

Summary
Although PD-1-blocking immunotherapies demonstrate significant therapeutic promise, a subset of the patients could develop hyperprogressive disease (HPD) with accelerated tumor growth after anti-PD1 immunotherapy. To elucidate the underlying mechanisms, we compared the mutational and transcriptional landscapes between the pre- and post-therapy tumors of two patients developing HPD after anti-PD-1 immunotherapy. In post-therapy HPD tumors, somatic mutations were found in known cancer genes, including tumor suppressor genes such as TSC2 and VHL, along with transcriptional upregulation of oncogenic pathways, including IGF-1, ERK/MAPK, PI3K/AKT, and TGF-β. We found that post-therapy HPD tumors were less immunogenic than pre-therapy tumors, concurrent with an increased presence of ILC3 cells, a subset of innate lymphoid cells. We also developed a gene expression signature predictive of HPD. In summary, we identified the genomics and immune features associated with HPD, which may help identify patients at risk of adverse clinical outcome after anti-PD-1 immunotherapy.
Subject Areas: Physiology (170590663/189723279, ), Immunology (186131996Physiology, Biotechnology, Cell Biology, Omics
Graphical Abstract
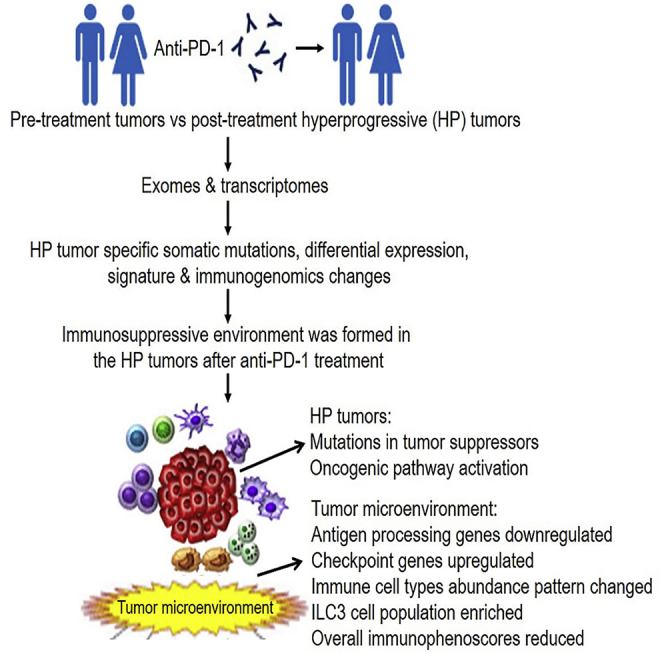
Highlights
-
•
Mutations/expression changes occur in hyperprogressive tumors after anti-PD-1 therapy
-
•
Immune cell population abundance pattern changed in the hyperprogressive tumors
-
•
ILC3 cells may be enriched in the hyperprogressive tumors after anti-PD-1 therapy
-
•
Post-therapy hyperprogressive tumors were less immunogenic than pre-therapy tumors
Physiology (170590663/189723279); Immunology (186131996Physiology; Biotechnology; Cell Biology; Omics
Introduction
Immune checkpoint therapies including those targeting PD-1, or its primary ligand PD-L1, have demonstrated therapeutic responses across a broad range of cancer types (Sharma and Allison, 2015). Anti-PD-1 therapy blocks the interaction of PD-1, an inhibitory receptor on tumor-infiltrating T cells, with its ligands PD-L1 and PD-L2 that are predominantly expressed on tumor cells and antigen-presenting cells (APCs), respectively (Topalian et al., 2012). Despite the success of anti-PD-1 immunotherapy in approximately 20%–30% of patients with cancer, the majority of patients do not respond to this treatment (Sharma et al., 2017). In addition, increasing clinical evidence suggests that a significant subset of nonresponsive patients may experience acceleration of disease progression after treatment with anti-PD-1, a phenomenon known as hyperprogressive disease (HPD). Although accurate identification of the frequency of patients developing HPD has been limited by variability in diagnostic criteria, conservative estimates suggest that HPD may occur in as many as 10% of patients treated with anti-PD-1 (Champiat et al., 2017, Kato et al., 2017, Saada-Bouzid et al., 2017).
In contrast to identifying factors that predict responsiveness to PD-1-blocking therapies such as tumor expression of PD-L1, high tumor mutational burden, and the presence of tumor-infiltrating CD8+ T cells, little is known about the mechanisms underlying HPD. Although a pilot study suggested that some patients with MDM2 family amplification or EGFR aberrations developed HPD after treatment with PD-1 or PD-L1 inhibitors (Kato et al., 2017), it is likely that alterations beyond those identified in that study are important in facilitating accelerated disease progression.
To comprehensively examine the mechanisms of HPD, we performed whole-exome sequencing (WES) and RNA sequencing (RNA-seq) analyses of formalin-fixed paraffin-embedded (FFPE) samples of tumors before and after anti-PD-1 therapy in patients with clinical evidence of HPD. We identified individual somatic mutations and mutation clusters associated with clonal evolution that may contribute to the accelerated tumor growth observed in HPD. We also identified characteristic decreases in HPD tumor immunogenicity. Finally, we identified a gene signature that may be predictive of HPD development. These changes were HPD patient specific, and were not found in the tumors of anti-PD-1-treated patients without HPD phenotypes from previous studies. Overall, our study identified the genomics and immune features associated with HPD tumors after anti-PD-1 immunotherapy.
Results
Mutation Patterns Are Altered in HPD Tumors after Anti-PD-1 Treatment
This study included two patients who received anti-PD-1 blockade immunotherapy. Relevant characteristics of the four FFPE tumor samples are summarized in Table 1. Paired tumor samples before and after anti-PD-1 treatment were obtained from a male patient with esophageal squamous cell carcinoma metastatic to lymph nodes (Patient 1) and from a female patient with clear cell renal cell cancer (ccRCC) that had metastasized to the bone (shoulder) and pleura (Patient 2). Following anti-PD-1 treatment that consisted of pembrolizumab (Merck), these two patients demonstrated HPD, as defined by the accelerated tumor growth rate and clinical deterioration using existing criteria (Kato et al., 2017). Each patient demonstrated progression at first radiologic evaluation (less than 2 months after anti-PD-1 therapy initiation). Before enrollment, written informed consent was obtained from all patients to use their tumor samples for research purposes. The study was approved by the Medical College of Wisconsin Institutional Review Board in accordance with federal regulations.
Table 1.
