

Abstract
Androgens such as testosterone and dihydrotestosterone are a critical driver of prostate cancer progression. Cancer resistance to androgen deprivation therapies ensues when tumors engage metabolic processes that produce sustained androgen levels in the tissue. However, the molecular mechanisms involved in this resistance process are unclear, and functional imaging modalities that predict impending resistance are lacking. Here, using the human LNCaP and C4-2 cell line models of prostate cancer, we show that castration treatment–sensitive prostate cancer cells that normally have an intact glucuronidation pathway that rapidly conjugates and inactivates dihydrotestosterone and thereby limits androgen signaling, become glucuronidation deficient and resistant to androgen deprivation. Mechanistically, using CRISPR/Cas9-mediated gene ablation, we found that loss of UDP glucuronosyltransferase family 2 member B15 (UGT2B15) and UGT2B17 is sufficient to restore free dihydrotestosterone, sustained androgen signaling, and development of castration resistance. Furthermore, loss of glucuronidation enzymatic activity was also detectable with a nonsteroid glucuronidation substrate. Of note, glucuronidation-incompetent cells and the resultant loss of intracellular conjugated dihydrotestosterone were detectable in vivo by 18F-dihydrotestosterone PET. Together, these findings couple a mechanism with a functional imaging modality to identify impending castration resistance in prostate cancers.
Keywords: steroid, steroid hormone receptor, steroidogenesis, metabolism, metabolic regulation, androgen, uridine 5'-diphospho-glucuronosyltransferase (UDP-glucuronosyltransferase), castration-resistant prostate cancer, nuclear medicine, PET, prostate cancer
Introduction
The long-standing standard treatment for advanced prostate cancer is androgen deprivation therapy by way of medical or surgical castration (1). Therapeutic responses usually occur and tumors eventually develop resistance as castration-resistant prostate cancer (CRPC) 2 (2, 3). This resistant state is frequently first heralded by a rising prostate-specific antigen (PSA), which is an androgen-responsive gene and is indicative of a reinstatement of androgen receptor (AR) stimulation (4). CRPC is also accompanied by tumors developing the metabolic capability of regenerating their own potent androgens, i.e. testosterone and/or dihydrotestosterone (DHT), from extragonadal precursor steroids (5–7). The role and requirement for extragonadal androgen synthesis in advanced prostate cancer was recently bolstered by a profound prolongation of survival in clinical trials of patients treated upfront with extragonadal androgen synthesis inhibition in addition to castration (8, 9).
The metabolic mechanisms that enable sustained tissue concentrations of potent androgens in CRPC that occurs alongside the absence of gonadal testosterone in circulation remain poorly understood. Furthermore, imaging for prostate cancer is very limited and mainly focused on tumor localization. There is a complete absence of functional imaging approaches that are informative of the metabolic state of the disease that provides information on tumor susceptibility to systemic therapies in the clinical standard of care.
Glucuronidation is a major mechanism of androgen inactivation that normally limits AR stimulation and downstream signaling in peripheral tissues, including prostatic tissues (10). The 17β-OH group of testosterone and DHT make them substrates for UDP-glucuronosyltransferase (UGT) enzymes to form their respective inactivated glucuronide metabolites. In humans, testosterone is glucuronidated primarily by UGT2B7 and -17, and DHT is glucuronidated by UGT2B7, -15, and -17 (11, 12). We show here that a profound metabolic switch marked by loss of glucuronidation enzymatic activity occurs in CRPC, protects intratumoral testosterone and/or DHT from its normal mechanism of tissue inactivation, and allows androgens from extragonadal origins to be sustained, thus enabling persistent AR signaling. We also show that positron-emission tomography (PET) imaging can be used to distinguish glucuronidation proficient tumors from their glucuronidation incompetent and androgen deprivation-resistant descendants.
Results
Loss of glucuronidation activity occurs with a classical model of “androgen independence”
The LNCaP human cell line model of prostate cancer and its C4-2 derivative, which was developed as a resistant model by repeatedly propagating LNCaP through castrated mice (13), probably represent the most widely utilized human matched models of castration-sensitive (formerly known as “androgen-sensitive”) and castration-resistant (formerly known as “androgen-independent”) prostate cancer. Development of resistance by way of a truly androgen-independent mechanism would be expected to occur without the acquisition of an androgen-sustaining metabolic phenotype. In contrast, we found that although the LNCaP model normally rapidly inactivates DHT by glucuronidation, the C4-2 model has completely lost this enzymatic activity. For example, 3 h after DHT treatment, all DHT in media is glucuronidated in the LNCaP model and all detectable DHT remains free in the C4-2 model (Fig. 1A). This results in a marked prolongation of cellular exposure to DHT in media (Fig. 1B) and as well as in the intracellular space (Fig. 1C). UGT2B15 and UGT2B17 are the 2 major enzymes that are responsible for androgen glucuronidation in prostate cancer (10, 14). The transition from LNCaP to the resistant C4-2 model is accompanied by a profound loss of expression of the glucuronidation machinery (Fig. 1, D and E), which accounts for the apparent switch in metabolic phenotype (Fig. 1A).
Figure 1.
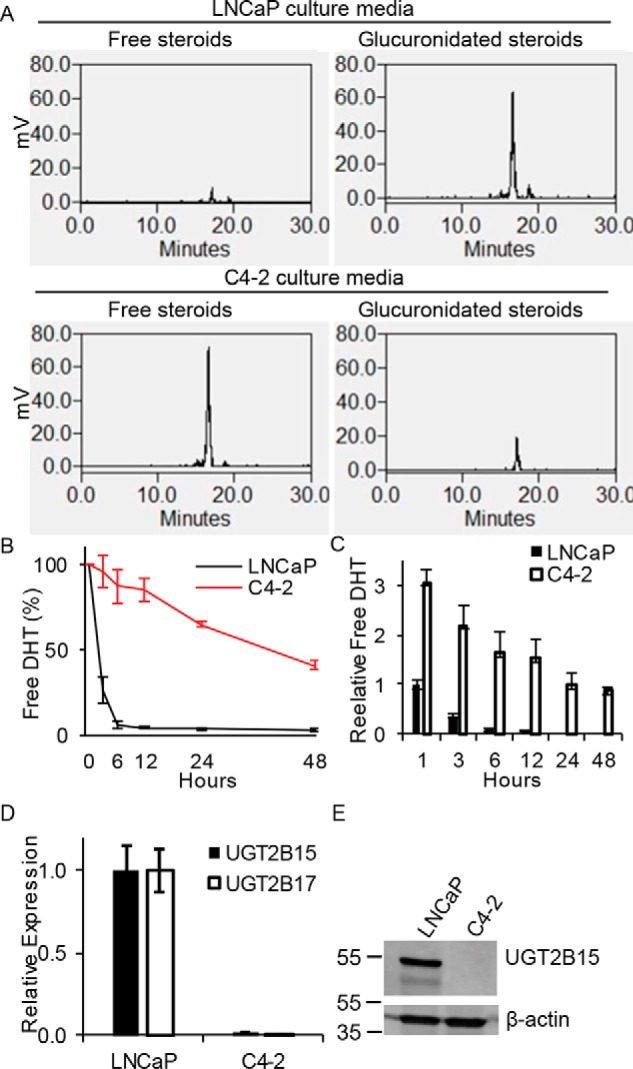
Development of CRPC is accompanied by a switch from androgen-glucuronidation competency to a glucuronidation-deficient metabolic phenotype and persistence of free DHT. A, the LNCaP prostate cancer cell line rapidly glucuronidates DHT, whereas C4-2, the castration-resistant derivative cell line, is deficient in glucuronidation activity. Cells were treated with [3H]DHT (10 nm) for 3 h, steroids were extracted from media, and analyzed by HPLC. Free DHT is quickly lost in the (B) extracellular and (C) intracellular space from incubations with LNCaP cells and in contrast has prolonged persistence in castration-resistant C4-2 cells. Cells were treated with [3H]DHT (10 nm) for the indicated incubation times, steroids were extracted, and analyzed by HPLC. Experiments in A–C were repeated 3 times and error bars represent the S.D. D, UGT2B15 and UGT2B17 transcript is lost in C4-2 cells. Expression was assessed by qRT-PCR in triplicate and normalized to RPLP0 and expression in LNCaP and error bars represent the S.D. E, UGT2B15 protein is lost in C4-2 cells. Experiments in D and E were repeated 3 times.
