

Abstract
Deleterious mutations in the serine hydrolase DDHD1 cause the SPG28 subtype of the neurological disease Hereditary Spastic Paraplegia (HSP), which is characterized by axonal neuropathy and gait impairments. DDHD1 has been shown to display PLA1-type phospholipase activity with a preference for phosphatidic acid. However, the endogenous lipid pathways regulated by DDHD1 in vivo remain poorly understood. Here we use a combination of untargeted and targeted metabolomics to compare the lipid content of brain tissue from DDHD1+/+ and DDHD1–/– mice, revealing that DDHD1 inactivation causes a substantial decrease in polyunsaturated lysophosphatidylinositol (lyso-PI) lipids and an corresponding increase in phosphatidylinositol (PI) lipids. Other phospholipids were mostly unchanged with the exception of decreases in select polyunsaturated lyso-phosphatidylserine (lyso-PS) and lyso-phosphatidylcholine (lyso-PC) lipids and a striking remodeling of PI phosphates (e.g., PIP, PIP2) in DDHD1–/– brain tissue. Biochemical assays confirmed that DDHD1 hydrolyzes PI/PS to lyso-PI/PS with sn-1-selectivity and accounts for a substantial fraction of the PI/PS lipase activity in mouse brain tissue. These data indicate that DDHD1 is a principal regulator of bioactive lyso-PI and other lysophospholipids, as well as PI phosphates, in the mammalian nervous system, pointing to a potential role for these lipid pathways in HSP.
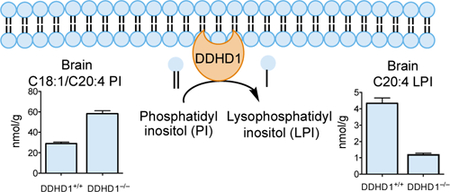
Graphical Abstract
Introduction
The enzymatic conversion of diacylated lipids to sn-2 monoacylated lipids furnishes diverse classes of chemical transmitters, including the endocannabinoid 2-arachidonoylglycerol1 and lysophospholipids, such lysophosphatidic acid (lyso-PA) and lysophosphatidylinositol (lyso-PI)2,3. The endogenous production of 2-AG is catalyzed by sn-1-selective diacylglycerol lipases, while 2-acylated lyso-PA and lyso-PI are thought to be generated by sn-1-selective phospholipases. One such candidate enzyme is DDHD14, an atypical serine hydrolase that was initially characterized as a phosphatidic acid-preferring phospholipase A1 and has been found to also accept phosphatidylinositol, phosphatidylcholine, and phosphatidylethanolamine as substrates in vitro5. Consistent with a role in regulating (lyso)phospholipid metabolism, DDHD1 has been shown, when overexpressed in human cells to produce 20:4 lyso-PA and lyso-PI6. Additionally, genetic disruption of a DDHD1 homologue in Caenorhabditis elegans results in sn-1 acyl chain remodeling of PI lipids7. The extent to which mammalian DDHD1 regulates lysophospholipid and phospholipid metabolism in vivo, however, remains unknown.
Understanding the endogenous metabolic function of DDHD1 is important for several reasons. First, deleterious mutations in human DDHD1 cause a rare neurological disorder termed hereditary spastic paraplegia (HSP) subtype 28 (SPG28)8. Symptoms of SPG28 include spastic gait, hyperreflexia and mildly penetrant peripheral neuropathy, cerebellar eye movements, and urinary incontenance9–11. Additional neurological disorders associated with DDHD1 mutations include juvenile ALS and neurodegeneration with brain iron accumulation12,13. Knowledge of DDHD1-regulated lipid pathways in the CNS may thus provide insights into the mechanistic basis for SPG28 and point to future treatment strategies for the disease. DDHD1 belongs to a small clade of sequence-related serine hydrolases termed DDHD proteins that also includes DDHD2 and SEC23IP, and the extent to which the DDHD enzymes perform shared versus distinct metabolic functions in vivo remains unknown. Notably, loss-of-function mutations in DDHD2 lead to a distinct form of HSP – SPG54 – that presents with both limb spasticity/weakness and intellectual disability14. We recently found that DDHD2 acts as a major brain triacylglycerol (TAG) lipase and that loss of this enzyme in mice leads to massive neuronal lipid droplet accumulation15. That genetic loss of either DDHD1 or DDHD2 leads to HSP might initially suggest shared lipid substrates for these enyzmes; however, SPG54 patients show a diagnostic lipid peak visible by brain magnetic resonance spectroscopy (consistent with TAG accumulation, as is observed in DDHD2–/– mice) that is not present in SPG28 patients10,14. Finally, as noted above, some of the proposed products of DDHD1 catalysis, such lyso-PA and lyso-PI, are important lipid mediators implicated in diverse physiological and pathological processes2,3. DDHD1 may thus represent an important target for pharmacologically controlling key lysophopholipid signaling pathways in vivo.
Here we sought to identify endogenous substrates and products of DDHD1 by performing a combination of untargeted and targeted lipidomics of tissues from DDHD1–/– mice. Considering that humans lacking DDHD1 present with neurological phenotypes, we focused our initial lipidomics efforts on brain tissue from DDHD1–/– mice. We found that genetic loss of DDHD1 leads to substantial reductions in polyunsaturated (e.g., C20:4, C22:6) lysophospholipids, including lyso-PI, lyso-PC, and lyso-PS, but not lyso-PA, lyso-PE, and lyso-PG, as well as concomitant changes in select PI, PIP, and PIP2 species. The changes in polyunsaturated lysophospholipids were primarily restricted to brain tissue in DDHD1–/– mice, pointing to a specialized role for DDHD1 in regulating these lipid transmitters in the nervous system. Also consistent with this hypothesis, we show that DDHD1 accounts for a substantial fraction of the total PI-PLA1 and PS-PLA1 lipase activity of the mouse brain. These findings, taken together, designate DDHD1 as a major biosynthetic enzyme for polyunsaturated lysophospholipid messengers that also modulates the acyl chain composition of PI phosphates in the mammalian nervous system, providing further support for deregulated lipid metabolism and signaling as a common basis for HSP-related syndromes in humans.
