

Abstract
Colorectal cancer (CRC) is one of the leading causes of cancer-related deaths worldwide. Similar to many other malignancies, CRC is a heterogeneous disease, making it a clinical challenge for optimization of treatment modalities in reducing the morbidity and mortality associated with this disease. A more precise understanding of the biological properties that distinguish patients with colorectal tumors, especially in terms of their clinical features, is a key requirement towards a more robust, targeted-drug design, and implementation of individualized therapies. In the recent decades, extensive studies have reported distinct CRC subtypes, with a mutation-centered view of tumor heterogeneity. However, more recently, the paradigm has shifted towards transcriptome-based classifications, represented by six independent CRC taxonomies. In 2015, the colorectal cancer subtyping consortium reported the identification of four consensus molecular subtypes (CMSs), providing thus far the most robust classification system for CRC. In this review, we summarize the historical timeline of CRC classification approaches; discuss their salient features and potential limitations that may require further refinement in near future. In other words, in spite of the recent encouraging progress, several major challenges prevent translation of molecular knowledge gleaned from CMSs into the clinic. Herein, we summarize some of these potential challenges and discuss exciting new opportunities currently emerging in related fields. We believe, close collaborations between basic researchers, bioinformaticians and clinicians are imperative for addressing these challenges, and eventually paving the path for CRC subtyping into routine clinical practice as we usher into the era of personalized medicine.
Keywords: colorectal cancer, heterogeneity, molecular subtyping, personalized medicine
1. Introduction
Colorectal cancer (CRC) is one of the leading causes of cancer-related mortality worldwide, with an estimated 50,260 deaths recorded in the United States alone in 2017[1]. Even with the recent advances in novel therapies, the 5-year overall survival from this malignancy remains ~50%, highlighting the need to develop early detection, prognostic and predictive biomarkers that can be utilized in routine clinical practice for reducing the morbidity and mortality associated with this disease [2]. It is now widely recognized that cancers are heterogeneous groups of diseases with distinct molecular properties, resulting in diverse clinical outcomes. In the recent decades, several cancer initiatives and consortia have generated large-scale multi-omic data profiles, enabling unsupervised classifications of various types of cancers followed by comprehensive characterizations. In this context, previous ‘mutation-centered’ categorization of cancers has now gradually shifted towards a more ‘transcriptome-based’ molecular subtyping. Several of such transcriptome-based classification systems have demonstrated superior associations with clinical outcomes, making them more attractive for their eventual translation into the clinic. For instance, in breast cancer patients, intrinsic molecular subtyping based on immunohistochemical staining for ER, PR and HER2 markers, is now widely accepted in routine clinical practice for stratifying these patients for appropriate treatments.
For CRC, six independent classification systems have been reported recently [3–8]. Due to inherent differences in the discovery cohort patients, gene expression profiling platforms, bioinformatic analyses, and data interpretation, each subtyping system have identified different numbers of CRC subtypes, which are seemingly discrepant. In order to elucidate potential overlaps and generate a more unified taxonomy, in 2014, a CRC Subtyping Consortium (CRCSC) was established, which involved six independent research groups and Sage Bionetworks as an additional evaluation team. Using a network-based meta-analysis of the six subtyping systems, four unique consensus molecular subtypes (CMSs) were identified, followed by comprehensive multi-omic and clinical characterization [9]. In spite of the tremendous early enthusiasm for this large community effort, the clinical translation of this CMS system has been challenging due to several potential limitations. Meanwhile, new opportunities are emerging to address these challenges, including recent advances in single-cell sequencing and big data sciences such as deep learning, new biological insights into intratumor heterogeneity and tumor microenvironment, as well as a growing number of clinical trials interrogating novel targeted therapies, combination therapies and immunotherapies.
In this article, we summarize genetic and epigenetic characteristics used for conventional CRC classification, transcriptome-based molecular subtyping, and the consensus molecular subtypes recently identified by the CRCSC. We will compare and discuss their contributions to the biological understanding of CRC heterogeneity and clinical associations, as well as their limitations. Furthermore, we highlight and discuss six major challenges preventing effective translation of CMSs into routine clinical practice, and possible solutions, alternative strategies and new opportunities. We stress that multidisciplinary collaborations are key to addressing these challenges, which is a prerequisite for more widespread embracement and implementation of molecular subtyping into the clinical management of patients with CRC.
2. Genetic and epigenetic characteristics in colorectal cancer
2.1. The three major pathways of colorectal cancer and clinical implications
Clinically, colorectal cancer is still categorized based on the histopathological features such as tumor size, grade and disease stage. Although these classification methods are in use for decades, identifying the true high-risk population post-surgery still remains a major clinical concern. Furthermore, they provide limited understanding of the underlying tumor biology, and are in many cases inadequate for decision-making about appropriate treatment regimens in CRC patients. Increasingly, it became clear that the heterogeneous response to various therapeutic modalities in CRC is largely due to various genotypic and phenotypic differences that exist in CRC patients. A typical view is that adenoma to carcinoma progression manifests through three major phenotypes: microsatellite instability (MSI) [10] chromosomal instability (CIN) [11] and the CpG island methylator phenotype (CIMP) [12].
Majority of CRC patients exhibit CIN (65–85%) [13,14], and are typically characterized to possess a ‘classical’ or ‘canonical’ type of cancer. Although infrequent, chromosomal abnormalities have been detected in adenomas, which suggests that CIN may arise early in the adenoma to CRC sequence [15]. Though the precise molecular mechanisms responsible for the development of CIN remain elusive, large chromosomal aberrations including duplication and aneuploidy, mutations in the TP53 gene and other checkpoint genes are believed to contribute in the formation of CIN cancers. Furthermore, it is shown that oncogene stress induced genomic instability, telomere erosion, and DNA hypomethylation could play a role in inducing CIN in CRCs [14,16–18]. In contrast to MSI patients, CIN patients have relatively unfavorable prognosis, especially in early stage disease [16,19].
MSI represents a hypermutator phenotype, caused by the loss of DNA mismatch repair (MMR) activity, and accounts for approximately 12–15% of all CRCs. Of these, 2–3% of the MSI-positive CRCs are associated with Lynch syndrome while the remainder represents the sporadic or acquired form of the disease. A major difference is that most sporadic MSI tumors evolve via hypermethylation of the MLH1 gene promoter, while Lynch syndrome tumors primarily acquire MSI through germline mutations in one of the MMR genes which include MLH1, MSH2, MSH6, and PMS2[20]. Recent studies have revealed that sporadic MSI CRCs are often associated with the serrated neoplasia pathway and frequently carry BRAF-V600E mutations, while germline Lynch syndrome patients do not harbor BRAF mutations [21]. MSI CRCs are generally mutually exclusive of CIN tumors as they typically display a near diploid karyotype and carry a set of unique gene mutations that are distinct from those seen in CIN CRCs [22]. The clinical relevance of MSI has been well characterized before. For instance, MSI was shown to be associated with better prognosis and reduced metastasis [23]. High-frequency microsatellite instability (MSI-H) or defective MMR (dMMR) was also predictive of response to Fluorouracil-based adjuvant therapy [24] as well as immunotherapy [25]. Recently, pembrolizumab, an anti-PD1 immunotherapy, has been approved by FDA for treatment of MSI-H or dMMR colorectal cancer.
Epigenetic instability caused by promoter CpG island hypermethylation is another well recognized pathway in CRCs. Aberrant DNA methylation is present in essentially all CRCs, however, ~10–20% of patients exhibit extremely high frequency of aberrantly methylated CpG loci, often characterized as having a CpG Island Methylator Phenotype (CIMP) [26]. According to the level of CIMP [26], CRC tumors can be divided into CIMP-high and CIMP-low CRC groups, associated with different precursor lesions [27–29]. Compared to CIMP-low patients with a CIMP-high phenotype often demonstrate worse prognosis. When MSI is taken into consideration, CIMP-high patients with MSI have a more favorable survival [30,31]. These patients also tend to associate with MSI and BRAF mutations, but lack KRAS and TP53 mutations [32–34]. However, the exact relationships between CIMP, MSI and BRAF remain elusive.
2.2. Mutation-centered CRC classifications
The pathogenesis of CRC involves both genetic and epigenetic changes that result in histologic differences, and is most clearly described in the model of “adenoma-carcinoma” sequence proposed by Fearon and Vogelstein [35,36]. In this model, CRC pathogenesis is illustrated as a stepwise process, involving accumulation of genetic and epigenetic alterations that contribute to the transformation of normal mucosa to an adenoma and the subsequent progression to a more malignant stage. Acquisition of APC inactivated mutations evolve first, subsequently mutations occur in KRAS, followed by further mutations in elements of TP53, PI3K, TGF-β and other pathways [37,38].
Along the “adenoma-carcinoma” sequence, mutations in several key genes such as KRAS and TP53 have demonstrated prognostic and predictive values. KRAS mutations are found in ~30–50% of CRC patients [39,40]. Of these, 90% of KRAS mutations occur in codons 12 and 13 within the second exon (G12/13 changed to valine) [41–43], causing constitutive activation of the mitogen-activated protein kinase (MAPK) cascade. There are conflicting reports on the association of KRAS mutations and CRC prognosis. However, in metastatic CRC, KRAS mutant patients are currently not considered for anti-EGFR based therapy [44]. Also prominent in CRC are mutations in the TP53 tumor suppressor gene, which play a crucial role in cell proliferation and apoptosis [45,46]. The loss of function of TP53 is a late event in CRC progression and has been identified in up to 60% of CRCs [47]. The presence of TP53 mutations are predictive of decreased cancer cell sensitivity to most chemotherapeutic agents, particularly 5-fluorouracil [48,49].
Besides, BRAF mutations have also been well studied for their prognostic and predictive roles in CRC. BRAF is an isoform of the RAF serine/threonine family downstream of KRAS in the MAPK/ERK pathway (also known as Ras-Raf-MEK-ERK pathway)[50] and is mutated in about 10% of CRC patients[51], with the primary mutations at the residue 600 which results in a valine to glutamate (V600E) substitution in up to 80% of BRAF mutant CRCs [52]. BRAF V600E mutations often occur in patients with MSI and high CIMP [39]. BRAF mutations have a modest effect on disease outcome, but mutant MSI patients show favorable prognosis [53]. BRAF inhibitors were inefficient in most CRCs with BRAF V600E mutations due to feedback activation of EGFR [54]. While the predictive role of KRAS mutation to cetuximab is well established, such a role of mutated BRAF was a subject of intense debate until recently. Latest studies have revealed that in RAS wild type patients, BRAF mutations act as negative predictors for anti-EGFR therapy [55].
While the abovementioned molecular characteristics have provided substantial insights into CRC initiation and progression, individual markers are not sufficient to explain the diverse patient outcomes [56]. There is a growing consensus that combining multiple characteristics may improve the clinical associations and offer promising prognostic or predictive markers. For instance, BRAF mutations coupled with MSI have been demonstrated to have a superior prognostic value [57]. As another example, although BRAF monotherapy is ineffective in CRC patients with V600E mutations due to the intrinsic EGFR-mediated resistance, combinations of BRAF, EGFR with or without MEK or PI3K inhibitors are becoming a more effective therapeutic strategy [58–64]. Even though, the conventional mutation-centered classification strategy cannot fully explain the diversity in patient outcomes, which may also be affected by the tumor microenvironment and epigenetic alterations. Therefore, a more systematic, unbiased methodology overcoming the limitations of mutation-based classifications is needed for better stratification of CRC patients.
3. Gene-expression based CRC classifications
Emerging evidence in the last decade has made it clear that intratumor heterogeneity is best captured at the transcriptomic level, as it provides a more comprehensive molecular landscape of the disease process. Not surprisingly, the paradigm of cancer subtyping has gradually shifted from mutation-centered to transcriptome-based approaches, and from supervised to unsupervised classifications. Different from histopathological subtyping and classical mutation-oriented stratification, transcriptome-based strategy employs whole genome expression profiles for unsupervised data analyses. This approach has been successfully applied to various malignancies such as breast, gastric, lung, brain and ovarian cancers [65]. Similar to other cancers, much effort has recently been dedicated to CRC subtyping. Within a short timespan between 2012 to 2014, the entire field of CRC research witnessed publication of 6 representative classification systems in high-impact journals[4,6–8,66,67]. In the following sections, we briefly summarize data, bioinformatic methods employed for molecular subtyping, the major findings, and the biological and clinical implications from each of the studies (Table 1).
Table 1.
