

Abstract
The ribosome translates nucleotide sequences of messenger RNA to proteins through selection of cognate transfer RNA according to the genetic code. To date, structural studies of ribosomal decoding complexes yielding high-resolution data have predominantly relied on experiments performed at cryogenic temperatures. New light sources like the X-ray free electron laser (XFEL) have enabled data collection from macromolecular crystals at ambient temperature. Here, we report an X-ray crystal structure of the Thermus thermophilus 30S ribosomal subunit decoding complex to 3.45 Å resolution using data obtained at ambient temperature at the Linac Coherent Light Source (LCLS). We find that this ambient-temperature structure is largely consistent with existing cryogenic-temperature crystal structures, with key residues of the decoding complex exhibiting similar conformations, including adenosine residues 1492 and 1493. Minor variations were observed, namely an alternate conformation of cytosine 1397 near the mRNA channel and the A-site. Our serial crystallography experiment illustrates the amenability of ribosomal microcrystals to routine structural studies at ambient temperature, thus overcoming a long-standing experimental limitation to structural studies of RNA and RNA–protein complexes at near-physiological temperatures.
Keywords: serial femtosecond X-ray crystallography, ribosome, decoding, ambient temperature, antibiotics
INTRODUCTION
The bacterial ribosome possesses universally conserved functional centers that are structurally dynamic and undergo local and large-scale conformational rearrangements during protein synthesis (Ogle et al. 2001; Petrov et al. 2011; Loveland et al. 2017). In particular, the small (30S) ribosomal subunit, which is responsible for decoding the messenger RNA sequence, undergoes rearrangement of the universally conserved monitoring residues A1492 and A1493 in the decoding center, as well as a large-scale domain closure that involves movement of the body of the whole 30S subunit closer to helix 44 (Yoshizawa et al. 1999; Ogle et al. 2001; Demirci et al. 2013a). The structural dynamics and allostery of the initial step of decoding has been mostly studied using 30S crystals (Wimberly et al. 2000), allowing observation of these conformational changes ranging from small-scale base flipping at the decoding center to medium- and large-scale domain motions upon soaking with translation factors, antibiotics, cognate and near-cognate decoding complexes without destroying the diffraction quality of the crystal (Carter et al. 2000, 2001; Wimberly et al. 2000; Ogle et al. 2001; Demirci et al. 2013a). Although such conformational changes during translation have been identified, current understanding of the structural dynamics of decoding remains incomplete (Ogle et al. 2003; Loveland et al. 2017).
X-ray crystallography performed at synchrotron light sources has revealed the structures of the ribosome complexes at high resolution (Ban et al. 2000; Schluenzen et al. 2000; Wimberly et al. 2000; Yusupov et al. 2001; Schuwirth et al. 2005; Korostelev et al. 2006; Selmer et al. 2006; Yusupova et al. 2006; Ben-Shem et al. 2011). A key aspect to this important achievement was the development of procedures to collect data at cryogenic temperature (Haas and Rossmann 1970; Evers et al. 1994), which enabled the acquisition of data sets from single crystals that contained sufficient data to build detailed atomic models (Warkentin et al. 2014). Electron microscopy performed at cryogenic temperature has added even more information about the ribosome during several steps of protein synthesis, at increasing resolution to an existing wealth of structural information spanning several species and conformational states (Mitra et al. 2005; Fischer et al. 2015; Natchiar et al. 2017; Razi et al. 2017). Cooling ribosomal samples to cryogenic temperature mitigates the propagation of radiation damage by reducing the movement of radicals produced by irradiation with electrons or X-rays, lowering the thermal fluctuations and conformational distributions of side- and main-chain residues, and rigidifying the structure, partly through dehydration by adding hygroscopic cryo-protectants (Warkentin et al. 2014). Another possible impact of cryo-cooling on protein structures includes limiting the breadth of conformational heterogeneity that may be observed (Keedy et al. 2014). Therefore, while it enables successful data collection, cryo-cooling can potentially mask important details about local and global conformational dynamics and the allosteric mechanisms at play in RNA and protein structures.
The flexibility of ribosomal RNA is of significant importance in our understanding of the functional relevance of nucleic acids (Noller 2013). Additionally, the structure and conformational heterogeneity of RNA molecules are determined by the composition and physico-chemical state of the surrounding electrolytic medium (Anthony et al. 2012; Lipfert et al. 2014; Allred et al. 2017). Structural studies of ribosomes at temperatures closer to the physiological range could potentially reveal previously obscured conformations and provide a means to evaluate their local dynamics and role in catalysis (Sierra et al. 2016).
One such method is the recent advent of X-ray free electron lasers (XFELs), a light source that generates pulses of X--rays spanning tens of femtoseconds in duration and exceeding the brightness of current synchrotrons (Chapman et al. 2011). The Linac Coherent Light Source (LCLS), one such XFEL, can produce X-ray pulses of 1012 photons at photon energies of 500 eV to 12.7 keV with a duration of a few to a few hundred femtoseconds (Emma et al. 2010; Hunter et al. 2016). Serial femtosecond X-ray crystallography (SFX) harnesses these pulses to probe crystals at ambient temperature and is emerging as a promising method to complement synchrotron-based crystallography studies (Chapman et al. 2011; Fromme and Spence 2011; Schlichting and Miao 2012; Bogan 2013; Helliwell 2013). One typical approach is to deliver crystals flowing in a liquid suspension to the interaction point, at which the extremely short and brilliant X-ray pulses produce diffraction patterns before Coulomb explosion of the crystal (DePonte et al. 2009; Barty et al. 2012; Sierra et al. 2012; Weierstall et al. 2012). The ability of the “diffract-before-destroy” approach to obtain high-resolution data was first demonstrated by the 1.9 Å resolution structure of lysozyme and the 2.1 Å resolution structure of cathepsin B (Neutze et al. 2000; Boutet et al. 2012; Redecke et al. 2013). The potential of this approach for the study of large macromolecular complexes has also shown great promise with the analysis of photosystem I, photosystem II, and ribosome microcrystals (Chapman et al. 2011; Kern et al. 2012, 2013; Demirci et al. 2013b). Recent SFX studies of an adenine riboswitch aptamer domain mixed with its substrate immediately prior to probing captured dynamics of the reaction at ambient temperature and revealed conformational changes that also induced a conversion of the space group in crystallo (Stagno et al. 2017). Such findings indicate that SFX can offer opportunities to probe RNA or RNA–protein complexes using microcrystals, either as static structures or as they undergo biologically relevant reactions.