Characteristics of the Four FFPE Specimens from Two Patients, Consisting of Paired Pre- and Post-anti-PD-1 (Pembrolizumab) Treatment Samples
| Patient | Gender | Specimen | Cancer | Treatment | Other Clinical Phenotype | % Tumor |
|---|---|---|---|---|---|---|
| #1 | Male | S1624794 | Esophageal squamous cell carcinoma | Pre-anti-PD-1 | Metastatic to lymph node | 75 |
| S1707359 | Esophageal squamous cell carcinoma | Post-anti-PD-1 | Metastatic to lymph node | 75 | ||
| #2 | Female | M16248 | Clear cell renal cell carcinoma | Pre-anti-PD-1 | Metastatic to the pleura and shoulder | 50 |
| S1701860 | Clear cell renal cell carcinoma | Post-anti-PD-1 | Metastatic to the pleura and shoulder | 75 |
To understand the global changes that take place in HPD tumors after treatment with anti-PD-1, we performed mutational analysis on tumors obtained before and after treatment with pembrolizumab. We observed that Patient 1 had 195 somatic mutations before anti-PD-1 treatment and 338 somatic mutations after treatment; Patient 2 had 156 somatic mutations before treatment and 251 somatic mutations after treatment (Table S1). There were 154 and 124 common somatic mutations shared by the HPD and pre-therapy tumors for Patients 1 and 2, respectively (Figure S1). Our results were in line with another group's results showing increased tumor mutation load from baseline in PD (progressive disease) in patients with melanoma after anti-PD-1 therapy (nivolumab) initiation (Riaz et al., 2017). In the latter, the tumor mutation load was decreased in the responding patients (complete response/partial response) from baseline since nivolumab initiation, consistent with immunoediting (Riaz et al., 2017). We also analyzed the mutation profiles of these two patients in the context of known cancer genes based on a comprehensive list of cancer-related genes (downloaded from http://www.bushmanlab.org/links/genelists). There were 47 cancer genes mutated in at least one of the tumors from Patient 1 and 40 cancer genes mutated in at least one of the tumors from Patient 2 (Figure 1, Table S2). Four cancer genes (APH1A, ARHGEF12, GPER1, and KIF14) mutated in the pre-therapy tumor of Patient 1 were not mutated in the HPD tumors, suggesting that the tumor clones containing these four cancer genes were eliminated by anti-PD-1 treatment. However, the HPD tumor of Patient 1 had somatic mutations in 20 cancer genes, including IGFBP2, KMT2C, MAP3K4, MUC16, MUC2, NCOR2, and NOTCH4, which were not present in the pre-therapy tumors. Similar patterns were also observed for Patient 2. Four cancer genes (APC2, OBSCN, PHLPP1, and SATB1) that were mutated in the pre-therapy tumor of Patient 2 were not mutated in the post-treatment tumors, whereas the HPD tumor of Patient 2 had somatic mutations in 21 cancer genes, including IGFBP2, MUC4, NCOR2, NFE2L2, TSC2, and VHL, which were not present in the pre-therapy tumors. The identified mutations in these genes were not present in the tumors of non-HPD patients after anti-PD-1 treatment when compared with previous studies (Biton et al., 2018, Gong et al., 2017, Hanna et al., 2018, Hugo et al., 2016, Miao et al., 2018, Riaz et al., 2017, Rizvi et al., 2015, Teo et al., 2018, Yoshikawa et al., 2017, Zaretsky et al., 2016). These data indicate that the mutational landscape of tumors was significantly altered after anti-PD-1 therapy in patients who demonstrated hyperprogression after anti-PD-1 treatment.
Figure 1.

Profiles of Mutated Cancer Genes (Nonsilent Somatic Mutations) in the Pre- and Post-anti-PD-1 Treatment Tumor Samples
(A) Mutation pattern of Patient 1.
(B) Mutation pattern of Patient 2. Indel: insertions or deletions.
For comparison in the context of corresponding cancer populations, we analyzed the numbers of somatic mutations of the esophageal carcinoma (ESCA, n = 184) and kidney renal clear cell carcinoma (KIRC, n = 384) samples from The Cancer Genome Atlas (TCGA). The numbers of nonsilent somatic mutations were in the range of 4–1,763 for ESCA and 15–1,349 for KIRC. The lower quartile, median, and upper quartile were 85, 110, and 168 for ESCA and 54, 77, and 109 for KIRC, respectively (Figure S2). The numbers of nonsilent somatic mutations in the before and after anti-PD-1 therapy tumors of the two HPD patients in this study were 195 and 338 for the patient with ESCA and 156 and 251 for the patient with KIRC. Therefore, they were all above the upper quartiles of TCGA ESCA and KIRC datasets, which suggested that these two patients have an exceptionally high number of somatic mutations compared with the TCGA esophageal cancer (ESCA) and ccRCC (KIRC).
HPD Tumors Contain Deleterious Mutations and Significantly Activated Oncogenic Signaling Pathways
To determine if certain genes were altered in both patients with HPD tumors, we searched for gene mutations that were common for the HPD tumors of both patients. Four genes were mutated in the post-treatment tumors of both patients: NCOR2, GXYLT1, ZFPM1, and IGFBP2 (Figure 2A). There were 96 and 64 subject-specific nonsilent somatic mutations from 154 genes in post-treatment tumors of Patients 1 and 2, respectively (Figures 2B and 2C). The detailed information of these mutations are given in Table S3.
Figure 2.

Mutation Signatures of Post-anti-PD-1 Treatment Hyperprogressor Tumors
(A–C) (A) Commonly mutated genes in the two hyperprogressor tumors, (B) specific mutated genes in Patient 1's hyperprogressor tumor, and (C) specific mutated genes in Patient 2's hyperprogressor tumor. See also Figure S1, Tables S1 and S3.
Bioinformatics analyses of these 161 mutations led to the identification of 11 potentially deleterious somatic variants in the HPD tumors, which were predicted to be “deleterious” by SIFT, “probably damaging” by PolyPhen-2, and “potentially associated with cancer” by FATHMM (Table 2). The 11 genes having these deleterious mutations were TRPC4, POTEE, FBN2, KMT2C, FUT10, PQBP1, TSC2, MFSD6, CYP2D6, VHL, and RAD54B. Of the 11 mutations, 10 were located at evolutionarily conserved sites, as predicted by GERP++ (scores >2; Table 2). IPA (Ingenuity Pathway Analysis, Qiagen Inc., MD, USA), based on the 11 genes with the deleterious somatic mutations, identified a network involving these mutated genes that contributes to suppression of the TP53 tumor suppressor and activation of MYC, CCND1, and VEGF oncogenes (Figure S3). The mutated TSC2 gene carrying a missense mutation, p.Y1611S, was in the center of this network and is linked to inhibition of the TP53 pathway and activation of the MYC, CCND1, and VEGF pathways (Figure S3). TSC2 (also known as TUBERIN) is a tumor suppressor that negatively regulates cellular signaling networks that control cellular growth and proliferation (Dang et al., 2017). The MuPIT interactive protein mutation analysis (Niknafs et al., 2013) showed that the pY1611S mutation is located in the Rap/ran-GAP domain of the TSC2 protein, which is critical for the biological function of TSC2 (Figure S4). Previous studies showed that TSC2 knockdown transforms mouse and human renal epithelial cells into neoplastic stem cells that can serially propagate upon re-inoculation in mice (Dang et al., 2017). Together, it is reasonable to hypothesize that the deleterious p.Y1611S mutation could result in the loss of function of the TSC2 protein, which in turn will lead to uncontrolled proliferation of cancer cells in the HPD tumors that survive anti-PD-1 treatment.
Table 2.
Characteristics of the 11 Deleterious Somatic Mutations in the HPD Tumors after Anti-PD-1 Treatment
| Gene | Genomic Positiona | Genomic Mutation | Exon | Protein Alteration | Predicted Effect of Somatic Mutation |
snp137 | ESP MAFf | |||
|---|---|---|---|---|---|---|---|---|---|---|
| SIFTb | PolyPhen-2c | FATHMMd | GERP++e | |||||||
| TRPC4 | chr13: 38211734 | c.G2045A | 10 | p.R682H | Deleterious (0.00) | Probably damaging (0.999) | Potentially associated with cancer (−2.83) | 6.06 | NA | NA |
| POTEE | chr2: 132021334 | c.A2306T | 15 | p.Y769F | Deleterious (0.00) | Probably damaging (0.997) | Potentially associated with cancer (−4.69) | NA | NA | NA |
| FBN2 | chr5: 127666313 | c.C4297T | 33 | p.R1433C | Deleterious (0.00) | Probably damaging (0.983) | Potentially associated with cancer (−2.9) | 4.21 | NA | 7.70 × 10−5 |
| KMT2C | chr7: 151932981 | c.G2690C | 16 | p.R897P | Deleterious (0.00) | Probably damaging (0.995) | Potentially associated with cancer (−2.21) | 5.1 | NA | NA |
| FUT10 | chr8: 33246817 | c.G876T | 4 | p.K292N | Deleterious (0.00) | Probably damaging (1.00) | Potentially associated with cancer (−4.75) | 3.42 | NA | NA |
| PQBP1 | chrX: 48759773 | c.C256T | 4 | p.P86S | Deleterious (0.00) | Probably damaging (0.996) | Potentially associated with cancer (−1.13) | 5.02 | NA | NA |
| TSC2 | chr16: 2137907 | c.A4832C | 37 | p.Y1611S | Deleterious (0.02) | Probably damaging (0.997) | Potentially associated with cancer (−3.16) | 4.59 | NA | NA |
| MFSD6 | chr2: 191301728 | c.G973A | 3 | p.G325R | Deleterious (0.00) | Probably damaging (0.998) | Potentially associated with cancer (−2.42) | 6.07 | NA | NA |
| CYP2D6 | chr22: 42522990 | c.C1025T | 7 | p.T342M | Deleterious (0.00) | Probably damaging (0.996) | Potentially associated with cancer (−2.26) | 4.06 | NA | NA |
| VHL | chr3: 10191479 | c.C349G | 2 | p.L117V | Deleterious (0.00) | Probably damaging (0.994) | Potentially associated with cancer (−6.95) | 3.07 | NA | NA |
| RAD54B | chr8: 95411747 | c.T721G | 6 | p.F241V | Deleterious (0.01) | Probably damaging (0.996) | Potentially associated with cancer (−3.01) | 5.55 | NA | NA |
ESP, NHLBI Exome Sequencing Project; NA, not available. See also Figure S3.