The six independent CRC classification systems used for identification of the four consensus molecular subtypes
| Classification system | Discovery dataset(s) | Validation dataset(s) | Clustering method | Statistic for cluster count selection | Classification method | Subtypes |
|---|---|---|---|---|---|---|
| Schlicker et al.[4] | 62 samples | 1643 samples (15 datadsets) 74 cell lines | Iterative NMF-based consensus clustering | Cophenetic correlation coefficient | Two-step hierarchical clustering | Subtype 1.1: mesenchymal, even distribution of MSI and MSS Subtype 1.2: mesenchymal, more female Subtype 1.3: mesenchymal, high expression of transporter genes Subtype 2.1: epithelial, enriched for MSS Subtype 2.2: epithelial |
| Marisa et al.[7] | 443 samples(multi-center) | 1029 samples (7 datasets) | Classical consensus clustering | Area under CDF curve | Standard centroid-based classifier | C1 (21 %, CINImmuneDown): CIN+ C2(19%, dMMR): dMMR, CIMP+, enriched for BRAF mutations C3(13%, KRASm): enriched for KRAS mutations C4(10%, CSC): EMT activation, poor prognosis C5(27%, CINWntUp): CIN+, enriched for TP53 mutations C6(10%, CINnormL): CIN+, poor prognosis Goblet-like: high expression of MUC2, TFF3, good prognosis |
| Sadanandam et al.[8] | 445 samples(2 datasets) | 744 samples (7 datasets) 51 cell lines | NMF-based consensus clustering | Cophenetic correlation coefficient | PAM | Enterocyte: high expression of enterocyte-specific genes, intermediate DFS Stem-like: high expression of stem cell, poor prognosis |
| De Sousa e Melo et al.[3] | 90 samples | 1074 samples (6 datasets) | Classical consensus clustering | Gap statistic | PAM | Inflammatory: high expression of chemokines, enriched for MSI, intermediate DFS Cetuximab-sensitive transit-amplifying: good prognosis Cetuximab-resistant transit-amplifying: poor prognosis CCS1(49%): enriched for KRAS and TP53 mutations, CIN+, left sided, good prognosis CCS2(24%): enriched for MSI, CIMP+, right-sided, intermediate prognosis |
| Budinska et al.[5] | 1113 samples | 720 samples | Classical consensus clustering | Dynamic cut tree | Multiclass linear discriminant (LDA) | CCS3(27%): heterogenous, poor prognosis, refractory to anti-EGFR therapy Surface crypt-like: well differentiated and normal like Lower crypt-like SCNAs, better prognosis CIMP-H-like: enriched for MSI and BRAF mutations, poor OS Mesenchymal: high expression of EMT genes, poor OS Mixed: high expression of EMT genes, enriched for TP53 mutations |
| Roepman et al.[6] | 188 sampels | 543 samples | Hierarchical clustering | N/A | Single sample centroid-based classifier | TypeA(22%): MMR-deficient epithelial, MSI, best prognosis TypeB(62%): Proliferative epithelial, MSS, intermediate prognosis TypeC(16%): Mesenchymal, enriched for BRAF mutations, worst survival |
3.1. Schlicker et al. 2012
Using gene expression profiles of 62 primary CRCs as the discovery cohort, Schlicker et al. employed iterative non-negative matrix factorization (iNMF) for unsupervised classification [4]. These researchers identified two main clusters (Types 1 and 2) in the first iteration, which were further split into five subtypes in the second iteration [4]. Subsequently, using 15 public gene expression datasets which included data from more than 1600 patients, they were able to validate their findings [4].
- Type 1: characterized by activation of pathways related to epithelial-mesenchymal transition (EMT); associated with poor disease free survival (DFS).
-
○Subtype 1.1: strongly mesenchymal with an even distribution of MSI and MSS; upregulated genes in this subtype were related to calcium signaling, MAPK and TGF-β pathways.
-
○Subtype 1.2: mesenchymal and specifically featured with upregulated immune-system related pathways; the only subtype consisting of more tumor samples from female patients.
-
○Subtype 1.3: mesenchymal with high expression of transporter genes.
-
○
- Type 2: enriched for MSS; characterized by upregulated epithelial features such as Wnt pathway and cell cycle signature genes.
-
○Subtype 2.1: featured with upregulated immune system-related genes and stress response genes.
-
○Subtype 2.2: characterized by upregulated cell cycle and amino acid synthesis; highly expressed genes on chromosome 13q and 20q.
-
○
Furthermore, differential response to targeted inhibitors was observed based on analysis of pharmacological data from cell lines classified into different subtypes, suggesting a potential clinical utility of this classification system for directing more targeted therapy for CRC patients [4].
3.2. Marisa et al. 2013
Marisa and co-authors performed gene expression profiling in fresh-frozen tissue specimens from a large, multi-center cohort of 566 CRC patients (CIT cohort); with a well-balanced discovery (443 samples) and a validation dataset (123 samples) [7]. Using the discovery set, they performed consensus clustering analysis and revealed six CRC subtypes with distinct molecular characteristics and corresponding clinical relevance [7]. Their findings were further confirmed using the validation dataset and other 7 independent public datasets totaling 1,029 CRC patients.
C1 (21%, CINImmuneDown): High frequency of CIN; downregulated immune system and EMT.
C2 (19%, dMMR): High frequency of dMMR; high frequency CIMP; significant enrichment for BRAF mutations; upregulated immune system and proliferation related pathways; downregulated WNT pathways.
C3 (13%, KRASm): very significant enrichment for KRAS mutations; downregulated immune system and EMT.
C4 (10%, CSC): very significant enrichment for up-regulated genes in stem cell-like and serrated-CC like signatures; upregulated EMT; down regulated proliferation pathway.
C5 (27%, CINWntUp): high frequency of CIN; significant enrichment for TP53 mutations; upregulated WNT pathway.
C6 (10%, CINnormL): high frequency of CIN; very significant enrichment for up-regulated genes in Normal-like signature; upregulated EMT; downregulated proliferation pathways.
Furthermore, an additional highlight was that CRCs with C2, C3, C4 and C6 may arise from the serrated neoplasia pathway, while C1 and C5 subtypes of cancers primarily developed through conventional adenomas. In addition, C4 and C6 cancers tended to have worse prognosis (relapse-free survival) compared to others, while the combined C4 and C6 subtypes were independent prognostic indicators [7].
3.3. Sadanandam et al. 2013
Sadanandam et al. identified six CRC subtypes using consensus non-negative matrix factorization (NMF) clustering on gene expression profiles for 445 patients merged from two public datasets [8]. Based on similarities between gene expression profiles and known gene signatures calculated using nearest template prediction (NTP) algorithm [68], they proposed that the six subtypes were associated with distinct cell types in normal colon crypt. These findings were further validated in 7 independent public datasets as well as 51 cell lines.
Goblet-like: highly expressed goblet-specific genes MUC2 and TFF3; associated with crypt top signatures; good prognosis.
Enterocyte: highly expressed enterocyte-specific genes; intermediate DFS.
Stem-like: highly expressed WNT signaling targets genes and stem cells; markedly associated with crypt base signature; shortest DFS.
Inflammatory: highly expressed chemokines and interferon-related genes; highly enriched for MSI; intermediate DFS.
- Transit-amplifying: highly enriched for MSS; a heterogeneous subtype which was further divided into two groups according to differential drug response:
-
○Cetuximab-sensitive transit-amplifying (CS-TA); good prognosis.
-
○Cetuximab-resistant transit-amplifying (CR-TA); poor prognosis.
-
○
Based on comprehensive drug response data analysis, the authors proposed a potential subtype-guided therapy strategy for CRC. More specifically, patients classified to stem-like, inflammatory and enterocyte subtypes were deemed to be best treated with FOLFIRI in an adjuvant setting. In the metastatic setting, stem-like tumors were shown to benefit more from FOLFIRI than others, while cetuximab and cMET inhibitors were effective in CS-TA and CR-TA tumors, respectively.
3.4. De Sousa E Melo et al. 2013
De Sousa E Melo and colleagues revealed 3 colon cancer subtypes (CCSs) using consensus clustering on microarray data for 90 stage II fresh frozen specimens[3]. The authors found that the two subtypes, CCS1 and CCS2, essentially corresponded to CIN and MSI subgroups, while CCS3 was a newly identified subtype associated with worst prognosis. Their findings were validated in six independent datasets and cell lines involving more than 1100 CRC patients.
CCS1 (49%): enriched for KRAS and TP53 mutation; CIN+; mainly left-sided; good prognosis.
CCS2 (24%): enriched for MSI; CIMP+; mainly right-sided; intermediate prognosis.
CCS3 (27%): heterogeneous with respect to characteristic mutations, CIMP and MSI; upregulated EMT and matrix remodeling; evenly distributed through the colon; markedly poor prognosis; high recurrence risk; refractory to anti-EGFR therapy.
Comparing CCS1 and CCS3, they observed a significantly differential response to cetuximab, a popular anti-EGFR monoclonal therapy. More specifically, patients with CCS3 showed significant resistance regardless of KRAS mutation, which is predictive of anti-EGFR efficacy in CCS1 [69]. Intriguingly, by comparing gene expression profiles of adenomas and CRCs, CCS3 and CCS1 tumors were specifically associated with sessile serrated adenomas and tubular adenomas, respectively. More interestingly, the genes associated with metastasis were already upregulated in serrated adenomas, suggesting the malignancy potential of CCS3 CRC patients is primed at an early stage of adenomas.
3.5. Budinska et al. 2013
In contrast to the traditional molecular classification based on original gene expression profiling, Budinska et al. used meta-gene profiles derived from 54 gene modules generated by hierarchical clustering approach to perform the classical consensus-based clustering, which they claimed could be effectively resistant to noise [5]. As a result, they identified 5 major subtypes on the discovery dataset containing data from 1113 patient specimens and successfully validated their results in an independent dataset consisting of 720 CRC patients.
Surface crypt-like: low expression of EMT/stroma gene module; histologically related to serrated and papillary phenotype; well differentiated; had a similar gene expression pattern with normal colon mucosa.
Lower crypt-like: low expression of EMT/stroma gene module; histologically related to complex tubular phenotype; higher copy number gain in chromosome 20q; better prognosis.
CIMP-H-like: high expression of immunity-associated gene module and low expression of gut development gene module; histologically related to solid/trabecular and mucinous phenotype; enriched for MSI and BRAF mutations; poor overall survival (OS).
Mesenchymal: high expression of EMT/stroma gene module and low expression of lipid synthesis genes; histologically related to desmoplastic phenotype; poor OS.
Mixed: high expression of EMT/stroma gene module; histologically related to complex tubular phenotype; enriched for TP53 mutations.
A unique perspective from gene modules of this classification system added insights into a new layer of CRC heterogeneity on top of the traditional classifications, which largely focused on the molecular features and clinical factors.
3.6. Roepman et al. 2014
Based on a their previous approach for establishing the prognostic classifier, ColoPrint[70], Roepman et al. further characterized three intrinsic subtypes using 188 tissue specimens from all stages of CRC patients, followed by validation of their results in 543 stage II and III patients[6].
TypeA (22%): MMR-deficient epithelial; enriched for BRAF mutations and MSI; best prognosis.
TypeB (62%): proliferative epithelial; absence of BRAF mutations; high enrichment of MSS; intermediate prognosis; benefit from chemotherapy.
TypeC (16%): mesenchymal; upregulation of mesenchymal signatures; worst survival; resistant to chemotherapy.
Intriguingly, the three intrinsic subtypes were consistent with the three CCS subtypes identified by De Sousa E Melo et al.[3], although the two subtyping studies were based on two independent discovery data sets.
4. The consensus molecular subtypes of colorectal cancer
The six CRC subtyping systems differ in terms of the methodological approaches used for specific analysis, but follow the same single-omic classification workflow (Figure 1a). The strategy often started with a training/discovery cohort of primary tissue samples subjected for whole-genome gene expression profiling followed by normalization, as well as non-biological batch effect detection and correction in cases where multiple data sets were merged together. For the identification of specific molecular subgroups, most of the studies employed either classical consensus clustering [71] or the extended method based on Non-negative matrix factorization (NMF) [72]. Consensus clustering quantifies the stability for varying cluster counts by calculating how frequently two samples are clustered together in hundreds of subsamples randomly drawn from the training dataset [71,72]. Determination of the optimal cluster count is based on area under the empirical cumulative distribution (CDF) curve for the classical method [71], cophenetic correlation coefficient for the NMF method [72], or gap statistic for within-cluster dispersion measurement [73]. To facilitate classification of other independent datasets for validation, classifiers are built using conventional Linear Discriminant Analysis (LDA) [5], centroid-based approach [74], as well as Prediction Analysis for Microarrays (PAM) [75]. Different from the other five classification systems, Schlicker et al. assigned samples to subtypes using a two-step hierarchical clustering approach [4].
Figure 1. Six CRC subtyping systems derived from a single-omic classification workflow and two major strategies for integrative analysis. (a).
