

Abstract
Formins direct the elongation of unbranched actin filaments that are incorporated into a diverse set of cytoskeletal structures. Elongation of formin-bound filaments occurs along two parallel pathways. The formin homology 2 (FH2) pathway allows actin monomers to bind directly to barbed ends bound by dimeric FH2 domains. The formin homology 1 (FH1) pathway involves transfer of profilin-bound actin to the barbed end from polyproline tracts located in the disordered FH1 domains. Here, we used a total internal reflection fluorescence (TIRF) microscopy-based fluorescence approach to determine the fraction of actin subunits incorporated via the FH1 and FH2 pathways during filament elongation mediated by two formins. We found that the fraction of filament elongation that occurs via each pathway directly depends on the efficiency of the other pathway, indicating that these two pathways compete with each other for subunit addition by formins. We conclude that this competition allows formins to compensate for changes in the efficiency of one pathway by adjusting the frequency of subunit addition via the other, thus increasing the overall robustness of formin-mediated actin polymerization.
Keywords: formin, actin, profilin, fluorescence, cytoskeleton, actin polymerization, Bni1, Cdc12, formin homology domain, TIRF microscopy
Introduction
Cells rely on their dynamic cytoskeleton to perform essential functions such as growth, division, signaling, and the establishment of polarity. To support these cellular tasks, the formin family of proteins mediates the assembly of unbranched actin filaments that are incorporated into a number of specialized cytoskeletal structures, including cytokinetic rings (1), filopodia (2), stress fibers (3), and adhesion junctions (4). The formin family is diverse; each of the 3 fission yeast and 15 mammalian formin isoforms possesses unique actin assembly properties and plays a specific role in cells (1, 5–8). Mutations in formin genes are associated with a number of diseases, including preleukemic disorders, microcephaly, dilated cardiomyopathy, the neurological disorder Charcot–Marie–Tooth disease, and the kidney disease focal segmental glomerulosclerosis (8–12), underscoring the importance of formin isoforms in promoting cellular viability and organismal health.
Formins are large multidomain proteins that mediate actin filament assembly via their formin homology 2 (FH2) 2 domains (13–16), which associate in a head-to-tail orientation (17) to form donut-shaped dimers that encircle filament barbed ends and nuclei (18). Once bound, FH2 dimers fluctuate rapidly between an “open” conformation that is polymerization-competent and a “closed” conformation that is polymerization-incompetent (Fig. 1A) (19–21). The fraction of time an FH2 dimer spends in the open state is unique to each formin isoform and is called the “gating factor.” When an FH2 dimer is in the open state, an actin monomer can bind to the barbed end and be incorporated into the filament via FH2 “stepping,” which requires one FH2 monomer to dissociate from its barbed end-binding site to bind the new subunit (Fig. 1A). Because of gating, FH2-mediated filament elongation tends to be slow compared with the elongation of filaments polymerizing in the absence of formins (22, 23).
Figure 1.

Pathways for formin-mediated actin filament elongation. A and B, schematic representation of the mechanism of actin filament (gray circles) elongation mediated by a formin. Open and closed conformations of the FH2 domain are represented in green and red. An incoming actin monomer (orange) can either bind directly to the barbed end (top row in A; FH2 pathway in B) or bind to a polyproline tract (purple ovals) in the FH1 domain via profilin (blue circle) for delivery to the barbed end (bottom row in A; FH1 pathway in B). C, the dependence of the fraction of total (orange open circles) and profilin-bound (purple filled symbols) fluorescently labeled actin monomers on the concentration of profilin. The total fluorescent fraction of actin is 0.33, independent of profilin concentration (open orange circles). The fluorescent fractions of profilin-bound actin were calculated using published binding affinities of actin for S. cerevisiae and S. pombe profilin (2.9 and 0.2 μm, respectively) (32, 33) and the effect of fluorescent labeling at cysteine 374 on these affinities (31).
The formin FH1 domain, located directly N-terminal to the FH2 domain, enables formins to overcome the limit on elongation imposed by gating by providing an alternative route for subunit incorporation. FH1 domains are unstructured regions (24, 25) that contain multiple polyproline tracts, which bind profilin, a protein that also binds actin monomers. Upon binding a polyproline tract, profilin–actin is directly transferred to the barbed end via diffusional translation of the FH1 domain and subsequent FH2 stepping (Fig. 1A) (20). In this manner, formins mediate filament elongation via two parallel pathways: an FH2 pathway, along which actin monomers slowly and directly bind to formin-bound barbed ends, and an FH1 pathway, which requires the presence of profilin (Fig. 1B).
Vavylonis et al. (21) described the mechanism of formin-mediated filament elongation in a computational model that accounted for subunit addition through both the FH1 and FH2 pathways and successfully captured the effects of variations in the concentrations of actin, profilin and profilin–actin on the rate of elongation. This model predicted that the rate of subunit addition through the FH2 pathway decreases as the concentration of profilin–actin (and thus the activity of the FH1 domain) increases, suggesting that polymerization through each pathway depends on the availability of the other as an alternative route to the barbed end. This prediction suggests that formins can respond to changes in reaction conditions by altering the relative flux of actin subunits through each pathway to maximize the polymerization rate.