MATERIALS/EXPERIMENTAL DETAILS
Materials
The following lipids were purchased from Avanti Polar Lipids: C17:1 LPG, Cat. No. 110712; C17:1 LPS, 858141P; C17:1 LPI, Cat. No. 850103P; C17:0 LPA, Cat. No. 110679; C17:0 LPC, Cat. No. 110686, C17: 1 LPE, Cat. No. 110699; C17:0/C17:0 PS, Cat. No. 840028P; C16:0 D31 /C18:1 PI, Cat. No. 110923; C17:0/C20:4 PI(3)P, Cat. No. LM1900; C17:0/C20:4 PI(4,5)P2, Cat. No. LM1904; C17:0/C20:4 PI(3,4,5)P3, Cat. No. LM1906; C16:0 D31/C18:1 PG, Cat. No. 110919; C14:0/C14:0 BMP, Cat. No. 857131; C16:0 D31/C18:1 PC; Cat. No. 110918; C12:0/C12:0 PC, Cat. No. 850335; C16:0 D31/C18:1 PE, Cat. No. 110921; C12:0/C12:0 PE, Cat. No. 850702; C16:0 D31/C18:1 PA, Cat. No. 110920. The following lipids were purchased from Cayman Chemical: C16:0/C16:0 PI, Cat. No. 10007710; C18:0/C20:4 D8 DAG, Cat. No. 10009872. The following lipids were purchased from Nu-Chek Prep: C17:1/C17:1/C17:1 TAG, Cat. No. T-404, C18:1/C18:1/C18:1 TAG, Cat. No. T-235, C15:0 MAG, Cat. No. M-149.
Confirmation of loss of DDHD1 in DDHD1–/– mice
DDHD1–/– mice on C56BL/6N inbred strain were recovered from cryopreserved embryos at the KOMP Repository (allele DDHD1tm1a). In brief,these animals were generated as follows. Homologous recombination between the DDHD1 targeting vector PGS00049_B_D06 and the DDHD1 locus of JM8.N4 ES cells (C57BL/6N strain) resulted in clone 13035 that was injected into embryos to generate chimeras. Germline transmission was performed by crossing chimeric offspring with C57BL/6N mice. Tail biopsies from 4-week old mice were digested with proteinase K solution and DNA was extracted for PCR genotyping. The DDHD1 specific primers 5’-AGGGCCGGTCAGTCACTAGAGG-3’, 5’-GGGATCTCATGCTGGAGTTCTTCG-3’ and 5’-GATCAATTCTTTTCTTTCTTCCTCTCC-3’ were used to amplify sequences of 270 and 558 base pairs corresponding to the WT and targeted allele, respectively.
RNA was isolated from snap-frozen brain tissue using the RNeasy Mini Kit (Quiagen, Cat. No. 74104). cDNA synthesis according to iScript Reverse Transcription Kit (Biorad, Cat. No. 1708891) manufacturer instructions yielded RT-PCR template DNA. DDHD1 specific primers 5’-TTTCTAAAGCCTCCAGCAGTG-3’ and 5’-TGATCGCCATTCAACAGGCA-3’ were used to amplify DDHD1 following instructions for 2× SYBR Select Master Mix (Applied Biosystems, Cat. No. 4472908). Data were normalized to GAPDH using primers 5’-AGGTCGGTGTGAACGGATTTG-3’ and 5’-TGTAGACCATGTAGTTGAGGTCA-3’ and the delta delta CT method. ABPP reductive demethylation of brain proteomes was performed as described in15. Gel-based ABPP was performed as in15.
Preparation of lipid extracts from cells and tissues
Organic soluble metabolites were extracted from cells or tissues as described in15. Briefly, biological samples were suspended in 1 volume of cold PBS (1 mL for cells, 2 mL for tissues). To this, 3 volumes of 2:1 (v/v) CHCl3:MeOH with internal standards were added. Tissues were homogenized with 10 strokes of a Dounce homogenizer and transferred to 2-dram vials, and cells were lysed by vortexing the extraction mixture. Extracts were centrifuged for 3 min, 1,400 xg to separate the upper aqueous phase and lower organic phase. The organic phase was transferred to 1 dram vials and the aqueous phase was reextracted with 2 volumes of CHCl3 acidified with 10 μL of formic acid or 0.2 volumes of 3N HCl to increase recovery of LPI/PIP/PIP2. Combined organic phases were dried under N2 and solubilized in 125 μl 2:1 (v/v) CHCl3: MeOH. Samples were stored at −80 °C before analysis.
Untargeted lipidomic analysis
Untargeted metabolite analysis was performed to identify significantly altered features between two groups of samples15,16. Chromatographic separation of lipids was performed using a previously described gradient15. Mobile Phase A was 95:5 (v/v) H2O:MeOH and Mobile Phase B was 60:35:5 (v/v/v) i-PrOH:MeOH:H2O supplemented with 0.1% (v/v) formic acid, 10 mM NH4HCO2 or 0.1% (v/v) 28% NH4OH in positive and negative ionization modes, respectively. Lipid extracts were injected onto a Gemini C18 column (Phenomenex, 50 × 4.6 mm, 5 μm diameter particles) coupled to a SecurityGuard C18 pre-column (Phenomenex, C18, 4 × 3 mm). MS analysis was performed with an Agilent 6520 Q-TOF by scanning a 200–1500 m/z mass range with electrospray ionization. The drying gas was set at 350 °C and 11 L/min, nebulizer pressure was 45 psi, fragmentor voltage was 100 V and capillary voltage was 4000 V. Data files were analyzed by XCMSOnline17,18 to align retention times among LC-MS runs, integrate peaks and perform statistical analysis to identity features that are significantly different between experimental and control groups.
