

Abstract
Transpositions transfer DNA segments between different loci within a genome; in particular, when a transposition is found in a sample but not in a reference genome, it is called a non-reference transposition. They are important structural variations that have clinical impact. Transpositions can be called by analyzing second generation high-throughput sequencing datasets. Current methods follow either a database-based or a database-free approach. Database-based methods require a database of transposable elements. Some of them have good specificity; however this approach cannot detect novel transpositions, and it requires a good database of transposable elements, which is not yet available for many species. Database-free methods perform de novo calling of transpositions, but their accuracy is low. We observe that this is due to the misalignment of the reads; since reads are short and the human genome has many repeats, false alignments create false positive predictions while missing alignments reduce the true positive rate. This paper proposes new techniques to improve database-free non-reference transposition calling: first, we propose a realignment strategy called one-end remapping that corrects the alignments of reads in interspersed repeats; second, we propose a SNV-aware filter that removes some incorrectly aligned reads. By combining these two techniques and other techniques like clustering and positive-to-negative ratio filter, our proposed transposition caller TranSurVeyor shows at least 3.1-fold improvement in terms of F1-score over existing database-free methods. More importantly, even though TranSurVeyor does not use databases of prior information, its performance is at least as good as existing database-based methods such as MELT, Mobster and Retroseq. We also illustrate that TranSurVeyor can discover transpositions that are not known in the current database.
INTRODUCTION
A transposition is a type of structural variation (SV), which transfers a DNA fragment from one locus into another. Although not as frequent as other variations such as single-nucleotide variant (SNVs) and small indels (each individual is expected to have one million of SNVs and hundreds of thousands of small indels (1)), they account for a number of genetic diseases. For example, they have been observed to occur in diseases related to the central nervous system (2), in hemophilia (3) and cancers such as colon (4), colorectal (5), gastrointestinal (6) and many others (7). For more information on genomic elements that commonly cause transpositions and their classification, see (8–10).
Transposition events can be classified into ancient and immobile (i.e. reference insertions) or active and polymorphic (i.e. non-reference insertions). Reference transpositions appear in the reference genome but not in the analyzed sample, while non-reference appear in the sample but not in the reference. SV callers normally predict the former as deletions, and the latter as insertions or transpositions. In this work, we focused on non-reference transpositions and we will simply refer to them as transpositions.
Transpositions can be predicted from high-throughput sequencing. There are two strategies: third generation and second generation sequencing. Third generation sequencing, such as single-molecule real-time sequencing by Pacific BioSciences that enables us to sequence reads of length up to tens of thousands of base pairs, and transpositions can be discovered by aligning such reads on the reference genome (11). However, their usage is limited by problems such as a higher amount of DNA required, a more difficult library preparation step, a higher error rate and a higher cost (12).
Another strategy is to discover transpositions by aligning short paired-end reads generated from second generation sequencing (i.e. Illumina sequencing). Second generation sequencing is cheap, high-throughput and robust. It is routinely used to discover SNVs and small indels; unfortunately, since the reads are short and more than half of the human genome is made of repeats (10,13), many false positive transpositions are predicted while many true transpositions are missed. This motivates us to study the possibility of improving transposition calling using short paired-end reads.
The problem of calling transpositions using second generation sequencing has been well-studied in the last decade. Current methods follow either a database-based (DB-based) or a database-free (DB-free) approach.
DB-based methods predict transpositions with the help of prior knowledge. Most existing methods require a known list of transposable elements or the target site duplication sequences. These methods include: PoPoolation TE (14), TE-locate (15), RetroSeq (16), TEMP (17), Mobster (18), TIF (19), ITIS (20) and MELT (21). Ewing (22) gave a survey of 20 such methods.
Although DB-based methods can call known transpositions accurately, they have severe drawbacks since (i) the database of transposable elements is unknown for some species and (ii) even when a database is available, they may miss novel transpositions.
To solve the problem, we use a DB-free approach. DB-free methods call transpositions by solely analyzing the sequencing dataset; such methods include general SV callers. Classical SV callers include PEM (23), break-dancer (24) and CREST (25). Currently, Lumpy (26) and Delly (27) are popular methods; recent surveys on the topic can be found in (28,29). To the best of our knowledge, DD_DETECTION (30) is the only DB-free specialized transposition caller. Unlike general SV callers, DD_DETECTION only predicts the insertion sites, but does not identify the source of the transposition nor the inserted sequence.
The performance of existing DB-free methods (i.e. DD_DETECTION and the existing SV callers) is not good in practice, as we show in the ‘Results’ section using the benchmark datasets from HX1 (31) and the Genome in a bottle (GIAB) project (32). The major problem is on the alignment of reads: as transposable elements have multiple copies in our genome, reads do not align properly, which causes low accuracy.
In this paper, we develop an improved DB-free transposition caller. We propose two new techniques. First, we propose a realignment strategy that corrects the alignments of reads in interspersed repeats. Second, we propose a SNV-aware filter that removes some incorrectly aligned reads in repeat regions. By combining these two techniques with other techniques like clustering and positive-to-negative ratio, we propose an accurate transposition caller, TranSurVeyor. Among all DB-free transposition callers tested, TranSurVeyor shows at least 3.1-fold improvement over the second best method in terms of F1-score. Even when compared to DB-based methods such as MELT, Mobster and Retroseq, TranSurVeyor’s F1-score is better in all tested datasets.