Genomic positions are given according to the UCSC Genome Browser hg19 reference assembly.
SIFT scores range from 0 to 1. The amino acid substitution is predicted to be damaging if the score is ≤0.05 and tolerated if the score is >0.05.
PolyPhen-2 scores 0.85–1 are interpreted as probably damaging, scores 0.2–0.85 are possibly damaging, and sores 0–0.2 are benign.
Predictions with FATHMM scores less than 0.75 indicate that the mutation is potentially associated with cancer; otherwise the mutation is not associated with cancer.
There is an indication of evolutionary conservation if a given site shows a GERP++ score >2.
MAFs are according to the NHLBI GO Exome Sequencing Project (ESP6500SI-V2 release) Exome Variant Server v.0.0.21 (August 2013).
Based on the differentially expressed genes, IPA identified four significantly activated oncogenic signaling pathways in the HPD tumors after anti-PD-1 therapy compared with the pre-therapy tumors (p value <0.01, Z score >2, Figure 3A). They were the insulin growth factor (IGF)-1, extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK), Phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/AKT, and transforming growth factor (TGF)-β signaling pathways. A large number of genes in these oncogenic pathways were upregulated in the HPD tumors (Figure 3B). Such concerted gene expression changes may synergistically contribute to the generation of the HPD tumors after anti-PD-1 immunotherapy.
Figure 3.

Activation of Oncogenic Pathways in HP Tumors after Anti-PD-1 Therapy
(A and B) (A) Four oncogenic pathways were activated in the HP tumors. (B) The differentially expressed genes in these oncogenic pathways. Most of the genes were upregulated in the HP tumors after anti-PD-1 therapy. HP, hyperprogressive.
Clonal Evolution Was Detected in HPD Tumors after Anti-PD-1 Therapy
The generation of WES data allowed us to quantify the mutant allele frequencies in all cases. Based on mutation clustering results, we inferred the identity of three clones having distinct sets of mutations (clusters) in pre-therapy tumors when compared with post-therapy HPD tumors of the two patients. Multiple mutation clusters (n = 3) were present in each of the pre-therapy tumors of the two HPD patients. In Patient 1, the post-anti-PD-1 treatment HPD tumor was associated with the outgrowth of new clone(s) represented by mutations in cancer-associated genes including KMT2C, NCOR2, COL28A1, ING3, CAMKK2, and CARD8 (Figures 4A and 4C). The pre-therapy tumor clone(s) characterized by mutations in APH1A, ARHGEF12, GPER1, and KIF14 genes was eliminated by anti-PD-1 treatment (Figures 4A and 4C). The clone(s) represented by mutations in the cancer genes EP400, CUBN, SPP1, PHLPP2, PALB2, ERCC1, TFRC, MARK4, and MDM4 remained stable under the selection pressure of anti-PD-1 treatment (Figures 4A and 4C). In Patient 2, the post-anti-PD-1 treatment HPD tumor was associated with the evolution of new clone(s) represented by mutations in the cancer genes including BAP1, CARD11, CBFA2T3, CYP2D6, PBRM1, TSC2, and VHL (Figures 4B and 4D), whereas the pre-therapy tumor clone with mutations in APC2, CDC27, OBSCN, PHLPP1, and SATB1 was not detectable after anti-PD-1 treatment (Figures 4B and 4D). Other clones, including those represented by mutations in COL4A3, TTC40, NPHS1, UGT2A3, RYR1, AGGF1, and LANCL1, remained stable before and after anti-PD-1 treatment (Figures 4B and 4D).
Figure 4.

Illustration of Clonal Evolution of the Tumors before and after Anti-PD-1 Immunotherapy of the Two Patients with HPD
(A–D) (A) Tumor clonal evolution in Patient 1 and (B) tumor clonal evolution in Patient 2. The gray area denotes the tumor clones unaffected by anti-PD-1 therapy, the green and blue areas denote the tumor clones diminishing and appearing due to anti-PD-1 therapy. The dynamics of these clones represented by changes in the variant allele frequency between the pre- and post-therapy tumors was plotted for (C) Patient 1 and (D) Patient 2. See also Figures S5 and S6.
The tumor clonal evolution pattern associated with anti-PD-1 treatment was further validated by analyzing an independent dataset from a previous study, which conducted WES of paired baseline and relapsed tumors (before and after anti-PD-1 treatment) of four patients with melanoma (Zaretsky et al., 2016). As can be seen from Figures S5 and S6, all four melanoma cases demonstrated allele clusters after anti-PD-1 therapy. Variant allele frequencies (VAFs) of the Cluster 1 mutations were not significantly changed by PD-1 blockade; Cluster 2 mutations had reduced VAFs but were still prevalent in the relapsing tumor after PD-1 blockade; Cluster 3 mutations represented the newly evolved tumor clone(s) in the relapsing tumor after PD-1 blockade; Cluster 4 mutated genes represented the tumor clone(s) that diminished to undetectable levels after PD-1 blockade. These data are consistent with our own analysis of tumors from HPD patients before and after anti-PD-1 therapy.
HPD Tumors Demonstrate Decreased Immunogenicity Relative to Pre-therapy Tumors
Since anti-PD-1 treatment renders its effects on tumors in a manner completely dependent on immunity, we investigated whether HPD tumors demonstrated changes in their capacity to elicit productive immune reactions using an in silico immunophenogram approach (Charoentong et al., 2017). The results showed that HPD tumors had much smaller immunophenoscores compared with the pre-therapy tumors for both patients (Figure 5). Expression of HLAs (human leukocyte antigens) was downregulated in the post-therapy HPD tumors compared with the pre-therapy tumors, whereas checkpoint genes were upregulated in the HPD tumors (Figure 5). These changes resulted in the overall reduction of immunophenoscores in HPD tumors. Consistent with results from the immunophenogram analysis, the differential expression analysis showed that seven genes involved in antigen processing were downregulated in the HPD tumors, i.e., B2M, HLA-B, HLA-DPA1, HLA-DPB1, HLA-DRA, HLA-E, and HLA-F (Figure 6A). In addition, eight genes encoding immune checkpoints or modulators were upregulated in the HPD tumors, i.e., CTLA4, KDR, CD96, CD70, TNFRSF18, TNFRSF25, BTNL2, and TNFRSF8 (Figure 6A). Changes in expression of these immune-related genes were likely contributors to the weakened immunogenicity of the HPD tumors.
Figure 5.

Immunophenoscores of the Hyperprogressor versus Non-hyperprogressor Tumors of the Two Patients Subject to Anti-PD-1 Immunotherapy
HLAs were downregulated in the HPD tumors compared with the pre-therapy tumors (shown in the upper left quadrant termed MHC), whereas checkpoints were upregulated in the HPD tumors (shown in the lower left quadrant termed CP). These changes resulted in the overall reduction of immunophenoscores in the HPD tumors resistant to anti-PD-1 immunotherapy. See also Figure S14.
Figure 6.

Changes in the Expression of Critical Immune-Related Genes and the Activity of Immune Cell Populations Contribute to Decreased Immunogenicity in the Post-α-PD-1 HPD Tumors
(A) Seven genes involved in antigen processing were downregulated, whereas eight genes encoding immune checkpoints or modulators were upregulated in hyperprogressor tumors.
(B) The activity of eight immune cell populations were weakened and three were strengthened, as detected by GSVA method.
See also Figure S14.