Targeted lipidomic analysis
Phospholipids were quantified using multiple reaction monitoring (MRM) on an Agilent 6410 QQQ LC/MS instrument. The source temperature was 350 °C, capillary voltage was 3500 V and −3500V for positive and negative mode, respectively, gas flow was 9 L/min, nebulizer pressure was 35 psi. Chromatographic separations were performed using the same mobile phases and column as for discovery metabolite profiling. The following gradient was used: 0% B for 5 min, increase to 100% B over 19 min, hold 100% B for 4 min, switch to 0% B, hold 0%B for 3 min. Data were analyzed with MassHunter Quantitative Analysis software (Agilent) and signals for endogenous lipids were normalized to the concentration of appropriate internal standards (C17:1 LPG, C17:1 LPS, C17:1 LPI, C17:0 LPA, C17:0 LPC, C17: 1 LPE, C17:0/C17:0 PS, C16:0 D31 /C18:1 PI, C17:0/C20:4 PI(3)P, C17:0/C20:4 PI(4,5)P2, C17:0/C20:4 PI(3,4,5)P3, C16:0 D31/C18:1 PG, C14:0/C14:0 BMP, C12:0/C12:0 PC, C12:0/C12:0 PE, C16:0 D31/C18:1 PA, C17:1/C17:1/C17:1 TAG, C18:0/C20:4 D8 DAG),and wet weight of tissues or number of cells. See supplemental methods for detailed MRM parameters for each phospholipid class.
Substrate assay
Substrate assays were performed as described previously15 with minor modifications. Lipid substrates (150 μM) were dispersed in assay buffer (25 mM HEPES, 150 mM NaCl, 2 mM EDTA, 0.1% n-Decyl-β-D-Maltopyranoside). To 100 μL prepared lipids was added 50 μL proteome (2 μg for DDHD1-overexpressing HEK293T, 200 μg for tissues). Reactions were incubated at 37 °C for 5 min or 60 min for HEK293T cells or tissues, respectively. The reaction was quenched with 300 μL of 2:1 CHCl3:MeOH (v/v) containing internal standards for appropriate lysophospholipids or FFA. Data were analyzed on an Agilent 6100 single quadrupole mass spectrometer and reaction products were separated by HPLC using the same mobile phases and C18 column as for discovery metabolite profiling.
RESULTS
Targeted disruption of DDHD1 in mice
DDHD1–/– mice have been previously described, but this past study mainly focused on male fertility-related phenotypes in the animals19, and the lipid content of tissues from these animals was not assessed, leaving open the question of the endogenous substrates and products of DDHD1. We obtained DDHD1+/– mice from the Knockout Mouse Project (KOMP) Repository (https://www.komp.org/) that possess a targeted lacZ insertion sequence between exon 5 and 6 of the Ddhd1 gene20 (Figure 1A). These mice were bred to generate DDHD1–/– mice, and we verified the genetic insertion into the Ddhd1 locus by PCR genotyping (Figure 1B). We found that brain tissue from DDHD1–/– mice showed negligible DDHD1 mRNA transcript (Figure 1C) or protein (Figure 1D) as determined by quantitative RT-PCR and western blotting, respectively. For the western blotting study, we also analyzed DDHD1 expression in lipopolysaccharide-stimulated macrophages, which have been reported to show high expression of DDHD1 (www.biogps.org; Figure S1), and confirmed the absence of DDHD1 in these cells from DDHD1–/– mice (Figure 1D). We finally aimed to confirm DDHD1 deletion and the preservation of other serine hydrolase activities in brain tissue from DDHD1–/– mice by activity-based protein profiling (ABPP)15,21. We first attempted to visualize DDHD1 using the general serine hydrolase probe fluorophosphonate-rhodamine (FP-Rh) coupled with gel-based ABPP; however, we could not detect an FP-Rh-labeled protein at the predicted molecular weight for DDHD1 (~100 kDa) that differed in brain tissue proteomes from DDHD1+/+ and DDHD1–/– mice (Figure S2). We interpret this result to indicate that DDHD1 activity was either below the limit of detection of gel-based ABPP or visualization of the enzyme was obfuscated by co-migrating serine hydrolase activities. We therefore instead verified loss of DDHD1 activity by performing quantitative mass spectrometry (MS)-based proteomics with the serine hydrolase-directed activity-based probe fluorophosphonate-biotin (FP-biotin)21 which revealed that DDHD1 activity was absent in DDHD1–/– brains, while other serine hydrolases, including the sequence-related enzyme DDHD2, were unaffected (Figure 1E). These data indicate that DDHD1 protein and activity are absent, as expected, in DDHD1–/– mice and that other serine hydrolase activities, including the related enzyme DDHD2, are not affected by DDHD1 loss.
Figure 1.

Generation and initial characterization of DDHD1–/– mice.
(A) Map of whole body knockout allele of the Ddhd1 gene with conditional potential. Homologous recombination between Ddhd1 and 5’ and 3’ homology arms of a targeting vector resulted in the insertion of a lacZ/neo insert between exons 5 and 6 of Ddhd1. LoxP sites are introduced flanking exon 6 of Ddhd1. The catalytic serine of DDHD1 is downstream of the insertion. Half arrows represent annealing sites of genotyping primers. Map is not drawn to scale. (B) Genotyping of tail DNA of mice with various Ddhd1 genotypes. The targeted allele (knockout, or KO) generates a larger 558 bp product whereas the wild-type (WT) allele produces a 270 bp product. (C) Quantitative RT-PCR of mouse brain cDNA reveals a profound reduction of Ddhd1 mRNA in DDHD1–/– mice. N = 4, Error bars represent mean ± SEM. (D) Western blot of brain and macrophage lysates from DDHD1+/+ and DDHD1–/– mice. An immunoreactive band at the predicted molecular weight of DDHD1 is absent in DDHD1–/– tissues and cells (mac, macrophages). (E) Quantitative MS-based ABPP of serine hydrolase activities in brain tissue shows complete and selective loss of DDHD1 activity in DDHD1–/– mice. Error bars represent mean ± SEM of median isotopic ratios for tryptic peptides from each indicated serine hydrolase (data are from two independent experiments).
