

Abstract
Autosomal dominant polycystic kidney disease (ADPKD), the most common monogenic cause of ESKD, is characterized by relentless development of kidney cysts, hypertension, and destruction of the kidney parenchyma. Over the past few years, major advancements in diagnosing, prognosticating, and understanding the pathogenesis and natural course of the disease have been made. Currently, no kidney disease is more suitable for nephron-protective strategies. Early nephrology referral and implementation of these strategies may have a substantial effect. Total kidney volume is a good prognostication marker and allows stratification of patients into slow or rapid progressing disease, with implications for their management. Measurement of total kidney volume, disease stratification, and prognostication are possible using readily available tools. Although some patients require only monitoring and basic optimized kidney protective measures, such as rigorous BP control and various lifestyle and dietary changes, others will benefit from disease-modifying treatments. Vasopressin V2 receptor antagonists, a likely disease-modifying treatment, has been approved in several countries and recently by the US Food and Drug Administration; other therapies, such as somatostatin analogs and other novel agents, are currently in clinical trials. The purpose of this article is to present our views on the optimal management to delay kidney disease progression in ADPKD.
Keywords: ADPKD; Dietary Sodium; vasopressin; Polycystic Kidney, Autosomal Dominant; blood pressure; Receptors, Vasopressin; United States Food and Drug Administration; Somatostatin; Kidney Failure, Chronic; Antidiuretic Hormone Receptor Antagonists; Nephrons; hypertension; Referral and Consultation; Disease Progression; Cysts
Introduction
Pierre Rayer wrote in 1841 that when the cystic degeneration of the kidneys (polycystic kidney disease was coined by Felix Lejars in 1888) can be suspected during life, it is an illness without cure (1). This view of autosomal dominant polycystic kidney disease (ADPKD) prevailed during most of the 20th century. Research accelerated after PKD1 and PKD2 were identified in 1994 and 1996. The Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) was established in 1999 to better understand its development and progression. Currently, no kidney disease is more suitable for nephron-protective strategies. Early nephrology referral and implementation of these strategies may have a substantial effect. The purpose of this article is to present our views on the optimal management to delay kidney disease progression in ADPKD (Figure 1).
Figure 1.

Basic universal management should be recommended to all patients with ADPKD. The first step is to confirm ADPKD diagnosis, then classify the patient on the basis of imaging features and total kidney volume. Basic optimized management should be recommended to all patients with ADPKD. BMI, body mass index, CT, computed tomography, MRI, magnetic resonance imaging.
Novel Tools to Assess Prognosis and Disease Progression
Imaging in ADPKD was, until recently, limited to screening for the disease and diagnosing its complications. The CRISP study, a longitudinal study of 15- to 46-year-old patients with creatinine clearance ≥70 ml/min, characterized the relationship between total kidney volume (TKV) and GFR (1). Magnetic resonance images at baseline illustrated its huge phenotypic variability (Figure 2). Follow-up images showed TKV growth to be quasi-exponential, unique to and variable among patients. Whereas TKV changed significantly year after year, GFR started declining years later. TKV was the best predictor of eGFR decline (2). A collaborative effort including the PKD Foundation, US Food and Drug Administration (FDA), Critical Path Institute, academic centers, and Pharma, led to the FDA and European Medicines Agency qualification of TKV, together with age and eGFR, as a prognostic biomarker for risk of GFR decline (3).
Figure 2.

Imaging of different patients with ADPKD who enrolled in the CRISP study (creatinine clearance ≥70 ml/min), showing the large spectrum of disease severity from very mild to very severe disease at the baseline visit. These examples depict the large phenotypic variability and emphasize the importance of prognostication using imaging. htTKV, height adjusted total kidney volume (ml/m).
Measuring TKV for use as a prognostic biomarker does not require high precision. Measurements by the ellipsoid equation and various imaging modalities can be used to inform patients about their prognosis and determine eligibility for clinical trials or novel therapies when available. Its value is limited in atypical cases with markedly asymmetric or coexisting ischemic disease. The Mayo Clinic imaging classification facilitates its use (4). It uses criteria to exclude atypical cases and stratify typical cases into five classes (A–E) on the basis of growth rates (<1.5%, 1.5%–3%, 3%–4.5%, 4.5%–6%, or >6% per year) estimated from patient age and a theoretical starting height-adjusted TKV (150 ml/m) (Figure 3, Table 1). A model on the basis of the classification can predict future GFR decline with reasonable accuracy (http://www.mayo.edu/research/documents/pkd-center-adpkd-classification/doc-20094754).
Figure 3.

Mayo Clinic classification diagram (modified from Nephrology, Dialysis and Transplantation, in press). HtTKV, height adjusted total kidney volume (ml/m).
Table 1:
Classification of ADPKD patients by imaging
| Class 1: Typical | Bilateral and diffuse distribution, with mild, moderate, or severe replacement of kidney tissue by cysts, where all cysts contribute similarly to TKV. | ||
| Measure TKV (Could be done by nephrologist or radiologist in few minutes) http://www.mayo.edu/research/documents/pkd-center-adpkd-classification/doc-20094754 | |||
| Estimated eGFR slope (ml/min/1.73m2 per year) | Disease outcome and recommendation | ||
|---|---|---|---|
| Male | Female | ||
| Class 1 A | −0.23 | 0.03 | Likely will not reach ESRD; reassure patient |
| Class 1 B | −1.33 | −1.13 | Slowly progressing disease; monitor patient |
| Class 1 C | −2.63 | −2.43 | Rapidly progressing disease; propose treatment |
| Class 1 D | −3.48 | −3.29 | Rapidly progressing disease; propose treatment |
| Class 1 E | −4.78 | −4.58 | Rapidly progressing disease; propose treatment |
| Class 2: Atypical | |||
| Class 2 A: | Unilateral | Diffuse cystic involvement of one kidney causing marked kidney enlargement with a normal contralateral kidney defined by a normal kidney volume (<275 ml in men; <244 ml in women) and having no or only 1–2 cysts | |
| Segmental | Cystic disease involving only one pole of one or both kidneys and sparing the remaining kidney tissue | ||
| Asymmetric | Diffuse cystic involvement of one kidney causing marked kidney enlargement with mild segmental or minimal diffuse involvement of the contralateral kidney defined by a small number of cysts (>2 but <10) and volume accounting for <30% of TKV | ||
| Lopsided | Bilateral distribution of kidneycysts with mild replacement of kidney tissue with atypical cysts where ≤5 cysts account for ≥50% TKV (the largest cyst diameter is used to estimate individual cyst volume) | ||
| Class 2 B: | Bilateral presentation with acquired unilateral atrophy | Diffuse cystic involvement of one kidney causing moderate to severe kidney enlargement with contralateral acquired atrophy. | |
| Bilateral presentation with bilateral kidney atrophy | Impaired kidney function (serum creatinine≥1.5 mg/dl) without significant enlargement of the kidneys, defined by an average length <14.5 cm, and replacement of kidney tissue by cysts with atrophy of the parenchyma. | ||
TKV has been used as an end point in clinical trials. Measurements for this purpose require high precision, achievable using planimetry (the gold standard) or stereology. Stereology is faster than planimetry but does not segment the kidney, which is a requirement for advanced image analysis. Fast, automatic segmentations, including a deep learning-based, fully automated approach capable to replace humans for the task of segmenting polycystic kidneys, have been developed (5,6). Once segmented, advanced magnetic resonance image processing and analysis, such as texture analysis, may be superior and/or complementary to TKV in predicting or measuring disease progression (7).