In this work, we present an ambient-temperature structure of a 30S ribosomal decoding complex through a serial femtosecond X-ray crystallography (SFX) experiment. Using 40-femtosecond pulses, we obtain diffraction from microcrystals prior to the onset of radiation damage induced by the X-ray beam using the Coherent X-ray Imaging (CXI) instrument at LCLS (Liang et al. 2015). The microcrystals contained ribosomal subunits bound to a cognate mRNA-anticodon stem–loop (ASL) complex and were introduced to the X-ray beam in a liquid suspension with an electrokinetic sample injector (Sierra et al. 2016). We then compared our structure to two analogous structures solved through synchrotron X-ray diffraction collected at cryogenic temperature (Ogle et al. 2001; Demirci et al. 2013a) to identify any potentially noteworthy differences.
RESULTS AND DISCUSSION
SFX workflow for microcrystals of 30S ribosomal subunits
A brief outline of the experimental methods used for data collection follows, along with references to the pertinent subsections within the Materials and Methods. Microcrystals of the 30S ribosomal subunit were soaked with 80 µM paromomycin, 200µM mRNA, and 200 µM phenylalanine tRNA anticodon stem–loop, ASLPhe oligonucleotide, resulting in a slurry of 2 × 2 × 4 µm3 size 30S decoding complex microcrystals (see “Preparation and crystallization of 30S ribosomal subunits for SFX crystallography at an XFEL” section). They were delivered to the X-ray beam at the Coherent X-ray Imaging (CXI) instrument (Liang et al. 2015) of the Linac Coherent Light Source (LCLS) via a concentric electrokinetic liquid injector (see sections “coMESH construction” and “Operation of the coMESH”), as previously used for crystalline ribosome samples (Fig. 1A; Sierra et al. 2016) (see sections “Selecting a sister liquor for the ribosome microcrystalline slurry” and “Ribosome microcrystalline sample injection with coMESH”). During a six-hour “protein crystal screening” beam-time, we collected a complete data set extending to 3.45 Å resolution (Table 1). A total of 1,731,280 detector frames were collected (corresponding to 240 min of net data collection time), of which 165,954 contained diffraction data. Of these frames, 19,374 patterns were indexed and merged into the final data set. (see sections “Data collection and analysis for SFX studies at LCLS” and “Ambient temperature 30S ribosomal subunit SFX structure refinement”).
FIGURE 1.

Approach to serial femtosecond X-ray (SFX) crystallography studies of a 30S ribosomal subunit decoding complex. (A) Diagram of the concentric-flow MESH injector setup at the CXI instrument of the LCLS. The liquid jet, comprising microcrystals and their mother liquor (colored in yellow), flows in the continuous inner capillary (100 µm × 160 µm × 1.5 m; colored in gray). The sister liquor (colored in green) is charged by a high voltage power supply (0–5000 V) for electro-focusing of the liquid jet. A mixer (indicated within the dashed orange rectangle) joins the two capillaries (colored in gray) concentrically. The sample reservoir containing ribosome microcrystals is mounted on an anti-settling device, which rotates, at an angle, about the capillary axis to keep the microcrystals suspended homogenously in the slurry. The liquid jet and the LCLS pulses interact at the point indicated by the orange circle. (B) Comparison of T. thermophilus 30S-ASL-mRNA-paromomycin complex structures. Superposition of 16S rRNA backbones from cryo-cooled structures colored in cyan and slate (PDB IDs: 4DR4 and 1IBL, respectively) with the ambient-temperature structure colored in salmon. The positions of the major 30S domains are indicated. All X-ray crystal structure figures are produced with PyMOL (http://www.schrodinger.com/pymol).
TABLE 1.
Data collection and refinement statistics

Decoding complex structure at ambient temperature largely similar to prior structures
The electron density map indicated good mRNA and ASLPhe density quality in Fo-Fc difference electron density maps (Fig. 2A). The crystal structure of the 30S decoding complex at ambient temperature adopted the canonical decoding conformation, with the h44 residues A1492 and A1493 flipped out toward the minor groove of ASL and mRNA pair, consistent with the cryogenic data (Fig. 2B). Overall, the ambient-temperature decoding crystal structure was found to be very similar to its equivalents at cryogenic temperature (PDB IDs: 1IBL and 4DR4) (Ogle et al. 2001; Demirci et al. 2013a), with a notable exception for an alternate conformation of mRNA channel residue C1397 (further discussed below). A least-squares alignment of all 17,056 16S rRNA atoms in the 30S structures showed an overall root-mean-square deviation (RMSD) of 0.45 and 0.62 Å between the new structure and the cryogenic structures 4DR4 and 1IBL, respectively (Fig. 1B). The minimal binding differences observed between the cryogenic and ambient temperature 30S decoding complex structures (Figs. 1B, 2B–D) suggest that ribosomal decoding complexes can be probed at cryogenic temperature and may still be representative of what occurs at ambient temperature. However, we identified small differences within the mRNA channel in the region between the S4–S5 protein interface and the 30S acceptor A-site.
FIGURE 2.
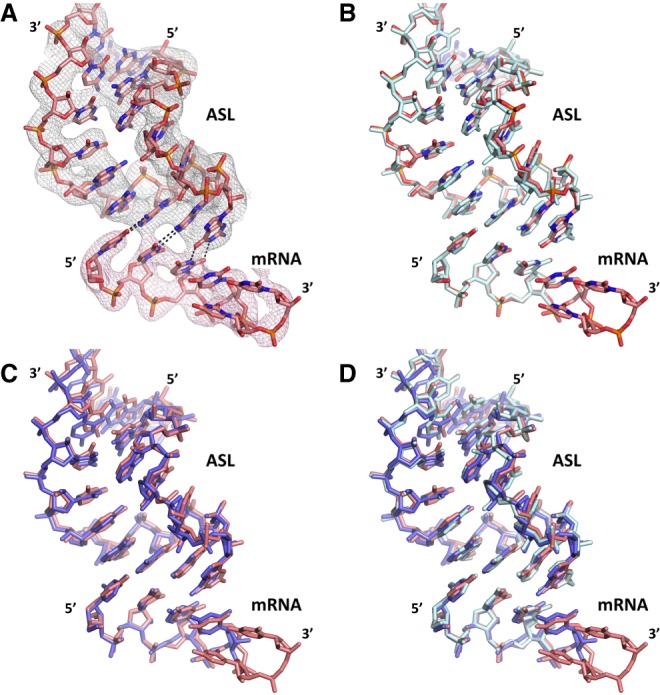
Structural comparison of ambient- and cryogenic-temperature decoding complexes. (A) Final unbiased Fo-Fc simple omit ambient temperature electron-density map of mRNA and ASL contoured at the 3σ level, colored in gray and shown at 3 Å. (B) Superposition of the ambient (salmon) and cryo (cyan) temperature structures from our group showing the agreement between them. (C) Superposition of our ambient temperature structure (salmon) and the identical cryo temperature structure obtained by another laboratory (slate; PDB ID: 1IBL) showing the agreement between them. (D) Superposition of our ambient temperature structure (salmon) with the two cryo-cooled structures (cyan and slate).