Behavioral characterization of DDHD1–/– mice
A previous report described male infertility in DDHD1–/– mice19, but CNS-related behavioral phenotypes of these animals have not, to our knowledge, been reported. Breeding of DDHD1+/– mice produced DDHD1–/– offspring that were born at the predicted Mendelian frequency and were outwardly healthy and largely indistinguishable from their DDHD1+/+ and DDHD1+/– littermates. In an assay of gait measurement, DDHD1+/+ and DDHD1–/– mice had similar stride length in all limbs (Figure S3A). Locomotor activity, as measured by counting beam breaks caused by movement over a two hour period, was also similar between DDHD1+/+ and DDHD1–/– mice, and no genotype differences were observed in ambulation or rearing activity (Figure S3B). DDHD1+/+ and DDHD1–/– mice further performed equivalently in the hanging wire (Figure S3C) and rotarod (Figure S3D) tests of coordination and motor function. These findings, taken together, indicate that DDHD1 disruption in mice does not cause a strong defect in motor function, in contrast to the SPG28 phenotypes observed in humans with deleterious mutation in DDHD1.
We assessed additional neurobehavioral phenotypes in DDHD1–/– mice and found that these animals did not differ from DDHD1+/+ mice in the Y-maze test that assesses a willingness to explore novel environments (Figure S3E). In assays of basal nociception, DDHD1–/– mice showed reduced mechanical, but not thermal nociception (Figure S3F–H).
DDHD1 regulates brain lysophospholipid and PI content in vivo
We next sought to map lipid pathways regulated by DDHD1 in vivo by performing untargeted LC-MS analysis of lipid extracts from brain tissue of DDHD1+/+ and DDHD1–/– mice (2 months old). In brief, the organic solvent-extracted fractions of brain tissue from DDHD1+/+ and DDHD1–/– mice were separately analyzed by LC-MS and the resulting chromatograms compared using the XCMS algorithm22,23 to identify altered metabolites. The largest fold changes corresponded to m/z values of 619.3 and 643.3 displaying a retention time of 24 min and a substantial reduction in signal intensity in DDHD1–/– brain tissue (Figure 2A, Table S1). Based on calculated molecular weights, we hypothesized that the 619.3 and 643.3 features corresponded to C20:4 LPI and C22:6 LPI, respectively, and confirmed these assignments by matching the retention time and fragmentation pattern of endogenous C20:4 LPI to a purchased C20:4 LPI standard (Figure S4A, B). Other m/z values predicted to match additional lysophospholipids (e.g., C20:4 LPC, C22:6 LPC) were also decreased in DDHD1–/– brain tissue (Figure 2A, Table S1). Multiple lipids with m/z values matching those predicted for PI lipids were conversely found to increase in DDHD1–/– brain tissue (Figure 2A, Table S1), and we confirmed these assignments by comparison to a commercially available PI lipid standard (Figure S4C, D).
Figure 2.

Metabolomic analysis identifies phospholipid changes in tissues from DDHD1–/– mice.
A) Volcano plot showing relative abundance of metabolites (x-axis) versus significance of the observed changes (y-axis) in brain tissue from DDHD1–/– and DDHD1+/+ mice as measured by untargeted metabolomics. Structural assignments for significantly changing metabolites (P<0.001) with log2 transformed fold changes of <−1 or >1 are shown next to data points. For integrated peak areas of all extracted metabolites see (Table S1). (B) Targeted LC-MS-based quantification of the indicated lysophospholipids containing C20:4 or C22:6 acyl chains in DDHD1+/+ or DDHD1–/– brains. (C-E) Targeted LC-M-based quantification of the indicated LPS (C) and LPI (D) and PI (E) species in DDHD1+/+ and DDHD1–/– brains. (F, G) Targeted LC-M-based quantification of C20:4 LPI (F) and C18:1/C20:4 PI (G) in the indicated tissues from DDHD1+/+ and DDHD1–/– mice. For additional lysophospholipid and PI data, see Figures S5 and S6, respectively. For overall lipid data, see Table S2. N = 4, error bars represent mean ± SEM. ** P<0.01, *** P<0.001, **** P<0.0001, DDHD1+/+ vs. DDHD1–/–.
Using a separate cohort of mice (1.5–3 months old), we quantified tissue phospholipids by targeted LC-MS using multiple reaction monitoring (MRM) in which collision induced dissociation generates fragment ions that indicate the acyl chain composition of each lipid. These experiments verified the decreases in polyunsaturated LPI and LPC in DDHD1–/– brain and also supported similar reductions in C22:6 LPS (Figure 2B and Table S2). In contrast, no changes in polyunsaturated LPE or LPG were observed and a contrasting increase in C22:6 LPA occurred in DDHD1–/– brains (Figure 2B and Table S2). The alterations in LPS and LPI lipids were restricted to polyunsaturated species, as saturated and monounsaturated members of these lysophospholipid classes were generally unaltered in DDHD1–/– brain (Figure 2C, D and Figure S5 and Table S2). The MRM experiments also confirmed elevations in a subset of PI lipids bearing C20:4 acyl chains in DDHD1–/– brain (Figure 2E and Figure S6 and Table S2).
Most the lysophospholipid changes observed in DDHD1–/– mice were restricted to central nervous system tissues (brain, spinal cord) and not observed in various peripheral organs, including those that express high levels of DDHD1 (e.g., testis) (Figure 2F and Figures S5 and S7 and Table S2). On the other hand, select peripheral tissues (testis, spleen) from DDHD1–/– mice displayed increases in various C20:4 PI lipids that were also altered in brain and spinal cord (Figure 2G and Figures S6 and S7 and Table S2). Finally, the alterations in (lyso)phospholipid content in DDHD1–/– mice were not generally observed in tissues from DDHD1+/– mice, which more closely resembled DDHD1+/+ tissues in lipid composition (Figure S5 and S6 and Table S2).