Glucuronidation deficiency and the metabolic phenotype that sustains active DHT is experimentally inducible with castration
To determine whether the loss of glucuronidation activity that occurs in the C4-2 model that allows the persistence of free DHT is attributable to the development of castration-resistance, LNCaP xenografts were grown in eugonadal or castrated mice and xenograft tissues were interrogated for DHT glucuronidation activity (Fig. 2A). A subset of these tumors lose the capability of rapidly inactivating DHT, which otherwise consistently occurs with LNCaP xenografts grown in eugonadal mice. Expression of UGT2B15 protein was assessed and is lost in tumors with metabolic phenotypes that permit high free DHT compared with those that restrict DHT maintenance (Fig. 2B). A tumor subclone was grown from a xenograft selected for the absence of an ability to inactivate free DHT (Fig. 2A) and stably displayed glucuronidation incompetence (Fig. 2C) that is attributable to loss of UGT2B15 and UGT2B17 expression (Fig. 2D). Together, these results suggest that coordinate UGT2B15 and UGT2B17 loss commonly occurs in the transition to CRPC.
Figure 2.
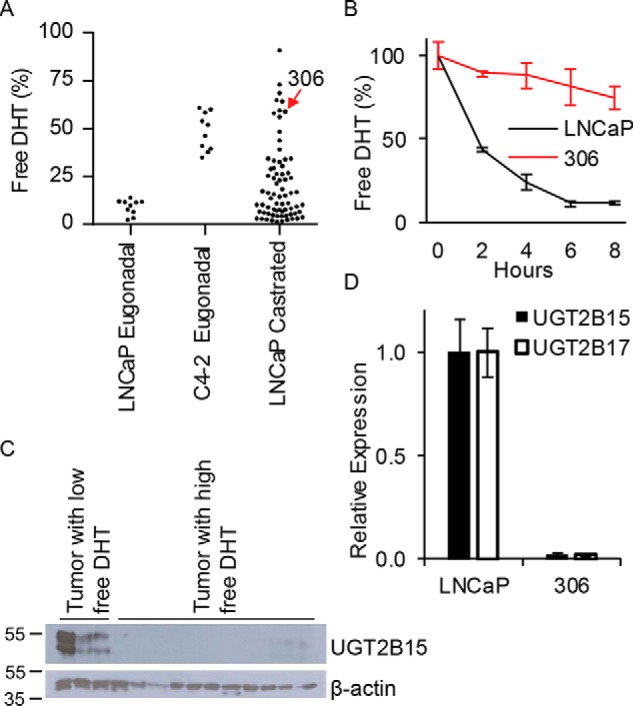
Prostate cancer growth in castrated mice selects for clones that lose expression of enzymes required for DHT glucuronidation. A, sustained LNCaP growth in castrated mice leads to development of subclones that sustain free DHT. LNCaP and C4-2 xenografts were grown in eugonadal mice. LNCaP xenografts were separately passaged in castrated mice over 14 months, 40 mg of xenograft tissue was incubated with [3H]DHT for 4 h and free DHT was assessed by HPLC. B, tumors acquiring the capability of sustaining free DHT lose UGT2B15 protein expression. Immunoblot was performed in proteins extracted from tumors that sustained low (left) or high (right) free DHT. C, a subcultured cell line derived from LNCaP grown in castrated mice gains the capability of sustaining free DHT. Experiments were repeated 3 times and error bars represent the standard deviation. D, a subcultured cell line gaining the ability to sustain free DHT does so by abolishing expression of UGT2B15 and UGT2B17. Expression was assessed by qRT-PCR in triplicate and normalized to RPLP0 and expression in LNCaP. Experiments were repeated 3 times.
Genetic ablation of UGT2B15 and UGT2B17 is sufficient to allow androgen persistence in cells treated directly or with precursors of testosterone and DHT
To determine whether the androgen-glucuronidation incompetence that occurs in the transition from LNCaP to C4-2 cells is attributable to loss of UGT2B15 and UGT2B17, CRISPR-Cas was employed to genetically ablate these two genes, resulting in loss of transcript and protein expression (Fig. 3, A and B). Loss of UGT2B15 and UGT2B17 in LNCaP is sufficient to block glucuronidation and maintain persistence of free testosterone and DHT in media to an extent that is comparable with that which occurs in C4-2 cells (Fig. 3C). UGT2B15 and UGT2B17 loss also enables sustained intracellular free DHT, as assessed with radioactive signal emitted from [3H]DHT (Fig. 3D) and with the DHT concentration assessed by LC tandem-MS (LC-MS/MS; Fig. 3E). In contrast, glucuronidated androgens predominate to a much greater extent in Ctrl compared with UGT2B15 and UGT2B17 knockout cells (Fig. S1). Furthermore, the concentration of intracellular testosterone and DHT is also augmented in cells that are UGT2B15 and UGT2B17 deficient when treated with DHEA, a major adrenal precursor steroid that is converted to more potent androgens by steroidogenic enzymes (Fig. 3F).
Figure 3.