MATERIALS AND METHODS
A major challenge in transposition calling
Although many DB-free callers that predict transpositions using paired-end short reads exist, they show poor performance when they are applied to real datasets. One reason for this is the fact that for most transpositions the inserted sequence is present in multiple copies throughout the genome.
It is known that the inserted sequences of many transpositions are repetitive. For instance, mobile elements (13), which are frequently transposed, have many similar copies in the reference genome. Another example is interspersed duplications of low-complexity regions, which may also be present in many copies in the reference.
In the following, we discuss why DB-free callers have difficulty calling transpositions whose inserted sequences have multiple copies: the reason is due to misalignment. DB-free callers normally expect a number of ‘discordant’ read pairs supporting the event, i.e. pairs having one read mapped near the insertion site of the transposition and the other mapped to the source of the inserted sequence (see ‘Overview of the method’ and ‘Discordant read pairs and clipped reads identification’ sections for a technical definition of discordant pair). However, when a repetitive sequence is transposed into a unique region of the genome, we will have some discordant read pairs where one end is confidently mapped to the unique region (we call this the stable end of the pair), while the other end is randomly mapped to one of many possible loci (we call this the unstable end) where the repetitive inserted sequence appears in the reference genome. This situation is illustrated as an example in Figure 1, where a transposition transfers a segment from chr2 to chr1 (see Figure 1A). Three discordant pairs should link chr1 and chr2. However, while chr1 has three reads (stable ends) aligned on it, chr2 only has one. The other two unstable ends can map equally well on different loci, so one of them was randomly chosen by the aligner.
Figure 1.

A simple example of one-end remapping. (A) A sequence is inserted from chromosome 2 into chromosome 1. The same sequence appears, with some differences, in chr11; furthermore, the first (blue) half of the sequence appears also in chr7. (B) Although all the reverse mapping reads come from chr2, the gray read maps equally well to chr7, so it is randomly placed there; similarly the green read is mapped to chr11. (C) Assume for the sake of simplicity all reads are 100 bp long, and we award 2 points for a match and −2 for a mismatch. All reads align perfectly to chr2, so they get 200 points; on the other end, the green read cannot align to chr7 (near-zero score) while the black read only partially aligns to chr7 (140 points); all reads align to chr11, but the gray and the black reads do so with mismatches (188 and 192 points respectively). (D) Since it yields the best overall alignment, chr2 is chosen.
Specialized DB-based tools are normally designed to deal with this. However, DB-free callers are not, and these incorrectly aligned discordant read pairs will support false transposition events (e.g. chr7 in Figure 1B). At the same time, the mis-alignment also reduces the number of discordant read pairs that support the true transposition events.
This may cause DB-free callers to fail detecting the transpositions. In ‘DB-free callers miss transpositions whose inserted sequences appear in multiple copies in the reference’ section we experimentally verify that most transpositions missed by the tested DB-free SV callers are indeed present in multiple copies in the reference.
Overview of the method
We propose a DB-free method, TranSurVeyor, that finds transpositions given the alignments of second generation reads to a reference genome. Figure 2 illustrates the flow of TranSurVeyor. Given a BAM file, TranSurVeyor performs five steps: (i) Discordant read pairs and clipped reads identification, (ii) Mismatch filter, (iii) One-end remapping, (iv) Predicting candidate transpositions by clustering and (v) Positive-to-negative ratio filter.
Discordant read pairs and clipped reads identification: this step identifies read pairs and reads that aligned abnormally. These reads potentially support the transpositions.
Mismatch filter: some of the discordant read pairs identified from step (i) may be noise. This step filters the read pairs based on whether the reads display the same SNVs at the same positions.
One-end remapping: as shown in previous section, for some discordant read pairs, one of their ends may be aligned incorrectly. By a technique named one-end remapping, we realign these read pairs.
Predicting candidate transpositions by clustering: this step groups discordant read pairs if they support the same transposition. We group the discordant read pairs by a simple clustering algorithm; then, candidate transpositions are produced.
Positive-to-negative ratio filter: this filtering step scores every candidate transposition and determine if we report it. Precisely, for each candidate transposition, we determine if we accept it by computing a positive-to-negative ratio.
Figure 2.

The steps of TranSurVeyor.
Below, we detail these five steps.
Discordant read pairs and clipped reads identification
Discordant read pairs are pairs of reads that map inconsistently to the library preparation parameters, i.e. they map significantly outside the expected range of insert size distribution or on two different chromosomes. They potentially support the presence of transpositions. Given the BAM file, we find these discordant read pairs in two phases.
In the first phase, we extract discordant read pairs directly from the BAM file. For the purpose of detecting transpositions, we identify a subset of discordant read pairs such that (i) the two mates map on different chromosomes or (ii) the distance between the mapping loci of the two mates is greater than δ (by default δ = 100 kbp).
In the second phase, we examine clipped reads, i.e. reads that are only partially aligned onto the reference genome. First, clipped reads are clustered by their clip positions; precisely, the clipped reads are partitioned into left-clipped (i.e. their left part is clipped) and right-clipped (their right part is clipped). Then, left-clipped (respectively, right-clipped) reads are clustered so that their left (respectively, right)-end coordinates are within 10 bp from each other. We call such cluster the anchor of the clip. The consensus clip is then computed by simply piling up all the clipped portions of the reads in each cluster and taking the consensus sequence. Second, for each cluster, the consensus clip is realigned on the reference genome using BWA-MEM (33). If the realignment is successful, an artificial discordant read pair is created such that (i) one mate is the realigned consensus clip and (ii) one mate is the anchor of the clip.