Structural perturbation in the mRNA channel
In the structure presented here, the hydrogen bonding interactions of codon residues 1–3 of the hexauridine in the mRNA-ASL complex were markedly similar to those observed in the cryogenic temperature crystal structure, as shown by the clear positive difference density for each base pair in the omit Fo-Fc electron density maps at 3.45 Å resolution (Fig. 3A–F). On the other hand, residues 4–6 of the hexa-uridine were disordered and as a result not modeled in the cryogenic structure, while a well-defined electron density was observed in the ambient-temperature structure (Fig. 2A). The 16S rRNA residue C1397 was captured in a different conformation compared to the prior structure, pointing away from the disordered hexauridine in the cryogenic data set and involved in a stabilizing interaction with the mRNA phosphate group of residue 4 in the ambient-temperature structure (Fig. 4A–D).
FIGURE 3.

X-ray crystallography structures of the decoding mRNA-ASL minihelix. (A) Simple Fo-Fc omit maps of the decoding complex individual base pair mRNA(U1)-ASL(A36) contoured at the 3σ level, colored in gray and shown at 3 Å. (B) Same as A rotated around x-axis by 90°. (C) Simple Fo-Fc omit maps of the decoding complex individual base pair mRNA(U2)-ASL(A35) contoured at the 3σ level, colored in gray and shown at 3 Å. (D) Same as C rotated around x-axis by 90°. (E) Simple Fo-Fc omit maps of the decoding complex individual base pair mRNA(U3)-ASL(G34) contoured at the 3σ level, colored in gray and shown at 3 Å. (F) Same as E rotated around the x-axis by 90°.
FIGURE 4.
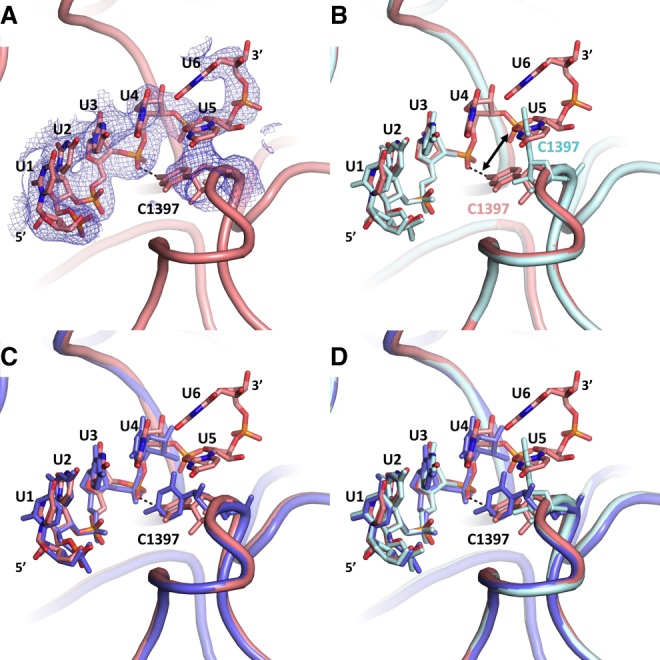
Temperature dependence of C1397 conformational dynamics. (A) In the decoding region of the ambient-temperature mRNA model, C1397 engages in H-bonding with U4 which demarcates the boundary between the A-site mRNA codon and codon-1. A final unbiased Fo-Fc simple omit ambient temperature electron-density map of mRNA and C1397 contoured at the 3σ level is shown in blue and at 3 Å. (B) Superposition of the ambient temperature 30S-decoding complex with cryogenic structure shows disorder in the cryogenic mRNA structure as well as the alternate conformation of C1397. (C) Superposition of our ambient temperature structure (salmon) and the identical cryo temperature structure obtained by another laboratory (slate; PDB ID: 1IBL) showing the disorder in the cryogenic structure. (D) Superposition of our ambient temperature structure (salmon) with the two cryo-cooled structures (cyan and slate).
The movement of the messenger RNA is controlled during the transition from pre- to post-translocation (Moazed and Noller 1989; Achenbach and Nierhaus 2015). The universally conserved bases C1397 and A1503 of the 16S rRNA head domain located on top of large secondary rRNA structures (Zhou et al. 2013; Achenbach and Nierhaus 2015) respectively intercalate between nucleotide pairs +9 and +10 and −1 and −2 of the mRNA, exclusively in the intermediate states of translocation. Those two residues are thought to prevent a back-sliding of the mRNA during back-rotation of the 30S head, thus exerting a pawl function. In the post-translation state, however, the two intercalating nucleotides do not touch the mRNA. Residue C1397 has been described as being able to adopt multiple conformations in several crystal structures, in response to the presence of a tRNA at the A-site (Jenner et al. 2010; Zhou et al. 2013). The comparison of our ambient and cryogenic temperature structures therefore illustrates the intrinsic flexible nature of C1397 even in the presence of tRNA at the A-site. This observed conformational plasticity of C1397 at ambient temperature may also explain the unexpected disorder in the noncovalently bound mRNA substrate of cryogenic structure. Presumably, the conformation of C1397 adopted at ambient temperature stabilizes the interaction of the 3′ end of the mRNA oligo and as a result decreases the disorder in this region.