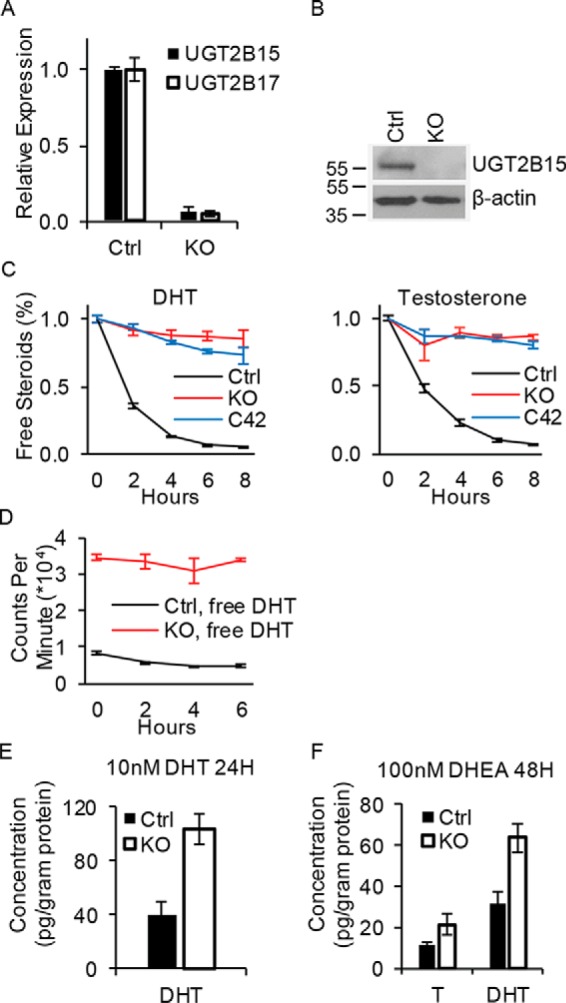
UGT2B15 and UGT2B17 double deletion permits sustained levels of free and active androgens. A and B, CRISPR-Cas knockout ablates expression of UGT2B15 and UGT2B17 mRNA (A) and (B) protein. Transcript expression by qRT-PCR was assessed in triplicate and normalized to RPLP0 and expression in Ctrl cells. Experiments were repeated 3 times and error bars represent the S.D. C, UGT2B15 and UGT2B17 KO in LNCaP reverses rapid loss of free DHT and testosterone that otherwise occurs in Ctrl LNCaP and is comparable with persistence of free androgens in C4-2 cells. Cells were treated with [3H]DHT (10 nm) or [3H]testosterone (100 nm) for the indicated incubation times, steroids were extracted from media and analyzed by HPLC in triplicate. D, intracellular free [3H]DHT is sustained in glucuronidation-deficient cells. Ctrl and KO cells were treated with [3H]DHT (10 nm) and the radioactive signal was assessed in the organic extract from treated cells in triplicate. E and F, UGT2B15 and UGT2B17 ablation allows for sustained free intracellular DHT with direct treatment (10 nm) (E) and sustains free intracellular DHT and testosterone (T) when treated with the adrenal precursor steroid DHEA (100 nm) (F). Cells were treated for the indicated incubation time in triplicate and intracellular steroids were quantitated by MS. Experiments were repeated three times in E and twice in F. Error bars represent the S.D. in C–F.
Absence of DHT inactivation machinery spurs sustained AR signaling, cell proliferation, and more rapid development of CRPC
Castration-resistant growth of C4-2 cells is generally thought to be androgen independent (15–18). We sought to determine whether the loss of androgen-glucuronidation activity in this model allows for exaggerated and more sustainable responses to a single treatment with a low concentration of DHT that would provide an alternative explanation to androgen-independent mechanisms. C4-2 cells treated with DHT (10 nm) indeed have sustained induction of DHT-stimulated androgen-responsive gene expression (PSA, FKBP5, and TMPRSS2) compared with the original LNCaP model (Fig. 4A). Genetic elimination of UGT2B15 and UGT2B17 expression from LNCaP recapitulates sustained induction of androgen-regulated transcripts relative to expression in glucuronidation-competent cells (Fig. 4B) and results in augmented responses to direct treatment over a concentration range of DHT (Fig. 4C) as well as the adrenal precursor DHEA (Fig. 4D). Unimpeded exposure to DHEA, testosterone, and DHT, due to loss of glucuronidation activity enables rapid cell proliferation (Fig. 4E) and hastened development of CRPC in vivo (Fig. 4F). Reinstatement of glucuronidation activity reverses prostate cancer accessibility to sustained active DHT (Fig. 4G) and restricts castration-resistant growth (Fig. 4H).
Figure 4.

Genetic loss of UGT2B15 and UGT2B17 promotes sustained androgen-responsive gene expression, cell proliferation, and promotes CRPC growth. A, castration-resistant C4-2 cells have persistent induction of androgen-responsive gene expression with DHT stimulation. Cells were treated with CSS media for 72 h, treated with DHT (10 nm) for the designated incubation times, and expression of PSA, FKBP5, and TMPRSS2 was assessed in triplicate by qPCR and normalized to RPLPO. Compared with WT LNCaP (Ctrl), combined loss of UGT2B15 and UGT2B17 (LNCaP KO) recapitulates sustained androgen-responsive gene expression with (B) DHT (10 nm) treatment over time and in (C) DHT and (D) DHEA concentration-dependent manners. Expression of PSA, FKBP5, and TMPRSS2 was assessed in triplicate by qPCR and normalized to RPLPO and vehicle treatment. In B, expression in KO cells is also normalized to Ctrl cells. Error bars represent the S.D. E, loss of UGT2B15 and UGT2B17 augments androgen-responsive cell proliferation. Cells were treated with vehicle, DHEA (100 nm), testosterone (100 nm), and DHT (10 nm), in triplicate, and counted after treatment for the indicated number of days. F, castration-resistant growth in vivo is hastened in KO (n = 10 mice) compared with Ctrl (n = 10) tumors (p = 0.0002). Three days after subcutaneously injecting cells into NSG mice, mice were surgically orchiectomized and implanted with DHEA pellets to mimic human adrenal physiology. The significance of the difference in progression-free survival between groups, as assessed by tumor volume >300 mm3, was assessed by log rank test. G, UGT2B15 expression in C4-2 cells reinstates enzymatic DHT glucuronidation. C4-2 cells were stably transfected with control vector (Ctrl) or UGT2B15 (2B15) and expression was assessed by immunoblot. Next, cells were treated with [3H]DHT (10 nm) for 6 h and free and glucuronidated DHT was assessed by HPLC in triplicate. Error bars represent the S.D. H, restoration of DHT glucuronidation activity suppresses castration-resistant xenograft growth. Ctrl and 2B15 cells (n = 7 mice per group) were subcutaneously injected in orchiectomized mice, progression-free survival was assessed as tumor volume >300 mm3, and the significance of the difference between groups was compared with log rank test; p = 0.0035.