Mismatch filter
From the previous step, a set of discordant read pairs is identified. Some discordant read pairs may be noise, which can create false positives. This step aims to determine if a discordant read pair is noise by checking if its SNVs are compatible with the SNVs of other reads in the same region.
Our basic assumption is that most reads are mapped correctly. In order to decide if a read r is misaligned or not, we compare it to the reads mapped in the same area. More specifically, for each base b in r we compare it to the bases mapped to the same genomic position on the reference; if b agrees with less than p% of them, we call it an error. If a read r has more than ε · length(r) errors then we filter it. p must be set to account for the ploidy of the input; by default, it is set to 40 to handle diploid inputs. ε is the maximum error rate of the sequencing platform; the default is set to 0.04 for Illumina platform. Figure 3 gives an example to illustrate this idea.
Figure 3.

Suppose we set a maximum sequencing error rate (platform dependant) of 4%, and the reads are 100 bp long. Read 3 has 1 base (T), which is different from the consensus (A), hence, Read 3 has 1 mistake out of 100 bases (i.e. error rate = 1%) and we keep Read 3. On the other hand, Read 1 has 5 mistakes out of 100 bases (i.e. error rate = 5%); this is higher than our maximum accepted error rate, and we remove Read 1.
One-end remapping
From the ‘A major challenge in transposition calling’ section, we know that discordant read pairs supporting a transposition event often share one of their ends (stable ends), i.e. one of their ends map close to each other, while the other ends map to distant loci (unstable ends). During the clustering step, because of the unstable ends, these pairs are not clustered together and create two possible problems: either many events are called, creating false positives, or the support is so spread-out that no event has enough support to be called. The aim of the one-end remapping is to find a common locus where the majority of the unstable ends can be remapped.
Let μ and σ be the mean and the standard deviation of the insert (also called fragment) size distribution of the library, and let maxIS be μ + 3σ (maxIS stands for maximum insert size). Assuming the insert size distribution is normal, we expect that the vast majority of the read pairs will have insert size less than or equal to maxIS and it is unlikely that two reads distant more than maxIS can support a same breakpoint. This is why maxIS will normally be used as the maximum distance for two reads to be clustered together.
The input to this step is a set of discordant read pairs. The algorithm proceeds as follows. First, we label as stable the read in a pair having the highest mapping quality; this simple strategy classifies correctly over 90% of the pairs in HX1 and HG001, and was the most effective among the strategies we tried (see Supplementary S4). Second, we cluster the pairs by their stable ends; this produces a set of clusters 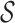 , such that for each cluster S in
, such that for each cluster S in 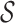 , all stable reads of the read pairs in S must be on the same chromosome, strand and within maxIS bp from each other. Third, for each cluster S in
, all stable reads of the read pairs in S must be on the same chromosome, strand and within maxIS bp from each other. Third, for each cluster S in 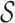 , let
, let 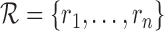 be the set of unstable reads in S. (For example, in Figure 1B), the three unstable reads are in chr 2, 7 and 11.) We remap the unstable reads as follows. For each ri, we extract the genomic region Ri surrounding it, and we call it a candidate region for remapping for S (see Figure 1C).
be the set of unstable reads in S. (For example, in Figure 1B), the three unstable reads are in chr 2, 7 and 11.) We remap the unstable reads as follows. For each ri, we extract the genomic region Ri surrounding it, and we call it a candidate region for remapping for S (see Figure 1C).
We define score(Ri, r) as the score of a Smith–Waterman local alignment between Ri and a read r. The score of the region Ri is 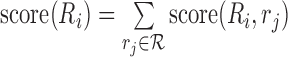 . We choose the region with the maximum score and call it Rbest. (For example, in Figure 1C), the chr2’s region is Rbest.)
. We choose the region with the maximum score and call it Rbest. (For example, in Figure 1C), the chr2’s region is Rbest.)
At last, we consider a special candidate region, the base region Rbase, which is the region around the stable end of S. If score(Rbase) ≥ α · score(Rbest), 0 ≤ α ≤ 1, it means that it is possible to realign the unstable reads near their stable mate with a tolerable score loss; in such case, we assume that there is no transposition, i.e. we remove all the reads in S from the discordant set. α = 0.9 by default. Otherwise, we realign all unstable reads to Rbest: all pairs in S now support a single transposition event.
When 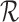 is too large, the above solution may be slow. In the actual implementation, we randomly sample a subset of reads
is too large, the above solution may be slow. In the actual implementation, we randomly sample a subset of reads 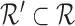 (by default |R′| = 15), and we compute an approximate score
(by default |R′| = 15), and we compute an approximate score 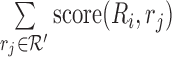 for each candidate region Ri. We choose the three regions with the highest approximate score, and at last, we align all reads to these three regions and identify Rbest.
for each candidate region Ri. We choose the three regions with the highest approximate score, and at last, we align all reads to these three regions and identify Rbest.
Clustering
After we correct the alignments of the unstable reads, we cluster the discordant read pairs to identify candidate transpositions. Clustering of discordant paired reads has been well-studied and many solutions have been proposed (see (34) for a review); we adapted the Delly (27) approach since it offers a good balance between simplicity and performance. Below we give a brief description of the method.
The clustering step repeatedly merges the two closest clusters, according to a distance measure, which will be explained shortly. The process is repeated until no clusters can be further merged.