Ambient-temperature studies build upon previous cryo-crystallography data
The low RMS deviation between the crystal structures collected in this work and its equivalents studied at cryogenic temperature suggests that previous structural data have captured the 30S ribosomal subunit in a representative conformation, although the causes of the specific discrepancies between data sets identified here are not immediately apparent. The broader lack of conformational differences is not likely due to crystal lattice contacts restricting the ribosome to specific conformations as it is known that this crystal form can exhibit numerous allosteric conformational changes such as at hinge region h28 and large scale conformational changes throughout the 30S subunit structure including domain closure and engaged disengaged conformational equilibrium in h44–h45 region (Ogle et al. 2001; Demirci et al. 2013a; Mohan et al. 2014; Sierra et al. 2016). In the absence of a bound mRNA-ASL pair, a prior ambient-temperature structure of the 30S subunit (bound to paromomycin) displayed significant conformational differences in the phosphate backbone of the helix 28 region up to 3.6 Å compared to its cryogenic counterpart (Sierra et al. 2016). This decoding-complex structure surprisingly did not exhibit conformational changes in rRNA bases or the backbone suggesting that the influence of the bound substrates on the observed ribosomal conformation is greater than that of the crystal-lattice contacts.
Our demonstration of data collection at ambient temperature indicates that the long-standing limitation of relying on cryogenic temperatures to study ribosome decoding complexes at high resolution is surmountable with current approaches. This latest effort builds upon prior attempts by our group to obtain a full data set at an XFEL. One factor we felt to be important to the success in our data collection was the use of an electrokinetic injector, the “concentric MESH” (Sierra et al. 2016). This sample injector delivers microcrystals to the X-ray beam at a low flow-rate by relying on electric charge rather than pressure and allowed us to simplify the sample delivery apparatus to the components illustrated (Fig. 1A). The time needed for data collection, four hours, suggests that a full data set can be collected within one beam-time shift at any XFEL (i.e., 12 h). With additional improvements in real-time data processing and the use of high-repetition-rate sources such as European XFEL, the amount of time needed could potentially decrease.
A major promise of structural studies performed at ambient temperature is the ability to probe biological macromolecules at near-physiologic temperatures in liquid suspension. Time-resolved SFX is an emerging approach that builds on the successes of SFX in determining static structures and has been demonstrated for photo-sensitive targets (Tenboer et al. 2014; Pande et al. 2016) with a time resolution as short as 100 fsec. Experimental approaches to mix enzymes with their substrates on relevant enzymatic time scales in real-time prior to their delivery to the X-ray beam are under development and would enable the probing of structural intermediates of these targets (Kupitz et al. 2017). The first demonstration of time-resolved structural enzymology at an XFEL has been performed on a riboswitch and its substrate with a mixing time of 10 sec (Stagno et al. 2017), indicating opportunities for future studies on ribonucleoprotein complexes, particularly the ribosome. For example, a stream of empty 30S ribosomal subunit microcrystals could be mixed with a tRNA ASL minihelix substrate and the complex probed by intense femtosecond XFEL pulses at various time points. Thus, key steps in the initation of translation including the initial binding of the ASL minihelix to the ribosome, the flipping of A1492 and A1493 and domain closure may be visualized using crystallography in a way not possible with large cryo-cooled crystals. These SFX studies could provide new insights into the structural basis for the initial steps of decoding and thereby refine our understanding of the mechanisms of translation.
MATERIALS AND METHODS
Preparation and crystallization of 30S ribosomal subunits for SFX crystallography at an XFEL
30S ribosomal subunits from T. thermophilus HB8 (ATCC27634) (Yoshizaki et al. 1971) were prepared as previously described (Wimberly et al. 2000; Demirci et al. 2010). Purified 30S ribosomal subunits were crystallized at 4°C by the hanging drop method using a mother liquor solution containing 17% (v/v) 2-methyl-2,4-pentanediol (MPD), 15 mM magnesium acetate, 200 mM potassium acetate, 75 mM ammonium acetate, and 100 mM 2-(N-morpholino) ethanesulfonic acid (MES)-KOH (pH 6.5). The mRNA fragment (with codon sequences underlined), 5′UUUUUU3′ and the anticodon stem–loop ASLPhe (with anticodon sequence underlined) GGGGAUUGAAAAUCCCC, were purchased from Dharmacon (GE Life Sciences). Microcrystals 2 × 2 × 4 µm3 in size were harvested in the same mother liquor composition, pooled (total volume of 500 µL loaded) and supplemented with 200 µM of mRNA oligomer, ASLPhe, and 80 µM paromomycin for 48 h before data collection. 30S ribosomal subunit microcrystals are prescreened at the SSRL beamline 12–2 for their diffraction quality. The crystal concentration was approximated to be 1010–1011 particles per mL based on light microscopy and nanoparticle tracking analysis (NanoSight LM10-HS with corresponding Nanoparticle Tracking Analysis [NTA] software suite) (Malvern Instruments).
coMESH construction
The concentric microfluidic electrokinetic sample holder (coMESH), which is the liquid injector used to deliver the microcrystals to the X-ray beam, is comprised of two capillaries: one large and one small. The central, smaller capillary carrying the microcystals of the 30S decoding complex was a continuous fused silica capillary of 100 µm inner diameter, 160 µm outer diameter, and 1.5 m length (Polymicro). This capillary directly connected the sample reservoir with the X-ray interaction region, passing through a vacuum feedthrough and then through a microfluidic “Interconnect Cross” (C360-204, LabSmith; C360-203 “Interconnect Tee” may also be used with no need to plug the fourth channel) (Fig. 1A). The uninterrupted, straight capillary was free of in-line filters, unions or any other connections, thus minimizing the risk of sample clogs and leaks, which most often occur at flow channel transitions. A similar uninterrupted approach was used for the larger capillary. The fitting at the top of the “cross” in Figure 1A was an adapter fitting (T116-A360 as well as T116-100, LabSmith) which compressed a 1/16 in (1.6 mm) OD, 0.007 in (178 µm) ID polymer (FEP) tubing sleeve (F-238, Upchurch) onto the sample capillary. This connection method was necessary to properly seal the capillary to the cross. The sheath line at the bottom of the figure was a short 5 cm fused silica capillary with an outer diameter of 360 µm and an inner diameter of 180, 200, or 250 µm depending on the desired flow rate of the sheath liquid at a given driving pressure. This outer capillary was connected to the cross, and the sheath liquid was supplied from the third port of the cross (the left port in the figure) through a compatible microfluidic tubing; we typically used silica capillaries with 360 µm outer diameters and 200 µm inner diameters for optimum flow rate. For simplicity, the injector has been designed to operate at relatively low backing pressures (up to a few atmospheres). For this experiment, the sheath liquid line was connected to a syringe filled with the appropriate sister liquor which contains the cryoprotection solution, driven by a syringe pump (PHD Ultra, 703006, Harvard Apparatus). We electrically charged the sheath liquid between 0 and 5000 V potential using an in-line conductive charging union (M-572, Upchurch) connected to a SRS PS350 (Stanford Research Systems) high voltage source.