Tumors permissive for unimpeded androgen exposure are noninvasively distinguishable with PET imaging using 18F-DHT
We set out to explore alternative approaches that might be feasible for noninvasively distinguishing between androgen-glucuronidation-proficient and androgen-glucuronidation-deficient tumors by exploiting changes in the balance between free versus conjugated substrates of glucuronidation. We reasoned that under certain circumstances, nonsteroid substrates of glucuronidation might be advantageous as alternatives to steroid substrates. SCH23390 is a dopamine D1 receptor antagonist that is also a substrate for hepatic glucuronidation (19). Our results demonstrate that [3H]SCH23390 is clearly glucuronidated by prostate cancer cells, although it appears to be a less effective substrate compared with DHT (Fig. 5A). Similar to androgen glucuronidation, SCH23390 glucuronidation is genetically reversible with loss of UGT2B15 and UGT2B17 (Fig. 5B). However, our attempts to image prostate cancer xenografts with [11C]SCH23390 resulted in very little tumor radiotracer uptake. Next, we reasoned that a pulse-chase approach using [3H]DHT would mimic in vivo conditions for PET imaging with 18F-DHT. [3H]DHT treatment followed by a chase yields higher total radioactivity in glucuronidation-proficient cells compared with glucuronidation-deficient cells that is greatest at earliest time points and diminishes over the washout time (Fig. 5C). Cellular radioactivity residing in the aqueous (glucuronidated) and organic (free) phases of steroid extraction demonstrate that the increase in cellular signal in the glucuronidation-proficient cells is attributable to glucuronidated DHT (Fig. 5D). It should be noted that these data differ from Fig. 3D, which is instead done with continuous DHT treatment, as opposed to a pulse-chase approach. To determine whether the intracellular glucuronidated fraction could be further selectively increased in glucuronidation-competent cells, we performed similar experiments with cyclosporin A (CSA) co-treatment, which is known to inhibit membrane transporters that normally export intracellular substrates and would therefore be expected to increase cellular glucuronidated DHT retention (20). CSA selectively increases radioactivity attributable to [3H]DHT in cells with intact glucuronidation activity (Fig. 5E) and only augments the glucuronidated DHT fraction without any change in intracellular free DHT (Fig. S2). Finally, we sought to determine whether preferential retention is demonstrable in glucuronidation-competent prostate cancer with 18F-DHT and PET imaging. Matched xenografts that are glucuronidation-competent (Ctrl) and UGT2B15/UGT2B17-defective LNCaP (KO) were grown in mice and treated with CSA immediately prior to administration of 18F-DHT and microPET-CT scanning (Fig. 5F). Control tumors demonstrated higher maximum tracer uptake and more rapid washout compared with KO tumors. There was no difference in AR protein expression between Ctrl and KO cells, supporting a model in which the increase in 18F-DHT uptake in Ctrl tumors occurs independent of AR content (Fig. S3). Together, these results demonstrate that the glucuronidation-deficient metabolic phenotype, which occurs in the transition to castration resistance is detectable with a timed PET imaging approach that assesses the tumor's functional metabolic status.
Figure 5.

Prostate cancer glucuronidation enzymatic activity is captured in vitro by SCH23390 and in vivo by PET imaging with 18F-DHT. A, SCH23390, a nonsteroid substrate of glucuronidation, is a surrogate for DHT glucuronidation in prostate cancer. LNCaP cells were treated with [3H]SCH23390 (100 nm) for 6 h, extracted from media and quantitated by HPLC. Glucuronidated [3H]SCH23390 was freed by β-glucuronidase treatment prior to analysis. B, UGT2B15 and UGT2B17 KO allows persistence of free SCH23390 compared with Ctrl. Cells were treated with [3H]SCH23390 (100 nm) and quantitation was with HPLC. Experiments in A and B were repeated 3 times and error bars represent the S.D. C, cellular DHT content is augmented in glucuronidation-competent prostate cancer. Cells were treated in triplicate with [3H]DHT (10 nm) for the indicated incubation times and intracellular radioactivity was assessed. Experiments were repeated 3 times and error bars represent the S.D. D, cellular DHT content in glucuronidation-proficient cells predominantly reflects glucuronidated androgens. Experiments were done similar to C, 5 min after the chase step, except radioactivity was assessed in both the aqueous (glucuronidated) and organic (free steroid) extracted intracellular fractions. E, cyclosporin A selectively increases DHT retention in cells that are glucuronidation competent. Pulse-chase experiments were performed with pretreatment with or without cyclosporin A (10–100 μm), followed by treatment with [3H]DHT, and analysis of the radioactive signal in cells. Experiments were performed in triplicate and error bars represent the S.D. Experiments were repeated three times. F, glucuronidation-competent and -deficient prostate cancers are distinguishable with PET imaging using 18F-DHT. Matched Ctrl and KO xenografts were grown in the flanks of mice, which were castrated 1 week before imaging. Mice were injected with cyclosporin A 45–60 min prior to 18F-DHT administration and PET-CT scan was performed. Imaging was performed when tumor volume reached 500 mm3. Images are displayed at 10 min post-injection, showing significantly higher uptake in Ctrl tumors versus KO tumors.
Discussion
The past few years has witnessed a dizzying pace of advances in the characterization of prostate cancer genomics (21–23). One of the themes that has emerged from these rich studies is that although there is an incredible level of genetic heterogeneity on one hand, there is also a requirement for a common set of pathways that drive tumor progression and resistance to systemic therapies. Another theme that has developed is the dynamic nature of the repertoire of somatic genetic aberrations, which evolves through response and resistance to systemic treatments (24–26). Long missing have been imaging modalities that capture aspects of the biologic state of the disease in a way that reflects on tumor susceptibility to response or resistance to systemic therapies. Functional imaging modalities are required to capture the dynamic evolution of metastatic tumors that are spatially and temporally separated. No such imaging modalities are currently employed in standard clinical practice, which are usually instead employed to determine tumor size and/or localization (27, 28).
As currently used, 18F-DHT does not offer any advantages over 18F-deoxyglucose for metastatic prostate cancer localization and neither are routinely used for clinical management (29, 30). PET imaging with 18F-DHT was mainly developed as a high-affinity ligand for localization and expression of AR ligand binding in prostate cancer (31). In our study, we found no increase in AR protein expression in glucuronidation-proficient tumors (Fig. S3), thus excluding the possibility that the increase in 18F-DHT signal that we show in these tumors is due to the known effect and affinity of 18F-DHT for AR (30, 31). In fact, a decrease in free 18F-DHT and AR binding occurring in glucuronidation-proficient tumors probably blunts the detectable difference in PET signal with glucuronidation-defective prostate cancer (Fig. 5). We would anticipate that development of a nonsteroidal glucuronidation substrate may therefore in fact exhibit an accentuated difference in tracer retention between these tumor types. Although we demonstrated that the prostatic glucuronidation machinery that normally physiologically inactivates androgens also conjugates the (nonsteroid) substrate SCH23390, tumor uptake was negligible. Nonetheless, this approach with nonsteroid substrates should be explored further in future studies.