In our approach, an anchor a is defined as a genomic region, which is characterized by a chromosome a.chr, a strand a.strand, a start a.start and an end a.end. Consider two anchors a1 and a2. We define the distance between these two anchors as max(a1.end, a2.end) − min(a1.start, a2.start). Two anchors a1 and a2 are compatible if (i) they are on the same chromosome and strand and (ii) their distance is less than maxIS. We also define a merging operation over two compatible anchors a1 and a2, which creates a third anchor am having the same chromosome and strand as a1 (and a2), and am.start = min(a1.start, a2.start) and am.end = max(a1.end, a2.end).
Every cluster c is composed of two anchors, c.a1 and c.a2. Two clusters c1 and c2 are compatible if the anchors are pairwise compatible (i.e. c1.a1 and c2.a1 are compatible and c1.a2 and c2.a2 are compatible).
The distance between two compatible clusters is defined as the sum of (i) the distance between c1.a1 and c2.a1 and (ii) the distance between c1.a2 and c2.a2
Let C be the set of clusters. Initially, each discordant read pair is a singleton cluster, and the two reads in the pair are the two anchors of the cluster. C contains one cluster for each discordant read pair. The clustering proceeds as follows. At each step, a pair of compatible clusters c1, c2 in C with minimum distance is selected and we create a new cluster c by merging the two pairs of compatible anchors, i.e. we merge c1.ai and c2.ai into c.ai for i = 1, 2. We remove c1 and c2 from C and we insert c into it. When no pairs of compatible clusters are in C, the procedure terminates.
At last, each cluster in C corresponds to one candidate transposition. Each anchor in the cluster generates a breakpoint: if the anchor is on the forward strand, its rightmost coordinate will be breakpoint; otherwise, it will be the leftmost coordinate.
Positive-to-negative ratio filter
This step computes a score for each candidate transposition. If the score is small, we will assume it is noise; otherwise, we report the transposition.
Each candidate transposition has two breakpoints (see Figure 4). For each breakpoint, we count the positive and negative evidences. The positive evidence count p is the number of discordant read pairs in the cluster that support the transposition. The negative evidence count n is the number of concordant (i.e. not discordant) read pairs crossing the breakpoint; note that concordant read pairs are evidence against the transposition event. We compute the positive-to-negative ratio of a breakpoint as
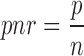 |
The higher the value of pnr, the higher the confidence that the breakpoint exists. If both breakpoints have pnr-value below a threshold β, we filter the transposition. The reason why we require that both breakpoints have low-confidence is because in copy-and-paste transpositions one breakpoint may have a low pnr-value, but still be real. By default 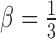 .
.
Figure 4.

An example of how the positive-to-negative ratio works. In this case, for each breakpoint we have three pieces of positive evidence and four pieces of negative information, which yields a positive-to-negative ratio of 0.75. This profile is compatible with a heterozygous transposition, for which we expect the ratio to be close to 1; in practice, as discordant pairs may be more difficult to align, or one haplotype may be more represented than the other, they may sometimes have lower values. We suggest a cut-off value of 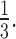
Details of software and the simulated and real datasets
Software
Many SV calling and transposition calling tools exist, and testing them all was impractical. Three SV calling tools were selected to represent the main SV calling approaches: Delly (27) (discordant pair clustering), Socrates (35) (remapping of soft-clipped reads) and Lumpy (26) (combination of both). We tried several other methods, but they either provided much poorer results, or could not finish a single sample in 1 week.
For transposition callers, we relied on a recent survey paper (36), which benchmarked and compared seven tools. We selected the best three tools according to this comparison, namely MELT, Mobster and Retroseq. At last, we also included DD_DETECTION as a DB-free caller.
We downloaded the tested programs from their original repositories and ran them with their default suggested parameters (Supplementary S5). We benchmarked the tools first using simulated datasets, and then using real human WGS datasets.
Simulated datasets
Simulated datasets were generated as follows. We used the RepeatMasker annotation for hg38 to classify the transpositions provided in the HX1 benchmark. Among the 10 most represented categories, seven belong to the SINE AluY family (85% of total transpositions), while the remaining three are LINE L1 repeats. We selected 8770 SINE and LINE repeats from the RepeatMasker annotation, maintaining as much as possible the proportions between categories found in HX1, and we inserted them into random locations in hg38; this way we obtained a sample genome with simulated transpositions.
We then used pIRS (37) to simulate three paired-end read datasets from the sample genome, with sequencing depth 5×, 10× and 20×, respectively. The insert sizes of all generated paired-end reads have mean 500 bp and standard deviation 100 bp. The read length was 100 bp. At last, all datasets were mapped against the reference hg38 by BWA MEM 0.7.10 (33).
Real-world datasets
We considered five samples: a Chinese genome for the HX1 project (31) and the HG001–HG004 datasets from the GIAB (32). From the literature, benchmark lists of insertions are available for these samples. Insertions can be classified into tandem duplications, transpositions and novel insertions; Supplementary S1 describes our approach to isolate transpositions from tandem duplications and novel insertions. Only benchmark insertions classified as transpositions were used to assess the sensitivity of the tools. The precision for a dataset was measured using its whole set of insertions. In other words, when measuring the sensitivity of a tool, a true positive is defined as a benchmark transposition correctly called by the tool. When measuring precision, a true positive is defined as a prediction from the tool, which corresponds to a benchmark insertion. We also removed insertions into centromeric regions and alternative chromosomes, as well as chromosomes X and Y (see Supplementary S2). The details of the benchmarks follow.
HX1: A total of 9125 insertions (after post-processing) were discovered by local assembly (38), and they were reported based on hg38. They were used to measure precision. We classified 2155 of them as transpositions, which were used as the ground truth to assess sensitivity.