Immune Cell Signatures in HPD Tumors Are Predominately Immunosuppressive
Previous studies have characterized the signature genes of 28 immune cell populations critical to immune responses across multiple cancers (Angelova et al., 2015, Charoentong et al., 2017). Using GSVA (Gene Set Variation Analysis) (Hanzelmann et al., 2013), we evaluated the immune cell landscape in the HPD tumors from our two patients. We identified that the activities of eight immune cell populations were significantly decreased in the HPD tumors after anti-PD-1 treatment (Figure 6B). These populations were monocytes, central memory CD4 T cells, immature dendritic cells, CD56dim NK (natural killer) cells, NK cells, gamma-delta (γδ) T cells, activated dendritic cells, and follicular helper T cells, most of which are linked to functional tumor clearance. In addition, the activities of three immune cell populations, i.e., neutrophils, activated B cells, and neutrophil-like myeloid-derived suppressor cells (MDSC), were upregulated in the hyperprogressors (Figure 6B). These data suggest that the depletion of monocytes, certain types of T cells, NK cells, and dendritic cells may contribute to the ability of HPD tumors to escape immune surveillance. Furthermore, the upregulated neutrophil population as well as the neutrophil-like MDSC (i.e., the MDSC subpopulation with neutrophil signature gene expression) (Zhang et al., 2017) may also contribute to the immune evasion of HPD tumors since these cell populations have been implicated in generating a milieu that attenuates immune responses in the tumor microenvironment (Galdiero et al., 2013, Mishalian et al., 2013, Sagiv et al., 2015, Tuting and de Visser, 2016, Zhang et al., 2017).
ILC3 Innate Lymphocytes Are Upregulated in HPD Tumors
Recent studies have revealed the importance of innate lymphoid cells (ILCs) in homeostasis and inflammation of tumors (Bjorklund et al., 2016, Wallrapp et al., 2017). Although three main populations of ILCs, ILC1, ILC2, and ILC3, have been categorized based on their transcription factor profiles and secreted cytokines (Spits et al., 2013), little is known about their roles in carcinogenesis and immunotherapy resistance. To evaluate ILCs in HPD tumors, we analyzed the transcriptional levels of the marker genes characteristic of the ILC1, ILC2, and ILC3 populations (Bjorklund et al., 2016, Wallrapp et al., 2017). GSEA (Subramanian et al., 2005) showed that the ILC3 marker genes were significantly enriched among the top upregulated genes in the HPD tumors after anti-PD-1 treatment (Figures 7A and 7B). In contrast, the ILC1 and ILC2 marker genes were not enriched in either the up- or downregulated genes in the HPD tumors (Figure S7). These data suggest that the ILC3 population is activated in HPD tumors. To validate this finding, we analyzed the RNA-seq data from other studies that evaluated tumor changes in response to anti-PD-1 therapies. Analysis of the transcriptomes of responding (n = 15) and nonresponding (n = 13) pre-treatment melanoma tumors from the patients subject to PD-1 blockade (Hugo et al., 2016) showed that ILC3 marker genes were commonly upregulated in the melanoma tumors resistant to anti-PD-1 therapy (Figure 7C). Based on the RNA-seq data of the KrasG12D mouse model, we also found that there were a large number of ILC3 marker genes significantly upregulated in murine lung adenocarcinoma tumors that were resistant to anti-PD-1 therapy when compared with untreated tumors (Koyama et al., 2016) (Figure 7D). These results are concordant with our HPD RNA-seq data, suggesting that enrichment of the ILC3 population in the HPD tumors may be a characteristic feature of tumors that are insensitive to anti-PD-1. This finding is consistent with the previous report that ILC3 lymphocytes contribute to the initiation and progression of cancers (Fung et al., 2017). The mechanistic connection between ILC3 population and anti-PD-1 therapy effect is unknown. However, it was reported that ILC3 may promote the growth of mutant tumor cells that express the receptors needed for oncogenic pathways (Fung et al., 2017, Kirchberger et al., 2013). Our and others' data (Riaz et al., 2017) suggested that anti-PD-1 therapy increased tumor mutation burden in patients with cancer with hyperprogressive or progressive tumor phenotype. Therefore, activated ILC3 cell population may be required for the promotion of the growth of more mutant cells in the patients with cancer with HPD or PD subjected to anti-PD-1 therapy.
Figure 7.

The ILC3 Population Was Activated in the HPD Tumors after Anti-PD-1 Therapy
(A–D) (A) GSEA showed that ILC3 marker genes were significantly enriched in the top upregulated genes in HPD tumors resistant to anti-PD-1 therapy. (B) Most of the differentially expressed ILC3 marker genes in the HPD tumors resistant to anti-PD-1 treatment were upregulated. (C) A higher percentage of ILC3 marker genes were upregulated in the nonresponding melanoma tumors resistant to anti-PD-1 therapy based on the analysis of data from an independent study in humans. (D) Upregulation of ILC3 marker genes comparing anti-PD-1-treatment-resistant mouse tumors with untreated tumors in the KrasG12D mouse model.
Pro-inflammatory Pathways Were Activated in the Pre-therapy Tumors of Patients with HPD and Further Activated by Anti-PD-1 Therapy
PD-1 has been demonstrated to inhibit excessive inflammatory responses during infection in mouse models (Lazar-Molnar et al., 2010). To identify the inflammatory changes in HPD tumors, we evaluated changes in inflammatory-related genes included in the “hallmark inflammatory” gene set (Liberzon et al., 2011, Liberzon et al., 2015). To characterize the inflammation activity in post-anti-PD-1 treatment HPD tumors versus pre-treatment tumors, we again utilized GSVA, which identified four founder datasets of inflammation pathways that were significantly enhanced in the HPD tumors after anti-PD-1 treatment (Figure 8A). In each of these four pro-inflammatory datasets, many more genes were up- than downregulated (Figures 8B–8E), suggesting an overall pro-inflammatory trend after anti-PD-1 treatment.
Figure 8.

Activation of Inflammatory Pathways in the HPD Tumors after Anti-PD-1 Treatment
(A) GSVA identified the activation of four founder datasets of inflammation pathways.
(B) Differentially expressed genes in the inflammatory signature of RESPONSE_TO_CHEMICAL_STIMULUS; (C) Differentially expressed genes in the inflammatory signature of KEGG_CHEMOKINE_SIGNALING_PATHWAY; (D) Differentially expressed genes in the inflammatory signature of INFLAMMATORY_RESPONSE; (E) Differentially expressed genes in the inflammatory signature of CHEMOKINE_ACTIVITY. In each of the four pro-inflammatory datasets from (B–E), there were much more upregulated than downregulated genes. See also Figure S8.
For comparison, we analyzed the gene expression data of tumor samples from the GSE52562 dataset before anti-PD-1 treatment (Westin et al., 2014). This dataset included two potential HPD patients whose progression-free survival (PFS) was less than 2 months post-pidilizumab treatment (SAMPLE.25 and SAMPLE.5 in Table S4) and four responsive patients whose PFS was more than 2 years (24 months) after treatment (SAMPLE.23, SAMPLE.19, SAMPLE.13, and SAMPLE.17 in Table S4). This analysis showed that the tumors of HPD patients have elevated inflammation pathway activity (mainly chemokine activity) even before anti-PD-1 therapy when compared with tumors from non-HPD patients (Figure S8). These and our data collectively suggested that anti-PD-1 therapy further boosts the pre-existing high levels of inflammation in patients who subsequently develop HPD in ways that are not conducive to promoting tumor rejection.
HPD-Associated Gene Expression Signature
Based on the pre-therapy tumor expression data of Dataset_1 (See Transparent Methods), we developed a 121-gene set to differentiate HPD patients from non-HPD patients (Figure S9, Table S5). The effectiveness of this 121-gene classifier in the identification of HPD patients was tested using the pre-therapy tumor expression data from Dataset_2 (See Transparent Methods). This classifier had an area under curve (AUC) value of 0.91 (95% confidence interval [CI], 0.87–0.96), a sensitivity of 71% (95% CI, 51%–87%), and a specificity of 93% (95% CI, 80%–99%) in predicting HPD patients in Dataset_2 (Figure 9A). Kaplan-Meier analysis of TCGA data showed that the 121-gene expression signature can significantly separate low-risk group from high-risk group in the 13 major types of cancers including melanoma (SKCM), glioma, and carcinomas of the esophagus (ESCA), stomach (STAD), breast (BRCA), kidney (KIRC), bladder (BLCA), liver (LIHC), head and neck (HNSC), lung (LUAD and LUSC), colon (COAD), and pancreas (PAAD) (Figures 9B–9D and S10–S12). This panel was able to identify extremely high-risk groups in ESCA, COAD, and PAAD (Figures 9B–9D).
