson AD, Perdew GH, Aryl hydrocarbon receptor ligands in cancer: friend and foe, Nat. Rev. Cancer, 14 (2014) 801–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jin UH, Lee SO, Pfent C, Safe S, The aryl hydrocarbon receptor ligand omeprazole inhibits breast cancer cell invasion and metastasis, BMC Cancer, 14 (2014) 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jin UH, Lee SO, Safe S, Aryl hydrocarbon receptor (AHR)-active pharmaceuticals are selective AHR modulators in MDA-MB-468 and BT474 breast cancer cells, J. Pharmacol. Exp. Ther, 343 (2012) 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jin UH, Kim SB, Safe S, Omeprazole inhibits pancreatic cancer cell invasion through a nongenomic aryl hydrocarbon receptor pathway, Chem. Res. Toxicol, 28 (2015) 907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL, TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions, Genome Biol, 14 (2013) R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L, Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation, Nat. Biotechnol, 28 (2010) 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol, 15 (2014) 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dong B, Matsumura F, The conversion of rapid TCCD nongenomic signals to persistent inflammatory effects via select protein kinases in MCF10A cells, Mol. Endocrinol, 23 (2009) 549–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tomkiewicz C, Herry L, Bui LC, Metayer C, Bourdeloux M, Barouki R, Coumoul X, The aryl hydrocarbon receptor regulates focal adhesion sites through a non-genomic FAK/Src pathway, Oncogene, 32 (2013) 1811–1820. [DOI] [PubMed] [Google Scholar]
- [21].Li W, Wu J, Li Z, Zhou Z, Zheng C, Lin L, Tan B, Huang M, Fan M, Melatonin induces cell apoptosis in Mia PaCa-2 cells via the suppression of nuclear factor-kappaB and activation of ERK and JNK: A novel therapeutic implication for pancreatic cancer, Oncol. Rep, 36 (2016) 2861–2867. [DOI] [PubMed] [Google Scholar]
- [22].Okada M, Kuramoto K, Takeda H, Watarai H, Sakaki H, Seino S, Seino M, Suzuki S, Kitanaka C, The novel JNK inhibitor AS602801 inhibits cancer stem cells in vitro and in vivo, Oncotarget, 7 (2016) 27021–27032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, Davis RJ, Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms, Science, 267 (1995) 682–685. [DOI] [PubMed] [Google Scholar]
- [24].Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, Bar-Sagi D, Davis RJ, The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase, Mol. Cell. Biol, 22 (2002) 4929–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tappenden DM, Hwang HJ, Yang L, Thomas RS, Lapres JJ, The Aryl-Hydrocarbon Receptor Protein Interaction Network (AHR-PIN) as Identified by Tandem Affinity Purification (TAP) and Mass Spectrometry, J. Toxicol., 2013 (2013) 279829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dutta S, Roy S, Polavaram NS, Stanton MJ, Zhang H, Bhola T, Honscheid P, Donohue TM Jr., Band H, Batra SK, Muders MH, Datta K, Neuropilin-2 Regulates Endosome Maturation and EGFR Trafficking to Support Cancer Cell Pathobiology, Cancer Res, 76 (2016) 418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nair S, Bora-Singhal N, Perumal D, Chellappan S, Nicotine-mediated invasion and migration of non-small cell lung carcinoma cells by modulating STMN3 and GSPT1 genes in an ID1-dependent manner, Mol. Cancer, 13 (2014) 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kahlert C, Weber H, Mogler C, Bergmann F, Schirmacher P, Kenngott HG, Matterne U, Mollberg N, Rahbari NN, Hinz U, Koch M, Aigner M, Weitz J, Increased expression of ALCAM/CD166 in pancreatic cancer is an independent prognostic marker for poor survival and early tumour relapse, Br. J. Cancer, 101 (2009) 457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chen WC, Wang CY, Hung YH, Weng TY, Yen MC, Lai MD, Systematic Analysis of Gene Expression Alterations and Clinical Outcomes for Long-Chain Acyl-Coenzyme A Synthetase Family in Cancer, PLoS One, 11 (2016) e0155660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yamamura M, Noguchi K, Nakano Y, Segawa E, Zushi Y, Takaoka K, Kishimoto H, Hashimoto-Tamaoki T, Urade M, Functional analysis of Zyxin in cell migration and invasive potential of oral squamous cell carcinoma cells, Int. J. Oncol, 42 (2013) 873–880. [DOI] [PubMed] [Google Scholar]