The capillary assembly was loaded into a vacuum chamber with 1 × 1 µm2 focused XFEL beam and the standard load-lock system of the CXI instrument. A grounded, conical counter electrode with a 1 cm opening was placed ∼5 mm below the capillary tip; the capillaries and opening of the cone were coaxial. The angle of the cone and its distance from the tip were set to enable a diffraction cone with a 45° 2q scattering angle. All capillaries were fed through vacuum flanges with 1/16 in (1.6 mm) Swagelok bulkhead fittings using appropriately sized polymer sleeves. The sheath reservoir was a 1 mL Gastight Hamilton syringe with a PTFE Luer tip (1001 TLL SYR). The ribosome microcrystalline sample was supplied from a 500 µL Hamilton Gastight syringe.
Selecting a sister liquor for the ribosome microcrystalline slurry
The coMESH incorporates a “sister liquor” as part of its sample delivery design which is used to protect the microcrystals and their mother liquor from the dehydrative effects of the sample chamber vacuum (10−5 Torr). This fluid travels in the larger capillary and, after reaching the cross, peripherally to the sample line until the terminus of the capillary within the sample chamber. The choice of fluid may reflect the composition of the crystals’ mother liquor but need not strictly match it. For delivery of ribosome microcrystals using coterminal capillaries (Sierra et al. 2016), the compatibility between the sister liquor and the ribosome crystals was not considered since fluid interaction occurred immediately prior to reaching the X-ray interaction region, allowing minimal time, if any, for the fluids to mix. Current and prior experimental experience suggest that a 26% MPD-containing sister liquor seems to be an ideal choice for coterminal sample delivery. The buffer has proven to be quite reliable, surviving injection into vacuum with minimal issues especially with respect to buildup of dehydrated precipitates, which can occur with salts or polyethylene glycols and is problematic for sample delivery due to its adherence to (and thus, blockage of) the capillary tip. Other factors influencing the choice of fluid include availability of the fluid and its viscosity. The lower viscosity allows the sister liquor to be pumped easily by a standard syringe pump through the more resistive cross manifold or concentric annular flow before the exit region of the capillary.
Operation of the coMESH
Before connecting the central sample line, the sister liquor was loaded, flowed, and electrically focused. Once a stable jet was achieved, the central sample line carrying ribosome slurry was connected. Notably, the sister liquor never fully stabilized because of the entrained air from the disconnected sample line continuously introducing bubbles. The central sample line had much less fluidic resistance compared to the outer line; connecting it first with a vacuum sensitive sample will cause immediate jet clogging and blockages; for this reason the outer line should always be on while operating in a vacuum environment. In general, the sister liquor was set to flow at or near the flow rate of the mother liquor. If diffraction hits were not observed, the sister liquor flow was reduced and/or the mother liquor flow was increased slowly (to ensure stable jetting) until diffraction patterns reappeared.
Ribosome microcrystalline sample injection with coMESH
For the study of 30S-mRNA-ASL-paromomycin complex crystals, the inner sample line contained unfiltered crystals in their native mother liquor containing 17% (v/v) 2-methyl-2,4-pentanediol (MPD). The size distribution of the 30S crystals was uniform owing to their controlled slower growth at 4°C (Demirci et al. 2013b). Occasional larger-sized 30S crystals were discarded by repeated gentle differential settling without centrifugation. The outer sister liquor was the same buffer, with no crystals, as the mother liquor in that the original substituent concentrations remained constant while having increased the MPD to 26% (v/v) to aid in-vacuum injection.
Data collection and analysis for SFX studies at LCLS
The serial femtosecond X-ray crystallography (SFX) experiment was performed with the Coherent X-ray Imaging instrument (CXI) of the Linac Coherent Light Source (LCLS; SLAC National Accelerator Laboratory) under beam-time ID: cxil1416. The LCLS X-ray beam with a pulse duration of ∼50 fsec was focused using X-ray optics in a Kirkpatrick-Baez geometry to a beam size of 1.3 × 1.3 µm2 full width at half maximum (FWHM) at a pulse energy of 2.9 mJ, a photon energy of 9.5 keV (1.29 Å), and a repetition rate of 120 Hz.
A total of 1,731,280 images were collected with the Cornell-SLAC Pixel Array Detector (CSPAD) (Pietrini and Nettelblad 2017) from crystals of the 30S-mRNA-ASL-paromomycin complex. The total beam-time used for this data set was 240 min (out of an available 360 min). Crystal hits were defined as frames containing more than 30 Bragg peaks with a minimum signal-to-noise ratio greater than 4.5 on each detector image, for a total of 165,954 hits. The detector distance was set to 223 mm, with an achievable resolution of 3.08 Å at the edge of the detector (2.6 Å in the corner).
After the detection of crystal hits and following the conversion of individual diffraction patterns to the HDF5 format by CHEETAH (Barty et al. 2014), the CrystFEL software suite (White et al. 2016) was used for crystallographic analysis. The information of peak positions was used for the indexing of individual, randomly oriented crystal diffraction patterns using FFT-based indexing approaches. The detector geometry was refined using an automated algorithm to match found and predicted peaks to subpixel accuracy (Yefanov et al. 2015). The integration step was performed using a built-in Monte Carlo algorithm to estimate accurate structure factors from thousands of individually measured Bragg peaks (Kirian et al. 2010). After the application of per-pattern resolution cutoff, frames that did not match to an initial merged data set with a Pearson correlation coefficient of less than 0.4 were excluded from the final data set. Diffraction intensities from a total of 19,374 indexed diffraction patterns (11.8 % indexing rate) were merged into a final data set (overall CC* = 0.93; 3.45 Å cutoff) for further analysis (P41212, unit cell: a = b = 402.3 Å; c = 176.4 Å; α = β = γ = 90°). The final resolution cutoff was estimated to be 3.45 Å using a combination of CC1/2 (Karplus and Diederichs 2012) and other refinement parameters. The final data set had an overall Rsplit = 63.3% and CC* = 0.36 in the highest resolution shell.