Figure 9.

Performance of the 121-Gene Set Classifier in the Validation Dataset and Its Effectiveness in the Prognosis of Worse Survival Outcome in the TCGA Datasets
(A–D) (A) Receiver operating characteristic (ROC) curves shown for separating HPD patients from non-HPD patients in the validation dataset (Dataset_2, 21 HPD versus 30 non-HPD patients, AUC = 0.91 [95% CI, 0.87–0.96]); Kaplan-Meier analysis showed that the 121-gene set classifier can separate significantly low- and high-risk groups in all of the 13 major TCGA cancers, of which the top three cancers with greatest hazard ratios (HRs) were shown in (B) ESCA (HR = 100.1, 95% CI, 23.1–433.6); (C) COAD (HR = 17.5, 95% CI, 7.4–40.9), and (D) PAAD datasets (HR = 16.0, 95% CI, 8.8–29.0). See also Figures S9–S12.
Discussion
Checkpoint blockade with anti-PD-1 antibodies has resulted in excellent responses in a subset of patients with cancer. However, there is a sizable proportion of patients with cancer who do not respond to anti-PD-1 treatment, with a subset of these patients developing hyperprogression with accelerated tumor growth after anti-PD-1 immunotherapy (Champiat et al., 2017, Kato et al., 2017). Currently, there is a lack of systematic genome studies to identify the genes or immune factors that predict resistance to immune checkpoint inhibition or HPD in response to anti-PD-1 treatment. In this study, we utilized WES and RNA-seq approaches to identify the mutation spectrum and gene expression profiling changes in HPD tumors when compared with pre-therapy tumors. We also performed pathway and tumor immunogenicity analyses based on the RNA-seq data. Finally, we combined our data with publicly available datasets and developed an HPD gene expression signature capable of predicting patients unlikely to respond to anti-PD-1.
The mutation analysis highlighted 11 genes with deleterious mutations in the HPD tumors after anti-PD-1 therapy (Table 2). Most of these genes have not been adequately studied in the context of cancer before. However, a query of this 11 mutated gene set in the cBioPortal website (http://www.cbioportal.org/) (Cerami et al., 2012, Gao et al., 2013) showed that this gene set has somatic mutations or copy number aberrations (CNAs) in 8,887 (22%) of the 41,320 sequenced patients. The alterations of these 11 genes were most frequent in the six major cancer types with an alteration frequency >30% (Figure S13), i.e., prostate cancer (70.8% tumor samples had mutations or CNAs in at least one of the 11 genes), melanoma (50.2% altered), renal cell carcinoma (45.3% altered), brain cancer (33.3% altered), breast cancer (31.1% altered), and colorectal adenocarcinoma (31.0% altered). These data support the cancer linkage to these 11 genes, the mutations of which could contribute to the tumor hyperprogressive phenotype.
Among the 11 genes, some have tumor suppressive properties, good examples being TSC2 and VHL. Inactivating mutations in TSC2 that encode the protein tuberin lead to constitutive activation of mTOR kinase through the Rheb-GTP signaling axis (Menon et al., 2014, Zoncu et al., 2011), which in turn induces cell growth, motility, invasion, and development of tumors (Goncharova et al., 2004, Goncharova et al., 2006). These outcomes were consistent with our observation that the deleterious pY1611S mutation in the key Rap/ran-GAP domain of the TSC2 protein (Table 2, Figure S4) occurred in the hyperprogressive tumors after anti-PD-1 therapy. We also found that the VHL gene had a deleterious mutation—pL117V—in the ccRCC hyperprogressive tumors after anti-PD-1 treatment (Table 2). VHL, located on chromosome 3p25, is a major tumor suppressor gene involved in ccRCC oncogenesis (Gossage et al., 2015). Interestingly, a recent study found that PD-L1 expression was associated with dense PD-1 expression and wild-type VHL ccRCC, but not with mutated/inactivated VHL ccRCC (Kammerer-Jacquet et al., 2017). Therefore, only the patients with ccRCC with wild-type VHL may benefit from immunotherapies inhibiting PD-L1/PD-1 (Kammerer-Jacquet et al., 2017). In our case, we found that only the post-anti-PD-1 therapy hyperprogressive ccRCC tumor had detectable deleterious VHL mutation, but the pre-therapy ccRCC tumor did not. This suggested that the selection pressure of anti-PD-1 therapy eliminated most of the wild-type VHL ccRCC cells but had little effect on cells with mutated VHL ccRCC, such that these mutated cells were highly enriched in the post-therapy HPD tumors. This has significant implications in that it suggests that ccRCC cells with an altered/mutated VHL gene may be a key factor leading to HPD after anti-PD-1 therapy.
The pre- and post-treatment tumors in this study were acquired through biopsy from the primary lesion. After anti-PD-1 therapy, the initial minor subclones of somatic mutations could be boosted by the treatment and expanded in the tumor samples of the two HPD patients as shown in Figure 4, which contributed to the tumor heterogeneity that may account for changes in the mutational and/or expression landscape. Clonal evolution analysis (Figure 4) indicates that HPD tumor-specific mutations in TSC2 and VHL along with mutations in a number of other cancer genes including KMT2C, NCOR2, COL28A1, ING3, CAMKK2, CARD8, BAP1, CARD11, CBFA2T3, CYP2D6, and PBRM1 could be significant to the progression of nonaggressive pre-therapy tumors to the hyperprogressive state after anti-PD-1 treatment. Figure S3 showed that the mutated KMT2C, TSC2, VHL, and CYP2D6 genes were involved in the gene-gene interaction network leading to suppression of the TP53 pathway activity. Previous studies showed that KMT2C (MLL3) co-activates TP53, whereas KMT2C levels decrease during cancer progression, which correlates with distinct clinical stages (Ford and Dingwall, 2015, Lee et al., 2009, Rabello et al., 2018). These results are consistent with our observations in HPD tumors after anti-PD-1 treatment.
Our RNA-seq data revealed that the IGF-1, ERK/MAPK, PI3K/AKT, and TGF-β signaling pathways were activated in the HPD tumors after anti-PD-1 therapy (Figure 3). Recent studies have found that TGF-β signaling may play an important role in resistance to immunotherapy. For example, Mariathasan et al. reported that lack of response to anti-PD-L1 antibody was associated with TGF-β signaling in fibroblasts and the exclusion of CD8+ T cells, indicating that TGF-β-mediated stromal remodeling restricts T cell infiltration to suppress antitumor immunity and that TGF-β inhibition may enhance the efficacy of immune checkpoint blockade (Mariathasan et al., 2018). In parallel, Tauriello et al. found that single-agent PD-1/PD-L1 inhibition had little effect, but co-targeting TGF-β produced a robust antitumor immune response that could prevent the development of metastasis and eliminate established metastases in a mouse model (Tauriello et al., 2018). Collectively, these studies indicate that inhibiting TGF-β could significantly improve the efficacy of anti-PD-1/anti-PD-L1 treatment (Mariathasan et al., 2018, Tauriello et al., 2018). Herein, our data suggest that enhanced TGF-β signaling could also contribute to the development of HPD after anti-PD-1 therapy. Therefore, inhibiting TGF-β signaling may also help prevent the development of HPD in response to anti-PD-1 treatment. Another interesting finding is the activation of PI3K/AKT in HPD tumors. A recent study demonstrated that the activity of PI3K/AKT signaling was crucial for lymphomas with PD-1 deletion (Wartewig et al., 2017). Therefore, when the tumors are exposed to anti-PD-1 therapy, elevated PI3K/AKT signaling may be another important mechanism for the survival, progression, or even hyperprogression of the tumor cells.