- [31].Amsellem V, Kryszke MH, Hervy M, Subra F, Athman R, Leh H, Brachet-Ducos C, Auclair C, The actin cytoskeleton-associated protein zyxin acts as a tumor suppressor in Ewing tumor cells, Exp. Cell Res, 304 (2005) 443–456. [DOI] [PubMed] [Google Scholar]
- [32].Zienert E, Eke I, Aust D, Cordes N, LIM-only protein FHL2 critically determines survival and radioresistance of pancreatic cancer cells, Cancer Lett, 364 (2015) 17–24. [DOI] [PubMed] [Google Scholar]
- [33].Verset L, Feys L, Trepant AL, De Wever O, Demetter P, FHL2: a scaffold protein of carcinogenesis, tumour-stroma interactions and treatment response, Histol. Histopathol, 31 (2016) 469–478. [DOI] [PubMed] [Google Scholar]
- [34].Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, Jugold M, Guillemin GJ, Miller CL, Lutz C, Radlwimmer B, Lehmann I, von Deimling A, Wick W, Platten M, An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor, Nature, 478 (2011) 197–203. [DOI] [PubMed] [Google Scholar]
- [35].Gramatzki D, Pantazis G, Schittenhelm J, Tabatabai G, Kohle C, Wick W, Schwarz M, Weller M, Tritschler I, Aryl hydrocarbon receptor inhibition downregulates the TGF-beta/Smad pathway in human glioblastoma cells, Oncogene, 28 (2009) 2593–2605. [DOI] [PubMed] [Google Scholar]
- [36].Meyer BK, Pray-Grant MG, Vanden Heuvel JP, Perdew GH, Hepatitis B virus X-associated protein 2 is a subunit of the unliganded aryl hydrocarbon receptor core complex and exhibits transcriptional enhancer activity, Mol. Cell. Biol, 18 (1998) 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Berg P, Pongratz I, Two parallel pathways mediate cytoplasmic localization of the dioxin (aryl hydrocarbon) receptor, J. Biol. Chem, 277 (2002) 32310–32319. [DOI] [PubMed] [Google Scholar]
- [38].Kazlauskas A, Poellinger L, Pongratz I, The immunophilin-like protein XAP2 regulates ubiquitination and subcellular localization of the dioxin receptor, J. Biol. Chem, 275 (2000) 41317–41324. [DOI] [PubMed] [Google Scholar]
- [39].Petrulis JR, Kusnadi A, Ramadoss P, Hollingshead B, Perdew GH, The hsp90 Co-chaperone XAP2 alters importin beta recognition of the bipartite nuclear localization signal of the Ah receptor and represses transcriptional activity, J. Biol. Chem, 278 (2003) 2677–2685. [DOI] [PubMed] [Google Scholar]
- [40].Ikuta T, Kobayashi Y, Kawajiri K, Phosphorylation of nuclear localization signal inhibits the ligand-dependent nuclear import of aryl hydrocarbon receptor, Biochem. Biophys. Res. Commun, 317 (2004) 545–550. [DOI] [PubMed] [Google Scholar]
- [41].Caruso JA, Laird DW, Batist G, Role of HSP90 in mediating cross-talk between the estrogen receptor and the Ah receptor signal transduction pathways, Biochem. Pharmacol, 58 (1999) 1395–1403. [DOI] [PubMed] [Google Scholar]
- [42].Lai SW, Sung FC, Lin CL, Liao KF, Use of proton pump inhibitors correlates with increased risk of pancreatic cancer: a case-control study in Taiwan, Kuwait Med. J, 46 (2014) 44–48. [Google Scholar]
- [43].Kearns MD, Boursi B, Yang YX, Proton pump inhibitors on pancreatic cancer risk and survival, Cancer Epidemiol., 46 (2017) 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Spugnini E, Fais S, Proton pump inhibition and cancer therapeutics: A specific tumor targeting or it is a phenomenon secondary to a systemic buffering?, Semin. Cancer Biol, 43 (2017) 111–118. [DOI] [PubMed] [Google Scholar]
- [45].Udelnow A, Kreyes A, Ellinger S, Landfester K, Walther P, Klapperstueck T, Wohlrab J, Henne-Bruns D, Knippschild U, Wurl P, Omeprazole inhibits proliferation and modulates autophagy in pancreatic cancer cells, PLoS One, 6 (2011) e20143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.