Ambient temperature 30S ribosomal subunit SFX structure refinement
We determined the ambient temperature structure of the 30S-mRNA-ASL-paromomycin complex using the automated molecular replacement program PHASER (McCoy et al. 2007) with the previously published cryo-cooled 30S ribosomal subunit-mRNA-ASL-paromomycin complex synchrotron structure as a search model (PDB entries 4DR4 and 1IBL) (Ogle et al. 2001; Demirci et al. 2013a). The resulting structure was refined with rRNA modifications, which are mostly located near the decoding center. This choice of starting search model minimized experimental variations between the two structures such as sample preparation, crystal growth, data processing, and model registry. Coordinates of the 4DR4 with additional RNA and protein modifications were used for the initial rigid body refinement with the PHENIX software package (Adams et al. 2010). During the initial refinement of ambient temperature XFEL structure, the entire mRNA and ASL components were omitted and the new models were built into unbiased difference density. After simulated-annealing refinement, individual coordinates, three group B factors per residue, and TLS parameters were refined. Potential positions of magnesium or potassium ions were compared with those in the high-resolution (2.5 Å) 30S subunit structure (PDB accession code 2VQE)7 in program COOT (Emsley and Cowtan 2004), and positions with strong difference density were retained. All magnesium atoms were replaced with magnesium hexahydrate. Water molecules located outside of significant electron density were manually removed. The Ramachandran statistics for this data set (most favored/additionally allowed/generously allowed/disallowed) are 87.3/11.7/0.7/0.2%, respectively. The structure refinement statistics are summarized in Table 1. Structure alignments were performed using the alignment algorithm of PyMOL (www.schrodinger.com/pymol) with the default 2σ rejection criterion and five iterative alignment cycles. All X-ray crystal structure figures were produced with PyMOL.
DATA DEPOSITION
The coordinates have been deposited in the Protein Data Bank with the accession code 6CAO.
ACKNOWLEDGMENTS
The authors thank Michelle Young for her support and critical reading of the manuscript. Portions of this research were carried out at the Linac Coherent Light Source (LCLS) and Stanford Synchrotron Radiation Lightsource at the SLAC National Accelerator Laboratory. Use of the LCLS and SSRL is supported by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences (OBES) under contract no. DE-AC02-76SF00515. The LCLS is acknowledged for beam-time access under experiment no. cxil1416. E.H.D., R.G.S., and H.D. acknowledge the support of OBES through the AMOS program within the CSGB and the DOE through the SLAC Laboratory Directed Research and Development Program. E.H.D. acknowledges financial support from the Stanford University Dean of Research. H.D. acknowledges support from National Science Foundation (NSF) Science and Technology Centers grant NSF-1231306 (Biology with X-ray Lasers, BioXFEL). F.P., N.C., and F.P.A. are supported by National Institutes of Health (NIH) grant R35GM122543. Parts of the sample injector used at LCLS for this research were funded by the National Institutes of Health, P41GM103393, formerly P41RR001209. C.G. acknowledges SLAC and the DOE for financial support through the Panofsky fellowship. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (P41GM103393). C.D.L. acknowledges Stiftelsen JC Kempes Minnes Stipendiefond for financial support. We thank A. Fry's continuous support for LCLS intern students who contributed to the portions of the experiments carried out at LCLS. H.D. acknowledges valuable discussions with A. Takeuchi, K. Dursuncan, and E. Satunaz. We thank G. Stewart of SLAC for excellent technical assistance with creating graphics for Figure 1A.
Author contributions: H.D. and E.H.D. designed the project. H.D., E.H.D., Y.R., H.C., and F.A. prepared the samples for SFX studies. R.G.S., H.D., A.M., P.M., B.H., T.O., C.D.L., and E.H.D. built the coMESH injectors. F.P. and C.G. executed the SFX data reduction. N.C. and F.P.A. provided realtime XFEL data analysis. H.D. refined the structure. The experiments were executed by H.D., E.H.D., R.G.S., F.P., N.C., F.P.A., M.H., M.L., M.S., F.A., H.I.C., L.Z., and Y.R. Data were analyzed by H.D., E.H.D., F.P., S.W., C.G., and R.G.S. The manuscript was prepared by H.D., E.H.D., F.P., C.G., S.W., and R.G.S. with input from all the coauthors.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.067660.118.
Freely available online through the RNA Open Access option.