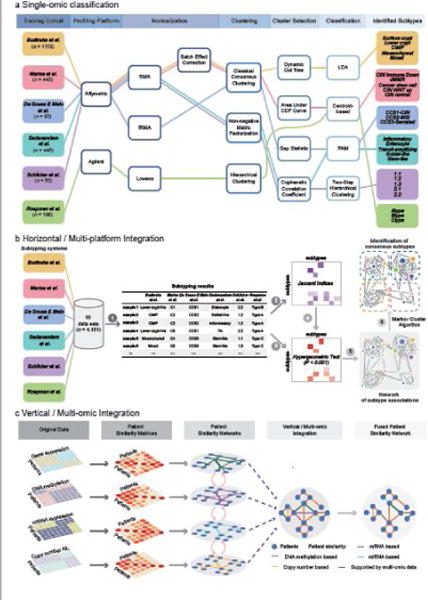
The workflow for six independent CRC subtyping systems based on single-omic classification strategy. The six CRC subtyping studies employed different training cohorts, gene expression profiling platforms, clustering and classification methods, yielding discrepant subtyping results. (b) We proposed a network-based approach for multi-platform (horizontal) integration, involving several major steps: (1) classifying 18 data sets totalling over 4000 samples using each of the six subtyping systems; (2) calculating a matrix of Jaccard indices quantifying the association between each pair of subtypes; (3) evaluating the statistical significance of the association between each pair of subtypes using hypergeometric tests; (4) filtering Jaccard indices by the p-values derived from hypergeometric tests to retain only significant associations (P < 0.001); (5) constructing a network of subtype associations; (6) partitioning the network into consensus molecular subgroups using Markov cluster algorithm (MCL) [82]. More technical details can be found in [9]. (c) Multi-omic (vertical) integration for cancer classification using similarity network fusion (SNF) [80]. Two or more types of omic data such as mRNA expression, miRNA expression, DNA methylation and copy number profiles can be integrated for more comprehensive dissection of cancer heterogeneity occurring at multiple omic levels of gene regulations.
Single-omic classification has been applied for cancer subtyping not only in CRC, but also many other cancers [65]. As powerful as it might be deemed for the discovery of individual subtypes, this methodology was criticized for having a poor performance in identifying true cluster numbers based on simulation datasets with a known structure in a previous study [76]. More critically, the single-omic classification strategy employed by all six subtyping systems has two major limitations:
The single-omic classification workflow involves multiple technical steps, and calibration of parameters in each step is needed to achieve a statistically optimized classifier. The use of different patient discovery cohorts, bioinformatic approaches, parameter settings, as well as biological interpretations, can all influence the eventual subtyping results. In fact, the six CRC subtyping studies identified various numbers of subtypes (ranging from 3 to 6; Table 1, Figure 1a), which caused a lot of confusion, preventing further mechanistic studies and their efficient translation into the clinic. To overcome these limitations, we proposed a ‘horizontal’ integration strategy, which can effectively reconcile different subtyping systems (Figure 1b). Our network-based approach can not only elucidate the association between subtypes identified by various classification systems, but also identify consensus molecular subtypes (CMSs) based on network partition [9]. Recently, a similar approach has been used for identification of consensus subtypes of pancreatic cancer [77]. The detailed application to CRC subtyping will be introduced in the following paragraphs.
Although all six studies gained new biological insights, these were based on transcriptomic profiles only, ignoring potential heterogeneity occurring at other omic levels. To generate a more comprehensive biological overview of diseases, methods such as iCluster [78] and Multiple Dataset Integration (MDI) [79] have been proposed for ‘vertical’ integration of multi-omic data. Another representative approach recently published is similarity network fusion (SNF), which constructs networks of patient samples using each available type of omic data, followed by network fusion for classification [80] (Figure 1c). SNF has been demonstrated to outperform other single-omic classification methods in the identification of more biologically coherent and clinically relevant subtypes in multiple cancer types [80]. Although multi-omic cancer classification methods such as SNF provide a promising solution to overcome the limitations of the current CRC subtyping systems, more comprehensive biological interpretations and clinical characterizations and comparison with the consensus molecular subtypes (CMSs) are still needed.
In this review, we specifically focus on multi-platform/’horizontal’ integration, which provides an efficient methodology to elucidate the associations between various classification systems. ‘Horizontal’ integration is based on the hypothesis that major data substructures are consistent across multiple classification systems, though variations may occur due to differences in training data, sample size, bioinformatic analysis, etc. Indeed, despite the seemingly discrepant conclusions, the six CRC subtyping results actually have some obvious overlaps and correlations. For instance, almost all classifications recapitulate CIN and MSI subtypes that have already been well characterized explicitly. All taxonomies identified and highlighted a mesenchymal/stem-like subtype which is associated with poor prognosis. Nevertheless, it was still unclear how other subtypes could potentially relate to each other, without systematic meta-analysis. In 2014, to clarify the discordant conclusions, Sadanandam et al. and De Sousa E Melo et al. jointly agreed that CRCAssigner and CCS are actually highly related to each other [81]. They used their own classifiers to classify common datasets, and statistically evaluated the sample-wise associations between the 5 subtypes of CRCAssigner and the 3 subtypes of CCS. They concluded that: (1) the CCS1-CIN subtype encompasses the TA and enterocyte subtypes; (2) the inflammatory and goblet-like subtypes are subsets of the CCS2-MSI subtype; and (3) the CCS3-serrated subtype is essentially the stem-like subtype [81].
This reconciliation was brought to the attention of other relevant research groups, who realized that elucidating the inter-relationships between the six subtyping systems was essential and urgent for facilitating their implementation in clinical practice and prospective trials. Accordingly, in 2014, the CRC Subtyping Consortium (CRCSC) was established in an effort to unify these data and potentially reach a consensus on CRC subtyping. This CRCSC represented one of the largest communities for international collaborative research on CRC, involving six participating groups who developed the six CRC classification systems, and an ‘evaluation group’ (Sage Bionetworks) who provided Sage Synapse platform for data sharing and analysis. Central to the reconciliation challenge was the question “how to develop an analytical method for unbiased meta-analysis (or secondary analysis) for the inter-relations between the 6 established classification systems without reinventing the wheel from scratch”? i.e. reclassifying merged datasets used by all groups using a unified bioinformatic approach. The CRCSC overcame this challenge using the network-based approach we proposed for multi-platform integration (Figure 1b): (1) classifying 18 datasets including a total of 4151 patients using each of the six subtyping systems; (2) calculating a matrix of Jaccard indices to quantify the association between each pair of subtypes; (3) evaluating the statistical significance of the association between each pair of subtypes using hypergeometric tests; (4) filtering Jaccard indices by the p-values derived from hypergeometric tests to retain only significant associations (P < 0.001); (4) constructing a network of subtype associations; (5) partitioning the network into four consensus molecular subgroups using MCL (Markov cluster algorithm) [82]. For classification, we assigned CRC samples to the four CMSs using hypergeometric tests, and subsequently trained a robust random forest classifier [9].
Following such a network-based meta-analysis, based on comprehensive multi-omic characterizations, the CRCSC observed that the four CMSs are indeed associated with distinct molecular properties and clinical characteristics (Figure 2):
CMS1-MSI immune subtype (~14%): characterized by MSI, CIMP high, diffuse immune infiltrate and BRAF V600E mutations; associated with worse survival after relapse.
CMS2-canonical subtype (~37%): characterized by epithelial features, CIN, activated WNT and MYC signaling pathways;
CMS3-metabolic subtype (~13%): characterized by deregulation of metabolic pathways, KRAS mutations, low level of CIMP and CIN, and mixed MSI status.
CMS4-mesenchymal subtype (~23%): characterized by upregulation of EMT, TGF-β activation, angiogenesis, stromal infiltration; associated with worse relapse-free and overall survival.
Figure 2. The CRC consensus molecular subtyping system.
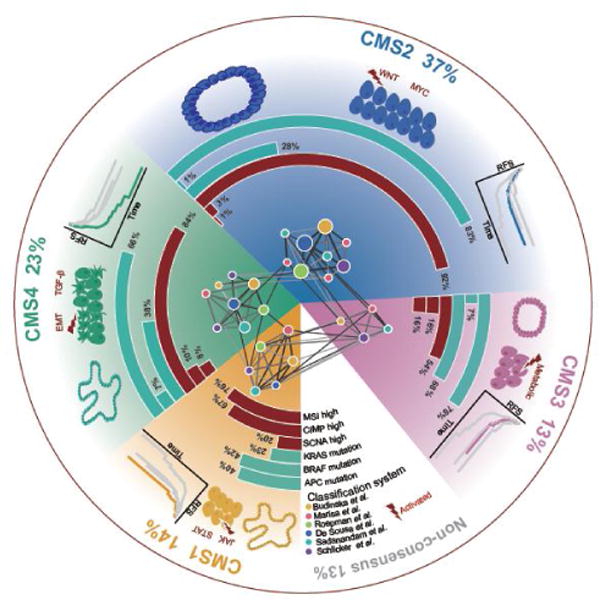
Network-based meta-analysis on six representative classification systems identified four consensus molecular subtypes of CRC. Each subtype shows distinct molecular characteristics and clinical associations. CMS1 (MSI Immune) tumors are characterized by CIMP high status, BRAF V600E mutations, diffuse immune infiltration, and are associated with worse survival after relapse. CMS2 (canonical) tumors are characterized by high somatic copy number alterations (SCNAs), overrepresented APC mutations and activated WNT and MYC signaling pathways. CMS3 (metabolic) tumors are largely CIMP low, enriched for KRAS mutations, and characterized by deregulation of metabolic pathways. CMS4 (mesenchymal) tumors display a high level of SCNAs, and are characterized by upregulation of EMT, TGF-β activation, stromal infiltration and worse relapse-free and overall survival. CMS2 and CMS3 tumors are more likely to be developed from tubular adenomas, while CMS1 and CMS4 tumors are potentially derived from serrated adenomas [142].
Apart from the four major CMSs, ~13% of CRC patient specimens remained unassigned to a specific CMS, indicating a mixed phenotype of multiple CMSs or intratumoral heterogeneity. Overall, this was a marathon effort, and in spite of certain limitations, this consensus molecular subtyping of CRC impacted the field in several aspects:
It represented the first example of joint effort of a community of experts through international collaborations identifying and advocating for a single reproducible model for colon cancer subtyping by effectively unifying previous taxonomies.
It clarified previous confusions, elucidated the inter-relations between various subtypes defined in the 6 pre-existing taxonomies and pieced together interpretations from different perspectives by different research groups for more comprehensive dissection of CRC inter-tumor heterogeneity.
Last but not the least, it provided thus far the most robust classification system for CRC, with clear molecular basis and clinical associations, which not only laid a foundation for further mechanistic studies of subtype-specific biological mechanisms, but also pushed forward clinical translation for more optimized therapy and design of novel targeted agents.
5. New challenges and emerging opportunities
Despite the massive community effort and encouraging progress of consensus molecular subtyping of CRC, it has been somewhat unfortunate that its true translational potential in CRC patients remains to be harnessed. As it stands currently, there is still a large gap between these exciting molecular subtypes and what role, if any, these findings may play in the clinical management of patients with CRC. To accomplish the eventual goal of personalized management of CRC patients, we propose a putative roadmap (Figure 3) involving four major phases.
Collection of pre- and post-surgical biopsies, surgical tissues, and/or serum/plasma from CRC patients for the isolation of DNA, RNA, miRNA and proteins for molecular profiling analysis Thus far, almost all CRC subtyping studies were based on fresh tissue specimens, and it remains questionable whether such a CMS taxonomy could be directly applied to other types of specimens that are readily available in the clinic. In fact, it has been previously shown that the CMS classifier developed by CRCSC had a poor performance in FFPE and biopsy specimens [9,83]. We and others have recently proposed that miRNA signatures are capable of identifying CMS4 subtype patients quite robustly, and further work on miRNA classifiers can be highly rewarding as it might pave the path for a non-invasive classification systems [84,85]. Moreover, recent years have seen growing interest to develop non-invasive biomarkers based on circulating tumor DNA (ctDNA), cell-free RNA, miRNAs, proteins and lipids extracted from serum, plasma or whole blood, as well as exosomal DNA and miRNAs [86–90]. These exciting ‘liquid biopsy’ technologies provide an opportunity to further explore the possibility of non-invasive identification of CMS subtypes.
Performing single-omic or multi-omic profiling using various high- and/or low-throughput platforms that are currently available, as well as the methods that are under development The current CMS classifier employs a signature involving hundreds of genes [9], which is difficult to implement in clinical settings. Various alternative strategies are available, and the optimal solution for a specific clinical application depends on the availability of techniques, data throughput, cost efficiency and performance. On the one hand, high-throughput platforms such as RNA-Seq and gene expression microarrays provide genome-wide information content, which is ideal for research purposes, but inadequate for clinical applications due to high cost, poor accessibility and slow turnaround times. On the other hand, traditional techniques such as qPCR, immunohistochemistry and ELISA are easily accessible, but are not suitable for profiling hundreds of genes currently required for CRC classification. To partly overcome such challenges, Nanostring-based custom arrays and targeted-sequencing could be potentially viable options [91]. Furthermore, the current CMS approach was established based on bulk tumor-derived RNA, which in retrospect, may be inadequate as it fails to address the issue of inter-tumor heterogeneity. Hence, by combining the emerging single-cell sequencing-based research, together with the established CMS taxonomy, may collectively help elucidate a more refined and robust subtype-specific CRC cell origin, its development and evolution.