PBHoney: For the GIAB datasets, using the SV caller PBHoney (39) on PacBio reads, 5120, 6737, 4944 and 4654 insertions were provided for HG001, HG002, HG003 and HG004, respectively, on hg19 (after post-processing). They were used to measure precision. We classified 1939, 2277, 1860 and 1754 insertions as transpositions, respectively; these were used to measure the sensitivity of the tools. The notes associated to the GIAB annotation files state that ‘Post-Processing was performed to generate the most specific call set, which is what’s been provided’. Thus, these benchmarks are likely incomplete. This is ideal for testing sensitivity, but it is likely to underestimate precision.
Pooled: For HG001, (40) obtained another list of insertions (on hg19) by merging the calls of different PacBio SV calling methods. These insertions are likely to be less specific but more complete. The total number of insertions is 22 199. Unfortunately, it was not possible to distinguish transpositions from other types of insertions in this dataset, because the inserted sequences were not provided. Here, we used this dataset to estimate the precision of different callers.
Second generation sequencing datasets are also available for these five samples. We downloaded these five real datasets from NCBI. Accession for HX1 was SRX1423751. As for GIAB, we downloaded runs from SRR2052337 to SRR2052356 for HG001, SRR1766442 to SRR1766486 for HG002, SRR1766542 to SRR1766648 for HG003 and SRR1766755 to SRR1766872 for HG004. This way we obtained five datasets of ∼50× coverage each. For comparison with the benchmark lists, HX1 was mapped to the hg38 human reference using BWA MEM 0.7.10, while the GIAB datasets were mapped to hg19.
Performance measures
We used three performance measures to compare the different callers: sensitivity, precision and F1-score. We computed such measures for each caller on each dataset using the following definitions. Let TP be the number of true positives detected by a caller, i.e. the number of transpositions in the benchmark correctly called. Let FP the number of false positives, i.e. the number of called transpositions, which have no confirmation in the benchmark. Let FN be the number of false negatives, i.e. the number of transpositions in the benchmark, which are missed by the caller. Then we define:
Sensitivity: also called ‘recall’, it is defined as 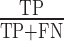 .
.
Precision: it is defined as 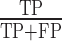 .
.
F1-score: it is a commonly used measure that summarizes both sensitivity and precision. It is defined as the harmonic mean between sensitivity and precision, i.e. 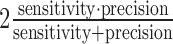 .
.
RESULTS
DB-free callers miss transpositions whose inserted sequences appear in multiple copies in the reference
As stated in ‘A major challenge in transposition calling’ section, the transpositions whose inserted sequences appear in multiple copies in the reference genome pose a challenge to existing DB-free callers. This section experimentally verifies this by studying the datasets HX1 and HG001-004.
We isolated the transpositions in HX1 that no caller among Delly (27), Lumpy (26) and Socrates (35) was able to discover, which are 1868. We also did the same for HG001, and we obtained 1434 missed transpositions.
Next, for each missed transposition in the HX1 benchmark, we mapped its inserted sequence onto the reference genome. Nearly 75% of them are present in the reference in multiple copies (Figure 5A), and around 40% in very large numbers (>100). In HG001 (Figure 5B), this behavior is even more evident, with <15% of the inserted sequences having a single copy in the reference. We also checked the hypothesis on HG002, HG003 and HG004 (see Supplementary Figure 1), and the same behavior was observed.
Figure 5.

Transpositions in the benchmark of (A) HX1 and (B) HG001 were partitioned into categories according to how many times the transposed (i.e. inserted) sequence appears in the reference. Most of them appear multiple times.
Performance comparison with DB-free callers
In this section the accuracy of DD DETECTION, Delly, Lumpy, Socrates and TranSurVeyor are compared. Remarkably, DD_DETECTION only reports the insertion sites of the transposition events, reporting neither the source of the event nor the inserted sequence. Thus, in order to perform a fair comparison of the tools, we compared the benchmark and the predicted events only by insertion site. Precisely, a predicted transposition was deemed to be the same as a benchmark insertion site if their positions are close. See Supplementary S3 for a more in-depth description of the comparison criteria.
Figure 6 shows the sensitivity, precision and F1-score of different transposition callers on the simulated datasets. Only DD_DETECTION was more sensitive than TranSurVeyor (although this was not the case at 5× coverage), but this comes at expenses of precision; while all other tools achieved near-perfect precision, DD_DETECTION performed much worse. When using 20× coverage, DD_DETECTION’s precision was 0.489, while the other methods had >0.997. TranSurVeyor was the overall winner for all coverages when it came to F1-score.
Figure 6.

Comparing transposition callers using simulated datasets. Histograms show (A) sensitivity, (B) precision and (C) F1-score on transposition calling for DD_DETECTION, Delly, Lumpy, Socrates and TranSurVeyor. A prediction is deemed to be correct if the insertion site is in the benchmark.
Results on real datasets (Figure 7) confirmed the trend seen in simulation, but the difference between TranSurVeyor and the other methods was much more marked.
Figure 7.

Comparing transposition callers of five real datasets using HX1 benchmark and the PBHoney benchmark. Histograms show (A) sensitivity, (B) precision and (C) F1-score on transposition calling for DD_DETECTION, Delly, Lumpy, Socrates and TranSurVeyor. A prediction is deemed to be correct if the insertion site is in the benchmark.