Other advanced noncontrast magnetic resonance image acquisition techniques, such as four-dimensional flow magnetic resonance imaging (for automation and standardization of kidney blood flow measurements), blood oxygen level dependent, diffusion-weighted and magnetization transfer magnetic resonance imaging, arterial spin labeling, and magnetic resonance elastography, are being investigated and may be informative at very early stages of the disease.
Aside from imaging-based prognostic strategies, the Predicting Renal Outcome in Polycystic Kidney Disease (PROPKD) score, which uses clinical and genetic data, can be used to stratify the risk for disease progression (8). The PROPKD score can be used from 35 years of age or earlier in patients with both hypertension and urologic complications before this age. In other cases, the genetic information alone can be used for prognosis, with PKD1 truncating, PKD1 nontruncating, and PKD2 mutations being associated to most severe, intermediate, and least severe (average age at onset of ESKD of 56, 68, and 79 years, respectively) disease, respectively. Recently, several other genes have been associated to ADPKD and/or autosomal dominant polycystic liver disease (9,10). In addition to information on prognosis, genetic testing may be indicated when the diagnosis is not clear, in atypical patients, and for preimplantation genetic diagnosis.
Treatment of Hypertension
Hypertension is strongly associated with cardiovascular morbidity and mortality and risk for ESKD in ADPKD. Many pathogenic mechanisms, including impaired production of nitric oxide, contribute to its development and cardiovascular morbidity. Although in the past few decades cardiovascular and cerebrovascular mortalities have decreased, likely because of more frequent diagnosis and treatment of hypertension, age at onset of ESKD has not changed (11). Earlier and more intensive BP control may be needed to delay kidney disease progression.
Optimal BP treatment in the general population and particularly in patients at high risk for cardiovascular morbidity, diabetes mellitus, or CKD has been controversial. Although there is agreement that lowering systolic BP to <150 mm Hg reduces cardiovascular morbidity and mortality, the benefits of lowering it to <130 mm Hg have been debated. The Systolic Blood Pressure Intervention Trial (SPRINT) compared two systolic BP targets, 120 (intensive) versus 140 (standard) mm Hg, in hypertensive patients aged ≥50 years old with cardiovascular disease (CKD with eGFR 20–60 ml/min per 1.73 m2, 10-year Framingham risk score ≥15%, or age ≥75 years) (12). Patients with ADPKD, diabetes, prior stroke, symptomatic heart failure or left ventricular ejection fraction <35%, cardiovascular event or procedure within 3 months, or proteinuria >1 g/d were excluded. The trial was stopped early because of significantly lower rates of the primary composite cardiovascular outcome and all-cause mortality in the intensive-treatment group. Hypotension, syncope, electrolyte abnormalities, and AKI occurred more frequently in the intensive-treatment group. Results in the CKD subset were similar to those in the whole cohort. Patients with CKD in the intensive-treatment group were more likely to experience a 30% eGFR decline primarily due to the acute hemodynamic effect of lowering BP (13). Faster eGFR decline in the intensive-treatment group after month 6 (−0.47 versus −0.32 ml/min per 1.73 m2 per year) was considered clinically insignificant. Recent meta-analyses of clinical trials have supported the SPRINT results (14,15). Translation to clinical practice should take into account that 120 mm Hg systolic BP using automated, protocoled measurements in the SPRINT trial probably equates to 130 mm Hg BP in the doctor’s office, and that adverse events are likely detected and treated earlier in clinical trials than in clinical practice.
Compared with the SPRINT trial, hypertension treatment decisions in patients with ADPKD are made in younger individuals. Few studies have ascertained the effect of intensive compared with standard BP control in patients with ADPKD.
A small, 7-year trial in 20- to 69-year-old hypertensive patients with ADPKD with left ventricular hypertrophy and creatinine clearance >30 ml/min, showed that intensive (120/80 mm Hg) was more effective than standard (135–140/85–90 mm Hg) BP control in decreasing left ventricular mass but had no effect on creatinine clearance decline (16).
The Modification of Diet in Renal Disease (MDRD) trial compared intensive (mean arterial pressure <92 mm Hg for age ≤60 years old or <98 mm Hg for age >60 years old) and standard (mean arterial pressure ≤107 mm Hg for age ≤60 years old or ≤113 mm Hg for age >60 years old) control in patients with GFR 13–55 ml/min. Two hundred out of 840 participants had ADPKD (17). A subgroup analysis of these patients showed that GFR declined faster with intensive compared with standard treatment during the first 4 months (4.6 versus 2.8 ml/min; P=0.03) but not after month 4 (5.1 versus 5.4 ml/min per year) in those with a baseline GFR 25–55 ml/min. In patients with GFR 13–24 ml/min, GFR declined faster with intensive treatment (4.9 versus 3.9 ml/min). After completion of the 3-year trial, ESKD occurrence 7 years later was less frequent in the intensive-treatment group compared with the standard-treatment group (hazard ratio, 0.68; 95% confidence interval [95% CI], 0.57 to 0.82; P<0.001) (18).
The HALT PKD Study A trial compared intensive (95–110/60–75 mm Hg) and standard (120–130/70–80 mm Hg) BP control in 15- to 49-year-old patients with eGFR>60 ml/min/1.73 m2 (19). Intensive treatment lowered the percent TKV increase by 14.2% (P=0.006) without affecting the overall eGFR decline (2.9 and 3.0 ml/min per 1.73 m2 per year). During the first 4 months, eGFR declined faster with intensive treatment, likely because of the hemodynamic effect of lowering BP. After month 4, eGFR decline was marginally slower with intensive treatment. Because GFR declines late in ADPKD, benefits from intensive treatment could be easier to detect in patients with rapid progression. When stratified by disease severity by the imaging classification, patients with class D and E ADPKD assigned to intensive treatment had slower annual TKV increase (6.4% versus 7.8%, P=0.03), and eGFR decline after month 4 (−3.36 versus −4.44; P=0.011) and arguably overall (−3.57 versus −4.37; P=0.05) (20). Frequencies of cardiovascular or kidney events, hyperkalemia, and AKI events were not different.
Treatment of hypertension should be individualized (Table 2). We recommend a rigorous BP target (≤110/75 mm Hg) if tolerated in young hypertensive adults with an eGFR>60 ml/min per 1.73 m2, particularly those with severe kidney disease (class C–E by the imaging classification) or cardiovascular associations such as intracranial aneurysms or valvular heart disease. In other patients with ADPKD, a target ≤130/85 mm Hg is appropriate.
Table 2.