Our approach in some ways mimics the strategy used for the development and use of [18F]fluorodeoxyglucose. Both strategies have in common the use of a tracer that undergoes intracellular modification to a polar and membrane-impermeable metabolite that may be trapped in cells. Although we demonstrate a proof of principle approach for noninvasive detection of a switch in enzymatic steroid inactivation, we acknowledge that our approach may benefit from further optimization.
Finally, we highlight the discovery of a novel metabolic phenotype that enables the sustenance of elevated intratumoral androgens in androgen deprivation therapy resistance, specifically by way of a mechanism downstream of biologically active androgens that blocks intratumoral androgen loss. These studies complement known mechanisms upstream of biologically active androgens that increase synthesis from extragonadal androgen precursor steroids.
Experimental procedures
Cloning and CRISPR cell generations
UGT2B15 cDNA plasmid was purchased from GE Dharmacon (MHS6278-213245438) and cloned into the lentiviral pCMV vector. LentiCRISPR v2 was used for knockout of UGT2B15 and -2B17. LentiCRISPR v2 was a gift from Feng Zhang (Addgene plasmid no. 52961). Guide RNA sequence for targeting hUGT2B15 5′-GAATCGAAGACTGTACAGAAAGG-3′, hUGT2B17 5′-GATTAACGGCATCTGCCAGAAGG-3′, and nontargeting control 5′-GTTCCGCGTTACATAACTTA-3′ were designed, cloned, and virus was produced using the LentiCRISPRv2 protocol (32). To generate the UGT2B15 and UGT2B17 double knockout cell line, LNCaP cells were infected with both viruses simultaneously.
Cells and culture conditions
The LNCaP human prostate cancer cell line was purchased from American Type Culture Collection (ATCC) and C4-2 cells were generously provided by Dr. Leland Chung (Cedars-Sinai Medical Center, CA). Cells were maintained in RPMI 1640 (Sigma) with 10% fetal bovine serum (Gemini). All cell lines were incubated in a 5% CO2 humidified incubator and 1% penicillin/streptomycin was added to all culture media. All cell lines were authenticated prior to use and routinely tested for mycoplasma contamination.
Media and intracellular UGT activity studies
Radiolabeled steroids were purchase from PerkinElmer. 1 × 106 cells were treated in phenol red-free media with charcoal-stripped serum (CSS; Gemini) spiked with 10 μCi of [3H]DHT, [3H]testosterone, or [3H]SCH23390 (PerkinElmer Life Sciences), and 10 nm cold testosterone, 10 nm DHT, or 100 nm SCH23390 (Sigma). Culture media and cell pellets were harvested after the indicated incubation time. For MS analysis, 2 × 107 cells were seeded in 15-cm dishes with CSS media 48 h prior to treatment. Media was aspirated and 20 ml of fresh CSS media spiked with DHT (10 nm) or DHEA (100 nm) was added. Cells were collected, centrifuged, weighed, and frozen at −80 °C.
As previously described (33, 34), media samples were extracted with 1:1 ethyl acetate:isooctane and cell pellets were re-suspended in 600 μl of PBS and subjected to 3 freeze-thaw cycles before extraction. For analysis of free steroids using HPLC, the organic layer was collected and the solvent evaporated under nitrogen gas in glass tubes. To analyze glucuronidated steroids or SCH23390, the aqueous phase after the first extraction was washed 3 times by ethyl acetate:isooctane, and treated with β-glucuronidase at 37 °C for 4 h (for steroids) or overnight (for SCH23390) to remove the glucuronide conjugate. The glucuronidation substrate was extracted again with ethyl acetate:isooctane and analyzed by HPLC. Dried samples were reconstituted in 50% methanol and injected on a Breeze 1525 system equipped with a model 717 plus autoinjector (Waters Corp.) and a Kinetix 100 × 2.1-mm, 2.6 μmol/liter of C18 reverse-phase column (Phenomenex) and methanol:water gradients at 30 °C. The column effluent was analyzed using a b-RAM model 3 in-line radioactivity detector (IN/US Systems, Inc.) using Liquiscint scintillation mixture (National Diagnostics). Steroid metabolites were identified using tritiated standards of AD, T, DHEA, and DHT (PerkinElmer Life Sciences). For detection using a scintillation counter, samples were extracted with the same volume of ethyl acetate:isooctane. 100 μl of the organic layer or aqueous layer was loaded into scintillation tubes with 3 ml of Liquiscint scintillation mixture and scanned by scintillation counter.
Transcript and protein expression
mRNA expression and immunoblot were used. Briefly, total RNA was harvested using the RNeasy kit (Qiagen), and 1 μg of RNA was used in a reverse-transcriptase reaction with the iScript cDNA synthesis kit (Bio-Rad). Quantitative PCR (qPCR) analysis was performed in triplicate with the following primer sets for PSA (forward: 5′-GCATGGGATGGGGATGAAGTAAG-3′; reverse: 5′-CATCAAATCTGAGGGTTGTCTGGA-3′), FKBP5 (forward: 5′-AAAAGGCCACCTAGCTTTTTGC-3′; reverse: 5′-CCCCCTGGTGAACCATAATACA-3′), TMPRSS2 (forward: 5′-CCATTTGCAGGATCTGTCTG-3′; reverse: 5′-GGATGTGTCTTGGGGAGCAA-3′), UGT2B15 (forward: 5′-GTGTTGGGAATATTATGACTACAGTAAC-3′; reverse: 5′-GGGTATGTTAAATAGTTCAGCCAGT-3′), UGT2B17 (forward: 5′-TTTTGTCGCAGGAAAAAGGAAA-3′; reverse: 5′-AAGCCTGAAGTCGAATGACCAA-3′) and the housekeeping gene encoding large ribosomal protein P0 (RPLP0) (forward: 5′-CGAGGGCACCTGGAAAAC-3′; reverse: 5′-CACATTCCCCCGGATATGA-3′). The iTaq Fast SYBR Green Supermix with ROX kit (Bio-Rad) was used for the thermocycling reaction in an ABI-7500 Real-Time PCR machine (Applied Biosystems). Accurate quantitation of each mRNA was determined by normalizing the sample values to RPLP0 and to control cells (for knockout) or to vehicle-treated cells (for steroid treated cells). To assess androgen-regulated genes, cells were starved with CSS media 48 h prior to androgen treatment.
For Western blot analysis, cells were lysed using 10% SDS. Protein was resolved by 10% SDS-PAGE and incubated with a rabbit anti-UGT2B15 (Abcam), rabbit anti-AR (Sigma), and mouse anti-β-actin (Sigma) antibodies.