DD_DETECTION reported more than 100 000 events for each dataset, with a peak of 210 390 for HG001, which is the reason for the unnaturally high sensitivity, and the extremely low precision. Such method is hardly useful in practice, and comes out as the worst of the lot in terms of F1-score. Other callers had a much more reasonable number of calls, and our method showed dramatically higher sensitivity and precision, with at least 3.1-fold improvement over the second best method (Delly) in terms of F1-score. According to our experiments, TranSurVeyor is a substantial improvement over all the DB-free tools we tested.
Notably, TranSurVeyor was also the most precise caller when precision was assessed using the Pooled benchmark (Figure 8).
Figure 8.

Precision of DD_DETECTION, Delly, Lumpy, Socrates and TranSurVeyor using the Pooled benchmark.
Performance comparison with DB-based callers
Methods exploiting prior information have severe drawbacks compared to DB-free methods. They require a reliable database of transposable elements, which is not available for many species, and even in human they are not able to detect novel transpositions, i.e. transpositions of elements not previously annotated. In this section, we show that TranSurVeyor is able to achieve results comparable, if not superior, to the state-of-the-art DB-based tools. Note that MELT, Mobster and Retroseq were also tested in (36) on HG001, and the outcome of our benchmarks are compatible with theirs.
In the simulated dataset (Figure 9), TranSurVeyor was the most sensitive tool for all coverages. Precision was near-perfect for all tools, and was always >0.997; we note that TranSurVeyor seems to have a slight advantage, although very small. As a result of the better sensitivity, TranSurVeyor had the highest F1-score.
Figure 9.

Comparing transposition callers using the simulated datasets. Histograms show (A) sensitivity, (B) precision and (C) F1-score on transposition calling for MELT, Mobster, Retroseq and TranSurVeyor. A prediction is deemed to be correct if the insertion site is in the benchmark.
In the real data (Figure 10), TranSurVeyor was still the most sensitive tool of the lot in all datasets. Three hundred ninety-four transpositions in HG001 with support from PBHoney are only predicted by TranSurVeyor. We mapped their inserted sequences to MELT and Mobster databases using Bowtie 2 (41); for MELT, only 27 found at least 1 hit, while for Mobster only 112. This indicates that TranSurVeyor can find many novel transpositions, which are missing in the existing databases.
Figure 10.

Comparing transposition callers of 5 real datasets using HX1 benchmark and the PBHoney benchmark. Histograms show (A) sensitivity, (B) precision and (C) F1-score on transposition calling for MELT, Mobster, Retroseq and TranSurVeyor. A prediction is deemed to be correct if the insertion site is in the benchmark.
However, it was also the least precise when using the PBHoney annotations; note that only MELT seems to have a clear advantage over our method, while the precision of the other methods is often comparable. Overall, TranSurVeyor had the best F1-score.
As we noted in section Read-world datasets, the PBHoney benchmark is very conservative and it was tuned for specificity, therefore it may underestimate the precision; this is especially unfair for tools that call more aggressively. Hence, we also computed the precision for the Pooled benchmark (Figure 11), which gives an alternative point of view on the precision of the methods: while MELT was still the most precise tool (precision = 0.95), TranSurVeyor displayed the second highest precision (precision = 0.9). In general, all of the DB-based methods performed well and had precision greater than 0.8. In summary, even though TranSurVeyor does not use any database, its performance is comparable with DB-based methods.
Figure 11.

Precision of MELT, Mobster, Retroseq and TranSurVeyor using the Pooled benchmark.
A stricter comparison criteria
Previous section just compared the performance of calling insertion sites. Here, we employed a stricter comparison criteria. A transposition was deemed to be correctly predicted if both the insertion site and the source of the transposition are correct (see Supplementary S3). DD_DETECTION and the DB-based methods were excluded from this comparison since they report neither the source of the transposition nor the inserted sequence.
Figure 12 shows the results using this new comparison criteria. TranSurVeyor holds a larger advantage over the other callers in this comparison: its F1-score was at least 3.8 times Delly’s F1-score, the second best tool. Furthermore, the sensitivity and precision loss over the more relaxed comparison (comparing only insertion sites) was minimal. This means that (i) TranSurVeyor rarely predicts an incorrect source for the transposition and (ii) TranSurVeyor predicts the correct source of the transposition more often than the other tools.
Figure 12.

(A) Sensitivity, (B) precision and (C) F1-score on transposition for Delly, Lumpy, Socrates and TranSurVeyor. The experiment is done on five real datasets. A prediction is deemed to be correct if both the inserted sequence and the insertion site are correct.
Running time and memory comparison
We limited multi-threading to eight threads, when available, since this is what a modern personal computer can normally handle. We ran all software on an Intel Xeon E5-2620 processor, Red Hat Enterprise Linux Server release 6.3.
Table 1 reports the running time and peak memory usage for all the tools in processing the HG001 dataset, 50×. Only Socrates was faster than TranSurVeyor; however, as we have amply shown in the previous sections, TranSurVeyor produces vastly superior predictions. All the other tools were several times slower than TranSurVeyor, from a minimum of three times for Mobster to 32 for DD_DETECTION. TranSurVeyor also used less memory than all the other tools with the exception of Delly and Retroseq.
Table 1.