Basic optimized management of adult patients with ADPKD
| Intervention | Goal | Methods to Achieve Goal | Evidencea |
|---|---|---|---|
| Intensive BP control | ≤110/75 mm Hg in: | Early detection is essentialb | Grade 1B |
| 1. 18–50-year-olds | By order of preference: | ||
| 2. eGFR>60 ml/min per 1.73 m2 | 1. ACEI/ARB | ||
| 3. Particularly: | 2. α/β or cardioselective β-blocker | ||
| Mayo Clinic class 1 C–E | 3. Dihydropyridine CCB | ||
| Intracranial aneurysm | 4. Diuretic | ||
| Valvular heart disease | Dietary Approaches to Stop Hypertension (DASH)-like diet at early stages | ||
| ≤130/85 mm Hg in: | |||
| 1. Other adult hypertensives | |||
| Sodium | Moderate restriction (2.3–3 g/d) | Counseling | Grade 1C |
| Adjust for extrarenal losses (hot climate, runners, sauna, bowel disease) if appropriate | Dietitian follow-up | ||
| Monitor 24-h urine sodium | |||
| Hydration | Moderately enhanced hydration spread out over 24 h (during the day, at bedtime, and at night if waking up) | Counseling | Grade 1C |
| Maintain urine osmality ≤280 mOsm/kg | Monitor first morning urine osmality, plasma copeptin if available | ||
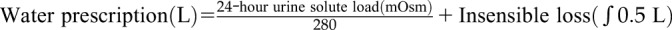 |
|||
| Protein | 0.8–1.0 g/kg of ideal body wt | Dietitian | Grade 1C |
| Monitor protein intake: | |||
| 6.25×(urine urea nitrogen in g/d+[0.03×weight in kilogram]) | |||
| Phosphorus | Moderate diet phosphate restriction (800 mg/d) | Dietician | Grade 2C |
| Read food labels and watch for food additives containing phosphates | |||
| Use of phosphate binders not different from other advanced CKD when needed | |||
| Acid base | Maintain plasma bicarbonate within the normal range (≥22 mEq/L) | Increase fruits/vegetables (2–4 cups/d) | Grade 2B |
| Oral sodium bicarbonate if needed | |||
| Caloric intake | Maintain normal BMI | Dietitian follow-up | Grade 1C |
| Moderation in caloric intake | Regular exercise | ||
| Lipid control | Aim for serum LDL ≤100 mg/dl | Dietician | Grade 2B |
| Regular exercise | |||
| Statin if needed (ezetimibe if intolerant to statin) |
ADPKD, autosomal dominant polycystic kidney disease; ACEI, angiotensin-converting enzyme inhibitors; ARB, angiotensin receptor blockers; CCB, calcium channel blocker; BMI, body mass index.
Grading of levels of evidence is provided in Supplemental Table 1.
Screen children at risk every 3 years starting at age 5 years. Children with hypertension should be referred and managed by experts in pediatric hypertension.
Angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers increase kidney blood flow at early stages of the disease and are the first-line therapy for ADPKD (Table 2) (21,22). Dual renin-angiotensin blockade had no beneficial effect compared with ACE inhibition alone (19). It is not clear which antihypertensive drug class should be used as second line. Combined α/β blockers have good metabolic and hemodynamic profiles and are well tolerated. β-blockers or α1 adrenergic antagonists are preferred for patients with comorbid conditions, such as angina or benign prostatic hyperplasia. Calcium channel blockers and diuretics should be used cautiously because of hypothetical concerns that they might worsen disease progression.
Dietary Sodium and Potassium
Most current guidelines for the general population and for patients with hypertension or CKD recommend a daily sodium intake of <2.3 g (<100 mmol of sodium, <6 g of salt). Although it is accepted that high dietary sodium increases BP, urine albumin excretion, and cardiovascular mortality in the general population, and proteinuria and rate of GFR decline in patients with CKD, there is no consensus on whether modest increases in dietary sodium or very low levels of dietary sodium are also detrimental. Some studies have suggested a U-shaped relationship between salt intake and risk for cardiovascular and kidney events (23). A systemic review including 1787 patients with CKD found consistent associations of high sodium intake (>4.6 g/d) with GFR decline, but no evidence that low intake (<2.3 g/d) was better than moderate intake (2.3–4.6 g/d) (24). The Chronic Renal Insufficiency Cohort study in 3757 patients with CKD stages 2–4 showed similar results (25). In two randomized, clinical trials of renin-angiotensin blockade for nondiabetic or diabetic CKD, higher dietary sodium was associated with higher risk for doubling serum creatinine or ESKD and for kidney and cardiovascular events, respectively (26,27). On the other hand, a combination of strict sodium restriction and ACE inhibition can induce tubulointerstitial damage under certain experimental conditions (28).
Whether dietary sodium affects the progression of ADPKD has been less studied. In the CRISP study, average daily dietary sodium was approximately 4.3 g, remained constant over time despite dietary instructions, and was associated with TKV increase (29). Daily dietary sodium in the HALT PKD study was 4.1 g at baseline; declined by 5.7 mg/mo in Study A and by 9.4 mg/mo in Study B; and was 3.8 g in Study A, 3.5 g in study B, and >2.3 g in >80% of study participants at the last follow-up (30). Dietary sodium was significantly associated with rates of TKV increase in Study A and of eGFR decline and risk for a composite end point of 50% eGFR reduction, ESKD, or death in Study B.
Higher urine potassium excretion in the HALT PKD study was associated with the rate of kidney growth in Study A and with better preservation of eGFR and reduced risk for the composite end point in Study B (30). Although these associations may indicate reverse causality (patients with declining kidney function may eat less or have been instructed to avoid foods high in potassium) or just reflect the effect of a heathier diet including higher potassium content, a reduced risk of CKD has also been associated to higher dietary potassium in other studies.
Taken together, these studies support a recommendation for moderate dietary sodium restriction (2.3–3 g) and, at early stages of ADPKD, a Dietary Approaches to Stop Hypertension (DASH)-like diet (Table 2).
Hydration
A large body of evidence suggests that vasopressin is important not only in the development of cystic disease in ADPKD but also in the progression of CKD in general (31). Therefore, lowering vasopressin concentration by increasing water intake is an intriguing treatment option. Protective effects of enhanced hydration have been found in a rat model of PKD, but not in mouse models orthologous to human ADPKD (32). Currently available information in patients with ADPKD is limited to post hoc analytical studies, short-term interventional trials without control groups, and a small, observational study of 12 months duration that paradoxically showed nonsignificant trends toward faster eGFR decline and TKV growth associated with increased fluid intake from 7 am to 6 pm (33). Two hydration trials are ongoing: PREVENT-ADPKD is a 3-year controlled trial comparing fluid intake ad libitum with a reduced intake to reduce urine osmolality to ≤270 mOsm/L in patients with ADPKD with eGFR>30 ml/min per 1.73 m2 (34); and the Impact of Increased Water Intake in CKD trial is a 2-year controlled trial comparing increased or usual hydration in patients with CKD stage 3 with albuminuria or proteinuria (31). Until more information becomes available, our recommendation is for moderately enhanced hydration spread out over 24 hours (during the day, at bedtime, and at night if waking up) to maintain an average urine osmolality of ≤280 mOsm/L (Table 2). Patients who comply with moderate sodium and protein restrictions will have lower osmolar loads and therefore will need to drink less water. Measurement of first morning urine osmolality and/or plasma copeptin can be used to confirm adequate vasopressin suppression.