REFERENCES
- Achenbach J, Nierhaus KH. 2015. The mechanics of ribosomal translocation. Biochimie 114: 80–89. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allred BE, Gebala M, Herschlag D. 2017. Determination of ion atmosphere effects on the nucleic acid electrostatic potential and ligand association using AH+·C wobble formation in double-stranded DNA. J Am Chem Soc 139: 7540–7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony PC, Sim AY, Chu VB, Doniach S, Block SM, Herschlag D. 2012. Electrostatics of nucleic acid folding under conformational constraint. J Am Chem Soc 134: 4607–4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. 2000. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289: 905–920. [DOI] [PubMed] [Google Scholar]
- Barty A, Caleman C, Aquila A, Timneanu N, Lomb L, White TA, Andreasson J, Arnlund D, Bajt S, Barends TR, et al. 2012. Self-terminating diffraction gates femtosecond X-ray nanocrystallography measurements. Nat Photonics 6: 35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barty A, Kirian RA, Maia FR, Hantke M, Yoon CH, White TA, Chapman H. 2014. Cheetah: software for high-throughput reduction and analysis of serial femtosecond X-ray diffraction data. J Appl Crystallogr 47: 1118–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. 2011. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 334: 1524–1529. [DOI] [PubMed] [Google Scholar]
- Bogan MJ. 2013. X-ray free electron lasers motivate bioanalytical characterization of protein nanocrystals: serial femtosecond crystallography. Anal Chem 85: 3464–3471. [DOI] [PubMed] [Google Scholar]
- Boutet S, Lomb L, Williams GJ, Barends TR, Aquila A, Doak RB, Weierstall U, DePonte DP, Steinbrener J, Shoeman RL, et al. 2012. High-resolution protein structure determination by serial femtosecond crystallography. Science 337: 362–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. 2000. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature 407: 340–348. [DOI] [PubMed] [Google Scholar]
- Carter AP, Clemons WM Jr, Brodersen DE, Morgan-Warren RJ, Hartsch T, Wimberly BT, Ramakrishnan V. 2001. Crystal structure of an initiation factor bound to the 30S ribosomal subunit. Science 291: 498–501. [DOI] [PubMed] [Google Scholar]
- Chapman HN, Fromme P, Barty A, White TA, Kirian RA, Aquila A, Hunter MS, Schulz J, DePonte DP, Weierstall U, et al. 2011. Femtosecond X-ray protein nanocrystallography. Nature 470: 73–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirci H, Murphy Ft, Belardinelli R, Kelley AC, Ramakrishnan V, Gregory ST, Dahlberg AE, Jogl G. 2010. Modification of 16S ribosomal RNA by the KsgA methyltransferase restructures the 30S subunit to optimize ribosome function. RNA 16: 2319–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirci H, Murphy Ft, Murphy E, Gregory ST, Dahlberg AE, Jogl G. 2013a. A structural basis for streptomycin-induced misreading of the genetic code. Nat Commun 4: 1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirci H, Sierra RG, Laksmono H, Shoeman RL, Botha S, Barends TR, Nass K, Schlichting I, Doak RB, Gati C, et al. 2013b. Serial femtosecond X-ray diffraction of 30S ribosomal subunit microcrystals in liquid suspension at ambient temperature using an X-ray free-electron laser. Acta Crystallogr Sect F Struct Biol Cryst Commun 69: 1066–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePonte DP, Doak RB, Hunter M, Liu Z, Weierstall U, Spence JC. 2009. SEM imaging of liquid jets. Micron 40: 507–509. [DOI] [PubMed] [Google Scholar]
- Emma P, Akre R, Arthur J, Bionta R, Bostedt C, Bozek J, Brachmann A, Bucksbaum P, Coffee R, Decker FJ, et al. 2010. First lasing and operation of an ångstrom-wavelength free-electron laser. Nat Photonics 4: 641–647. [Google Scholar]
- Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- Evers U, Franceschi F, Boddeker N, Yonath A. 1994. Crystallography of halophilic ribosome: the isolation of an internal ribonucleoprotein complex. Biophys Chem 50: 3–16. [DOI] [PubMed] [Google Scholar]
- Fischer N, Neumann P, Konevega AL, Bock LV, Ficner R, Rodnina MV, Stark H. 2015. Structure of the E. coli ribosome-EF-Tu complex at <3 Å resolution by Cs-corrected cryo-EM. Nature 520: 567–570. [DOI] [PubMed] [Google Scholar]
- Fromme P, Spence JC. 2011. Femtosecond nanocrystallography using X-ray lasers for membrane protein structure determination. Curr Opin Struct Biol 21: 509–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas DJ, Rossmann MG. 1970. Crystallographic studies on lactate dehydrogenase at-75 degrees C. Acta Crystallogr B 26: 998–1004. [DOI] [PubMed] [Google Scholar]
- Helliwell JR. 2013. Biochemistry. How to solve protein structures with an X-ray laser. Science 339: 146–147. [DOI] [PubMed] [Google Scholar]
- Hunter MS, Yoon CH, DeMirci H, Sierra RG, Dao EH, Ahmadi R, Aksit F, Aquila AL, Ciftci H, Guillet S, et al. 2016. Selenium single-wavelength anomalous diffraction de novo phasing using an X-ray-free electron laser. Nat Commun 7: 13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner LB, Demeshkina N, Yusupova G, Yusupov M. 2010. Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nat Struct Mol Biol 17: 555–560. [DOI] [PubMed] [Google Scholar]
- Karplus PA, Diederichs K. 2012. Linking crystallographic model and data quality. Science 336: 1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keedy DA, van den Bedem H, Sivak DA, Petsko GA, Ringe D, Wilson MA, Fraser JS. 2014. Crystal cryocooling distorts conformational heterogeneity in a model Michaelis complex of DHFR. Structure 22: 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern J, Alonso-Mori R, Hellmich J, Tran R, Hattne J, Laksmono H, Glockner C, Echols N, Sierra RG, Sellberg J, et al. 2012. Room temperature femtosecond X-ray diffraction of photosystem II microcrystals. Proc Natl Acad Sci 109: 9721–9726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern J, Alonso-Mori R, Tran R, Hattne J, Gildea RJ, Echols N, Glockner C, Hellmich J, Laksmono H, Sierra RG, et al. 2013. Simultaneous femtosecond X-ray spectroscopy and diffraction of photosystem II at room temperature. Science 340: 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirian RA, Wang X, Weierstall U, Schmidt KE, Spence JC, Hunter M, Fromme P, White T, Chapman HN, Holton J. 2010. Femtosecond protein nanocrystallography-data analysis methods. Opt Express 18: 5713–5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korostelev A, Trakhanov S, Laurberg M, Noller HF. 2006. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell 126: 1065–1077. [DOI] [PubMed] [Google Scholar]