Running times and memory usage on HG001 of the tested tools
| Tool | Time | Memory (GB) |
|---|---|---|
| DD_DETECTION | 2 days 16 h 33 min | 22 |
| Delly | 1 days 2 h 24 min | 1.4 |
| Lumpy | 10 h 44 min | 27 |
| MELT | 10 h 30 min | 35 |
| Mobster | 6 h 14 min | 38 |
| Retroseq | 1 days 4 h 31 min | 1.9 |
| Socrates | 1 h 9 min | 38 |
| TranSurVeyor | 2 h 6 min | 12 |
TranSurVeyor is much faster than any other tool except for Socrates; however, as reported in ‘Results’ section, TranSurVeyor predictions are far more accurate than Socrates. TranSurVeyor also uses considerably less memory than the other tools except for Delly and Retroseq.
DISCUSSION AND CONCLUSION
In this paper, we tackled the problem of transposition calling from high-throughput next-generation sequencing. We have shown that transposition of sequences appearing in multiple similar copies in the reference genome, such as mobile elements, pose a challenge to existing DB-free callers. This is because the reads belonging to the inserted sequence have multiple possible alignments, and it may be impossible to align them correctly by considering them individually. We addressed this problem by proposing a realignment strategy which groups reads supporting a same transposition, and uses them jointly to discover a best copy in the reference.
We implemented this strategy, along with a SNV-aware filter, in a DB-free transposition caller, TranSurVeyor, which vastly improves over its direct competitors, and is able to match and even improve over state-of-the-art methods, which rely on databases of known transposable elements. Using a database has serious drawbacks, since the transposable elements are not well characterized and annotated for many species (30,19), therefore it is crucial to develop reliable DB-free methods.
While TranSurVeyor represents a step forward compared to the other tested tools, there is still room for improvement. Across five real datasets, it was able to call approximately half of the transpositions discovered using PacBio reads. We identified two classes of transpositions, which were difficult to call using our current approach:
Transpositions of short sequences tend to have less inter-chromosomal pairs;
Transpositions where the insertion site is in a non-unique region tend to produce inter-chromosomal pairs where the stable end has multiple possible alignments; choosing the correct insertion site in this case is very challenging.
Techniques that can deal with these two difficulties could further improve transposition calling.
The source code, along with instructions can be found at https://github.com/Mesh89/TranSurVeyor.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the helpful discussion with Peiyong Guan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
MOE Tier2 Grant [R-252-000-664-112]. Funding for open access charge: Singapore MOE Tier 2 grant [R-252-000-664-112].
Conflict of interest statement. None declared.
REFERENCES
- 1. Jiang Y., Turinsky A.L., Brudno M.. The missing indels: an estimate of indel variation in a human genome and analysis of factors that impede detection. Nucleic Acids Res. 2015; 43:7217–7228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reilly M.T., Faulkner G.J., Dubnau J., Ponomarev I., Gage F.H.. The role of transposable elements in health and diseases of the central nervous system. J. Neurosci. 2013; 33:17577–17586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kazazian H.H., Wong C., Youssoufian H., Scott A.F., Phillips D.G., Antonarakis S.E.. Haemophilia a resulting from de novo insertion of l1 sequences represents a novel mechanism for mutation in man. Nature. 1988; 332:164–166. [DOI] [PubMed] [Google Scholar]
- 4. Miki Y., Nishisho I., Horii A., Miyoshi Y., Utsunomiya J., Kinzler K.W., Vogelstein B., Nakamura Y.. Disruption of the apc gene by a retrotransposal insertion of l1 sequence in a colon cancer. Cancer Res. 1992; 52:643–645. [PubMed] [Google Scholar]
- 5. Solyom S., Ewing A.D., Rahrmann E.P., Doucet T., Nelson H.H., Burns M.B., Harris R.S., Sigmon D.F., Casella A., Erlanger B. et al. Extensive somatic l1 retrotransposition in colorectal tumors. Genome Res. 2012; 22:2328–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ewing A.D., Gacita A., Wood L.D., Ma F., Xing D., Kim M.-S., Manda S.S., Abril G., Pereira G., Makohon-Moore A. et al. Widespread somatic l1 retrotransposition occurs early during gastrointestinal cancer evolution. Genome Res. 2015; 25:1536–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rodić N., Sharma R., Sharma R., Zampella J., Dai L., Taylor M.S., Hruban R.H., Iacobuzio-Donahue C.A., Maitra A., Torbenson M.S. et al. Long interspersed element-1 protein expression is a hallmark of many human cancers. Am. J. Pathol. 2014; 184:1280–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beck C.R., Garcia-Perez J.L., Badge R.M., Moran J.V.. Line-1 elements in structural variation and disease. Annu. Rev. Genomics Hum. Genet. 2011; 12:187–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cordaux R., Batzer M.A.. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009; 10:691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rishishwar L., Wang L., Clayton E.A., Mariño-Ramírez L., McDonald J.F., Jordan I.K.. Population and clinical genetics of human transposable elements in the (post) genomic era. Mob. Genet. Elements. 2017; 7:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ritz A., Bashir A., Sindi S., Hsu D., Hajirasouliha I., Raphael B.J.. Characterization of structural variants with single molecule and hybrid sequencing approaches. Bioinformatics. 2014; 30:3458–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lischer H.E.L., Shimizu K.K.. Reference-guided de novo assembly approach improves genome reconstruction for related species. BMC Bioinformatics. 2017; 18:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stewart C., Kural D., Strömberg M.P., Walker J.A., Konkel M.K., Stütz A.M., Urban A.E., Grubert F., Lam H.Y.K., Lee W.-P. et al. A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet. 2011; 7:e1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kofler R., Betancourt A.J., Schlötterer C.. Sequencing of pooled dna samples (pool-seq) uncovers complex dynamics of transposable element insertions in drosophila melanogaster. PLoS Genet. 