Dietary Protein
Experimental studies have shown that low-protein diets induce afferent arteriole constriction, lower intraglomerular pressure, and retard CKD progression, whereas high-protein diets cause afferent arteriole dilation and glomerular hyperfiltration. Low-protein diets slow disease progression in animal models of PKD. In MDRD Study A, 141 patients with ADPKD with GFR 25–55 ml/min were randomized to low protein and phosphorus (0.58 g and 5–10 mg/kg ideal body wt per day) or usual protein and phosphorus (1.30 g and 16–20 mg/kg ideal body wt per day) diets (17). During the first 4 months, GFR declined to 4.2 and 3.2 ml/min on the low and usual protein diets, respectively. After month 4, GFR declined by 5.0 and 5.5 ml/min per year in the low and usual protein diets, respectively. These differences were not statistically significant. Longer follow-up (mean follow-up was only 2.2 years) could have arguably revealed a beneficial effect. However, this was not observed 7 years after completion of the trial, during which protein intake was not controlled (35). In MDRD Study B, 59 patients with ADPKD with GFR 13–24 ml/min were randomized to low and very low protein diets. A trend toward slower GFR decline was observed in the very-low-protein diet compared with the low-protein diet group (17). In the CRISP study, estimated protein intake was associated with steeper TKV increase and GFR decline (univariable analyses only) during a period of observation of up to 6 years (29). Without more conclusive information supporting the prescription of a lower protein diet, recommending a daily protein intake of 0.8–1.0 g/kg ideal body wt seems appropriate (Table 2).
Dietary Phosphorus
Several studies have suggested that excess phosphate contributes to CKD progression and limits the renoprotective effect of ACE inhibition, possibly through stimulation of fibroblast growth factor 23 (FGF23) secretion and FGF23-mediated inhibition of nitric oxide production (36). This may be particularly important in patients with ADPKD because they have higher FGF23 levels compared with other patients with CKD (37). Therefore, we recommend a moderate daily phosphate restriction (800 mg) along with moderate protein restriction to our patients (Table 2). Usually, protein-restricted diets are already low in phosphorus, but patients should be educated to avoid processed foods containing readily absorbed inorganic phosphates.
Metabolic Acidosis
In a rat model of PKD with proximal tubular cysts, an acid load aggravated cyst development and an alkaline load inhibited cyst development. Both acid and alkaline loads aggravated cyst development in a rat model with collecting duct cysts (V.E. Torres, unpublished observations). The relevance of these observations to human ADPKD is uncertain. Nevertheless, acidosis has been linked to CKD progression (38) and bicarbonate supplementation (600 mg thrice daily, increased as necessary to maintain a bicarbonate level ≥23 mmol/L) reduced eGFR decline in a small trial of patients with CKD stage 4 with plasma bicarbonate of 16–20 mmol/L (35). On this basis, we recommend to treat metabolic acidosis maintaining a plasma bicarbonate level ≥22 mmol/L (Table 2).
Caloric Intake
Mild to moderate food restriction markedly reduced cystogenesis in animal models of PKD (39,40). In the HALT PKD trial, overweight body mass and particularly obesity are strongly and independently associated with rate of TKV growth and GFR decline in early-stage ADPKD (41). Obesity is also an established risk factor for CKD in general. Clinical trials to confirm the benefit of caloric restriction and weight control in patients with ADPKD have not been performed. Nevertheless, moderation in caloric intake and avoidance of weight gain should be recommended to patients with ADPKD and at-risk family members (Table 2).
Lipid Control
In the CRISP (29) and MDRD (17) trials, patients with ADPKD with lower serum HDL cholesterol had faster TKV growth and eGFR decline. Dyslipidemia likely increases the cardiovascular morbidity of ADPKD. A small, 3-year trial in children and young adults with ADPKD and normal kidney function showed that pravastatin lowered the increase in TKV (42). A recent retrospective analysis showed lower hazard ratios for doubling of serum creatinine and all-cause mortality in 858 patients with CKD using statins compared with 858 propensity score-matched patients with CKD not using statins, but only in those with an eGFR≥30 ml/min per 1.73 m2 (43). On the basis of these data, a recommendation for a serum LDL cholesterol level ≤100 mg/dl is appropriate for optimized treatment of ADPKD (Table 2).
Disease-Modifying Agents for High-Risk Patients with ADPKD (Rapid Progressors)
In addition to the basic optimized management of ADPKD patients, novel therapies may slow disease progression. Given the commitment of a long-term therapy and its associated burdens, such as extensive monitoring of side effects and costs, it is crucial to identify patients with ADPKD who will benefit the most from these therapies. The evaluation should be performed by a nephrologist with expertise in cystic diseases, preferably in a polycystic kidney disease center of excellence (44). During the evaluation, the diagnosis of typical patients with ADPKD can be confirmed and, if appropriate, a disease-modifying treatment offered to rapid progressors (Figure 1). Our preferred method to identify rapid progressors at early stages of the disease when GFR is still normal is the Mayo Clinic imaging classification (4). Measuring TKV in the clinic can be done in few minutes (Figure 4) and may, with increasing availability of automated image processing, become part of the radiology report. Prognostication on the basis of TKV will help inform management decisions (Figure 5). We recommend disease-modifying therapies for patients with ADPKD with CKD stages 1–3 and Mayo Clinic Class 1C, 1D, or 1E. Patients with higher GFR will benefit the most. Decisions to start treatment for patients with low GFR should weigh the burden of therapy against the potential benefit. Close monitoring will be essential to ensure safety and achieve optimal dosing. Prescribing physicians should educate patients on prevention and be vigilant in the surveillance and management of potential drug-related adverse events.
Figure 4.

Example depicting stepwise method to calculate TKV in the clinician’s office using the Mayo Clinic ADPKD classification online tool. The first three steps involve measuring the maximal sagittal, coronal, and axial width and depth of each kidney. These measurements can be done using the ruler tool available in most imaging viewing software. The following steps include plugging in the numbers to obtain TKV, classification, and estimation of future eGFR (see http://www.mayo.edu/research/documents/pkd-center-adpkd-classification/doc-20094754).
Figure 5.

Example of two patients with ADPKD with the same age and GFR, who have a substantial difference in their prognosis on the basis of their total kidney volume and predicted rate of decline of eGFR. Patient A is classified as Mayo Clinic class 1E and would benefit form a disease-modifying treatment. Patient B has better prognosis (class 1B) and would need monitoring.
V2-Receptor Antagonists
Water conservation in terrestrial animals promotes vasopressin-mediated cAMP increase that, in polycystic kidney disease, stimulates cyst formation and growth (45). Although vasopressin makes patients with ADPKD victims of evolution, it also makes the disease treatable by targeted therapy with V2-receptor (V2R) antagonists. Circulating levels of copeptin, a surrogate for vasopressin, are higher in patients with ADPKD (median, 6.8 pmol/L; interquartile range [IQR], 3.4–15.7 pmol/L) than in healthy kidney donors before (median, 3.8 pmol/L; IQR, 2.8–6.3 pmol/L) and after (median, 4.4 pmol/L; IQR, 3.6–6.1 pmol/L) kidney donation, and increase as the CKD stage advances from 1 to 2 (median, 6.4 pmol/L; IQR, 3.6–7.6 pmol/L) to CKD 3 (median, 9.2 pmol/L; IQR, 3.8–18.5 pmol/L) and 4 (median, 30.0 pmol/L; IQR, 13.0–45.2 pmol/L) (46,47).