The HPD tumors had reduced tumor immunogenicity when compared with the pre-therapy tumors. Such reduction may be caused by downregulation of antigen-processing genes, including several HLA genes and B2M, and upregulation of certain immune checkpoint or modulator genes other than PD-1/PD-L1 (Figures 5 and 6). In the context of studying 28 immune cell populations critical to pan-cancer immunogenomics (Angelova et al., 2015, Charoentong et al., 2017), we found that the activity of eight immune cell populations were weakened and two were strengthened in the HPD tumors. The weakened immune cell populations including monocytes, CD4 helper T cells, dendritic cells, and NK cells may contribute to the ability of HPD tumors to escape immune surveillance. The enhanced cell populations such as neutrophils are known to have a number of pro-tumor properties (Galdiero et al., 2013, Mishalian et al., 2013, Sagiv et al., 2015, Tuting and de Visser, 2016), thus the increase in neutrophil activity in HPD tumors was not surprising.
The two patients developed HPD after anti-PD-1 therapy, indicating the adverse immunity changes that may result in an immunosuppressive environment. The decreased portion of immune cell phenotypes after anti-PD-1 therapy led us to speculate whether anti-PD-1 therapy contributed to accelerated AICD (activation-induced cell death) in these two patients. To test this hypothesis, we applied the GSVA approach to the apoptosis gene sets collected in the MSigDB database (Liberzon et al., 2015). It can be seen that five apoptosis gene sets were activated in the two patients after anti-PD-1 therapy (Figure S14A), of which 27 apoptotic genes including marker genes in caspase/bcl2 pathways (CASP3, CASP7, BNIP2, and BNIP3L) were significantly upregulated (Figure S14B). This indicated that the accelerated AICD may occur in the anti-tumor activating lymphocytes, which accounted for the decreased portion of immune cell phenotypes and enhanced immunosuppressive environment after anti-PD-1 therapy.
So far, cancer immunotherapies have largely focused on T lymphocytes. However, ILCs could also play important roles in the immune response. ILCs were classified into cytotoxic ILCs, such as NK cells, and helper-like ILCs, such as the ILC1, ILC2, and ILC3 subsets. Much of the role of ILCs other than NK cells in cancer and immunotherapy remain elusive. ILCs might represent promising targets in the context of cancer therapy because they are endowed with potent immunomodulatory properties. In the present study, we analyzed the dynamic changes in the activity of ILC populations associated with anti-PD-1 therapy. This represents the first study analyzing the ILC populations in hyperprogressive tumors after anti-PD-1 therapy. Although ILC1 and ILC2 subsets did not show significant changes according to GSEA (Figure S7), the ILC3 population was activated in HPD tumors compared with pre-therapy tumors (Figure 7). Among the three subsets of ILCs, the role of ILC3 is gaining increased interest for its potential tumor-promoting activities. ILC3 that produces interleukin (IL)-22 has also been shown to promote tumor growth mediated via STAT3 activation (Kirchberger et al., 2013). Another study showed that ILC3 promoted lymphatic metastasis by modulating the local chemokine milieu of cancer cells (Irshad et al., 2017). ILC3 may also promote tumor formation and progression by suppressing T cell responses (van Beek et al., 2016). It had been shown that intestinal ILC3 cells limit T cell responses and induce T cell death via outcompeting T cells for IL-2 (Hepworth et al., 2015). We observed upregulated expression of ILC3 marker genes by anti-PD-1 immunotherapy in the two HPD patients, which may contribute to the suppression of T cell responses or the induction of T cell death. Our findings were in line with those of previous studies, indicating that inhibiting ILC3 may complement anti-PD-1 treatment to reduce the likelihood of developing hyperprogressive tumors after the therapy.
It is worth mentioning that IL-22 expression was not detected in the before and after anti-PD-1 treatment FFPE samples of the two patients, which may be due to the influence of the degradation of the RNA samples from the FFPE specimens on gene expression study. However, previous studies have defined a large group of marker genes whose expressions were characteristic of the ILC3 cell population (Bjorklund et al., 2016, Wallrapp et al., 2017). For example, the ILC3 cells were defined by using a repertoire of around 400 genes (Bjorklund et al., 2016, Wallrapp et al., 2017), which became the basis of our analyses on ILC3 cells. Therefore, we analyzed the expression pattern changes of these marker genes to study the dynamic changes of ILC cell populations in response to the anti-PD-1 immunotherapy in the tumors of the HPD patients (Figures 7 and S7).
Previous research showed that PD-1-deficient mice were extraordinarily sensitive to tuberculosis and had much shorter survival times compared with wild-type mice (Lazar-Molnar et al., 2010). This sensitivity results from the need for the PD-1 pathway to control excessive inflammatory responses to tuberculosis infection in the lungs of mice (Lazar-Molnar et al., 2010). This led us to hypothesize that the PD-1 pathway may also be required to control excessive inflammatory responses in patients susceptible to HPD. If anti-PD-1 therapy is administered to HPD patients, it may contribute to tumor growth by further upregulating inflammatory pathway activities. The analyses of our data and those of others (Westin et al., 2014) confirmed this hypothesis by showing that anti-PD-1 therapy can further boost the pre-existing high levels of inflammation in HPD patients, and thus contribute to the hyperprogressive phenotype (Figures 8 and S8).
On the basis of genome-wide expression data of tumors from our study, and two publicly available datasets (before anti-PD-1 therapy) (Riaz et al., 2017, Westin et al., 2014), we identified and validated a 121-gene expression signature that can distinguish HPD patients from non-HPD patients. This may have significant clinical predictive value to identify patients who are suitable for anti-PD-1/anti-PD-L1 immunotherapy. Having validated this gene set, we examined whether there exists any mechanism that might explain its association with HPD. Interestingly, most of these genes (70 of 121) belonged to gene sets that we identified as significant to different aspects of the HPD tumors in our samples. Specifically, these genes could be classified into the following six categories that were described above as important contributors to the HPD phenotype (Figure S9): (1) somatic mutated gene sets; (2) oncogenic pathways of IGF-1, ERK/MAPK, PI3K/AKT, and TGF-β; (3) immune checkpoint genes; (4) ILC3 population marker genes; (5) marker genes for other immune populations like monocytes, CD4 T cells, and dendritic cells; and (6) differentially expressed genes in post-anti-PD-1 HPD tumors versus pre-anti-PD-1 non-HPD tumors. Thus, a significant portion of these HPD signature genes could be involved in the critical biological processes important to tumor evolution, infiltrated immune cells, and tumor-microenvironment interactions. However, although we validated the 121-gene set, more patient cohorts subjected to anti-PD-1 therapy that contain HPD and non-HPD patients are needed for prospective validation.
To better define HPD, especially to differentiate HPD from intermediate and/or late tumor progression, we compared the mutational and gene expression of the two original samples in our study with the pre-treatment tumor samples of the four patients (#28, #9, #26, #38) who developed intermediate and/or late tumor progression (Table S6). Mutation analysis showed that 40 cancer genes had nonsilent somatic mutations in the original tumors of the HPD patients but no mutations in the tumors of the patients whose tumor progression was intermediate and/or late (Figure S15). These genes include, for example, MUC13, MUC6, APC2, ARID2, CDK4, EP400, MARK4, MDM4, MUC2, NOTCH1, and SLIT2. Previous research demonstrated that MDM4 alteration was significantly associated with hyperprogression in patients subjected to immunotherapy (Kato et al., 2017), which was consistent with our results. We tabulated the information of these 40 HPD-associated cancer genes in Table S7. At the transcriptome level, GSVA identified four gene sets from the MsigDB database that were significantly altered in the tumors of HPD patients compared with the patients with intermediate and/or late tumor progression. These gene sets were: HALLMARK_REACTIVE_OXIGEN_SPECIES_PATHWAY, HALLMARK_DNA_REPAIR, HALLMARK_ADIPOGENESIS, and SINGH_KRAS_DEPENDENCY_SIGNATURE. The first three pathways, i.e., the reactive oxygen species pathway, the DNA repair pathway, and the adipogenesis pathway, were significantly inhibited, whereas the KRAS signaling pathway was significantly activated in the tumors of HPD patients relative to the patients with intermediate and/or late tumor progression (Figure S16A). The corresponding gene expression changes of the above significantly altered pathways were also shown (Figure S16B). Together, these mutational and transcriptional changes of the tumors between the HPD and the intermediate/late tumor progression patients may contribute to the better characterization of the HPD condition.