Stratifying patients by using various biomarker assays tailored for specific clinical applications Although the CMS taxonomy is clinically relevant [9], more specific diagnostic, prognostic and predictive biomarkers are needed for patient stratification into more clinically meaningful subgroups for therapeutic decision making. The unique advantage of CRC subtyping associated biomarkers lies in their discovery using a more unsupervised approach and a broader biological basis, vis-à-vis, traditional biomarkers that are delineated from supervised analyses and are perhaps biased since the patients are identified by the clinical outcomes instead of the underlying molecular phenotype. However, the biomarker development in such an instance still follows a similar workflow as many others involving the following four major steps. Step-1: Identification of candidate biomarkers associated with CRC subtypes in pre-clinical or clinical studies. Step-2: Development of a diagnostic/prognostic/predictive model with statistical assessments. Step-3: Evaluation of inter-lab performance, reproducibility and robustness in multiple cohorts. Step-4: Validation of the biomarker assay in multi-center prospective studies. At each step, critical assessment of each assay is essential before it enters the subsequent step (Figure 3).
Implementation of subtyping in decision-making of CRC to address various clinical questions during disease progression. First, it is time to investigate whether the current ‘tissue-based’ classifier can be translated into a ‘non-invasive’ signature - be it an mRNA or a miRNA based approach, for appreciating its potential in early diagnosis, disease monitoring, as well as in predicting response to chemotherapy and molecularly-targeted drugs. Second, for stage II CRC patients, it is important to predict their risk for disease recurrence, and subsequently select true high-risk patients that are candidates for adjuvant therapy treatment. Although the CMS taxonomy is associated with disease-free survival, we must explore the potential of developing a more robust prognostic model based on CMS classification. Third, for patients with a more advanced disease, clinical management is centered on the selection of appropriate patient populations for various targeted-therapies, followed by disease monitoring using longitudinally collected blood samples. To this end, large-scale, multi-center, retrospective and prospective studies are needed to systematically evaluate the predictive value of CMS for such decision-making for a more personalized treatment modality in CRC patients. Fourth, the success of predictive markers largely relies on similarities between primary and the metastatic lesions. Therefore, it is important to fill this gap in knowledge by analyzing genomic expression profiles of tissues obtained from a large number of matched primary and metastatic sites. Furthermore, effective integration of CMS subtyping and tumor microenvironment-based classification will likely permit a more improved stratification of CRC patients, and yield more optimal therapeutic outcomes. Last but not the least, the regulatory mechanisms underlying specific subtypes remain largely unclear, particularly for the CMS3 (metabolic) and CMS4 (mesenchymal) subtypes. Elucidating subtype-specific regulatory mechanisms are pivotal to the identification of novel therapeutic targets and development of novel targeted therapies.
Figure 3. A putative roadmap to more personalized CRC management based on molecular subtyping.
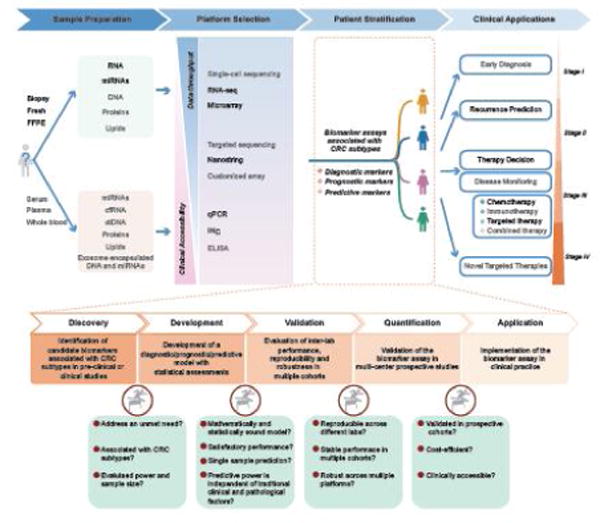
Implementing CMS subtyping for more personalized clinical management of CRC patients involves four major phases: (1) Collection of pre- or post-surgical biopsies, surgical tissues or whole blood/serum/plasma from CRC patients to isolate DNA/RNA/miRNA and proteins to perform molecular profiling. (2) Performing transcriptomic as well as multi-omic profiling using various high and low throughput platforms that are currently available as well as the methods that are under development. (3) Stratifying patients by various biomarker assays tailored for specific clinical applications. More specifically, establishing a robust biomarker associated with CRC subtyping involves multiple stages: biomarker discovery, model development, inter-lab validation as well as validation by prospective studies. At each stage, critical assessments of the assay are needed before entering the next stage. (4) Implementation of subtyping in decision making of CRC to address various clinical questions throughout the CRC progression. Multiple choices of sample sources, molecules and profiling platforms are available, yet largely unexplored (colored in gray), for developing an optimized biomarker assay for a specific clinical application.
The utmost importance right now remains to fully comprehend and overcome several of the prevailing challenges that are preventing translation of CMS subtyping into the clinic (Figure 4). Critically, tight collaborations between basic researchers, bioinformaticians and clinicians are key to addressing these challenges. We foresee that novel technologies, emerging big data science and the growing number of clinical trials will offer attractive opportunities for expediting the clinical translation of such a refined CMS approach in the near future. Among a number of unaddressed issues mentioned in the putative roadmap, we will specifically discuss in more detail about six challenges tightly related to CRC classification in the following sections.
Figure 4.
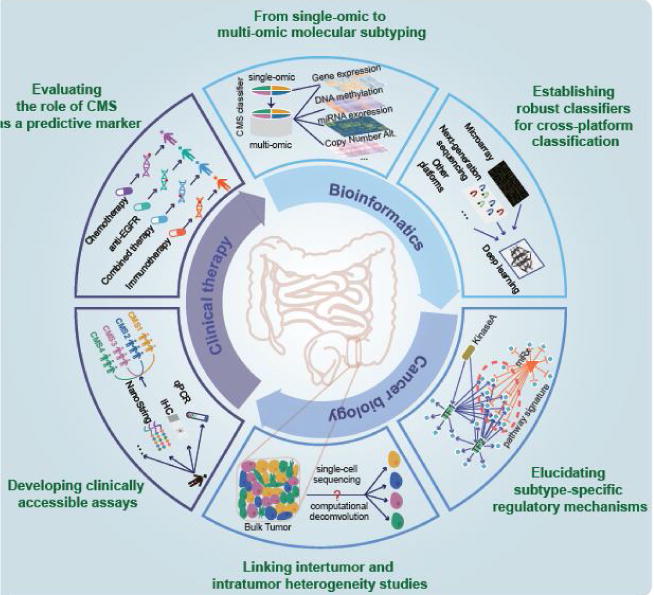
New challenges to clinical translation of CRC subtyping and emerging opportunities. Clockwise, from top to bottom to top are the six major challenges hampering the clinical translation of CRC subtyping as well as their corresponding new opportunities: (1) Transforming single-omic to multi-omic molecular subtyping by integrating other types of data such as DNA methylation and miRNA expression profiles; (2) Establishing more robust classifiers for cross-platform classification based on deep learning; (3) Elucidating subtype-specific regulatory mechanisms using a multi-dimensional network approach; (4) Linking inter-tumor and intra-tumor heterogeneity studies by single cell sequencing or computational deconvolution; (5) Developing clinically accessible assays using qPCR, IHC and NanoString to implement CMS taxonomy as routine clinical practice; (6) Evaluating the role of CMS as a predictive marker by integrative analysis of genomic, pharmacological and clinical data. Importantly, multidisciplinary collaborations between basic cancer research, bioinformatics and clinical research are key to addressing these urgent challenges preventing the clinical translation of CMS taxonomy.
5.1. From single-omic to multi-omic molecular subtyping
The vast majority of published work on molecular subtyping of cancers are based on unsupervised classification of single-omic data, especially transcriptomic data. However, molecular heterogeneity is present, both at genetic and epigenetic level, which may not be fully captured solely at the transcriptomic level. For example, CRC is also heterogenous at the proteomic level, as evidenced by the identification of 5 distinct proteomic subtypes previously identified by the Clinical Proteomic Tumor Analysis Consortium (CPTAC) [92]. A recent study by Wang et al. found that these proteomic subtypes can be recapitulated in CRC cell line proteomes, which better predicts drug sensitivity compared to genomic and transcriptomic profiles [93]. Hence, only relying on transcriptomic data can possibly fail to identify heterogeneity that exists on other omic levels such as epigenetic regulations represented by noncoding RNAs and aberrant DNA methylation [94]. For a more comprehensive dissection of tumor heterogeneity, multi-omic data integration will very likely provide a possible solution [95,96]. The immediate challenge, however, arises from the lack of sufficient data for discovery, as multi-omic data are still only available in consortiums such as TCGA and CRC cell lines [97]. More importantly, effective integration of multi-omic data for classification remains a bottleneck for bioinformaticians [98], mainly due to the difficulty of direct comparison, as they have different data scales and dimensionalities. Recently, several research groups have proposed novel multi-omic classification approaches with promising performance demonstrated [80,99,100]. For instance, Wang et al. developed a novel bioinformatic approach named “similarity network fusion (SNF)”, which fuses similarity networks constructed from each single-omic data type into a single similarity network using a nonlinear combination method [80]. SNF integrates mRNA expression, microRNA expression and DNA methylation data, and demonstrates better performance compared to a single-omic based approaches in cancer classification [80]. A recent study in bladder cancer also revealed that a multi-omics based classification actually worked better than the mRNA-based subtyping alone in guiding treatment decisions in muscle-invasive bladder cancer patients [101].
5.2. Establishing robust classifiers for cross-platform classification
A widely implemented strategy for cancer subtyping involves consensus clustering for determination of an optimal number of cancer subgroups, and classification with feature selection, i.e., selection of a list of signature genes [65]. However, the classification step suffers from several critical limitations. First, a signature gene-based approach places sole emphasis on the role of individual genes, without effective incorporation of biological knowledge such as the pathway activity, which often leads to poor interpretation [102–104]. Second, signature genes for classification are not always available due to unpaired gene annotation caused by platform differences, which hampers its portability and translational potential. Lastly, gene expression profiling is easily affected by factors such as technical platform variations and experimental protocols, leading to non-biological batch effects. Mathematical and statistical methods might be able to correct for such bias to some extent, however, such correction methods are not always suitable, especially in situations when the sources of bias are unclear. For instance, the correction power of existing methods such as ComBat [105] has been demonstrated to be limited to a balanced experimental design [106]. More critically for clinical implementation, the batch effects also prevent the development of gene expression signature-based classifiers for single sample classification.
Recent advances in the machine learning community have shown a greater promise for applying deep learning for improved cancer classification. For instance, deep convolutional neural networks (CNNs) have demonstrated to improve accuracy and reproducibility of tumor classification based on histopathological or radiographic images [107,108]. Deep learning-based frameworks such as D-GEX [109], DeepChrome [110] and DeepSEA [111] have also been developed for predicting gene expression or effects of noncoding variants based on high-dimensional genomic or epi-genomic profiles. Furthermore, several supervised and unsupervised deep learning-based classification methods have been proposed for cancer detection and diagnosis, and have demonstrated superior performance over classical methods such as support vector machines and random forests [112,113].
5.3. Elucidating subtype-specific regulatory mechanisms
Elucidating subtype-specific regulatory mechanisms is pivotal to the identification of driver pathways and master regulators, which are critical for prioritization of novel drug targets tailored for each CRC subtype. Based on large-scale multi-omic characterizations by the CRCSC, it is clear that each of the four CMSs are associated with distinct biological programs. What remains largely unclear, however, are the upstream regulatory mechanisms. For example, the mesenchymal CMS4 has no clear associations with specific mutations or conventional markers, which makes it difficult for their implementation in the clinical settings [9]. The upstream regulatory mechanism of CMS4 was not clear until the recent work by Fessler et al., who took a network-based approach and discovered that promoter hypermethylation of the miR-200 family is a major determinant of the upregulated epithelial-to-mesenchymal transition [114]. Recently the same group also reported that TGFβ plays an important role in directing sessile serrated adenomas to the mesenchymal CMS4 of CRC. More mechanistic studies of upstream regulatory signaling pathways are urgently needed to pinpoint the driving force for each particular subtype, so that more druggable targets can be identified. Furthermore, integrating multi-omics data and understanding the crosstalk between different omic classifiers will facilitate identification of synergistic targets that can lead to the development of combination therapies in CRC.
5.4. Linking inter-tumor and intra-tumor heterogeneity studies
Cancer molecular subtyping is primarily based on gene expression data derived from bulk tumors, which represent averaged transcriptomic profiles of various types of cells. However, stromal content is known to be one of the predominant sources of variation in transcriptomic profiles, which can significantly influence CRC classification [115,116]. Although it has been shown that CRC cell lines and patient-derived xenografts (PDXs) can be robustly classified to the four CMSs independent of stromal contribution [117], CRC intrinsic subtypes (CRISs) developed based on human-specific expression profiles from CRC PDX models demonstrated an overall better performance and robustness in the classification of resection and biopsy specimens [83,118]. Although bulk tumor-based gene expression profiles have been used to explore intratumor heterogeneity [119–121], it is still challenging to accurately characterize subpopulations of cancer cells. Two major strategies have been employed to dissect intratumor heterogeneity: single-cell sequencing and computational deconvolution of bulk-tumor gene expression profiles.