PI lipids are further metabolized by a range of lipid kinases to furnish PI phosphates that regulate diverse cellular processes in the nervous system24,25, at least in part by segregating to specific membrane compartments and targeting phosphoinositide-binding proteins to these respective subcellular locations26. We found that the PI phosphate content of brain tissue from DDHD1–/– mice was substantially altered, showing elevations in C38:5 (C18:1/C20:4) PI-monophosphate (PIP) and PIP-diphosphate (PIP2) and reductions in several saturated/monounsaturated PIP and PIP2 species (e.g., C32:0, C34:0, C34:1, C36:1) (Figure 3A, B).
Figure 3. Altered PI phosphate content of brain tissue from DDHD1–/– mice.

(A, B) Targeted LC-MS-based quantification of the indicated PIP (A) and PIP2 (B) species in brain tissue from DDHD1+/+ and DDHD1–/– mice. Data are normalized to PIP or PIP2 internal standards as described in the Supplemental Methods section. N = 4, error bars represent averages ± SEM. * P<0.05,** P<0.01,*** P<0.001, DDHD1+/+ vs. DDHD1–/–.
These data collectively demonstrate that DDHD1 regulates the endogenous content of polyunsaturated (lyso)PI/PS lipids in the mammalian CNS, and changes in these lipids following DDHD1 disruption extend to downstream metabolic products like PIP and PIP2. In contrast, other in vitro products of DDHD1 activity, such LPA lipids, were either unaltered or paradoxically increased in brain tissue from DDHD1–/– mice.
DDHD1 is a principal brain PI and PS lipase
Consistent with previous studies5,6, we found that recombinantly expressed DDHD1 (evaluated in transfected HEK293T-cell lysates) showed a marked preference for hydrolyzing phospholipids at the sn-1 position over the sn-2 position (PLA1 activity) and accepted a range of phospholipid substrates with the exception of those with positively charged head groups (Figure S8A). We should note that these in vitro substrate data do not entirely align with the changes in lysophospholipids observed in DDDH1–/– mice, as C22:6 lyso-PC was substantially reduced in brain tissue from these animals (Figure 2B), even though PC was not observed as a substrate of DDHD1 in transfected cell lysates (Figure S8A). These data may indicate the DDHD1 is capable of hydrolyzing a broader range of phospholipids in vivo than was captured with the in vitro assay conditions employed herein. Comparative analysis of brain tissue from DDHD1+/+ and DDHD1–/– mice demonstrated that DDHD1 accounts for a substantial fraction of the PI and PS lipase activity in soluble brain proteome, with the latter being specifically assigned to PLA1activity using a mixed C16:0/C18:1 PS substrate (Figure 4). We noted that the contribution of DDHD1 to total brain PI/PS activity was measurable with saturated/monounsaturated substrates, despite the unaltered content of these lipids in brain tissue from DDHD1–/– mice. We therefore asked whether DDHD1 had the capacity to regulate not only polyunsaturated, but also saturated/monounsaturated (lyso)phospholipids by measuring the lipid profiles of DDHD1-transfected HEK293T cells. These experiments revealed a broad array of changes in both polyunsaturated and saturated/monounsaturated (lyso)phospholipids in DDHD1-transfected cells (Figure S8B, C and Table S3), as well as elevations and reductions in some diacylglycerol (DAG) and triacylglycerol (TAG) species, respectively. The latter changes suggested that DDHD1 may also have the capacity to regulate TAGs in vivo, a physiological activity that has been assigned to the sequence-related enzyme DDHD215,27. However, unlike brain tissue from DDHD2–/– mice, which show substantial elevations in TAGs15, this class of neutral lipids, as well as DAGs and monoacylglycerols (MAGs), were unaltered in DDHD1–/– brain tissue (Figure S9).
Figure 4.

DDHD1 makes a major contribution to brain phospholipase A (PLA1) activity with PI and PS substrates. Soluble brain tissue lysates from DDHD1+/+ or DDHD1–/– mice were assessed for hydrolysis of C16:0/C18:1 PS or C16:0/C16:0 PI substrates as described in Methods. Heat-denatured (boiled) DDHD1+/+ soluble lysates served as an inactivated control sample. N = 3, error bars represent mean ± SEM. **** P<0.0001, DDHD1+/+ vs. DDHD1–/–.
These data, combined with our lipidomic analysis of DDHD1–/– brain tissue, indicate that, while DDHD1 is capable of hydrolyzing a range of phospholipids, and possibly neutral lipids as well, the enzyme only exerts physiological regulation over a restricted subset of these lipids bearing predominantly polyunsaturated acyl chains in vivo.
Discussion
Elucidating the physiological functions of metabolic enzymes and their mechanistic relationship to human disease requires knowledge of the endogenous substrates and biochemical pathways regulated by these enzymes. The SPG28 disease syndrome resulting from deleterious mutations in DDHD1 points to an important function for this enzyme in the central nervous system8–11. DDHD1 has been shown to display PLA1 activity with a range of phospholipids in vitro5,6 but the physiological substrates of this enzyme in the mammalian brain remain poorly understood. Here, we have used a combination of untargeted and targeted metabolomics applied to a DDHD1–/– mouse model to discover that DDHD1 regulates discrete brain (lyso)phospholipids in vivo. The coordinated decreases in polyunsaturated LPI and increases in polyunsaturated PI are consistent with the latter lipids serving as direct substrates for DDHD1. Indeed, PI lipids show an asymmetrical acyl chain distribution, with arachidonic acid being enriched in the sn-2 position28. The selective reductions in polyunsaturated LPI (and elevations in polyunsaturated PI) in DDHD1–/– brain tissue are thus consistent with DDHD1 maintaining its PLA1-restricted activity in vivo. It remains unclear why DDHD1 deletion did not equivalently affect all polyunsaturated PI lipids (e.g., C18:1/C20:4 was decreased in DDHD1–/– brain, while C18:0/C20:4 PI was unchanged), but these differences could reflect selective access of DDHD1 to certain PI substrates, or the contribution of other enzymes to the metabolism of PI lipids bearing distinct acyl chains in vivo.