Overall, our comprehensive analysis of HPD tumors after anti-PD-1 therapy and pre-therapy tumors identified the genomics and immune factors contributing to the hyperprogression phenotypes, such as deleterious somatic mutations in important tumor suppressors such as TSC2 and VHL, downregulated antigen-processing genes, and upregulated immune checkpoints or modulators other than PD-1/PD-L1. We also identified immune cell populations with significant activity changes in the HPD tumors; particularly the ILC subset, ILC3, was found to be activated in the HPD tumors after anti-PD-1 treatment. A gene expression signature for HPD tumors was also identified and validated using our samples and publicly available datasets. Our findings may contribute to understanding the mechanisms of the development of HPD after anti-PD-1 treatment, which is important to identify patients at high risk of developing HPD.
Limitations of Study
In this study, we analyzed the genomics, transcriptomics, and immunogenicity of two patients subjected to anti-PD-1 immunotherapy who developed hyperprogression after the treatment. We acknowledged that the patient sample size was small in this study. This is because the majority of the patients either did not develop hyperprogression or had the pseudo-hyperprogressive phenotype after checkpoint immunotherapy. Further larger patient samples involving more HPD patients treated with anti-PD-1 are needed to validate and extend our findings. Another limitation is that we only profiled HPD tumor sample one time upon hyperprogression after anti-PD-1 immunotherapy and did not collect post-hyperprogression tumor samples at later time points. This design rendered us unable to investigate whether the associated immunosuppressive profiles of HPD tumors remain as such even at later time points. However, our study is innovative in terms of analyzing both the before- and after-immunotherapy DNA/RNA samples of the HPD patients and serves as the starting point for similar studies that are lacking in the field.
Currently, the two outside datasets we used in the manuscript were the only ones that have the transcriptome-level gene expression data available publicly for us to develop a gene expression profile for HPD (Riaz et al., 2017, Westin et al., 2014). Based on the available data, we characterized a 121-gene expression profile to differentiate HPD patients from non-HPD patients in both the datasets (Riaz et al., 2017, Westin et al., 2014) with high AUC values and high sensitivity and specificity as described in the manuscript. More HPD patients with well-profiled transcriptome data and detailed clinical information related to the anti-PD-1 treatment are needed to verify our gene expression signature.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank the outside reviewers for manuscript suggestions and revisions. This work was supported by NIH grants N01CN201200015 and R01CA134682.
Author Contributions
M.Y. and Y.W. conceived the project and revised the manuscript. D.X. performed the experiment and all the data analyses and writing of the paper. A.K.S and A.C.M collected the samples for this study. B.G. recruited the patients and revised the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: November 30, 2018
Footnotes
Supplemental Information includes Transparent Methods, 16 figures, and 7 tables and can be found with this article online at https://doi.org/10.1016/j.isci.2018.10.021.
Data and Software Availability
The WES and RNA-seq raw sequence reads data from the before and after anti-PD-1 immunotherapy FFPE samples from the two cancer patients (4 FFPE samples) have been deposited in the Sequence Read Archive under accession number of PRJNA503522.
Supplemental Information
The list of cancer related genes was downloaded from http://www.bushmanlab.org/links/genelists.
References
- Angelova M., Charoentong P., Hackl H., Fischer M.L., Snajder R., Krogsdam A.M., Waldner M.J., Bindea G., Mlecnik B., Galon J. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015;16:64. doi: 10.1186/s13059-015-0620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biton J., Mansuet-Lupo A., Pecuchet N., Alifano M., Ouakrim H., Arrondeau J., Boudou-Rouquette P., Goldwasser F., Leroy K., Goc J. TP53, STK11 and EGFR mutations predict tumor immune profile and the response to anti-PD-1 in lung adenocarcinoma. Clin. Cancer Res. 2018;9:1–14. doi: 10.1158/1078-0432.CCR-18-0163. [DOI] [PubMed] [Google Scholar]
- Bjorklund A.K., Forkel M., Picelli S., Konya V., Theorell J., Friberg D., Sandberg R., Mjosberg J. The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat. Immunol. 2016;17:451–460. doi: 10.1038/ni.3368. [DOI] [PubMed] [Google Scholar]
- Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champiat S., Dercle L., Ammari S., Massard C., Hollebecque A., Postel-Vinay S., Chaput N., Eggermont A., Marabelle A., Soria J.C. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin. Cancer Res. 2017;23:1920–1928. doi: 10.1158/1078-0432.CCR-16-1741. [DOI] [PubMed] [Google Scholar]
- Charoentong P., Finotello F., Angelova M., Mayer C., Efremova M., Rieder D., Hackl H., Trajanoski Z. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017;18:248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- Dang H.X., White B.S., Foltz S.M., Miller C.A., Luo J., Fields R.C., Maher C.A. ClonEvol: clonal ordering and visualization in cancer sequencing. Ann. Oncol. 2017;28:3076–3082. doi: 10.1093/annonc/mdx517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford D.J., Dingwall A.K. The cancer COMPASS: navigating the functions of MLL complexes in cancer. Cancer Genet. 2015;208:178–191. doi: 10.1016/j.cancergen.2015.01.005. [DOI] [PubMed] [Google Scholar]
- Fung K.Y., Nguyen P.M., Putoczki T. The expanding role of innate lymphoid cells and their T-cell counterparts in gastrointestinal cancers. Mol. Immunol. 2017;11:1–9. doi: 10.1016/j.molimm.2017.11.013. [DOI] [PubMed] [Google Scholar]
- Galdiero M.R., Bonavita E., Barajon I., Garlanda C., Mantovani A., Jaillon S. Tumor associated macrophages and neutrophils in cancer. Immunobiology. 2013;218:1402–1410. doi: 10.1016/j.imbio.2013.06.003. [DOI] [PubMed] [Google Scholar]
- Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharova E., Goncharov D., Noonan D., Krymskaya V.P. TSC2 modulates actin cytoskeleton and focal adhesion through TSC1-binding domain and the Rac1 GTPase. J. Cell Biol. 2004;167:1171–1182. doi: 10.1083/jcb.200405130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharova E.A., Goncharov D.A., Lim P.N., Noonan D., Krymskaya V.P. Modulation of cell migration and invasiveness by tumor suppressor TSC2 in lymphangioleiomyomatosis. Am. J. Respir. Cell Mol. Biol. 2006;34:473–480. doi: 10.1165/rcmb.2005-0374OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong J., Wang C., Lee P.P., Chu P., Fakih M. Response to PD-1 blockade in microsatellite stable metastatic colorectal cancer harboring a POLE mutation. J. Natl. Compr. Canc Netw. 2017;15:142–147. doi: 10.6004/jnccn.2017.0016. [DOI] [PubMed] [Google Scholar]
- Gossage L., Eisen T., Maher E.R. VHL, the story of a tumour suppressor gene. Nat. Rev. Cancer. 2015;15:55–64. doi: 10.1038/nrc3844. [DOI] [PubMed] [Google Scholar]
- Hanna G.J., Lizotte P., Cavanaugh M., Kuo F.C., Shivdasani P., Frieden A., Chau N.G., Schoenfeld J.D., Lorch J.H., Uppaluri R. Frameshift events predict anti-PD-1/L1 response in head and neck cancer. JCI Insight. 2018;3:1–13. doi: 10.1172/jci.insight.98811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepworth M.R., Fung T.C., Masur S.H., Kelsen J.R., McConnell F.M., Dubrot J., Withers D.R., Hugues S., Farrar M.A., Reith W. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science. 2015;348:1031–1035. doi: 10.1126/science.aaa4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irshad S., Flores-Borja F., Lawler K., Monypenny J., Evans R., Male V., Gordon P., Cheung A., Gazinska P., Noor F. RORgammat(+) innate lymphoid cells promote lymph node metastasis of breast cancers. Cancer Res. 2017;77:1083–1096. doi: 10.1158/0008-5472.CAN-16-0598. [DOI] [PubMed] [Google Scholar]