Single-cell sequencing is a direct way to study intratumor heterogeneity, which will not only advance our knowledge about cancer development, but also shift the paradigm of clinical cancer practice by providing a high-resolution platform for real-time monitoring and early stage detection. For instance, a recent work by Li et al. based on single-cell transcriptomic sequencing found that EMT-related genes were upregulated in cancer-associated fibroblasts but not in tumor epithelial cells[122], confirming the previous conclusions made by Calon et al. and Isella et al. based on bulk-tumor data analysis[115,116]. Another recent study profiled a number of single cells from organoids derived from three CRC patients for WGS, RNA-Seq and methylome, and revealed a significant intratumoral heterogeneity highlighting the importance of these issues, prior to the implementation of CMS subtyping in clinical settings [123]. However, single-cell sequencing is expensive and demanding on the technology. Single-cell sequencing data is much noisier, posing challenges to bioinformatics. More importantly, additional studies are needed that delve into the associations between intratumor heterogeneity and clinical outcomes to exemplify the clinical utilities of single-cell sequencing.
Besides single-cell sequencing, another popular methodology for studying intratumor heterogeneity is to perform computational deconvolution of bulk-tumor gene expression profiles into cell-type specific profiles [124]. Using this approach, one can simultaneously obtain a global picture of the tumor and estimated internal cell compositions. More importantly, popular computational deconvolution approaches such as ESTIMATE [125], MCP-counter [126] and CIBERSORT [127] can be applied to different whole-genome gene expression data, which enables integrative analysis of multi-center multi-omic data and clinical data that are publicly available. Furthermore, bulk-tumor data deconvolution has also been coupled with unsupervised classification for molecular subtyping, leading to the identification of tumor and stroma-specific subtypes in pancreatic ductal adenocarcinoma [128]. For CRC, Etienne Becht and colleagues recently analyzed the compositions of immune, fibroblastic and angiogenic microenvironment using MCP-counter, and observed a high correlation between these microenvironmental signatures and molecular subtypes, highlighting the possibility to integrate them to guide targeted immunotherapies in the future [129]. As we already know that CRC pathogenesis involves accumulation of various mutations at different levels, further studies are pivotal in understanding the timing or establishment of CRCs in the pathogenesis of CRC, especially by profiling large number of CRC adenomas. Moreover, many recent studies revealed that the primary and metastatic tissues possess genetic heterogeneity [130–132]. However, there are not many convincing studies published at the transcriptomic level that can be availed for further analysis. Therefore, profiling primary, as well as synchronous and metachronous metastatic lesions at transcriptomic level is a must for the selection of patients for optimal treatment modalities.
To summarize, the two methodologies complement each other, and can be integrated for more comprehensive analysis linking inter-tumor and intra-tumor heterogeneity for a better understanding of CRC subtype-specific microenvironment and cancer evolution.
5.5. Developing clinically accessible assays
The six CRC subtyping systems as well as the consensus taxonomy, have all demonstrated prognostic and/or predictive values to certain extent [4,6–9,66,67]. Effective translation of these findings into routine clinical practice, however, is still quite difficult due to several major challenges. First, all of these taxonomies were developed based on gene expression profiles derived from microarray or RNA-seq platforms, which are largely impractical in actual clinical settings due to their exorbitant cost, long turnaround time, and reliance on bioinformatic expertise. Second, the discovery cohorts used for developing these classifiers contained mostly fresh frozen samples, and therefore, the accuracy and robustness of these classifiers on FFPE samples remains questionable. In fact, the CMS classifier had poor performance on several data sets derived from FFPE samples [7]. Therefore, in practice, the CMS classifier needs to be tailored to the specific type of clinical specimens used for analysis. Last but not the least, there is still a lack of biomarkers predictive of CMS that can be easily comprehensible and conducted by pathologists and clinicians. Instead, molecular subtyping of breast cancer has become a routine practice just because ER/PR/HER2 biomarker testing is highly predictive, easy to understand and process.
Addressing these challenges are prerequisites for widespread embracement and clinical applications of the CMS classification system. To replace microarray or RNA-seq based assays for CMS subtyping in the clinic, several alternative strategies may be worth exploring. First, would be to develop qPCR or IHC based assays based on optimized panel of markers selected from the full list of signature genes. This often necessitates feature selection to reduce the number of signature genes to a small panel while maintaining acceptable prediction accuracy. Substantial effort has already been made by several groups to develop more accessible classifiers using qPCR and/or IHC based assays [8,66,133]. For instance, an IHC-based assay was developed which employs staining of five markers (CDX2, FRMD6, HTR2B, ZEB1, and KER) selected from the total 146 signature genes of the CCS classifier, together with MSI status, for CCS-based CRC classification [133]. Another qRT-PCR study based on the expression of a two gene panel PDGFR/KIT showed high accuracy in detecting CMS4 subtype tumors amid the issue of intratumor heterogeneity [134]. Second, recent advances in biotechnologies have made it possible to develop diagnostic assays based on dozens or even hundreds of gene expression markers at relatively low costs. For instance, technologies, such as NanoString nCounter® gene expression system [135], are cost-effective and can be used for developing FDA-approved diagnostic kits. Targeted RNA sequencing has also been demonstrated to be a more sensitive methodology for identifying triple-negative breast cancers compared to conventional assays [136]. Lastly, liquid biopsies are transforming molecular diagnosis to a non-invasive promising technique for early detection of cancers [137]. Distinguishing different molecular subtypes of CRC at early stages is important for the selection of patients for optimized therapies. Towards this end, it is critical to identify subtype-specific cell-free tumor DNA (ctDNA) or RNA, proteins and lipids contained within exosomes.
5.6. Evaluating the role of CMS as a predictive marker
The ultimate goal of cancer subtyping is not only for more accurate prognosis, but also for precise decision-making of treatment. Although the associations of CRC subtypes with adjuvant chemotherapies or anti-EGFR therapies have been extensively studied elsewhere, the CRCSC did not discuss the predictive power of CMS explicitly [9]. Therefore, additional effort should be made for a comprehensive evaluation of the predictive role of CMS. In the last decades, various drug sensitivity studies have been conducted using CRC cell lines, xenografts and patients, which generated valuable transcriptomic data for molecular subtyping and corresponding pharmacological and clinical data for association studies [69,138–140]. Recently, Sveen et al. managed to translate this CMS system to preclinical models including a panel of cell lines and PDX models by first optimizing the CMSs classifier enriched for cancer cell-intrinsic signals and then integrating high-throughput in vitro drug screening [141]. They eventually confirmed CMS2 to be strongly responsive to EGFR and HER2 inhibitors, and found CMS1 and CMS4 to possess high sensitivity towards HSP90 inhibitors [141]. Furthermore, this study uncovered a potential for combination treatment with 5-fluorouracil and luminespib to alleviate chemoresistance in CMS4 patients [141].
Apart from the recent encouraging progress, many more ongoing clinical trials testing combined therapies (e.g., BRAF/MEK/EGFR combinatorial inhibitions) or immunotherapies (e.g., anti-PD1 and anti-PDL1 therapies) highlight new opportunities for evaluating the association of different CMS subgroups with novel CRC therapies. It is highly advocated that these genomic, pharmacological and clinical data can be made publicly available so that the entire research community can contribute and collaborate to unravel the urgent challenges in order to achieve the ultimate goal of translating molecular subtyping of CRC for personalized treatment in the clinic [97]. A non-invasive CMS tool would allow the clinicians in not only implementing individualized therapies, but will also aid in disease monitoring and anticipating secondary resistance. With the recent advances in technologies like NanoString, it is worth developing a plasma-based CMS classifier either using the mRNA or miRNA based strategy, which might also help to combat intratumor heterogeneity. As more and more studies are revealing that the drug treatment leads to emergence of multiple drug-resistant sub clones, each carrying distinct genetic alterations relative to the parent tumor cells, it is essential to elucidate whether a specific subtype switching occurs when such drugs are administered alone or in combination with other targeted therapies in metastatic CRC patients.
Taken together, the CMS has allowed us to reach a reasonable degree of consensus in better understanding the underlying biology in CRC patients. However, additional studies and data analytics must be undertaken to overcome some of the challenges described in this article, before these data can transition from the bench – to the bedside.
Acknowledgments
The present work was supported by a start-up grant for new faculty (7200455) and a VPRT grant (9610337) from the City University of Hong Kong, a grant from the Research Grants Council of the Hong Kong Special Administrative Region, China (Project No. CityU 21101115), as well as a grant from The Science Technology and Innovation Committee of Shenzhen Municipality (JCYJ20170307091256048) awarded to Xin Wang, and the CA72851, CA181572, CA184792, CA187956 and CA202797 grants from the National Cancer Institute, National Institute of Health; RP140784 from the Cancer Prevention Research Institute of Texas; grants from the Sammons Cancer Center and Baylor Foundation, as well as funds from the Baylor Scott & White Research Institute, Dallas, TX, USA awarded to Ajay Goel.
Abbreviations
- CRC
colorectal cancer
- CMSs
consensus molecular subtypes
- CRCSC
CRC Subtyping Consortium
- CIN
chromosomal instability
- MSI
microsatellite instability
- CIMP
CpG island methylator phenotype
- MMR
mismatch repair
- MSI-H
high-frequency microsatellite instability
- dMMR
defective DNA mismatch repair
- MAPK
mitogen-activated protein kinase
- V600E
mutations at the residue 600 from valine to glutamate
- iNMF
iterative non-negative matrix factorization
- EMT
epithelial-mesenchymal transition
- DFS
disease free survival
- NMF
non-negative matrix factorization
- NTP
nearest template prediction
- CS-TA
cetuximab-sensitive transit-amplifying
- CR-TA
cetuximab-resistant transit-amplifying
- CCSs
colon cancer subtypes
- OS
overall survival
- CDF
cumulative distribution
- LDA
Linear Discriminant Analysis
- PAM
Prediction Analysis for Microarrays
- MDI
Multiple Dataset Integration
- SNF
similarity network fusion
- MCL
Markov cluster algorithm
- CPTAC
the Clinical Proteomic Tumor Analysis Consortium
- CNNs
convolutional neural networks
- CRISs
colorectal cancer intrinsic subtypes
- ctDNA
circulating tumor DNA
- PDXs
patient-derived xenografts
- SCNAs
somatic copy number alterations
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
Authors declare no conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.O’Connell JB, Maggard MA, Ko CY. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J Natl Cancer Inst. 2004;96:1420–1425. doi: 10.1093/jnci/djh275. [DOI] [PubMed] [Google Scholar]
- 3.De Sousa E Melo F, Wang X, Jansen M, Fessler E, Trinh A, de Rooij LPMH, Jong JH de, de Boer OJ, Leersum R van, Bijlsma MF, Rodermond H, van der Heijden M, van Noesel CJM, Tuynman JB, Dekker E, Markowetz F, Medema JP, Vermeulen L. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med. 2013;19:614–618. doi: 10.1038/nm.3174. [DOI] [PubMed] [Google Scholar]