Inositol phospholipids, including PI and its array of inositol-phosphorylated products, play fundamental roles in cell biology that include membrane-delineated signal transduction, as well as regulation of membrane trafficking and cytoskeletal dynamics29. That genetic disruption of DDHD1 alters not only PI, but also downstream PIP and PIP2 lipids, indicates a role for this enzyme in modulating broader PI phosphate function. Even though the changes in PI and lyso-PI in DDHD1–/– brain tissue were restricted to polyunsaturated lipids, the alterations in PIP and PIP2 were more pervasive and included several phospholipids comprised of saturated and mono-unsaturated acyl chains, possibly indicating that DDHD1 loss leads to general acyl chain remodeling among PI phosphates. Interestingly, other enzymes involved in regulating C20:47containing PI content, such as MBOAT7, when genetically disrupted, cause abnormal brain morphology in mice30 and autism in humans31. Several other enzymes involved in PI phosphate-metabolism are also implicated in neurological disorders32. The extent to which the SPG28 disease syndrome caused by DDHD1 loss is mediated by perturbations in PI phosphate function will require future investigation. One important area of investigation will be to map the sites of phosphorylation on the inositol ring of PIP and PIP2 species regulated by DDHD1, as the location of these phosphorylation events can impart different biological activities on PI phosphates29.
C20:4 LPI is also a signaling lipid, acting as an endogenous agonist for the G-protein-coupled receptor GPR5533. GPR55–/– mice appear generally healthy, but display impaired mechanical hyperalgesia in multiple inflammatory and neuropathic pain models34 as well as aberrant sensory axonal projection35, phenotypes that resemble the reduced mechanical nociception observed in DDHD1–/– animals (Figure S3F). These findings point to the existence of a DDHD1-regulated C20:4 LPI-GPR55 pathway regulating pain behavior in mammals. A general future challenge will be to discern the relative contributions of PI lipids versus C20:4 LPI to the biological functions of DDHD1 in vivo, and GPR55 antagonists would offer valuable chemical probes for this purpose36.
Finally, we believe that our findings underscore the value of unbiased metabolomics methods for elucidating endogenous substrates and products of disease-relevant enzymes. Indeed, one may have initially assumed that DDHD1 and DDHD2, given their sequence-relatedness and shared human disease relevance (mutations in either enzyme cause HSP in humans), performed similar metabolic functions in vivo. On the contrary, we have found that each enzyme regulates a distinct class of lipids in the mammalian nervous system – TAGs for DDHD2 and PI/PS lipids for DDDH1. These differences, so far elucidated in mice, are likely relevant to humans as well, given that the genetic loss of DDHD2, but not DDHD1, produces a lipid MRI peak in the human brain10,14 that is consistent with TAG accumulation. We do not yet understand why DDHD2–/– mice appear to better replicate the neurological phenotypes of HSP15 compared to DDHD1–/– mice, which, despite substantial changes in brain phospholipid metabolism, were largely normal in their performance in neurobehavioral assays. It is possible that DDHD1–/– mice will present with neurological phenotypes at later stages in life. From a translational perspective, our data suggest that identifying enzymes within the PI metabolic network that can rectify metabolic alterations caused by DDHD1 loss could lead to a therapeutic strategy for HSP28. Conversely, DDHD1-selective inhibitors may provide a way to pharmacologically control PI phosphate signaling and its contribution to various physiological and disease processes29.
Supplementary Material
ACKNOWLEDGMENTS
The vector, ES cell(s), and/or mouse strain used for this research project was generated by the trans-NIH Knock-Out Mouse Project (KOMP) and obtained from the KOMP Repository (www.komp.org). NIH grants to Velocigene at Regeneron Inc (U01HG004085) and the CSD Consortium (U01HG004080) funded the generation of gene-targeted ES cells for 8500 genes in the KOMP Program and archived and distributed by the KOMP Repository at UC Davis and CHORI (U42RR024244). For more information or to obtain KOMP products go to www.komp.org or email service@komp.org.
Funding Sources
This work was supported by a Spastic Paraplegia Foundation grant (J.M.I.) and National Institutes of Health Grant R01 DA037660 and NS092980 (B.F.C.)
ABBREVIATIONS
- DDHD1
DDHD domain containing 1.
Footnotes
Publisher's Disclaimer: This document is confidential and is proprietary to the American Chemical Society and its authors. Do not copy or disclose without written permission. If you have received this item in error, notify the sender and delete all copies.
Supporting Information.