Cysts derive mostly from the collecting duct and distal nephron-expressing V2Rs. Preclinical studies confirmed that V2R activation aggravated polycystic kidney disease in rodent models. Conversely, suppression of vasopressin production, release, or action reduced cyst burden, protected kidney function, and prolonged survival (48,49). This strong evidence led to the implementation of clinical trials of tolvaptan for ADPKD. The goal of treatment with V2R antagonists is to inhibit the action of vasopressin on V2Rs continuously. Urine hypotonicity relative to plasma (normally 275–295 mOsm/kg) in the first morning voiding before the administration of tolvaptan can be monitored to confirm that the goal is achieved. Initial studies showed that split, twice daily dosing is necessary for effective V2R inhibition (urine osmolality <300 mOsm/kg continuously for 24 hours) (50). Aquaresis was accompanied by a small and reversible reduction in GFR, likely due to activation of tubuloglomerular feedback (51,52).
The double-blind Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and its Outcomes (TEMPO) 3:4 trial was designed to enroll rapidly progressing patients with relatively preserved GFR (18–50 years old, estimated creatinine clearance ≥60 ml/min, TKV≥750 ml) (53). A total of 1445 patients were randomized to tolvaptan or placebo. Tolvaptan reduced the rate of TKV growth from 5.5% to 2.8% per year, and the eGFR decline over 3 years from 10.1 to 6.8 ml/min per 1.73 m2 (both P<0.001). Transaminase elevations of more than three times the upper limit of normal occurred in 4.4% of the tolvaptan group and 1% of the placebo group; two met the Hy law criteria for severe hepatocellular toxicity. On the basis of these results, tolvaptan was approved for rapidly progressive ADPKD in Japan, Canada, the European Union, Switzerland, Norway, South Korea, and Australia.
The Replicating Evidence of Preserved Renal function: an Investigation of Tolvaptan Safety and Efficacy (REPRISE) trial, a phase 3, randomized, withdrawal, multicenter, placebo-controlled, double-blind trial, recruited 1370 18- to 55-year-old or 56- to 65-year-old patients with an eGFR of 25–65 or 25–44 ml/min per 1.73 m2, respectively (54). After 1 year, eGFR decreased by 2.34 and 3.61 ml/min per 1.73 m2 in the tolvaptan and placebo groups (P<0.001). Reversible elevations in the alanine aminotransferase of more than three times the upper limit of normal occurred in 5.6% in the tolvaptan group and 1.2% in the placebo group. No patients met the Hy law criteria, likely because of more frequent monitoring and earlier discontinuation of tolvaptan. After the publication of these results, the FDA has approved tolvaptan for the treatment of patients at risk for rapidly progressive ADPKD.
The TEMPO 3:4 and REPRISE trials showed that tolvaptan slowed eGFR decline over a broad range of disease stages and could be used safely. Recently, an analysis of 97 patients continuously treated at a single center for 4.6 (range, 1.1–11.2) years showed that tolvaptan slowed eGFR decline in a way that is sustained, cumulative over time, and consistent with the goal of delaying the need for kidney replacement (55).
In the TEMPO 3:4 trial, higher plasma copeptin at baseline predicted faster TKV growth in placebo-treated patients and larger treatment effect on TKV (and to some extent eGFR) in tolvaptan-treated patients (56). Therefore, plasma copeptin levels may help to predict prognosis and possibly tolvaptan treatment efficacy in patients with ADPKD. Tolvaptan-induced aquaresis is accompanied by an increase in the circulating levels of vasopressin and copeptin. Appropriate hydration is essential to prevent thirst and hypernatremia and to limit the increase in plasma copeptin. Because of potential hepatotoxicity, close monitoring of liver function tests is mandated by the FDA as part of a risk evaluation and mitigation strategy.
Somatostatin Analogs
Somatostatin acts on five Gi protein-coupled receptors (SSTR1–5), present on cholangiocytes and kidney tubular epithelial cells, inhibiting cAMP generation. Because somatostatin has a very short t1/2 (3 minutes), more stable synthetic peptides (octreotide, lanreotide, and pasireotide) have been developed for clinical use. In preclinical studies, octreotide inhibited hepatic and kidney cyst expansion from PCK rats in vitro and in vivo (57).
Several small clinical trials have shown beneficial effects of octreotide and lanreotide in patients with severe polycystic liver disease, particularly in premenopausal women (58). The Long-Acting Somatostatin on Disease Progression in Nephropathy due to ADPKD (ALADIN) trial showed a favorable trend for octreotide slowing TKV increase and GFR decline compared with placebo (59). Larger trials in patients with more advanced ADPKD are ongoing or near completion: Developing Interventions to Halt Progression of ADPKD (clinicaltrials.gov; NCT01616927), Somatostatin in Patients with Autosomal Dominant Polycystic Kidney Disease and Moderate to Severe Renal Insufficiency (clinicaltrials.gov; NCT01377246), and the Lanreotide in Polycystic Kidney Disease Study (clinicaltrials.gov; NCT02127437), as well as a pasireotide trial for severe polycystic liver disease (clinicaltrials.gov; NCT01670110).
Other Potential Disease-Modifying Therapies
Clinical trials of rapalogs showed no clinical benefit, possibly because their toxicity prevented adequate dosing to affect the cystic disease in humans (60–63). A phase 2 trial of bosutinib, an Src/Abl kinase inhibitor, showed inhibition of kidney growth but was complicated by a high withdrawal rate due to adverse events (64). A phase 2 trial of tesevatinib, a tyrosine kinase inhibitor, is ongoing (clinicaltrials.gov; NCT03203642). Two phase 2 trials are investigating the effect of metformin, previously shown to reduce cystic enlargement in rapidly progressive mouse models (clinicaltrials.gov; NCT02656017, NCT02903511). Small clinical trials of pioglitazone (clinicaltrials.gov; NCT02697617) and nicotinamide (clinicaltrials.gov; NCT02140814) are ongoing. Drugs targeting glucosyl ceramide synthase (clinicaltrials.gov; NCT03523728) and microRNAs (65) and the vasopressin V2 receptor antagonist lixivaptan (clinicaltrials.gov; NCT03487913) are also in the clinical trial pipeline.
Disclosures
V.E.T. is a member of the steering committees for the Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and Its Outcomes 3:4 and Replicating Evidence of Preserved Renal Function: an Investigation of Tolvaptan Safety and Efficacy in ADPKD clinical trials, has received research support from Otsuka Pharmaceutical, and is a consultant for Vertex, Sanofi, and Palladio.
Supplementary Material
Acknowledgments
This study has been supported in part by the Mayo Clinic Robert M. and Billie Kelley Pirnie Translational Polycystic Kidney Disease Center and the National Institute of Diabetes and Digestive and Kidney Diseases grant DK090728.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
This article contains supplemental material online at http://cjasn.asnjournals.org/lookup/suppl/doi:10.2215/CJN.03960318/-/DCSupplemental.