- 4.Schlicker A, Beran G, Chresta CM, McWalter G, Pritchard A, Weston S, Runswick S, Davenport S, Heathcote K, Castro DA, Orphanides G, French T, Wessels LFA. Subtypes of primary colorectal tumors correlate with response to targeted treatment in colorectal cell lines. BMC Med Genomics. 2012;5:66. doi: 10.1186/1755-8794-5-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Budinska E, Popovici V, Tejpar S, D’Ario G, Lapique N, Sikora KO, Di Narzo AF, Yan P, Hodgson JG, Weinrich S, Bosman F, Roth A, Delorenzi M. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J Pathol. 2013;231:63–76. doi: 10.1002/path.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roepman P, Schlicker A, Tabernero J, Majewski I, Tian S, Moreno V, Snel MH, Chresta CM, Rosenberg R, Nitsche U, Macarulla T, Capella G, Salazar R, Orphanides G, Wessels LFA, Bernards R, Simon IM. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int J Cancer. 2014;134:552–562. doi: 10.1002/ijc.28387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marisa L, de Reyniès A, Duval A, Selves J, Gaub MP, Vescovo L, Etienne-Grimaldi MC, Schiappa R, Guenot D, Ayadi M, Kirzin S, Chazal M, Fléjou JF, Benchimol D, Berger A, Lagarde A, Pencreach E, Piard F, Elias D, Parc Y, Olschwang S, Milano G, Laurent-Puig P, Boige V. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453. doi: 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, Ostos LCG, Lannon WA, Grotzinger C, Del Rio M, Lhermitte B, Olshen AB, Wiedenmann B, Cantley LC, Gray JW, Hanahan D. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guinney J, Dienstmann R, Wang X, Reyniès A de, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, Bot BM, Morris JS, Simon IM, Gerster S, Fessler E, De Sousa E Melo F, Missiaglia E, Ramay H, Barras D, Homicsko K, Maru D, Manyam GC, Broom B, Boige V, Perez-Villamil B, Laderas T, Salazar R, Gray JW, Hanahan D, Tabernero J, Bernards R, Friend SH, Laurent-Puig P, Medema JP, Sadanandam A, Wessels L, Delorenzi M, Kopetz S, Vermeulen L, Tejpar S. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 11.Okugawa Y, Grady WM, Goel A. Epigenetic Alterations in Colorectal Cancer: Emerging Biomarkers. Gastroenterology. 2015;149:1204–1225.e12. doi: 10.1053/j.gastro.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pino MS, Chung DC. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology. 2010;138:2059–2072. doi: 10.1053/j.gastro.2009.12.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 14.Bunting SF, Nussenzweig A. End-joining, translocations and cancer. Nat Rev Cancer. 2013;13:443–454. doi: 10.1038/nrc3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grady WM. Genomic instability and colon cancer. Cancer Metastasis Rev. 2004;23:11–27. doi: 10.1023/a:1025861527711. [DOI] [PubMed] [Google Scholar]
- 16.Matsui A, Ihara T, Suda H, Mikami H, Semba K. Gene amplification: mechanisms and involvement in cancer. Biomol Concepts. 2013;4:567–582. doi: 10.1515/bmc-2013-0026. [DOI] [PubMed] [Google Scholar]
- 17.Roger L, Jones RE, Heppel NH, Williams GT, Sampson JR, Baird DM. Extensive telomere erosion in the initiation of colorectal adenomas and its association with chromosomal instability. J Natl Cancer Inst. 2013;105:1202–1211. doi: 10.1093/jnci/djt191. [DOI] [PubMed] [Google Scholar]
- 18.Gilad O, Nabet BY, Ragland RL, Schoppy DW, Smith KD, Durham AC, Brown EJ. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res. 2010;70:9693–9702. doi: 10.1158/0008-5472.CAN-10-2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watanabe T, Kobunai T, Yamamoto Y, Matsuda K, Ishihara S, Nozawa K, Yamada H, Hayama T, Inoue E, Tamura J, Iinuma H, Akiyoshi T, Muto T. Chromosomal instability (CIN) phenotype, CIN high or CIN low, predicts survival for colorectal cancer. J Clin Oncol. 2012;30:2256–2264. doi: 10.1200/JCO.2011.38.6490. [DOI] [PubMed] [Google Scholar]
- 20.Boland CR, Boland C Richard, Goel A. Microsatellite Instability in Colorectal Cancer. Gastroenterology. 2010;138:2073–2087.e3. doi: 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Cunningham JM, Winters JL, Guenther JC, French AJ, Boardman LA, Burgart LJ, McDonnell SK, Schaid DJ, Thibodeau SN. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res. 2003;63:5209–5212. [PubMed] [Google Scholar]
- 22.Goel A, Xicola RM, Nguyen TP, Doyle BJ, Sohn VR, Bandipalliam P, Rozek LS, Reyes J, Cordero C, Balaguer F, Castells A, Jover R, Andreu M, Syngal S, Boland CR, Llor X. Aberrant DNA methylation in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology. 2010;138:1854–1862. doi: 10.1053/j.gastro.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malesci A, Laghi L, Bianchi P, Delconte G, Randolph A, Torri V, Carnaghi C, Doci R, Rosati R, Montorsi M, Roncalli M, Gennari L, Santoro A. Reduced likelihood of metastases in patients with microsatellite-unstable colorectal cancer. Clin Cancer Res. 2007;13:3831–3839. doi: 10.1158/1078-0432.CCR-07-0366. [DOI] [PubMed] [Google Scholar]
- 24.Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, French AJ, Kabat B, Foster NR, Torri V, Ribic C, Grothey A, Moore M, Zaniboni A, Seitz JF, Sinicrope F, Gallinger S. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28:3219–3226. doi: 10.1200/JCO.2009.27.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA., Jr PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bae JM, Kim JH, Kang GH. Epigenetic alterations in colorectal cancer: the CpG island methylator phenotype. Histol Histopathol. 2013;28:585–595. doi: 10.14670/HH-28.585. [DOI] [PubMed] [Google Scholar]
- 28.Ang PW, Loh M, Liem N, Lim PL, Grieu F, Vaithilingam A, Platell C, Yong WP, Iacopetta B, Soong R. Comprehensive profiling of DNA methylation in colorectal cancer reveals subgroups with distinct clinicopathological and molecular features. BMC Cancer. 2010;10:227. doi: 10.1186/1471-2407-10-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sideris M, Papagrigoriadis S. Molecular biomarkers and classification models in the evaluation of the prognosis of colorectal cancer. Anticancer Res. 2014;34:2061–2068. [PubMed] [Google Scholar]
- 30.Dahlin AM, Palmqvist R, Henriksson ML, Jacobsson M, Eklof V, Rutegard J, Oberg A, Van Guelpen BR. The Role of the CpG Island Methylator Phenotype in Colorectal Cancer Prognosis Depends on Microsatellite Instability Screening Status. Clin Cancer Res. 2010;16:1845–1855. doi: 10.1158/1078-0432.CCR-09-2594. [DOI] [PubMed] [Google Scholar]
- 31.Ward RL, Cheong K, Ku S-L, Meagher A, O’Connor T, Hawkins NJ. Adverse prognostic effect of methylation in colorectal cancer is reversed by microsatellite instability. J Clin Oncol. 2003;21:3729–3736. doi: 10.1200/JCO.2003.03.123. [DOI] [PubMed] [Google Scholar]
- 32.Toyota M, Ohe-Toyota M, Ahuja N, Issa JPJ. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proceedings of the National Academy of Sciences. 2000;97:710–715. doi: 10.1073/pnas.97.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hawkins N, Norrie M, Cheong K, Mokany E, Ku S-L, Meagher A, O’Connor T, Ward R. CpG island methylation in sporadic colorectal cancers and its relationship to microsatellite instability. Gastroenterology. 2002;122:1376–1387. doi: 10.1053/gast.2002.32997. [DOI] [PubMed] [Google Scholar]
- 34.Samowitz WS, Albertsen H, Herrick J, Levin TR, Sweeney C, Murtaugh MA, Wolff RK, Slattery ML. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology. 2005;129:837–845. doi: 10.1053/j.gastro.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 35.Miliani de Marval PL, Zhang Y. The RP-Mdm2-p53 pathway and tumorigenesis. Oncotarget. 2011;2:234–238. doi: 10.18632/oncotarget.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pei D, Zhang Y, Zheng J. Regulation of p53: a collaboration between Mdm2 and Mdmx. Oncotarget. 2012;3:228–235. doi: 10.18632/oncotarget.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 38.Baker SJ, Preisinger AC, Jessup JM, Paraskeva C, Markowitz S, Willson JK, Hamilton S, Vogelstein B. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. 1990;50:7717–7722. [PubMed] [Google Scholar]
- 39.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andreyev HJ, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, Young J, Walsh T, Ward R, Hawkins N, Beranek M, Jandik P, Benamouzig R, Jullian E, Laurent-Puig P, Olschwang S, Muller O, Hoffmann I, Rabes HM, Zietz C, Troungos C, Valavanis C, Yuen ST, Ho JW, Croke CT, O’Donoghue DP, Giaretti W, Rapallo A, Russo A, Bazan V, Tanaka M, Omura K, Azuma T, Ohkusa T, Fujimori T, Ono Y, Pauly M, Faber C, Glaesener R, de Goeij AF, Arends JW, Andersen SN, Lövig T, Breivik J, Gaudernack G, Clausen OP, Angelis PD De, Meling GI, Rognum TO, Smith R, Goh HS, Font A, Rosell R, Sun XF, Zhang H, Benhattar J, Losi L, Lee JQ, Wang ST, Clarke PA, Bell S, Quirke P, Bubb VJ, Piris J, Cruickshank NR, Morton D, Fox JC, Al-Mulla F, Lees N, Hall CN, Snary D, Wilkinson K, Dillon D, Costa J, Pricolo VE, Finkelstein SD, Thebo JS, Senagore AJ, Halter SA, Wadler S, Malik S, Krtolica K, Urosevic N. Kirsten ras mutations in patients with colorectal cancer: the “RASCAL II” study. Br J Cancer. 2001;85:692–696. doi: 10.1054/bjoc.2001.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boeckx N, Peeters M, Van Camp G, Pauwels P, Op de Beeck K, Deschoolmeester V. Prognostic and Predictive Value of RAS Gene Mutations in Colorectal Cancer: Moving Beyond KRAS Exon 2. Drugs. 2015;75:1739–1756. doi: 10.1007/s40265-015-0459-x. [DOI] [PubMed] [Google Scholar]
- 42.Al-Shamsi HO, Alhazzani W, Wolff RA. Extended RAS testing in metastatic colorectal cancer-Refining the predictive molecular biomarkers. J Gastrointest Oncol. 2015;6:314–321. doi: 10.3978/j.issn.2078-6891.2015.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, Masi G, Stasi I, Canestrari E, Rulli E, Floriani I, Bencardino K, Galluccio N, Catalano V, Tonini G, Magnani M, Fontanini G, Basolo F, Falcone A, Graziano F. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer. 2009;101:715–721. doi: 10.1038/sj.bjc.6605177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lièvre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, Ychou M, Bouché O, Landi B, Louvet C, André T, Bibeau F, Diebold MD, Rougier P, Ducreux M, Tomasic G, Emile JF, Penault-Llorca F, Laurent-Puig P. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374–379. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 45.Nakamura Y. Isolation of p53-target genes and their functional analysis. Cancer Sci. 2004;95:7–11. doi: 10.1111/j.1349-7006.2004.tb03163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Slebos RJ, Lee MH, Plunkett BS, Kessis TD, Williams BO, Jacks T, Hedrick L, Kastan MB, Cho KR. p53-dependent G1 arrest involves pRB-related proteins and is disrupted by the human papillomavirus 16 E7 oncoprotein. Proceedings of the National Academy of Sciences. 1994;91:5320–5324. doi: 10.1073/pnas.91.12.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 48.Blandino G, Levine AJ, Oren M. Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene. 1999;18:477–485. doi: 10.1038/sj.onc.1202314. [DOI] [PubMed] [Google Scholar]
- 49.Almazov VP, Kochetkov DV, Chumakov PM. Use of p53 for therapy of human cancer. Mol Biol. 2007;41:863–877. [PMC free article] [PubMed] [Google Scholar]
- 50.Montagut C, Settleman J. Targeting the RAF–MEK–ERK pathway in cancer therapy. Cancer Lett. 2009;283:125–134. doi: 10.1016/j.canlet.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 51.Tejpar S, Bertagnolli M, Bosman F, Lenz HJ, Garraway L, Waldman F, Warren R, Bild A, Collins-Brennan D, Hahn H, Harkin DP, Kennedy R, Ilyas M, Morreau H, Proutski V, Swanton C, Tomlinson I, Delorenzi M, Fiocca R, Van Cutsem E, Roth A. Prognostic and predictive biomarkers in resected colon cancer: current status and future perspectives for integrating genomics into biomarker discovery. Oncologist. 2010;15:390–404. doi: 10.1634/theoncologist.2009-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JWC, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 53.Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer. 2011;11:309–309. doi: 10.1038/nrc2645. [DOI] [PubMed] [Google Scholar]
- 54.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 55.Pietrantonio F, Petrelli F, Coinu A, Di Bartolomeo M, Borgonovo K, Maggi C, Cabiddu M, Iacovelli R, Bossi I, Lonati V, Ghilardi M, de Braud F, Barni S. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer. 2015;51:587–594. doi: 10.1016/j.ejca.2015.01.054. [DOI] [PubMed] [Google Scholar]
- 56.Linnekamp JF, Wang X, Medema JP, Vermeulen L. Colorectal cancer heterogeneity and targeted therapy: a case for molecular disease subtypes. Cancer Res. 2015;75:245–249. doi: 10.1158/0008-5472.CAN-14-2240. [DOI] [PubMed] [Google Scholar]
- 57.Popovici V, Budinska E, Tejpar S, Weinrich S, Estrella H, Hodgson G, Van Cutsem E, Xie T, Bosman FT, Roth AD, Delorenzi M. Identification of a poor-prognosis BRAF-mutant-like population of patients with colon cancer. J Clin Oncol. 2012;30:1288–1295. doi: 10.1200/JCO.2011.39.5814. [DOI] [PubMed] [Google Scholar]
- 58.Elez E, Schellens J, van Geel R, Bendell J, Spreafico A, Schuler M, Yoshino T, Delord JP, Yamada Y, Lolkema M, Faris JE, Eskens F, Sharma S, Yaeger R, Lenz HJ, Wainberg Z, Avsar E, Chatterjee A, Jaeger S, Demuth T, Tabernero J. Ann Oncol. Vol. 26. 2015; LBA-08Results of a phase 1b study of the selective BRAF V600 inhibitor encorafenib in combination with cetuximab alone or cetuximab + alpelisib for treatment of patients with advanced BRAF-mutant metastatic colorectal cancer; pp. iv120–iv120. [Google Scholar]
- 59.Bendell JC, Atreya CE, André T, Tabernero J, Gordon MS, Bernards R, Van Cutsem E, Tejpar S, Sidhu R, Go WY, Allred A, Motwani M, Suttle BB, Wu Y, Hoos A, Orford KW, Corcoran RB, Schellens JHM. Efficacy and tolerability in an open-label phase I/II study of MEK inhibitor trametinib (T), BRAF inhibitor dabrafenib (D), and anti-EGFR antibody panitumumab (P) in combination in patients (pts) with BRAF V600E mutated colorectal cancer (CRC) J Clin Orthod. 2014;32:3515–3515. [Google Scholar]