Supporting figures (Figures S1–S9), Supplemental tables (Tables S1–S3) and supplemental methods (PDF)
REFERENCES
- (1). Bisogno T; Howell F; Williams G; Minassi A; Cascio MG; Ligresti A; Matias I; Schiano-Moriello A; Paul P; Williams E-J; Gangadharan U; Hobbs C; Di Marzo V; Doherty P Cloning of the First Sn1-DAG Lipases Points to the Spatial and Temporal Regulation of Endocannabinoid Signaling in the Brain. J. Cell Biol 2003, 163 (3), 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2). Yung YC; Stoddard NC; Chun J LPA Receptor Signaling: Pharmacology, Physiology, and Pathophysiology. J. Lipid Res 2014, 55 (7), 1192–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3). Yamashita A; Oka S; Tanikawa T; Hayashi Y; Nemoto-Sasaki Y; Sugiura T The Actions and Metabolism of Lysophosphatidylinositol, an Endogenous Agonist for GPR55. Prostaglandins Other Lipid Mediat. 2013, 107, 103–116. [DOI] [PubMed] [Google Scholar]
- (4). Higgs HN; Han MH; Johnson GE; Glomset JA Cloning of a Phosphatidic Acid-Preferring Phospholipase A1 From Bovine Testis. J. Biol. Chem 1998, 273 (10), 5468–5477. [DOI] [PubMed] [Google Scholar]
- (5). Higgs HN; Glomset JA Purification and Properties of a Phosphatidic Acid-Preferring Phospholipase A1 From Bovine Testis. Examination of the Molecular Basis of Its Activation. J. Biol. Chem 1996, 271 (18), 10874–10883. [DOI] [PubMed] [Google Scholar]
- (6). Yamashita A; Kumazawa T; Koga H; Suzuki N; Oka S; Sugiura T Generation of Lysophosphatidylinositol by DDHD Domain Containing 1 (DDHD1): Possible Involvement of Phospholipase D/Phosphatidic Acid in the Activation of DDHD1. Biochim. Biophys. Acta 2010, 1801 (7), 711–720. [DOI] [PubMed] [Google Scholar]
- (7). Imae R; Inoue T; Kimura M; Kanamori T; Tomioka NH; Kage-Nakadai E; Mitani S; Arai H Intracellular Phospholipase A1 and Acyltransferase, Which Are Involved in Caenorhabditis Elegans Stem Cell Divisions, Determine the Sn-1 Fatty Acyl Chain of Phosphatidylinositol. Mol. Biol. Cell 2010, 21 (18), 3114–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8). Tesson C; Nawara M; Salih MAM; Rossignol R; Zaki MS; Balwi, Al M; Schule R; Mignot C; Obre E; Bouhouche A; Santorelli FM; Durand CM; Oteyza AC; El-Hachimi KH; Drees, Al A; Bouslam N; Lamari F; Elmalik SA; Kabiraj MM; Seidahmed MZ; Esteves T; Gaussen M; Monin M-L; Gyapay G; Lechner D; Gonzalez M; Depienne C; Mochel F; Lavie J; Schols L; Lacombe D; Yahyaoui M; Abdulkareem, Al I; Zuchner S; Yamashita A; Benomar A; Goizet C; Durr A; Gleeson JG; Darios F; Brice A; Stevanin G Alteration of Fatty-Acid-Metabolizing Enzymes Affects Mitochondrial Form and Function in Hereditary Spastic Paraplegia. Am. J. Hum. Genet 2012, 91 (6), 1051–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9). Bouslam N; Benomar A; Azzedine H; Bouhouche A; Namekawa M; Klebe S; Charon C; Durr A; Ruberg M; Brice A; Yahyaoui M; Stevanin G Mapping of a New Form of Pure Autosomal Recessive Spastic Paraplegia (SPG28). Ann. Neurol 2005, 57 (4), 567–571. [DOI] [PubMed] [Google Scholar]
- (10). Liguori R; Giannoccaro MP; Arnoldi A; Citterio A; Tonon C; Lodi R; Bresolin N; Bassi MT Impairment of Brain and Muscle Energy Metabolism Detected by Magnetic Resonance Spectroscopy in Hereditary Spastic Paraparesis Type 28 Patients with DDHD1 Mutations. J. Neurol 2014, 261 (9), 1789–1793. [DOI] [PubMed] [Google Scholar]
- (11). Mignarri A; Rubegni A; Tessa A; Stefanucci S; Malandrini A; Cardaioli E; Meschini MC; Stromillo ML; Doccini S; Federico A; Santorelli FM; Dotti MT Mitochondrial Dysfunction in Hereditary Spastic Paraparesis with Mutations in DDHD1/SPG28. J. Neurol. Sci 2016, 362, 287–291. [DOI] [PubMed] [Google Scholar]
- (12). Wu C; Fan D A Novel Missense Mutation of the DDHD1 Gene Associated with Juvenile Amyotrophic Lateral Sclerosis. Front. Aging Neurosci 2016, 8, 913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13). Dard R; Meyniel C; Touitou V; Stevanin G; Lamari F; Durr A; Ewenczyk C; Mochel F Mutations in DDHD1, Encoding a Phospholipase A1, Is a Novel Cause of Retinopathy and Neurodegeneration with Brain Iron Accumulation. Eur. J. Med. Genet 2017, 60 (12), 639–642. [DOI] [PubMed] [Google Scholar]
- (14). Schuurs-Hoeijmakers JHM; Geraghty MT; Kamsteeg E-J; Ben-Salem S; de Bot ST; Nijhof B; van de Vondervoort IIGM; van der Graaf M; Nobau AC; Otte-Höller I; Vermeer S; Smith AC; Humphreys P; Schwartzentruber J; FORGE Canada Consortium; Ali BR; Al-Yahyaee SA; Tariq S; Pramathan T; Bayoumi R; Kremer HPH; van de Warrenburg BP; van den Akker WMR; Gilissen C; Veltman JA; Janssen IM; Vulto-van Silfhout AT; van der Velde-Visser S; Lefeber DJ; Diekstra A; Erasmus CE; Willemsen MA; Vissers LELM; Lammens M; van Bokhoven H; Brunner HG; Wevers RA; Schenck A; Al-Gazali L; de Vries BBA; de Brouwer APM Mutations in DDHD2, Encoding an Intracellular Phospholipase a(1), Cause a Recessive Form of Complex Hereditary Spastic Paraplegia. Am. J. Hum. Genet 2012, 91 (6), 1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15). Inloes JM; Hsu K-L; Dix MM; Viader A; Masuda K; Takei T; Wood MR; Cravatt BF The Hereditary Spastic Paraplegia-Related Enzyme DDHD2 Is a Principal Brain Triglyceride Lipase. Proc. Natl. Acad. Sci. U.S.A 2014, 111 (41), 14924–14929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16). Saghatelian A; Trauger SA; Want EJ; Hawkins EG; Siuzdak G; Cravatt BF Assignment of Endogenous Substrates to Enzymes by Global Metabolite Profiling †. Biochemistry 2004, 43 (45), 14332–14339. [DOI] [PubMed] [Google Scholar]