2012; 8:e1002487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Platzer A., Nizhynska V., Long Q.. Te-locate: a tool to locate and group transposable element occurrences using paired-end next-generation sequencing data. Biology. 2012; 1:395–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Keane T.M., Wong K., Adams D.J.. Retroseq: transposable element discovery from next-generation sequencing data. Bioinformatics. 2013; 29:389–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhuang J., Wang J., Theurkauf W., Weng Z.. Temp: a computational method for analyzing transposable element polymorphism in populations. Nucleic Acids Res. 2014; 42:6826–6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thung D.T., de Ligt J., Vissers L.E.M., Steehouwer M., Kroon M., de Vries P., Slagboom E.P., Ye K. Veltman J.A., Hehir-Kwa J.Y.. Mobster: accurate detection of mobile element insertions in next generation sequencing data. Genome Biol. 2014; 15:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nakagome M., Solovieva E., Takahashi A., Yasue H., Hirochika H., Miyao A.. Transposon insertion finder (tif): a novel program for detection of de novo transpositions of transposable elements. BMC Bioinformatics. 2014; 15:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang C., Chen C., Huang Z., Liu R., Verdier J.. Itis, a bioinformatics tool for accurate identification of transposon insertion sites using next-generation sequencing data. BMC Bioinformatics. 2015; 16:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gardner E.J., Lam V.K., Harris D.N., Chuang N.T., Scott E.C., Pittard W.S., Mills R.E. 1000 Genomes Project Consortium . 1000 Genomes Project Consortium. Devine S.E. The mobile element locator tool (melt): population-scale mobile element discovery and biology. Genome Res. 2017; 27:1916–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ewing A.D. Transposable element detection from whole genome sequence data. Mob. DNA. 2015; 6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Korbel J.O., Urban A.E., Affourtit J.P., Godwin B., Grubert F., Simons J.F., Kim P.M., Palejev D., Carriero N.J., Du L. et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007; 318:420–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen K., Chen K., Wallis J.W., McLellan M.D., Larson D.E., Kalicki J.M., Pohl C.S., McGrath S.D., Wendl M.C., Zhang Q. et al. Breakdancer: an algorithm for high-resolution mapping of genomic structural variation. Nat. Methods. 2009; 6:677–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang J., Mullighan C.G., Easton J., Roberts S., Heatley S.L., Ma J., Rusch M.C., Chen K., Harris C.C., Ding L. et al. Crest maps somatic structural variation in cancer genomes with base-pair resolution. Nat. Methods. 2011; 8:652–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Layer R.M., Chiang C., Quinlan A.R., Hall I.M.. Lumpy: a probabilistic framework for structural variant discovery. Genome Biol. 2014; 15:R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rausch T., Zichner T., Schlattl A., Stütz A.M., Benes V., Korbel J.O.. Delly: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012; 28:i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guan P., Sung W.-K.. Structural variation detection using next-generation sequencing data: a comparative technical review. Methods. 2016; 102:36–49. [DOI] [PubMed] [Google Scholar]
- 29. Tattini L., D’Aurizio R., Magi A.. Detection of genomic structural variants from next-generation sequencing data. Front. Bioeng. Biotechnol. 2015; 3:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kroon M., Lameijer E.W., Lakenberg N., Hehir-Kwa J.Y., Thung D.T., Slagboom P.E., Kok J.N., Ye K.. Detecting dispersed duplications in high-throughput sequencing data using a database-free approach. Bioinformatics. 2016; 32:505–510. [DOI] [PubMed] [Google Scholar]
- 31. Shi L., Guo Y. Dong C., Huddleston J., Yang H., Han X., Fu A., Li Q., Li N., Gong S. et al. Long-read sequencing and de novo assembly of a chinese genome. Nat. Commun. 2016; 7:12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zook J.M., Catoe D., McDaniel J., Vang L., Spies N., Sidow A., Weng Z., Liu Y., Mason C.E., Alexander N. et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci. Data. 2016; 3:160025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li H., Durbin R.. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010; 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sung W.-K. Algorithms for Next-generation Sequencing. 2017; Boca Raton: CRC Press. [Google Scholar]
- 35. Schröder J., Hsu A., Boyle S.E., Macintyre G., Cmero M., Tothill R.W., Johnstone R.W., Shackleton M., Papenfuss A.T.. Socrates: identification of genomic rearrangements in tumour genomes by re-aligning soft clipped reads. Bioinformatics. 2014; 30:1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rishishwar L., Mariño-Ramírez L., Jordan I.K.. Benchmarking computational tools for polymorphic transposable element detection. Brief. Bioinform. 2017; 18:908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hu X., Yuan J., Shi Y., Lu J., Liu B., Li Z., Chen Y., Mu D., Zhang H., Li N. et al. Pirs: profile-based illumina pair-end reads simulator. Bioinformatics. 2012; 28:1533–1535. [DOI] [PubMed] [Google Scholar]
- 38. Chaisson M.J.P., Huddleston J., Dennis M.Y., Sudmant P.H., Malig M., Hormozdiari F., Antonacci F., Surti U., Sandstrom R., Boitano M. et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature. 2015; 517:608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. English A.C., Salerno W.J., Reid J.G.. Pbhoney: identifying genomic variants via long-read discordance and interrupted mapping. BMC Bioinformatics. 2014; 15:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mt. Sinai School of Medicine. Annotations for SVs in HG001ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/NA12878/NA12878_PacBio_MtSinai. [Google Scholar]
- 41. Langmead B., Salzberg S.L.. Fast gapped-read alignment with bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.