- Kupitz C, Olmos JL Jr, Holl M, Tremblay L, Pande K, Pandey S, Oberthur D, Hunter M, Liang M, Aquila A, et al. 2017. Structural enzymology using X-ray free electron lasers. Struct Dyn 4: 044003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang M, Williams GJ, Messerschmidt M, Seibert MM, Montanez PA, Hayes M, Milathianaki D, Aquila A, Hunter MS, Koglin JE, et al. 2015. The coherent x-ray imaging instrument at the Linac coherent light source. J Synchrotron Radiat 22: 514–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipfert J, Doniach S, Das R, Herschlag D. 2014. Understanding nucleic acid-ion interactions. Annu Rev Biochem 83: 813–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveland AB, Demo G, Grigorieff N, Korostelev AA. 2017. Ensemble cryo-EM elucidates the mechanism of translation fidelity. Nature 546: 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40: 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra K, Schaffitzel C, Shaikh T, Tama F, Jenni S, Brooks CL III, Ban N, Frank J. 2005. Structure of the E. coli protein-conducting channel bound to a translating ribosome. Nature 438: 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moazed D, Noller HF. 1989. Intermediate states in the movement of transfer RNA in the ribosome. Nature 342: 142–148. [DOI] [PubMed] [Google Scholar]
- Mohan S, Donohue JP, Noller HF. 2014. Molecular mechanics of 30S subunit head rotation. Proc Natl Acad Sci 111: 13325–13330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natchiar SK, Myasnikov AG, Kratzat H, Hazemann I, Klaholz BP. 2017. Visualization of chemical modifications in the human 80S ribosome structure. Nature 551: 472–477. [DOI] [PubMed] [Google Scholar]
- Neutze R, Wouts R, van der Spoel D, Weckert E, Hajdu J. 2000. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 406: 752–757. [DOI] [PubMed] [Google Scholar]
- Noller HF. 2013. By ribosome possessed. J Biol Chem 288: 24872–24885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogle JM, Brodersen DE, Clemons WM Jr, Tarry MJ, Carter AP, Ramakrishnan V. 2001. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science 292: 897–902. [DOI] [PubMed] [Google Scholar]
- Ogle JM, Carter AP, Ramakrishnan V. 2003. Insights into the decoding mechanism from recent ribosome structures. Trends Biochem Sci 28: 259–266. [DOI] [PubMed] [Google Scholar]
- Pande K, Hutchison CD, Groenhof G, Aquila A, Robinson JS, Tenboer J, Basu S, Boutet S, DePonte DP, Liang M, et al. 2016. Femtosecond structural dynamics drives the trans/cis isomerization in photoactive yellow protein. Science 352: 725–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov A, Kornberg G, O'Leary S, Tsai A, Uemura S, Puglisi JD. 2011. Dynamics of the translational machinery. Curr Opin Struct Biol 21: 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrini A, Nettelblad C. 2017. Artifact reduction in the CSPAD detectors used for LCLS experiments. J Synchrotron Radiat 24: 1092–1097. [DOI] [PubMed] [Google Scholar]
- Razi A, Guarne A, Ortega J. 2017. The cryo-EM structure of YjeQ bound to the 30S subunit suggests a fidelity checkpoint function for this protein in ribosome assembly. Proc Natl Acad Sci 114: E3396–E3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redecke L, Nass K, DePonte DP, White TA, Rehders D, Barty A, Stellato F, Liang M, Barends TRM, Boutet S, et al. 2013. Natively inhibited Trypanosoma brucei cathepsin B structure determined by using an X-ray laser. Science 339: 227–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlichting I, Miao J. 2012. Emerging opportunities in structural biology with X-ray free-electron lasers. Curr Opin Struct Biol 22: 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluenzen F, Tocilj A, Zarivach R, Harms J, Gluehmann M, Janell D, Bashan A, Bartels H, Agmon I, Franceschi F, et al. 2000. Structure of functionally activated small ribosomal subunit at 3.3 Å resolution. Cell 102: 615–623. [DOI] [PubMed] [Google Scholar]
- Schuwirth BS, Borovinskaya MA, Hau CW, Zhang W, Vila-Sanjurjo A, Holton JM, Cate JH. 2005. Structures of the bacterial ribosome at 3.5 Å resolution. Science 310: 827–834. [DOI] [PubMed] [Google Scholar]
- Selmer M, Dunham CM, Murphy FVt, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. 2006. Structure of the 70S ribosome complexed with mRNA and tRNA. Science 313: 1935–1942. [DOI] [PubMed] [Google Scholar]
- Sierra RG, Laksmono H, Kern J, Tran R, Hattne J, Alonso-Mori R, Lassalle-Kaiser B, Glockner C, Hellmich J, Schafer DW, et al. 2012. Nanoflow electrospinning serial femtosecond crystallography. Acta Crystallogr D Biol Crystallogr 68: 1584–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra RG, Gati C, Laksmono H, Dao EH, Gul S, Fuller F, Kern J, Chatterjee R, Ibrahim M, Brewster AS, et al. 2016. Concentric-flow electrokinetic injector enables serial crystallography of ribosome and photosystem II. Nat Methods 13: 59–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagno JR, Liu Y, Bhandari YR, Conrad CE, Panja S, Swain M, Fan L, Nelson G, Li C, Wendel DR, et al. 2017. Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature 541: 242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenboer J, Basu S, Zatsepin N, Pande K, Milathianaki D, Frank M, Hunter M, Boutet S, Williams GJ, Koglin JE, et al. 2014. Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science 346: 1242–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warkentin M, Hopkins JB, Haber JB, Blaha G, Thorne RE. 2014. Temperature-dependent radiation sensitivity and order of 70S ribosome crystals. Acta Crystallogr D Biol Crystallogr 70: 2890–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weierstall U, Spence JC, Doak RB. 2012. Injector for scattering measurements on fully solvated biospecies. Rev Sci Instrum 83: 035108. [DOI] [PubMed] [Google Scholar]
- White TA, Mariani V, Brehm W, Yefanov O, Barty A, Beyerlein KR, Chervinskii F, Galli L, Gati C, Nakane T, et al. 2016. Recent developments in CrystFEL. J Appl Crystallogr 49: 680–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimberly BT, Brodersen DE, Clemons WM Jr, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. 2000. Structure of the 30S ribosomal subunit. Nature 407: 327–339. [DOI] [PubMed] [Google Scholar]
- Yefanov O, Mariani V, Gati C, White TA, Chapman HN, Barty A. 2015. Accurate determination of segmented X-ray detector geometry. Opt Express 23: 28459–28470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki F, Oshima T, Imahori K. 1971. Studies on phosphoglucomutase from an extreme thermophile, Flavobacterium thermophilum HB8. I. Thermostability and other enzymatic properties. J Biochem 69: 1083–1089. [DOI] [PubMed] [Google Scholar]
- Yoshizawa S, Fourmy D, Puglisi JD. 1999. Recognition of the codon-anticodon helix by ribosomal RNA. Science 285: 1722–1725. [DOI] [PubMed] [Google Scholar]
- Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. 2001. Crystal structure of the ribosome at 5.5 Å resolution. Science 292: 883–896. [DOI] [PubMed] [Google Scholar]
- Yusupova G, Jenner L, Rees B, Moras D, Yusupov M. 2006. Structural basis for messenger RNA movement on the ribosome. Nature 444: 391–394. [DOI] [PubMed] [Google Scholar]
- Zhou J, Lancaster L, Donohue JP, Noller HF. 2013. Crystal structures of EF-G–ribosome complexes trapped in intermediate states of translocation. Science 340: 1236086. [DOI] [PMC free article] [PubMed] [Google Scholar]