- Kammerer-Jacquet S.F., Crouzet L., Brunot A., Dagher J., Pladys A., Edeline J., Laguerre B., Peyronnet B., Mathieu R., Verhoest G. Independent association of PD-L1 expression with noninactivated VHL clear cell renal cell carcinoma-A finding with therapeutic potential. Int. J. Cancer. 2017;140:142–148. doi: 10.1002/ijc.30429. [DOI] [PubMed] [Google Scholar]
- Kato S., Goodman A., Walavalkar V., Barkauskas D.A., Sharabi A., Kurzrock R. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin. Cancer Res. 2017;23:4242–4250. doi: 10.1158/1078-0432.CCR-16-3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchberger S., Royston D.J., Boulard O., Thornton E., Franchini F., Szabady R.L., Harrison O., Powrie F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med. 2013;210:917–931. doi: 10.1084/jem.20122308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S., Akbay E.A., Li Y.Y., Herter-Sprie G.S., Buczkowski K.A., Richards W.G., Gandhi L., Redig A.J., Rodig S.J., Asahina H. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016;7:10501. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar-Molnar E., Chen B., Sweeney K.A., Wang E.J., Liu W., Lin J., Porcelli S.A., Almo S.C., Nathenson S.G., Jacobs W.R., Jr. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc. Natl. Acad. Sci. U S A. 2010;107:13402–13407. doi: 10.1073/pnas.1007394107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Kim D.H., Lee S., Yang Q.H., Lee D.K., Lee S.K., Roeder R.G., Lee J.W. A tumor suppressive coactivator complex of p53 containing ASC-2 and histone H3-lysine-4 methyltransferase MLL3 or its paralogue MLL4. Proc. Natl. Acad. Sci. U S A. 2009;106:8513–8518. doi: 10.1073/pnas.0902873106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A., Birger C., Thorvaldsdottir H., Ghandi M., Mesirov J.P., Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A., Subramanian A., Pinchback R., Thorvaldsdottir H., Tamayo P., Mesirov J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S., Turley S.J., Nickles D., Castiglioni A., Yuen K., Wang Y., Kadel E.E., III, Koeppen H., Astarita J.L., Cubas R. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S., Dibble C.C., Talbott G., Hoxhaj G., Valvezan A.J., Takahashi H., Cantley L.C., Manning B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell. 2014;156:771–785. doi: 10.1016/j.cell.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao D., Margolis C.A., Gao W., Voss M.H., Li W., Martini D.J., Norton C., Bosse D., Wankowicz S.M., Cullen D. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. 2018;359:801–806. doi: 10.1126/science.aan5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishalian I., Bayuh R., Levy L., Zolotarov L., Michaeli J., Fridlender Z.G. Tumor-associated neutrophils (TAN) develop pro-tumorigenic properties during tumor progression. Cancer Immunol. Immunother. 2013;62:1745–1756. doi: 10.1007/s00262-013-1476-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niknafs N., Kim D., Kim R., Diekhans M., Ryan M., Stenson P.D., Cooper D.N., Karchin R. MuPIT interactive: webserver for mapping variant positions to annotated, interactive 3D structures. Hum. Genet. 2013;132:1235–1243. doi: 10.1007/s00439-013-1325-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabello D.D.A., Ferreira V., Berzoti-Coelho M.G., Burin S.M., Magro C.L., Cacemiro M.D.C., Simoes B.P., Saldanha-Araujo F., de Castro F.A., Pittella-Silva F. MLL2/KMT2D and MLL3/KMT2C expression correlates with disease progression and response to imatinib mesylate in chronic myeloid leukemia. Cancer Cell Int. 2018;18:26. doi: 10.1186/s12935-018-0523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz N., Havel J.J., Makarov V., Desrichard A., Urba W.J., Sims J.S., Hodi F.S., Martin-Algarra S., Mandal R., Sharfman W.H. Tumor and microenvironment evolution during immunotherapy with Nivolumab. Cell. 2017;171:934–949.e15. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi N.A., Hellmann M.D., Snyder A., Kvistborg P., Makarov V., Havel J.J., Lee W., Yuan J., Wong P., Ho T.S. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saada-Bouzid E., Defaucheux C., Karabajakian A., Coloma V.P., Servois V., Paoletti X., Even C., Fayette J., Guigay J., Loirat D. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2017;28:1605–1611. doi: 10.1093/annonc/mdx178. [DOI] [PubMed] [Google Scholar]
- Sagiv J.Y., Michaeli J., Assi S., Mishalian I., Kisos H., Levy L., Damti P., Lumbroso D., Polyansky L., Sionov R.V. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015;10:562–573. doi: 10.1016/j.celrep.2014.12.039. [DOI] [PubMed] [Google Scholar]
- Sharma P., Allison J.P. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P., Hu-Lieskovan S., Wargo J.A., Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits H., Artis D., Colonna M., Diefenbach A., Di Santo J.P., Eberl G., Koyasu S., Locksley R.M., McKenzie A.N., Mebius R.E. Innate lymphoid cells–a proposal for uniform nomenclature. Nat. Rev. Immunol. 2013;13:145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauriello D.V.F., Palomo-Ponce S., Stork D., Berenguer-Llergo A., Badia-Ramentol J., Iglesias M., Sevillano M., Ibiza S., Canellas A., Hernando-Momblona X. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–543. doi: 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- Teo M.Y., Seier K., Ostrovnaya I., Regazzi A.M., Kania B.E., Moran M.M., Cipolla C.K., Bluth M.J., Chaim J., Al-Ahmadie H. Alterations in DNA damage response and repair genes as potential marker of clinical benefit from PD-1/PD-L1 blockade in advanced urothelial cancers. J. Clin. Oncol. 2018;36:1685–1694. doi: 10.1200/JCO.2017.75.7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian S.L., Drake C.G., Pardoll D.M. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 2012;24:207–212. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuting T., de Visser K.E. CANCER. How neutrophils promote metastasis. Science. 2016;352:145–146. doi: 10.1126/science.aaf7300. [DOI] [PubMed] [Google Scholar]
- van Beek J.J.P., Martens A.W.J., Bakdash G., de Vries I.J.M. Innate lymphoid cells in tumor immunity. Biomedicines. 2016;4:7–21. doi: 10.3390/biomedicines4010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallrapp A., Riesenfeld S.J., Burkett P.R., Abdulnour R.E., Nyman J., Dionne D., Hofree M., Cuoco M.S., Rodman C., Farouq D. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature. 2017;549:351–356. doi: 10.1038/nature24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartewig T., Kurgyis Z., Keppler S., Pechloff K., Hameister E., Ollinger R., Maresch R., Buch T., Steiger K., Winter C. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature. 2017;552:121–125. doi: 10.1038/nature24649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westin J.R., Chu F., Zhang M., Fayad L.E., Kwak L.W., Fowler N., Romaguera J., Hagemeister F., Fanale M., Samaniego F. Safety and activity of PD1 blockade by pidilizumab in combination with rituximab in patients with relapsed follicular lymphoma: a single group, open-label, phase 2 trial. Lancet Oncol. 2014;15:69–77. doi: 10.1016/S1470-2045(13)70551-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa S., Kiyohara Y., Otsuka M., Kondou R., Nonomura C., Miyata H., Iizuka A., Ohshima K., Urakami K., Nagashima T. Multi-omics profiling of patients with melanoma treated with Nivolumab in project HOPE. Anticancer Res. 2017;37:1321–1328. doi: 10.21873/anticanres.11450. [DOI] [PubMed] [Google Scholar]
- Zaretsky J.M., Garcia-Diaz A., Shin D.S., Escuin-Ordinas H., Hugo W., Hu-Lieskovan S., Torrejon D.Y., Abril-Rodriguez G., Sandoval S., Barthly L. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Xu X., Shi M., Chen Y., Yu D., Zhao C., Gu Y., Yang B., Guo S., Ding G. CD13(hi) Neutrophil-like myeloid-derived suppressor cells exert immune suppression through Arginase 1 expression in pancreatic ductal adenocarcinoma. Oncoimmunology. 2017;6:e1258504. doi: 10.1080/2162402X.2016.1258504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R., Efeyan A., Sabatini D.M. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The list of cancer related genes was downloaded from http://www.bushmanlab.org/links/genelists.