References
- 1.Grantham JJ, Torres VE, Chapman AB, Guay-Woodford LM, Bae KT, King BF Jr, Wetzel LH, Baumgarten DA, Kenney PJ, Harris PC, Klahr S, Bennett WM, Hirschman GN, Meyers CM, Zhang X, Zhu F, Miller JP; CRISP Investigators: Volume progression in polycystic kidney disease. N Engl J Med 354: 2122–2130, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Chapman AB, Bost JE, Torres VE, Guay-Woodford L, Bae KT, Landsittel D, Li J, King BF, Martin D, Wetzel LH, Lockhart ME, Harris PC, Moxey-Mims M, Flessner M, Bennett WM, Grantham JJ: Kidney volume and functional outcomes in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 7: 479–486, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perrone RD, Mouksassi MS, Romero K, Czerwiec FS, Chapman AB, Gitomer BY, Torres VE, Miskulin DC, Broadbent S, Marier JF: Total kidney volume is a prognostic biomarker of renal function decline and progression to end-stage renal disease in patients with autosomal dominant polycystic kidney disease. Kidney Int Rep 2: 442–450, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Irazabal MV, Rangel LJ, Bergstralh EJ, Osborn SL, Harmon AJ, Sundsbak JL, Bae KT, Chapman AB, Grantham JJ, Mrug M, Hogan MC, El-Zoghby ZM, Harris PC, Erickson BJ, King BF, Torres VE; CRISP Investigators: Imaging classification of autosomal dominant polycystic kidney disease: A simple model for selecting patients for clinical trials. J Am Soc Nephrol 26: 160–172, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kline TL, Korfiatis P, Edwards ME, Warner JD, Irazabal MV, King BF, Torres VE, Erickson BJ: Automatic total kidney volume measurement on follow-up magnetic resonance images to facilitate monitoring of autosomal dominant polycystic kidney disease progression. Nephrol Dial Transplant 31: 241–248, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kline TL, Korfiatis P, Edwards ME, Blais JD, Czerwiec FS, Harris PC, King BF, Torres VE, Erickson BJ: Performance of an artificial multi-observer deep neural network for fully automated segmentation of polycystic kidneys. J Digit Imaging 30: 442–448, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kline TL, Korfiatis P, Edwards ME, Bae KT, Yu A, Chapman AB, Mrug M, Grantham JJ, Landsittel D, Bennett WM, King BF, Harris PC, Torres VE, Erickson BJ; CRISP Investigators: Image texture features predict renal function decline in patients with autosomal dominant polycystic kidney disease. Kidney Int 92: 1206–1216, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cornec-Le Gall E, Audrézet MP, Rousseau A, Hourmant M, Renaudineau E, Charasse C, Morin MP, Moal MC, Dantal J, Wehbe B, Perrichot R, Frouget T, Vigneau C, Potier J, Jousset P, Guillodo MP, Siohan P, Terki N, Sawadogo T, Legrand D, Menoyo-Calonge V, Benarbia S, Besnier D, Longuet H, Férec C, Le Meur Y: The PROPKD score: A new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 27: 942–951, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cornec-Le Gall E, Torres VE, Harris PC: Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J Am Soc Nephrol 29: 13–23, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cornec-Le Gall E, Olson RJ, Besse W, Heyer CM, Gainullin VG, Smith JM, Audrézet MP, Hopp K, Porath B, Shi B, Baheti S, Senum SR, Arroyo J, Madsen CD, Férec C, Joly D, Jouret F, Fikri-Benbrahim O, Charasse C, Coulibaly JM, Yu AS, Khalili K, Pei Y, Somlo S, Le Meur Y, Torres VE, Harris PC; Genkyst Study Group; HALT Progression of Polycystic Kidney Disease Group; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease: Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet 102: 832–844, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spithoven EM, Kramer A, Meijer E, Orskov B, Wanner C, Caskey F, Collart F, Finne P, Fogarty DG, Groothoff JW, Hoitsma A, Nogier MB, Postorino M, Ravani P, Zurriaga O, Jager KJ, Gansevoort RT; ERA-EDTA Registry; EuroCYST Consortium; WGIKD; EuroCYST Consortium; WGIKD: Analysis of data from the ERA-EDTA Registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney Int 86: 1244–1252, 2014 [DOI] [PubMed] [Google Scholar]
- 12.Wright JT Jr, Williamson JD, Whelton PK, Snyder JK, Sink KM, Rocco MV, Reboussin DM, Rahman M, Oparil S, Lewis CE, Kimmel PL, Johnson KC, Goff DC Jr, Fine LJ, Cutler JA, Cushman WC, Cheung AK, Ambrosius WT; SPRINT Research Group: A randomized trial of intensive versus standard blood-pressure control. N Engl J Med 373: 2103–2116, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheung AK, Rahman M, Reboussin DM, Craven TE, Greene T, Kimmel PL, Cushman WC, Hawfield AT, Johnson KC, Lewis CE, Oparil S, Rocco MV, Sink KM, Whelton PK, Wright JT Jr, Basile J, Beddhu S, Bhatt U, Chang TI, Chertow GM, Chonchol M, Freedman BI, Haley W, Ix JH, Katz LA, Killeen AA, Papademetriou V, Ricardo AC, Servilla K, Wall B, Wolfgram D, Yee J; SPRINT Research Group: Effects of intensive BP control in CKD. J Am Soc Nephrol 28: 2812–2823, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malhotra R, Nguyen HA, Benavente O, Mete M, Howard BV, Mant J, Odden MC, Peralta CA, Cheung AK, Nadkarni GN, Coleman RL, Holman RR, Zanchetti A, Peters R, Beckett N, Staessen JA, Ix JH: Association between more intensive vs less intensive blood pressure lowering and risk of mortality in chronic kidney disease stages 3 to 5: A systematic review and meta-analysis. JAMA Intern Med 177: 1498–1505, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bundy JD, Li C, Stuchlik P, Bu X, Kelly TN, Mills KT, He H, Chen J, Whelton PK, He J: Systolic blood pressure reduction and risk of cardiovascular disease and mortality: A systematic review and network meta-analysis. JAMA Cardiol 2: 775–781, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schrier R, McFann K, Johnson A, Chapman A, Edelstein C, Brosnahan G, Ecder T, Tison L: Cardiac and renal effects of standard versus rigorous blood pressure control in autosomal-dominant polycystic kidney disease: Results of a seven-year prospective randomized study. J Am Soc Nephrol 13: 1733–1739, 2002 [DOI] [PubMed] [Google Scholar]
- 17.Klahr S, Breyer JA, Beck GJ, Dennis VW, Hartman JA, Roth D, Steinman TI, Wang SR, Yamamoto ME; Modification of Diet in Renal Disease Study Group: Dietary protein restriction, blood pressure control, and the progression of polycystic kidney disease. J Am Soc Nephrol 5: 2037–2047, 1995 [DOI] [PubMed] [Google Scholar]