- (17). Tautenhahn R; Cho K; Uritboonthai W; Zhu Z; Patti GJ; Siuzdak G An Accelerated Workflow for Untargeted Metabolomics Using the METLIN Database. Nat. Biotechnol 2012, 30 (9), 826–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18). Zhu Z-J; Schultz AW; Wang J; Johnson CH; Yannone SM; Patti GJ; Siuzdak G Liquid Chromatography Quadrupole Time-of-Flight Mass Spectrometry Characterization of Metabolites Guided by the METLIN Database. Nat. Protoc 2013, 8 (3), 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19). Baba T; Kashiwagi Y; Arimitsu N; Kogure T; Edo A; Maruyama T; Nakao K; Nakanishi H; Kinoshita M; Frohman MA; Yamamoto A; Tani K Phosphatidic Acid (PA)-Preferring Phospholipase A1 Regulates Mitochondrial Dynamics. J. Biol. Chem 2014, 289 (16), 11497–11511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20). Skarnes WC; Rosen B; West AP; Koutsourakis M; Bushell W; Iyer V; Mujica AO; Thomas M; Harrow J; Cox T; Jackson D; Severin J; Biggs P; Fu J; Nefedov M; de Jong PJ; Stewart AF; Bradley A A Conditional Knockout Resource for the Genome-Wide Study of Mouse Gene Function. Nature 2011, 474 (7351), 337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21). Liu Y; Patricelli MP; Cravatt BF Activity-Based Protein Profiling: the Serine Hydrolases. Proc. Natl. Acad. Sci. U.S.A 1999, 96 (26), 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22). Smith CA; Want EJ; O’Maille G; Abagyan R; Siuzdak G XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem 2006, 78 (3), 779–787. [DOI] [PubMed] [Google Scholar]
- (23). Tautenhahn R; Patti GJ; Rinehart D; Siuzdak G XCMS Online: a Web-Based Platform to Process Untargeted Metabolomic Data. Anal. Chem 2012, 84 (11), 5035–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24). Volpicelli-Daley LA; Lucast L; Gong L-W; Liu L; Sasaki J; Sasaki T; Abrams CS; Kanaho Y; De Camilli P Phosphatidylinositol-4-Phosphate 5-Kinases and Phosphatidylinositol 4,5-Bisphosphate Synthesis in the Brain. J. Biol. Chem 2010, 285 (37), 28708–28714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25). Idevall-Hagren O; De Camilli P Detection and Manipulation of Phosphoinositides. Biochim. Biophys. Acta 2015, 1851 (6), 736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26). Balla T Phosphoinositides: Tiny Lipids with Giant Impact on Cell Regulation. Physiol. Rev 2013, 93 (3), 1019–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27). Inloes JM; Kiosses WB; Wang H; Walther TC; Farese RV; Cravatt BF Functional Contribution of the Spastic Paraplegia-Related Triglyceride Hydrolase DDHD2 to the Formation and Content of Lipid Droplets. Biochemistry 2018, 57 (5), 827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28). Holub BJ; Kuksis A; Thompson W Molecular Species of Mono-, Di-, and Triphosphoinositides of Bovine Brain. J. Lipid Res 1970, 11 (6), 558–564. [PubMed] [Google Scholar]
- (29). Di Paolo G; De Camilli P Phosphoinositides in Cell Regulation and Membrane Dynamics. Nature 2006, 443 (7112), 651–657. [DOI] [PubMed] [Google Scholar]
- (30). Lee H-C; Inoue T; Sasaki J; Kubo T; Matsuda S; Nakasaki Y; Hattori M; Tanaka F; Udagawa O; Kono N; Itoh T; Ogiso H; Taguchi R; Arita M; Sasaki T; Arai H LPIAT1 Regulates Arachidonic Acid Content in Phosphatidylinositol and Is Required for Cortical Lamination in Mice. Mol. Biol. Cell 2012, 23 (24), 4689–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31). Johansen A; Rosti RO; Musaev D; Sticca E; Harripaul R; Zaki M; Çağlayan AO; Azam M; Sultan T; Froukh T; Reis A; Popp B; Ahmed I; John P; Ayub M; Ben-Omran T; Vincent JB; Gleeson JG; Abou Jamra R Mutations in MBOAT7, Encoding Lysophosphatidylinositol Acyltransferase I, Lead to Intellectual Disability Accompanied by Epilepsy and Autistic Features. Am. J. Hum. Genet 2016, 99 (4), 912–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32). McCrea HJ; De Camilli P Mutations in Phosphoinositide Metabolizing Enzymes and Human Disease. Physiology (Bethesda) 2009, 24 (1), 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33). Oka S; Toshida T; Maruyama K; Nakajima K; Yamashita A; Sugiura T 2-Arachidonoyl-Sn-Glycero-3-Phosphoinositol: a Possible Natural Ligand for GPR55. J. Biochem 2009, 145 (1), 13–20. [DOI] [PubMed] [Google Scholar]
- (34). Staton PC; Hatcher JP; Walker DJ; Morrison AD; Shapland EM; Hughes JP; Chong E; Mander PK; Green PJ; Billinton A; Fulleylove M; Lancaster HC; Smith JC; Bailey LT; Wise A; Brown AJ; Richardson JC; Chessell IP The Putative Cannabinoid Receptor GPR55 Plays a Role in Mechanical Hyperalgesia Associated with Inflammatory and Neuropathic Pain. Pain 2008, 139 (1), 225–236. [DOI] [PubMed] [Google Scholar]
- (35). Guy AT; Nagatsuka Y; Ooashi N; Inoue M; Nakata A; Greimel P; Inoue A; Nabetani T; Murayama A; Ohta K; Ito Y; Aoki J; Hirabayashi Y; Kamiguchi H Glycerophospholipid Regulation of Modality-Specific Sensory Axon Guidance in the Spinal Cord. Science 2015, 349 (6251), 974–977. [DOI] [PubMed] [Google Scholar]
- (36). Kotsikorou E; Sharir H; Shore DM; Hurst DP; Lynch DL; Madrigal KE; Heynen-Genel S; Milan LB; Chung TDY; Seltzman HH; Bai Y; Caron MG; Barak LS; Croatt MP; Abood ME; Reggio PH Identification of the GPR55 Antagonist Binding Site Using a Novel Set of High-Potency GPR55 Selective Ligands. Biochemistry 2013, 52 (52), 9456–9469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.