- 60.Hong DS, Morris VK, El Osta B, Sorokin AV, Janku F, Fu S, Overman MJ, Piha-Paul S, Subbiah V, Kee B, Tsimberidou AM, Fogelman D, Bellido J, Shureiqi I, Huang H, Atkins J, Tarcic G, Sommer N, Lanman R, Meric-Bernstam F, Kopetz S. Phase IB Study of Vemurafenib in Combination with Irinotecan and Cetuximab in Patients with Metastatic Colorectal Cancer with BRAFV600E Mutation. Cancer Discov. 2016;6:1352–1365. doi: 10.1158/2159-8290.CD-16-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tabernero J, Chan E, Baselga J, Blay JY, Chau I, Hyman DM, Raje NS, Wolf J, Sirzen F, Veronese ML, Mitchell L, Hidalgo M. VE-BASKET, a Simon 2-stage adaptive design, phase II, histology-independent study in nonmelanoma solid tumors harboring BRAF V600 mutations (V600m): Activity of vemurafenib (VEM) with or without cetuximab (CTX) in colorectal cancer (CRC) J Clin Orthod. 2014;32:3518–3518. [Google Scholar]
- 62.Yaeger R, Cercek A, O’Reilly EM, Reidy DL, Kemeny N, Wolinsky T, Capanu M, Gollub MJ, Rosen N, Berger MF, Lacouture ME, Vakiani E, Saltz LB. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21:1313–1320. doi: 10.1158/1078-0432.CCR-14-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Corcoran RB, Atreya CE, Falchook GS, Kwak EL, Ryan DP, Bendell JC, Hamid O, Messersmith WA, Daud A, Kurzrock R, Pierobon M, Sun P, Cunningham E, Little S, Orford K, Motwani M, Bai Y, Patel K, Venook AP, Kopetz S. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600–Mutant Colorectal Cancer. J Clin Orthod. 2015;33:4023–4031. doi: 10.1200/JCO.2015.63.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pietrantonio F, Oddo D, Gloghini A, Valtorta E, Berenato R, Barault L, Caporale M, Busico A, Morano F, Gualeni AV, Alessi A, Siravegna G, Perrone F, Di Bartolomeo M, Bardelli A, de Braud F, Di Nicolantonio F. MET-Driven Resistance to Dual EGFR and BRAF Blockade May Be Overcome by Switching from EGFR to MET Inhibition in BRAF-Mutated Colorectal Cancer. Cancer Discov. 2016;6:963–971. doi: 10.1158/2159-8290.CD-16-0297. [DOI] [PubMed] [Google Scholar]
- 65.Wang X, Markowetz F, De Sousa E Melo F, Medema JP, Vermeulen L. Dissecting cancer heterogeneity–an unsupervised classification approach. Int J Biochem Cell Biol. 2013;45:2574–2579. doi: 10.1016/j.biocel.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 66.De Sousa E Melo F, Wang X, Jansen M, Fessler E, Trinh A, de Rooij LPMH, de Jong JH, de Boer OJ, van Leersum R, Bijlsma MF, Rodermond H, van der Heijden M, van Noesel CJM, Tuynman JB, Dekker E, Markowetz F, Medema JP, Vermeulen L. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med. 2013;19:614–618. doi: 10.1038/nm.3174. [DOI] [PubMed] [Google Scholar]
- 67.Budinska E, Popovici V, Tejpar S, D’Ario G, Lapique N, Sikora KO, Di Narzo AF, Yan P, Hodgson JG, Weinrich S, Bosman F, Roth A, Delorenzi M. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J Pathol. 2013;231:63–76. doi: 10.1002/path.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoshida Y. Nearest Template Prediction: A Single-Sample-Based Flexible Class Prediction with Confidence Assessment. PLoS One. 2010;5:e15543. doi: 10.1371/journal.pone.0015543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, Wong TW, Huang X, Takimoto CH, Godwin AK, Tan BR, Krishnamurthi SS, Burris HA, 3rd, Poplin EA, Hidalgo M, Baselga J, Clark EA, Mauro DJ. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25:3230–3237. doi: 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
- 70.Salazar R, Roepman P, Capella G, Moreno V, Simon I, Dreezen C, Lopez-Doriga A, Santos C, Marijnen C, Westerga J, Bruin S, Kerr D, Kuppen P, van de Velde C, Morreau H, Van Velthuysen L, Glas AM, Van’t Veer LJ, Tollenaar R. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J Clin Oncol. 2011;29:17–24. doi: 10.1200/JCO.2010.30.1077. [DOI] [PubMed] [Google Scholar]
- 71.Monti S, Tamayo P, Mesirov J, Golub T. Consensus Clustering: A Resampling-Based Method for Class Discovery and Visualization of Gene Expression Microarray Data. Mach Learn. 2003;52:91–118. [Google Scholar]
- 72.Brunet JP, Tamayo P, Golub TR, Mesirov JP. Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci U S A. 2004;101:4164–4169. doi: 10.1073/pnas.0308531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tibshirani R, Walther G, Hastie T. Estimating the number of clusters in a data set via the gap statistic. J R Stat Soc Series B Stat Methodol. 2001;63:411–423. [Google Scholar]
- 74.Roepman P, Schlicker A, Tabernero J, Majewski I, Tian S, Moreno V, Snel MH, Chresta CM, Rosenberg R, Nitsche U, Macarulla T, Capella G, Salazar R, Orphanides G, Wessels LFA, Bernards R, Simon IM. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int J Cancer. 2014;134:552–562. doi: 10.1002/ijc.28387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A. 2002;99:6567–6572. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Șenbabaoğlu Y, Michailidis G, Li JZ. Critical limitations of consensus clustering in class discovery. Sci Rep. 2014;4:6207. doi: 10.1038/srep06207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Santiago I, Yau C, Middleton M, Dustin M, Markowetz F, Sivakumar S. Immunophenotypes of Pancreatic Ductal Adenocarcinoma: Metaanalysis of transcriptional subtypes, bioRxiv. 2017:198903. doi: 10.1101/198903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shen R, Olshen AB, Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009;25:2906–2912. doi: 10.1093/bioinformatics/btp543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kirk P, Griffin JE, Savage RS, Ghahramani Z, Wild DL. Bayesian correlated clustering to integrate multiple datasets. Bioinformatics. 2012;28:3290–3297. doi: 10.1093/bioinformatics/bts595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang B, Mezlini AM, Demir F, Fiume M, Tu Z, Brudno M, Haibe-Kains B, Goldenberg A. Similarity network fusion for aggregating data types on a genomic scale. Nat Methods. 2014;11:333–337. doi: 10.1038/nmeth.2810. [DOI] [PubMed] [Google Scholar]
- 81.Sadanandam A, Wang X, de Sousa E Melo F, Gray JW, Vermeulen L, Hanahan D, Medema JP. Reconciliation of classification systems defining molecular subtypes of colorectal cancer: interrelationships and clinical implications. Cell Cycle. 2014;13:353–357. doi: 10.4161/cc.27769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Alderdice M, Richman SD, Gollins S, Stewart P, Hurt C, Adams R, McCorry A, Roddy A, Vimalachandran D, Isella C, Medico E, Maughan T, McArt DG, Lawler M, Dunne PD. Prospective patient stratification into robust cancer-cell intrinsic subtypes from colorectal cancer biopsies. J Pathol. 2018 doi: 10.1002/path.5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cantini L, Isella C, Petti C, Picco G, Chiola S, Ficarra E, Caselle M, Medico E. MicroRNA-mRNA interactions underlying colorectal cancer molecular subtypes. Nat Commun. 2015;6:8878. doi: 10.1038/ncomms9878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fessler E, Jansen M, De Sousa E Melo F, Zhao L, Prasetyanti PR, Rodermond H, Kandimalla R, Linnekamp JF, Franitza M, van Hooff SR, de Jong JH, Oppeneer SC, van Noesel CJM, Dekker E, Stassi G, Wang X, Medema JP, Vermeulen L. A multidimensional network approach reveals microRNAs as determinants of the mesenchymal colorectal cancer subtype. Oncogene. 2016;35:6026–6037. doi: 10.1038/onc.2016.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10:472–484. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- 87.Witwer KW. Circulating microRNA biomarker studies: pitfalls and potential solutions. Clin Chem. 2015;61:56–63. doi: 10.1373/clinchem.2014.221341. [DOI] [PubMed] [Google Scholar]
- 88.Schwarzenbach H, Nishida N, Calin GA, Pantel K. Clinical relevance of circulating cell-free microRNAs in cancer. Nat Rev Clin Oncol. 2014;11:145–156. doi: 10.1038/nrclinonc.2014.5. [DOI] [PubMed] [Google Scholar]
- 89.Diaz LA, Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32:579–586. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cai X, Janku F, Zhan Q, Fan JB. Accessing Genetic Information with Liquid Biopsies. Trends Genet. 2015;31:564–575. doi: 10.1016/j.tig.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 91.Ragulan C, Eason K, Nyamundanda G, Patil Y, Poudel P, Fontana E, Del Rio M, Koo SL, Tan WS, Martineau P, Cunningham D, Tan I, Sadanandam A. A Low-Cost Multiplex Biomarker Assay Stratifies Colorectal Cancer Patient Samples into Clinically-Relevant Subtypes, bioRxiv. 2017:174847. doi: 10.1101/174847. [DOI] [Google Scholar]
- 92.Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, Davies SR, Wang S, Wang P, Kinsinger CR, Rivers RC, Rodriguez H, Townsend RR, Ellis MJC, Carr SA, Tabb DL, Coffey RJ, Slebos RJC, Liebler DC. NCI CPTAC, Proteogenomic characterization of human colon and rectal cancer. Nature. 2014;513:382–387. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang J, Mouradov D, Wang X, Jorissen RN, Chambers MC, Zimmerman LJ, Vasaikar S, Love CG, Li S, Lowes K, Leuchowius KJ, Jousset H, Weinstock J, Yau C, Mariadason J, Shi Z, Ban Y, Chen X, Coffey RJC, Slebos RJC, Burgess AW, Liebler DC, Zhang B, Sieber OM. Colorectal Cancer Cell Line Proteomes Are Representative of Primary Tumors and Predict Drug Sensitivity. Gastroenterology. 2017;153:1082–1095. doi: 10.1053/j.gastro.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bardhan K, Liu K. Epigenetics and colorectal cancer pathogenesis. Cancers. 2013;5:676–713. doi: 10.3390/cancers5020676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang S, Chaudhary K, Garmire LX. More Is Better: Recent Progress in Multi-Omics Data Integration Methods. Front Genet. 2017;8:84. doi: 10.3389/fgene.2017.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vucic EA, Thu KL, Robison K, Rybaczyk LA, Chari R, Alvarez CE, Lam WL. Translating cancer “omics” to improved outcomes. Genome Res. 2012;22:188–195. doi: 10.1101/gr.124354.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Berg KCG, Eide PW, Eilertsen IA, Johannessen B, Bruun J, Danielsen SA, Bjørnslett M, Meza-Zepeda LA, Eknæs M, Lind GE, Myklebost O, Skotheim RI, Sveen A, Lothe RA. Multi-omics of 34 colorectal cancer cell lines - a resource for biomedical studies. Mol Cancer. 2017;16:116. doi: 10.1186/s12943-017-0691-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bersanelli M, Mosca E, Remondini D, Giampieri E, Sala C, Castellani G, Milanesi L. Methods for the integration of multi-omics data: mathematical aspects. BMC Bioinformatics. 2016;17(Suppl 2):15. doi: 10.1186/s12859-015-0857-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang H, Zheng H, Wang J, Wang C, Wu F. Integrating Omic Data with a Multiplex Network-based Approach for the Identification of Cancer Subtypes. IEEE Trans Nanobioscience. 2016 doi: 10.1109/TNB.2016.2556640. [DOI] [PubMed] [Google Scholar]
- 100.Zhang Z, Huang K, Gu C, Zhao L, Wang N, Wang X, Zhao D, Zhang C, Lu Y, Meng Y. Molecular Subtyping of Serous Ovarian Cancer Based on Multi-omics Data. Sci Rep. 2016;6:26001. doi: 10.1038/srep26001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, Hinoue T, Laird PW, Hoadley KA, Akbani R, Castro MAA, Gibb EA, Kanchi RS, Gordenin DA, Shukla SA, Sanchez-Vega F, Hansel DE, Czerniak BA, Reuter VE, Su X, Carvalho B de Sa, Chagas VS, Mungall KL, Sadeghi S, Pedamallu CS, Lu Y, Klimczak LJ, Zhang J, Choo C, Ojesina AI, Bullman S, Leraas KM, Lichtenberg TM, Wu CJ, Schultz N, Getz G, Meyerson M, Mills GB, McConkey DJ, TCGA Research Network. Weinstein JN, Kwiatkowski DJ, Lerner SP. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. 2017;171:540–556.e25. doi: 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Haury AC, Gestraud P, Vert JP. The Influence of Feature Selection Methods on Accuracy, Stability and Interpretability of Molecular Signatures. PLoS One. 2011;6:e28210. doi: 10.1371/journal.pone.0028210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ein-Dor L, Kela I, Getz G, Givol D, Domany E. Outcome signature genes in breast cancer: is there a unique set? Bioinformatics. 2005;21:171–178. doi: 10.1093/bioinformatics/bth469. [DOI] [PubMed] [Google Scholar]
- 104.Drier Y, Domany E. Do Two Machine-Learning Based Prognostic Signatures for Breast Cancer Capture the Same Biological Processes? PLoS One. 2011;6:e17795. doi: 10.1371/journal.pone.0017795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 106.Lazar C, Meganck S, Taminau J, Steenhoff D, Coletta A, Molter C, Weiss-Solís DY, Duque R, Bersini H, Nowé A. Batch effect removal methods for microarray gene expression data integration: a survey. Brief Bioinform. 2013;14:469–490. doi: 10.1093/bib/bbs037. [DOI] [PubMed] [Google Scholar]
- 107.Kim BC, Sung YS, Suk HI. Deep feature learning for pulmonary nodule classification in a lung CT, in: 2016 4th International Winter Conference on Brain-Computer Interface (BCI), 2016: pp. 1–3 [Google Scholar]
- 108.Xu Y, Jia Z, Wang LB, Ai Y, Zhang F, Lai M, Chang EIC. Large scale tissue histopathology image classification, segmentation, and visualization via deep convolutional activation features. BMC Bioinformatics. 2017;18:281. doi: 10.1186/s12859-017-1685-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen Y, Li Y, Narayan R, Subramanian A, Xie X. Gene expression inference with deep learning. Bioinformatics. 2016;32:1832–1839. doi: 10.1093/bioinformatics/btw074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Singh R, Lanchantin J, Robins G, Qi Y. DeepChrome: deep-learning for predicting gene expression from histone modifications. Bioinformatics. 2016;32:i639–i648. doi: 10.1093/bioinformatics/btw427. [DOI] [PubMed] [Google Scholar]
- 111.Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods. 2015;12:931–934. doi: 10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Karabulut EM, Ibrikci T. Biomedical Research, 2017, Discriminative deep belief networks for microarray based cancer classification, Alliedacademies.org 2017. http://www.alliedacademies.org/articles/discriminative-deep-belief-networks-for-microarray-based-cancer-classification.html.