- 18.Sarnak MJ, Greene T, Wang X, Beck G, Kusek JW, Collins AJ, Levey AS: The effect of a lower target blood pressure on the progression of kidney disease: Long-term follow-up of the modification of diet in renal disease study. Ann Intern Med 142: 342–351, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Schrier RW, Abebe KZ, Perrone RD, Torres VE, Braun WE, Steinman TI, Winklhofer FT, Brosnahan G, Czarnecki PG, Hogan MC, Miskulin DC, Rahbari-Oskoui FF, Grantham JJ, Harris PC, Flessner MF, Bae KT, Moore CG, Chapman AB; HALT-PKD Trial Investigators: Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med 371: 2255–2266, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Irazabal MV, Abebe KZ, Bae KT, Perrone RD, Chapman AB, Schrier RW, Yu AS, Braun WE, Steinman TI, Harris PC, Flessner MF, Torres VE; HALT Investigators: Prognostic enrichment design in clinical trials for autosomal dominant polycystic kidney disease: The HALT-PKD clinical trial. Nephrol Dial Transplant 32: 1857–1865, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chapman AB, Johnson A, Gabow PA, Schrier RW: The renin-angiotensin-aldosterone system and autosomal dominant polycystic kidney disease. N Engl J Med 323: 1091–1096, 1990 [DOI] [PubMed] [Google Scholar]
- 22.Torres VE, Wilson DM, Burnett JC Jr, Johnson CM, Offord KP: Effect of inhibition of converting enzyme on renal hemodynamics and sodium management in polycystic kidney disease. Mayo Clin Proc 66: 1010–1017, 1991 [DOI] [PubMed] [Google Scholar]
- 23.Graudal N, Jürgens G, Baslund B, Alderman MH: Compared with usual sodium intake, low- and excessive-sodium diets are associated with increased mortality: A meta-analysis. Am J Hypertens 27: 1129–1137, 2014 [DOI] [PubMed] [Google Scholar]
- 24.Smyth A, O’Donnell MJ, Yusuf S, Clase CM, Teo KK, Canavan M, Reddan DN, Mann JF: Sodium intake and renal outcomes: A systematic review. Am J Hypertens 27: 1277–1284, 2014 [DOI] [PubMed] [Google Scholar]
- 25.Mills KT, Chen J, Yang W, Appel LJ, Kusek JW, Alper A, Delafontaine P, Keane MG, Mohler E, Ojo A, Rahman M, Ricardo AC, Soliman EZ, Steigerwalt S, Townsend R, He J; Chronic Renal Insufficiency Cohort (CRIC) Study Investigators: Sodium excretion and the risk of cardiovascular disease in patients with chronic kidney disease. JAMA 315: 2200–2210, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vegter S, Perna A, Postma MJ, Navis G, Remuzzi G, Ruggenenti P: Sodium intake, ACE inhibition, and progression to ESRD. J Am Soc Nephrol 23: 165–173, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lambers Heerspink HJ, Holtkamp FA, Parving HH, Navis GJ, Lewis JB, Ritz E, de Graeff PA, de Zeeuw D: Moderation of dietary sodium potentiates the renal and cardiovascular protective effects of angiotensin receptor blockers. Kidney Int 82: 330–337, 2012 [DOI] [PubMed] [Google Scholar]
- 28.Hamming I, Navis G, Kocks MJ, van Goor H: ACE inhibition has adverse renal effects during dietary sodium restriction in proteinuric and healthy rats. J Pathol 209: 129–139, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Torres VE, Grantham JJ, Chapman AB, Mrug M, Bae KT, King BF Jr, Wetzel LH, Martin D, Lockhart ME, Bennett WM, Moxey-Mims M, Abebe KZ, Lin Y, Bost JE; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP): Potentially modifiable factors affecting the progression of autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 6: 640–647, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torres VE, Abebe KZ, Schrier RW, Perrone RD, Chapman AB, Yu AS, Braun WE, Steinman TI, Brosnahan G, Hogan MC, Rahbari FF, Grantham JJ, Bae KT, Moore CG, Flessner MF: Dietary salt restriction is beneficial to the management of autosomal dominant polycystic kidney disease. Kidney Int 91: 493–500, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark WF, Sontrop JM, Huang SH, Moist L, Bouby N, Bankir L: Hydration and chronic kidney disease progression: A critical review of the evidence. Am J Nephrol 43: 281–292, 2016 [DOI] [PubMed] [Google Scholar]
- 32.Hopp K, Wang X, Ye H, Irazabal MV, Harris PC, Torres VE: Effects of hydration in rats and mice with polycystic kidney disease. Am J Physiol Renal Physiol 308: F261–F266, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Higashihara E, Nutahara K, Tanbo M, Hara H, Miyazaki I, Kobayashi K, Nitatori T: Does increased water intake prevent disease progression in autosomal dominant polycystic kidney disease? Nephrol Dial Transplant 29: 1710–1719, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong ATY, Mannix C, Grantham JJ, Allman-Farinelli M, Badve SV, Boudville N, Byth K, Chan J, Coulshed S, Edwards ME, Erickson BJ, Fernando M, Foster S, Haloob I, Harris DCH, Hawley CM, Hill J, Howard K, Howell M, Jiang SH, Johnson DW, Kline TL, Kumar K, Lee VW, Lonergan M, Mai J, McCloud P, Peduto A, Rangan A, Roger SD, Sud K, Torres V, Vliayuri E, Rangan GK: Randomised controlled trial to determine the efficacy and safety of prescribed water intake to prevent kidney failure due to autosomal dominant polycystic kidney disease (PREVENT-ADPKD). BMJ Open 8: e018794, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM: Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol 20: 2075–2084, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zoccali C, Ruggenenti P, Perna A, Leonardis D, Tripepi R, Tripepi G, Mallamaci F, Remuzzi G; REIN Study Group: Phosphate may promote CKD progression and attenuate renoprotective effect of ACE inhibition. J Am Soc Nephrol 22: 1923–1930, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pavik I, Jaeger P, Kistler AD, Poster D, Krauer F, Cavelti-Weder C, Rentsch KM, Wüthrich RP, Serra AL: Patients with autosomal dominant polycystic kidney disease have elevated fibroblast growth factor 23 levels and a renal leak of phosphate. Kidney Int 79: 234–240, 2011 [DOI] [PubMed] [Google Scholar]
- 38.Dobre M, Rahman M, Hostetter TH: Current status of bicarbonate in CKD. J Am Soc Nephrol 26: 515–523, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warner G, Hein KZ, Nin V, Edwards M, Chini CC, Hopp K, Harris PC, Torres VE, Chini EN: Food restriction ameliorates the development of polycystic kidney disease. J Am Soc Nephrol 27: 1437–1447, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kipp KR, Rezaei M, Lin L, Dewey EC, Weimbs T: A mild reduction of food intake slows disease progression in an orthologous mouse model of polycystic kidney disease. Am J Physiol Renal Physiol 310: F726–F731, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nowak KL, You Z, Gitomer B, Brosnahan G, Torres VE, Chapman AB, Perrone RD, Steinman TI, Abebe KZ, Rahbari-Oskoui FF, Yu ASL, Harris PC, Bae KT, Hogan M, Miskulin D, Chonchol M: Overweight and obesity are predictors of progression in early autosomal dominant polycystic kidney disease. J Am Soc Nephrol 29: 571–578, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cadnapaphornchai MA, George DM, McFann K, Wang W, Gitomer B, Strain JD, Schrier RW: Effect of pravastatin on total kidney volume, left ventricular mass index, and microalbuminuria in pediatric autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol 9: 889–896, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho EY, Myoung C, Park HS, Kim AJ, Ro H, Chang JH, Lee HH, Chung W, Jung JY: Efficacy of statin treatment in early-stage chronic kidney disease. PLoS One 12: e0170017, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harris T, Sandford R, de Coninck B, Devuyst O, Drenth JPH, Ecder T, Kent A, Gansevoort RT, Gorriz JL, Ong ACM, Pirson Y, Torres VE, Budde K, Clement D, Derchi LE, Eleftheroudi M, Levtchenko E, Peters D, Van Poppel H, Vanholder R: European ADPKD Forum multidisciplinary position statement on autosomal dominant polycystic kidney disease care: European ADPKD Forum and Multispecialist Roundtable participants. Nephrol Dial Transplant 33: 563–573, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Gastel MDA, Torres VE: Polycystic kidney disease and the vasopressin pathway. Ann Nutr Metab 70[Suppl 1]: 43–50, 2017 [DOI] [PubMed] [Google Scholar]