In vitro cell growth assays
Control and KO cells were starved with CSS media 48 h prior to splitting. 2 × 104 cells were seeded in each well in 12-well plate with 2 ml of CSS media. Ethanol, 100 nm DHEA, 100 nm testosterone, or 10 nm DHT were added at day 0. Cells were grown in triplicate in each group and were counted at the indicated number of days.
Mouse xenograft studies
All mouse studies were performed under a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the Cleveland Clinic Lerner Research Institute. All NSG male mice (6–8 weeks old) were purchased from the Jackson Laboratory and the number of mice used in this study was based on previously published mouse xenograft studies that determined effects of steroid pathway inhibition/augmentation on xenograft growth (33, 35, 36). To mimic human adrenal DHEA production in patients with CRPC, mice were surgically orchiectomized and 5 mg, 90-day, sustained-release DHEA pellets were implanted subcutaneously.
For evaluation of the role of UGT2B15/17 in tumor progression, 107 vector control or UGT2B15/17 KO cells were subcutaneously injected with 50% Matrigel (BD Biosciences) into 10 eugonadal, castrated, or castrated plus DHEA pellet mice. Xenografts were measured twice a week after tumors became palpable. Tumor progression curves were generated by Prism and the statistical significance were analyzed by log-rank (Mantel-Cox) test.
For testing the effect of UGT activity reintroduction, 1 × 106 UGT2B15- or vector-expressing C4-2 cells were subcutaneously injected into 7 eugonadal or castrated mice with 50% Matrigel. Tumor progression curves were generated by Prism and the statistical significance was analyzed by log-rank (Mantel-Cox) test.
Xenograft UGT activity assays and cell line subculture
After removal of necrotic regions, xenograft tumor tissues (0.03–0.04 g each, 3 pieces from each tumor under castration selection, or 1 piece from each eugonadal LNCaP or C4-2 tumor) were weighed and carefully minced in 1.5 ml of CSS media using scissors in a 12-well plate under sterile conditions. Each sample was incubated in CSS media spiked with 10 μCi of [3H]DHT and 10 nm cold DHT. Aliquots of media were obtained after 4 h of treatment and frozen in −20 °C for further analysis. To subculture cell lines from xenografts, 1/10 volume of media with minced tissues were transferred to another a 12-well plate with 1 ml of RPMI 1640 and 10% fetal bovine serum.
Liquid chromatography-tandem MS analysis of steroid content
Extraction protocol
Cell pellets were thawed from storage at −80 °C and subjected to 5 freeze-thaw cycles, after which each cell pellet sample was added to 200 μl of media and transferred into a glass tube. Samples were spiked with 10 μl of internal standard (50 ng/ml of [2,3,4-13C3]androstenedione; AD-13C3; Sigma), and briefly vortexed. After addition of 2 ml of methyl tert-butyl ether (Across), the glass tube was vortexed for 5 min using a multitube vortexer (Fisher) and centrifuged for 5 min at 2500 rpm at 4 °C. Samples were then placed on dry ice for 10 min, and the top organic fraction with extracted steroid was collected in a new glass tube. The collected organic layer was evaporated under nitrogen gas at 40 °C and then reconstituted in 110 μl of 50% methanol:water (v/v) and briefly vortexed again. The reconstituted samples were transferred into 1.5-ml microcentrifuge tubes, centrifuged at 13,000 rpm for 10 min at 4 °C, and the supernatants were collected for mass spectrometry injections in HPLC vials.
Instrumentation and data analysis
The extracted steroids from media or cell pellet samples were quantified using LC tandem MS (LC-MS/MS). The LC-MS/MS system consists of an ultra-pressure LC system (UPLC; Shimadzu Corporation, Japan), comprised of two LC-30AD pumps, a DGU-20A5R vacuum degasser, a CTO-30A column oven, SIL-30AC autosampler, and a system controller CBM-20A and coupled with an API Qtrap 5500 mass spectrometer (AB Sciex, Framingham, MA). Briefly, steroid extract was injected onto the Shimadzu UPLC system, and the steroids were separated through a C18 column (Zorbax Eclipse Plus C18 column 150 × 2.1 mm, 3.5 μm, Agilent, Santa Clara, CA) using a gradient starting from 20% solvent B (acetonitrile/methanol (90/10, v/v) containing 0.2% formic acid) over 4 min and then to 75% solvent B over 10 min, followed by 95% solvent B for 3 min. The steroids were quantified on a Qtrap 5500 mass spectrometer using ESI in positive ion mode and multiple reaction monitoring using characteristic parent → daughter ion transitions for the specific molecular species monitored. [2,3,4-13C3]Androstenedione was used as an internal standard for calibration of steroids in each sample. Data acquisition and processing were performed using MultiQuant (version 3.0.1) from AB Sciex. The peak area ratio of the analyte over the internal standard was used for quantification purposes. Each sample run included calibration curves with standards for data quantification using the analyte/internal standard peak area ratio. Steroid metabolite concentrations in cell pellet samples were quantified by LC/MS/MS analysis and normalized to cell pellet sample mass. Experiments were done in biological triplicate and the mean ± S.D. were calculated.
Pulse-chase assays
1 × 106 control and KO LNCaP cells were seeded in poly-dl-ornithine (Sigma)-coated plates with CSS media for 24 h prior to initiation of treatment. For the pulse step, culture media was aspirated and cells were treated with CSS media spiked with 50 μCi of [3H]DHT and 10 nm cold DHT for 5 min. Then the media was carefully removed and cells were washed once with CSS media with 10 nm cold DHT. For the chase step, cells were treated with CSS media with 10 nm cold DHT and then cell pellets were collect at the indicated time points. Radioactivity was determined by a scintillation counter.
In vivo PET-CT image and analysis
Mice were injected subcutaneously in each flank with 1 × 107 control and 7 × 106 KO cells (to compensate for more rapid growth of KO cells) mixed with 50% Matrigel. Mice were castrated 1 week before imaging. 0.6 mg of cyclosporin A dissolved in 50 μl of DMSO was injected in tail vein 45–60 min before scanning. Mice were anesthetized (2% isoflurane) and injected with 7.2–8.0 MBq of [18F]16β-fluoro-5α-dihydrotestosterone (diluted in 0.2 ml of saline) via tail vein. 5 min or 1 h after injection micro-PET scans were acquired using an Inveon microPET-CT scanner (Siemens), followed by micro-CT scans for attenuation correction and localization purposes. PET images were acquired in list-mode for 60 and 10 min, respectively. Static and dynamic PET images were reconstructed using the 3D maximum a posteriori iterative algorithm on the micro-PET work station (settings: 2 iterations 3DOSEM, 18 iterations maximum a posteriori, 1.5-mm regularization), using corresponding CT images for PET attenuation and scatter corrections. Image analysis software (PMOD Image Fusion, PMOD Technologies, Zurich, Switzerland) was used for PET measurements. Spherical volumes of interest (6 mm diameter) were centered on each tumor based on CT images to measure PET tracer uptake versus time. PET measurements were normalized to injected activity and mouse weight to compute standardized uptake value. The washout rate was calculated by determining the slope of the curve past the peak and dividing it by the maximum value.