- 113.Ibrahim R, Yousri NA, Ismail MA, El-Makky NM. Multi-level gene/MiRNA feature selection using deep belief nets and active learning. Conf Proc IEEE Eng Med Biol Soc. 2014;2014:3957–3960. doi: 10.1109/EMBC.2014.6944490. [DOI] [PubMed] [Google Scholar]
- 114.Fessler E, Jansen M, De Sousa E Melo F, Zhao L, Prasetyanti PR, Rodermond H, Kandimalla R, Linnekamp JF, Franitza M, van Hooff SR, de Jong JH, Oppeneer SC, van Noesel CJM, Dekker E, Stassi G, Wang X, Medema JP, Vermeulen L. A multidimensional network approach reveals microRNAs as determinants of the mesenchymal colorectal cancer subtype. Oncogene. 2016;35:6026–6037. doi: 10.1038/onc.2016.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, Mellano A, Senetta R, Cassenti A, Sonetto C, Inghirami G, Trusolino L, Fekete Z, De Ridder M, Cassoni P, Storme G, Bertotti A, Medico E. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. 2015;47:312–319. doi: 10.1038/ng.3224. [DOI] [PubMed] [Google Scholar]
- 116.Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, Sevillano M, Palomo-Ponce S, Tauriello DVF, Byrom D, Cortina C, Morral C, Barceló C, Tosi S, Riera A, Attolini CSO, Rossell D, Sancho E, Batlle E. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet. 2015;47:320–329. doi: 10.1038/ng.3225. [DOI] [PubMed] [Google Scholar]
- 117.Linnekamp JF, van Hooff SR, Prasetyanti PR, Kandimalla R, Buikhuisen JY, Fessler E, Ramesh P, Lee KAST, Bochove GGW, de Jong JH, Cameron K, van Leersum R, Rodermond HM, Franitza M, Nürnberg P, Mangiapane LR, Wang X, Clevers H, Vermeulen L, Stassi G, Medema JP. Consensus molecular subtypes of colorectal cancer are recapitulated in in vitro and in vivo models. Cell Death Differ. 2018;25:616–633. doi: 10.1038/s41418-017-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Isella C, Brundu F, Bellomo SE, Galimi F, Zanella E, Porporato R, Petti C, Fiori A, Orzan F, Senetta R, Boccaccio C, Ficarra E, Marchionni L, Trusolino L, Medico E, Bertotti A. Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nat Commun. 2017;8:15107. doi: 10.1038/ncomms15107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ha G, Roth A, Khattra J, Ho J, Yap D, Prentice LM, Melnyk N, McPherson A, Bashashati A, Laks E, Biele J, Ding J, Le A, Rosner J, Shumansky K, Marra MA, Gilks CB, Huntsman DG, McAlpine JN, Aparicio S, Shah SP. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res. 2014;24:1881–1893. doi: 10.1101/gr.180281.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Miller CA, White BS, Dees ND, Griffith M, Welch JS, Griffith OL, Vij R, Tomasson MH, Graubert TA, Walter MJ, Ellis MJ, Schierding W, DiPersio JF, Ley TJ, Mardis ER, Wilson RK, Ding L. SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput Biol. 2014;10:e1003665. doi: 10.1371/journal.pcbi.1003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zare H, Wang J, Hu A, Weber K, Smith J, Nickerson D, Song C, Witten D, Blau CA, Noble WS. Inferring clonal composition from multiple sections of a breast cancer. PLoS Comput Biol. 2014;10:e1003703. doi: 10.1371/journal.pcbi.1003703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, Kong SL, Chua C, Hon LK, Tan WS, Wong M, Choi PJ, Wee LJK, Hillmer AM, Tan IB, Robson P, Prabhakar S. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet. 2017;49:708–718. doi: 10.1038/ng.3818. [DOI] [PubMed] [Google Scholar]
- 123.Roerink SF, Sasaki N, Lee-Six H, Young MD, Alexandrov LB, Behjati S, Mitchell TJ, Grossmann S, Lightfoot H, Egan DA, Pronk A, Smakman N, van Gorp J, Anderson E, Gamble SJ, Alder C, van de Wetering M, Campbell PJ, Stratton MR, Clevers H. Intra-tumour diversification in colorectal cancer at the single-cell level. Nature. 2018;556:457–462. doi: 10.1038/s41586-018-0024-3. [DOI] [PubMed] [Google Scholar]
- 124.Shen-Orr SS, Gaujoux R. Computational deconvolution: extracting cell type-specific information from heterogeneous samples. Curr Opin Immunol. 2013;25:571–578. doi: 10.1016/j.coi.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, Carter SL, Getz G, Stemke-Hale K, Mills GB, Verhaak RGW. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, Selves J, Laurent-Puig P, Sautès-Fridman C, Fridman WH, de Reyniès A. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218. doi: 10.1186/s13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SGH, Hoadley KA, Rashid NU, Williams LA, Eaton SC, Chung AH, Smyla JK, Anderson JM, Kim HJ, Bentrem DJ, Talamonti MS, Iacobuzio-Donahue CA, Hollingsworth MA, Yeh JJ. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47:1168–1178. doi: 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Becht E, de Reyniès A, Giraldo NA, Pilati C, Buttard B, Lacroix L, Selves J, Sautès-Fridman C, Laurent-Puig P, Fridman WH. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin Cancer Res. 2016;22:4057–4066. doi: 10.1158/1078-0432.CCR-15-2879. [DOI] [PubMed] [Google Scholar]
- 130.Kreso A, O’Brien CA, Galen P van, Gan OI, Notta F, Brown AMK, Ng K, Ma J, Wienholds E, Dunant C, Pollett A, Gallinger S, McPherson J, Mullighan CG, Shibata D, Dick JE. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339:543–548. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vermaat JS, Nijman IJ, Koudijs MJ, Gerritse FL, Scherer SJ, Mokry M, Roessingh WM, Lansu N, de Bruijn E, van Hillegersberg R, van Diest PJ, Cuppen E, Voest EE. Primary colorectal cancers and their subsequent hepatic metastases are genetically different: implications for selection of patients for targeted treatment. Clin Cancer Res. 2012;18:688–699. doi: 10.1158/1078-0432.CCR-11-1965. [DOI] [PubMed] [Google Scholar]
- 132.Lee SY, Haq F, Kim D, Jun C, Jo HJ, Ahn SM, Lee WS. Comparative genomic analysis of primary and synchronous metastatic colorectal cancers. PLoS One. 2014;9:e90459. doi: 10.1371/journal.pone.0090459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Trinh A, Trumpi K, De Sousa E Melo F, Wang X, de Jong JH, Fessler E, Kuppen PJK, Reimers MS, Swets M, Koopman M, Nagtegaal ID, Jansen M, Hooijer GKJ, Offerhaus GJA, Kranenburg O, Punt CJ, Medema JP, Markowetz F, Vermeulen L. Practical and Robust Identification of Molecular Subtypes in Colorectal Cancer by Immunohistochemistry. Clin Cancer Res. 2017;23:387–398. doi: 10.1158/1078-0432.CCR-16-0680. [DOI] [PubMed] [Google Scholar]
- 134.Ubink I, Elias SG, Moelans CB, Laclé MM, van Grevenstein WMU, van Diest PJ, Borel Rinkes IHM, Kranenburg O. A Novel Diagnostic Tool for Selecting Patients With Mesenchymal-Type Colon Cancer Reveals Intratumor Subtype Heterogeneity. J Natl Cancer Inst. 2017;109 doi: 10.1093/jnci/djw303. [DOI] [PubMed] [Google Scholar]
- 135.Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, James JJ, Maysuria M, Mitton JD, Oliveri P, Osborn JL, Peng T, Ratcliffe AL, Webster PJ, Davidson EH, Hood L, Dimitrov K. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 136.Lee BI, Oades K, Vo L, Lee J, Landers M, Wang Y, Monforte J. NGS-based targeted RNA sequencing for expression analysis of patients with triple-negative breast cancer using a modulized, 96-gene biomarker panel. J Clin Orthod. 2012;30:56–56. [Google Scholar]
- 137.Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol. 2017;14:531–548. doi: 10.1038/nrclinonc.2017.14. [DOI] [PubMed] [Google Scholar]
- 138.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, Liu Q, Iorio F, Surdez D, Chen L, Milano RJ, Bignell GR, Tam AT, Davies H, Stevenson JA, Barthorpe S, Lutz SR, Kogera F, Lawrence K, McLaren-Douglas A, Mitropoulos X, Mironenko T, Thi H, Richardson L, Zhou W, Jewitt F, Zhang T, O’Brien P, Boisvert JL, Price S, Hur W, Yang W, Deng X, Butler A, Choi HG, Chang JW, Baselga J, Stamenkovic I, Engelman JA, Sharma SV, Delattre O, Saez-Rodriguez J, Gray NS, Settleman J, Futreal PA, Haber DA, Stratton MR, Ramaswamy S, McDermott U, Benes CH. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P, Jr, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Julien S, Merino-Trigo A, Lacroix L, Pocard M, Goéré D, Mariani P, Landron S, Bigot L, Nemati F, Dartigues P, Weiswald LB, Lantuas D, Morgand L, Pham E, Gonin P, Dangles-Marie V, Job B, Dessen P, Bruno A, Pierré A, De Thé H, Soliman H, Nunes M, Lardier G, Calvet L, Demers B, Prévost G, Vrignaud P, Roman-Roman S, Duchamp O, Berthet C. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res. 2012;18:5314–5328. doi: 10.1158/1078-0432.CCR-12-0372. [DOI] [PubMed] [Google Scholar]
- 141.Sveen A, Bruun J, Eide PW, Eilertsen IA, Ramirez L, Murumägi A, Arjama M, Danielsen SA, Kryeziu K, Elez E, Tabernero J, Guinney J, Palmer HG, Nesbakken A, Kallioniemi O, Dienstmann R, Lothe RA. Colorectal Cancer Consensus Molecular Subtypes Translated to Preclinical Models Uncover Potentially Targetable Cancer Cell Dependencies. Clin Cancer Res. 2018;24:794–806. doi: 10.1158/1078-0432.CCR-17-1234. [DOI] [PubMed] [Google Scholar]
- 142.Fessler E, Drost J, van Hooff SR, Linnekamp JF, Wang X, Jansen M, De Sousa E Melo F, Prasetyanti PR, IJspeert JE, Franitza M, Nürnberg P, van Noesel CJ, Dekker E, Vermeulen L, Clevers H, Medema JP. TGFβ signaling directs serrated adenomas to the mesenchymal colorectal cancer subtype. EMBO Mol Med. 2016;8:745–760. doi: 10.15252/emmm.201606184. [DOI] [PMC free article] [PubMed] [Google Scholar]