- 46.Zittema D, van den Berg E, Meijer E, Boertien WE, Muller Kobold AC, Franssen CF, de Jong PE, Bakker SJ, Navis G, Gansevoort RT: Kidney function and plasma copeptin levels in healthy kidney donors and autosomal dominant polycystic kidney disease patients. Clin J Am Soc Nephrol 9: 1553–1562, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boertien WE, Meijer E, de Jong PE, ter Horst GJ, Renken RJ, van der Jagt EJ, Kappert P, Ouyang J, Engels GE, van Oeveren W, Struck J, Czerwiec FS, Oberdhan D, Krasa HB, Gansevoort RT: Short-term effects of tolvaptan in individuals with autosomal dominant polycystic kidney disease at various levels of kidney function. Am J Kidney Dis 65: 833–841, 2015 [DOI] [PubMed] [Google Scholar]
- 48.Wang X, Wu Y, Ward CJ, Harris PC, Torres VE: Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 19: 102–108, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aihara M, Fujiki H, Mizuguchi H, Hattori K, Ohmoto K, Ishikawa M, Nagano K, Yamamura Y: Tolvaptan delays the onset of end-stage renal disease in a polycystic kidney disease model by suppressing increases in kidney volume and renal injury. J Pharmacol Exp Ther 349: 258–267, 2014 [DOI] [PubMed] [Google Scholar]
- 50.Shoaf SE, Chapman AB, Torres VE, Ouyang J, Czerwiec FS: Pharmacokinetics and pharmacodynamics of tolvaptan in autosomal dominant polycystic kidney disease: Phase 2 trials for dose selection in the pivotal phase 3 trial. J Clin Pharmacol 57: 906–917, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Irazabal MV, Torres VE, Hogan MC, Glockner J, King BF, Ofstie TG, Krasa HB, Ouyang J, Czerwiec FS: Short-term effects of tolvaptan on renal function and volume in patients with autosomal dominant polycystic kidney disease. Kidney Int 80: 295–301, 2011 [DOI] [PubMed] [Google Scholar]
- 52.Boertien WE, Meijer E, de Jong PE, Bakker SJ, Czerwiec FS, Struck J, Oberdhan D, Shoaf SE, Krasa HB, Gansevoort RT: Short-term renal hemodynamic effects of tolvaptan in subjects with autosomal dominant polycystic kidney disease at various stages of chronic kidney disease. Kidney Int 84: 1278–1286, 2013 [DOI] [PubMed] [Google Scholar]
- 53.Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS; TEMPO 3:4 Trial Investigators: Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 367: 2407–2418, 2012. 23121377 [Google Scholar]
- 54.Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Perrone RD, Koch G, Ouyang J, McQuade RD, Blais JD, Czerwiec FS, Sergeyeva O; REPRISE Trial Investigators: Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N Engl J Med 377: 1930–1942, 2017 [DOI] [PubMed] [Google Scholar]
- 55.Edwards M, Chebib F, Irazabal Mira MV, Ofstie TG, Bungum LA, Metzger AJ, Senum SR, Hogan MC, El-Zoghby Z, Kline TL, Harris PC, Czerwiec FS, Torres VE: Long-term effect of tolvaptan on renal function in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gansevoort R, van Gastel M, Chapman A, Blais J, Czerwiec F, Perrone R, Stade K, Torres V, Devuyst O: Copeptin, a surrogate for Vasopressin, predicts disease progression and Tolvaptan treatment efficacy in ADPKD. Results of the TEMPO 3:4 trial. J Am Soc Nephrol 27: 34A, 2016 [Google Scholar]
- 57.Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF: Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′,5′-cyclic monophosphate. Gastroenterology 132: 1104–1116, 2007 [DOI] [PubMed] [Google Scholar]
- 58.Neijenhuis MK, Gevers TJ, Nevens F, Hogan MC, Torres VE, Kievit W, Drenth JP: Somatostatin analogues improve health-related quality of life in polycystic liver disease: A pooled analysis of two randomised, placebo-controlled trials. Aliment Pharmacol Ther 42: 591–598, 2015 [DOI] [PubMed] [Google Scholar]
- 59.Caroli A, Perico N, Perna A, Antiga L, Brambilla P, Pisani A, Visciano B, Imbriaco M, Messa P, Cerutti R, Dugo M, Cancian L, Buongiorno E, De Pascalis A, Gaspari F, Carrara F, Rubis N, Prandini S, Remuzzi A, Remuzzi G, Ruggenenti P; ALADIN study group: Effect of longacting somatostatin analogue on kidney and cyst growth in autosomal dominant polycystic kidney disease (ALADIN): A randomised, placebo-controlled, multicentre trial. Lancet 382: 1485–1495, 2013 [DOI] [PubMed] [Google Scholar]
- 60.Walz G, Budde K, Mannaa M, Nürnberger J, Wanner C, Sommerer C, Kunzendorf U, Banas B, Hörl WH, Obermüller N, Arns W, Pavenstädt H, Gaedeke J, Büchert M, May C, Gschaidmeier H, Kramer S, Eckardt KU: Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med 363: 830–840, 2010 [DOI] [PubMed] [Google Scholar]
- 61.Serra AL, Poster D, Kistler AD, Krauer F, Raina S, Young J, Rentsch KM, Spanaus KS, Senn O, Kristanto P, Scheffel H, Weishaupt D, Wüthrich RP: Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Engl J Med 363: 820–829, 2010 [DOI] [PubMed] [Google Scholar]
- 62.Braun WE, Schold JD, Stephany BR, Spirko RA, Herts BR: Low-dose rapamycin (sirolimus) effects in autosomal dominant polycystic kidney disease: an open-label randomized controlled pilot study. Clin J Am Soc Nephrol 9: 881–888, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ruggenenti P, Gentile G, Perico N, Perna A, Barcella L, Trillini M, Cortinovis M, Ferrer Siles CP, Reyes Loaeza JA, Aparicio MC, Fasolini G, Gaspari F, Martinetti D, Carrara F, Rubis N, Prandini S, Caroli A, Sharma K, Antiga L, Remuzzi A, Remuzzi G; SIRENA 2 Study Group: Effect of Sirolimus on Disease Progression in Patients with Autosomal Dominant Polycystic Kidney Disease and CKD Stages 3b-4. Clin J Am Soc Nephrol 11: 785–794, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tesar V, Ciechanowski K, Pei Y, Barash I, Shannon M, Li R, Williams JH, Levisetti M, Arkin S, Serra A: Bosutinib versus Placebo for Autosomal Dominant Polycystic Kidney Disease. J Am Soc Nephrol 28: 3404–3413, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yheskel M, Patel V: Therapeutic microRNAs in polycystic kidney disease. Curr Opin Nephrol Hypertens 26: 282–289, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.