Statistical analysis
Numerical data are presented as mean ± S.E. Tumor progression curves were generated by Prism and the statistical significance was analyzed by log-rank (Mantel-Cox) test.
Author contributions
Z. Z. and N. S. conceptualization; Z. Z., Y.-M. C., O. S., V. K., M. P. B., J. L., H.-K. K., Z. Li, M. P., F. D., and Z. Lee investigation; Z. Z. visualization; Z. Z. and Y.-M. C. methodology; Z. Z., Z. Lee, and N. S. writing-original draft; Z. Z., Y.-M. C., V. K., F. D., Z. Lee, and N. S. writing-review and editing; V. K. data curation; Z. Lee and N. S. formal analysis; N. S. resources; N. S. project administration.
Supplementary Material
This work was supported by a Howard Hughes Medical Institute Physician-Scientist Early Career Award and a Prostate Cancer Foundation Challenge Award (to N. S.), National Institutes of Health NCI Grants R01CA168899, R01CA172382, and R01CA190289 (to N. S.), NCI Grant 5R01CA204373 (to Z. L.), and Congressionally Directed Medical Research Programs, Prostate Cancer Research Program Award PC141550 (to Z. Z.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
- CRPC
- castration-resistant prostate cancer
- PSA
- prostate-specific antigen
- AR
- androgen receptor
- DHT
- dihydrotestosterone
- UGT
- UDP-glucuronosyltransferase
- DHEA
- dehydroepiandrosterone
- CSA
- cyclosporin A
- CSS
- charcoal-stripped serum
- qPCR
- quantitative PCR
- CT
- computed tomography
- Ctrl
- control.
References
- 1. Huggins C., and Hodges C. V. (1941) Studies on prostate cancer: I: the effect of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1, 293–297 [Google Scholar]
- 2. Watson P. A., Arora V. K., and Sawyers C. L. (2015) Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 15, 701–711 10.1038/nrc4016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dai C., Heemers H., and Sharifi N. (2017) Androgen signaling in prostate cancer. Cold Spring Harb. Perspect. Med. 7, a030452 10.1101/cshperspect.a030452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Attard G., Parker C., Eeles R. A., Schröder F., Tomlins S. A., Tannock I., Drake C. G., and de Bono J. S. (2016) Prostate cancer. Lancet 387, 70–82 10.1016/S0140-6736(14)61947-4 [DOI] [PubMed] [Google Scholar]
- 5. Montgomery R. B., Mostaghel E. A., Vessella R., Hess D. L., Kalhorn T. F., Higano C. S., True L. D., and Nelson P. S. (2008) Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 68, 4447–4454 10.1158/0008-5472.CAN-08-0249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Titus M. A., Schell M. J., Lih F. B., Tomer K. B., and Mohler J. L. (2005) Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin. Cancer Res. 11, 4653–4657 10.1158/1078-0432.CCR-05-0525 [DOI] [PubMed] [Google Scholar]
- 7. Chang K. H., Li R., Kuri B., Lotan Y., Roehrborn C. G., Liu J., Vessella R., Nelson P. S., Kapur P., Guo X., Mirzaei H., Auchus R. J., and Sharifi N. (2013) A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell 154, 1074–1084 10.1016/j.cell.2013.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. James N. D., de Bono J. S., Spears M. R., Clarke N. W., Mason M. D., Dearnaley D. P., Ritchie A. W. S., Amos C. L., Gilson C., Jones R. J., Matheson D., Millman R., Attard G., Chowdhury S., Cross W. R., et al. (2017) Abiraterone for prostate cancer not previously treated with hormone therapy. N. Engl. J. Med. 377, 338–351 10.1056/NEJMoa1702900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fizazi K., Tran N., Fein L., Matsubara N., Rodriguez-Antolin A., Alekseev B. Y., Özgüroǧlu M., Ye D., Feyerabend S., Protheroe A., De Porre P., Kheoh T., Park Y. C., Todd M. B., Chi K. N., and Investigators L. (2017) Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N. Engl. J. Med. 377, 352–360 10.1056/NEJMoa1704174 [DOI] [PubMed] [Google Scholar]
- 10. Barbier O., and Bélanger A. (2008) Inactivation of androgens by UDP-glucuronosyltransferases in the human prostate. Best Pract. Res. Clin. Endocrinol. Metab. 22, 259–270 10.1016/j.beem.2008.01.001 [DOI] [PubMed] [Google Scholar]
- 11. Sten T., Bichlmaier I., Kuuranne T., Leinonen A., Yli-Kauhaluoma J., and Finel M. (2009) UDP-glucuronosyltransferases (UGTs) 2B7 and UGT2B17 display converse specificity in testosterone and epitestosterone glucuronidation, whereas UGT2A1 conjugates both androgens similarly. Drug Metab. Dispos. 37, 417–423 10.1124/dmd.108.024844 [DOI] [PubMed] [Google Scholar]
- 12. Turgeon D., Carrier J. S., Lévesque E., Hum D. W., and Bélanger A. (2001) Relative enzymatic activity, protein stability, and tissue distribution of human steroid-metabolizing UGT2B subfamily members. Endocrinology 142, 778–787 10.1210/endo.142.2.7958 [DOI] [PubMed] [Google Scholar]
- 13. Thalmann G. N., Anezinis P. E., Chang S. M., Zhau H. E., Kim E. E., Hopwood V. L., Pathak S., von Eschenbach A. C., and Chung L. W. (1994) Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 54, 2577–2581 [PubMed] [Google Scholar]
- 14. Bao B. Y., Chuang B. F., Wang Q., Sartor O., Balk S. P., Brown M., Kantoff P. W., and Lee G. S. (2008) Androgen receptor mediates the expression of UDP-glucuronosyltransferase 2 B15 and B17 genes. Prostate 68, 839–848 10.1002/pros.20749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Asim M., Siddiqui I. A., Hafeez B. B., Baniahmad A., and Mukhtar H. (2008) Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4–2 cells. Oncogene 27, 3596–3604 10.1038/sj.onc.1211016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krueckl S. L., Sikes R. A., Edlund N. M., Bell R. H., Hurtado-Coll A., Fazli L., Gleave M. E., and Cox M. E. (2004) Increased insulin-like growth factor I receptor expression and signaling are components of androgen-independent progression in a lineage-derived prostate cancer progression model. Cancer Res. 64, 8620–8629 10.1158/0008-5472.CAN-04-2446 [DOI] [PubMed] [Google Scholar]
- 17. Sarkar S., Brautigan D. L., Parsons S. J., and Larner J. M. (2014) Androgen receptor degradation by the E3 ligase CHIP modulates mitotic arrest in prostate cancer cells. Oncogene 33, 26–33 10.1038/onc.2012.561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu J. B., Yin L., Shi C., Li Q., Duan P., Huang J. M., Liu C., Wang F., Lewis M., Wang Y., Lin T. P., Pan C. C., Posadas E. M., Zhau H. E., and Chung L. W. (2017) MAOA-dependent activation of Shh-IL6-RANKL signaling network promotes prostate cancer metastasis by engaging tumor-stromal cell interactions. Cancer Cell 31, 368–382 10.1016/j.ccell.2017.02.003 [DOI] [PubMed] [Google Scholar]
- 19. Tephly T. R., Coffman B., Styczynski P., Rios G., Charkowski D. M., Vanrollins M., McQuade R. D., and Tedford C. E. (1994) Studies on the glucuronidation of dopamine D-1 receptor antagonists, SCH 39166 and SCH 23390, by human liver microsomes. Drug Metab. Dispos. 22, 713–718 [PubMed] [Google Scholar]
- 20. International Transporter Consortium, Giacomini K. M., Huang S. M., Tweedie D. J., Benet L. Z., Brouwer K. L., Chu X., Dahlin A., Evers R., Fischer V., Hillgren K. M., Hoffmaster K. A., Ishikawa T., Keppler D., Kim R. B., Lee C. A., et al. (2010) Membrane transporters in drug development. Nat. Rev. 9, 215–236 10.1038/nrd3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robinson D., Van Allen E. M., Wu Y. M., Schultz N., Lonigro R. J., Mosquera J. M., Montgomery B., Taplin M. E., Pritchard C. C., Attard G., Beltran H., Abida W., Bradley R. K., Vinson J., Cao X., et al. (2015) Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228 10.1016/j.cell.2015.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cancer Genome Atlas Research Network (2015) The molecular taxonomy of primary prostate cancer. Cell 163, 1011–1025 10.1016/j.cell.2015.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fraser M., Sabelnykova V. Y., Yamaguchi T. N., Heisler L. E., Livingstone J., Huang V., Shiah Y. J., Yousif F., Lin X., Masella A. P., Fox N. S., Xie M., Prokopec S. D., Berlin A., Lalonde E., et al. (2017) Genomic hallmarks of localized, non-indolent prostate cancer. Nature 541, 359–364 10.1038/nature20788 [DOI] [PubMed] [Google Scholar]
- 24. Carreira S., Romanel A., Goodall J., Grist E., Ferraldeschi R., Miranda S., Prandi D., Lorente D., Frenel J. S., Pezaro C., Omlin A., Rodrigues D. N., Flohr P., Tunariu N., S. de Bono, J. et al. (2014) Tumor clone dynamics in lethal prostate cancer. Sci. Transl. Med. 6, 254ra125 10.1126/scitranslmed.3009448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Romanel A., Gasi Tandefelt D., Conteduca V., Jayaram A., Casiraghi N., Wetterskog D., Salvi S., Amadori D., Zafeiriou Z., Rescigno P., Bianchini D., Gurioli G., Casadio V., Carreira S., Goodall J., et al. (2015) Plasma AR and abiraterone-resistant prostate cancer. Sci. Transl. Med. 7, 312re310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Beltran H., Prandi D., Mosquera J. M., Benelli M., Puca L., Cyrta J., Marotz C., Giannopoulou E., Chakravarthi B. V., Varambally S., Tomlins S. A., Nanus D. M., Tagawa S. T., Van Allen E. M., Elemento O., et al. (2016) Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 22, 298–305 10.1038/nm.4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harmon S. A., Perk T., Lin C., Eickhoff J., Choyke P. L., Dahut W. L., Apolo A. B., Humm J. L., Larson S. M., Morris M. J., Liu G., and Jeraj R. (2017) Quantitative assessment of early [18F]sodium fluoride positron emission tomography/computed tomography response to treatment in men with metastatic prostate cancer to bone. J. Clin. Oncol. 35, 2829–2837 10.1200/JCO.2017.72.2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gillessen S., Attard G., Beer T. M., Beltran H., Bossi A., Bristow R., Carver B., Castellano D., Chung B. H., Clarke N., Daugaard G., Davis I. D., de Bono J., Dos Reis R. B., Drake C. G., et al. (2017) Management of patients with advanced prostate cancer: the report of the Advanced Prostate Cancer Consensus Conference APCCC 2017. Eur. Urol. 73, 178–211 [DOI] [PubMed] [Google Scholar]
- 29. Feldman J. L., Fox J. J., Martinez D. F., Autio K. A., Durack J. C., Humm J., Schöder H., Gavane S. C., Curtis K. R., Balakirsky S. M., Cohen R. M., Scher H. I., Larson S. M., and Morris M. J. (2014) Pathologic correlation of 18F-16β-fluoro-5α-dihydrotestosterone (FDHT) and FDG PET in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 32, 5016–5016 [Google Scholar]
- 30. Larson S. M., Morris M., Gunther I., Beattie B., Humm J. L., Akhurst T. A., Finn R. D., Erdi Y., Pentlow K., Dyke J., Squire O., Bornmann W., McCarthy T., Welch M., and Scher H. (2004) Tumor localization of 16β-18F-fluoro-5α-dihydrotestosterone versus 18F-FDG in patients with progressive, metastatic prostate cancer. J. Nucl Med. 45, 366–373 [PubMed] [Google Scholar]
- 31. Liu A., Carlson K. E., and Katzenellenbogen J. A. (1992) Synthesis of high affinity fluorine-substituted ligands for the androgen receptor: potential agents for imaging prostatic cancer by positron emission tomography. J. Med. Chem. 35, 2113–2129 10.1021/jm00089a024 [DOI] [PubMed] [Google Scholar]
- 32. Sanjana N. E., Shalem O., and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li Z., Bishop A. C., Alyamani M., Garcia J. A., Dreicer R., Bunch D., Liu J., Upadhyay S. K., Auchus R. J., and Sharifi N. (2015) Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature 523, 347–351 10.1038/nature14406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li Z., Alyamani M., Li J., Rogacki K., Abazeed M., Upadhyay S. K., Balk S. P., Taplin M. E., Auchus R. J., and Sharifi N. (2016) Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy. Nature 533, 547–551 10.1038/nature17954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chang K. H., Li R., Papari-Zareei M., Watumull L., Zhao Y. D., Auchus R. J., and Sharifi N. (2011) Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 108, 13728–13733 10.1073/pnas.1107898108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li J., Alyamani M., Zhang A., Chang K. H., Berk M., Li Z., Zhu Z., Petro M., Magi-Galluzzi C., Taplin M. E., Garcia J. A., Courtney K., Klein E. A., and Sharifi N. (2017) Aberrant corticosteroid metabolism in tumor cells enables GR takeover in enzalutamide resistant prostate cancer. eLife 6, e20183 10.7554/eLife.20183 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.