Despite the mechanistic significance of the interdependence of the FH1 and FH2 pathways for polymerization mediated by formins, to date this prediction has not been tested in vitro. In this study, we used a microscopy-based fluorescence approach to measure the fractions of actin subunits that are incorporated via the FH1 and FH2 pathways during filament elongation mediated by two representative formins: the Schizosaccharomyces pombe formin Cdc12p and the Saccharomyces cerevisiae formin Bni1p. We confirmed that these pathways are competitive modes of polymerization, such that an increase in the rate of subunit incorporation through one pathway is compensated with a decrease in incorporation through the other pathway. Further, the relative contributions of the two pathways to the overall actin elongation rate depend on the polymerization conditions, the formin's gating factor, and the number of polyproline tracts in the FH1 domain. In this manner, formins can partially compensate for a decrease in the rate of FH1-mediated polymerization, which may arise from a decrease in the local concentration of profilin–actin or a change in conformational freedom when the formin is under force, by increasing the rate of FH2-mediated subunit addition.
Results
To measure the relative fractions of actin subunits that are incorporated via the FH1 and FH2 pathways during filament elongation, we used total internal reflection fluorescence (TIRF) microscopy to visualize actin filaments bound at their barbed ends and assembled by formins. To observe elongating filaments, we used actin monomers that were fluorescently labeled at cysteine 374 with Oregon Green 488 (26). Although cysteine 374 is solvent-accessible in both the monomeric and filamentous states of actin, it is located on the barbed end surface of the actin monomer (27, 28). In addition to providing a binding site for longitudinal actin–actin interactions during polymerization, the barbed end surface is also the site for profilin binding (29, 30). As a result, inclusion of a label at cysteine 374 reduces the binding affinity of actin for profilin 10-fold (31).
We included a 1:2 mixture of labeled and unlabeled actin in our reactions and measured filament elongation mediated by formins in a range of profilin concentrations. Because of the tighter affinity of profilin for unlabeled actin, only a small fraction of profilin-bound actin is fluorescent in the presence of concentrations of profilin below its Kd for labeled actin (Fig. 1C). Filament elongation through the FH1 pathway, which requires profilin-bound actin, therefore promotes biased incorporation of unlabeled actin monomers into formin-bound filaments. As a result, these filaments are less fluorescent than control filaments that are not polymerized by formins (22).
We identified filaments that were specifically assembled by formins in our polymerization reactions by measuring and comparing their elongation rates and fluorescence intensities to those of control filaments that had free (i.e. not formin-bound) barbed ends (19, 22, 24). In each reaction, we normalized the fluorescence of filaments assembled by formins to the fluorescence of filaments with free barbed ends. We considered differences between the fluorescence of formin-bound and control filaments to be directly proportional to differences in the fraction of labeled polymerized actin. Using published, isoform-specific affinities of profilin for labeled and unlabeled actin (32, 33), we then calculated the fractions of subunits incorporated through each pathway (see “Experimental procedures”).
To guide the interpretation of our experimental results, we made predictions of filament fluorescence based on three potential modes of formin-mediated actin filament assembly. First, if a formin assembles actin filaments exclusively through its FH2 pathway, the fluorescence intensities of these filaments will be equal to those of control filaments if gating is insensitive to labeled actin (Fig. 1C, orange open circles). Second, if a formin assembles actin filaments exclusively through its FH1 pathway, the fluorescence intensities of these filaments will be proportional to the fraction of profilin-bound actin that is fluorescent in the reaction. Thus, the profilin dependence of the fluorescence of filaments assembled by this formin will mirror the shape of the binding curve that describes the profilin dependence of the fluorescent fraction of profilin-bound actin (Fig. 1C, purple filled circles and squares). Because this fraction is always less than or equal to the total fraction of fluorescent actin, filaments polymerized through the FH1 pathway will be dimmer than control filaments at low concentrations of profilin. Third, if a formin mediates polymerization via a combination of both pathways, the fluorescence intensities of filaments assembled by this formin will fall in between the predicted curves for the FH1 and FH2 pathways. These filaments will also be dimmer than control filaments at low concentrations of profilin.
Cdc12p mediates filament assembly through a single pathway
To test whether our approach could accurately reveal the fraction of actin subunits that are polymerized through each pathway, we first performed experiments with an FH1FH2 construct of the S. pombe formin Cdc12p. This formin has a very low gating factor (∼0.05) (22), indicating that the pointed end of a filament bound by Cdc12p elongates twice as fast as the barbed end in the absence of profilin (34). Thus, the vast majority of polymerization likely proceeds through the FH1 pathway in the presence of profilin. We therefore hypothesized that the fluorescence intensities of filaments polymerized by Cdc12p would be identical to the fraction of fluorescent profilin-bound actin in each reaction (Fig. 1C, filled squares and circles).
In the absence of profilin, formins incorporate actin into growing filaments exclusively via the FH2 pathway (Fig. 1A). Consistent with this mode of elongation, we found that filaments polymerized by Cdc12p and filaments with free barbed ends had very similar fluorescence intensities in the absence of profilin (Fig. 2A). Upon introduction of 0.5 μm S. pombe profilin, the fluorescence intensity of Cdc12p-bound filaments decreased to ∼35% of the intensity of the control filaments, suggesting that Cdc12p incorporates a larger fraction of unlabeled actin subunits into filaments when profilin is present. As higher concentrations of profilin were introduced into the reactions, the fluorescence of Cdc12p-bound filaments increased and, at concentrations exceeding 5 μm, ultimately reached a plateau that was approximately equal in intensity to that of control filaments (Fig. 2B).
Figure 2.
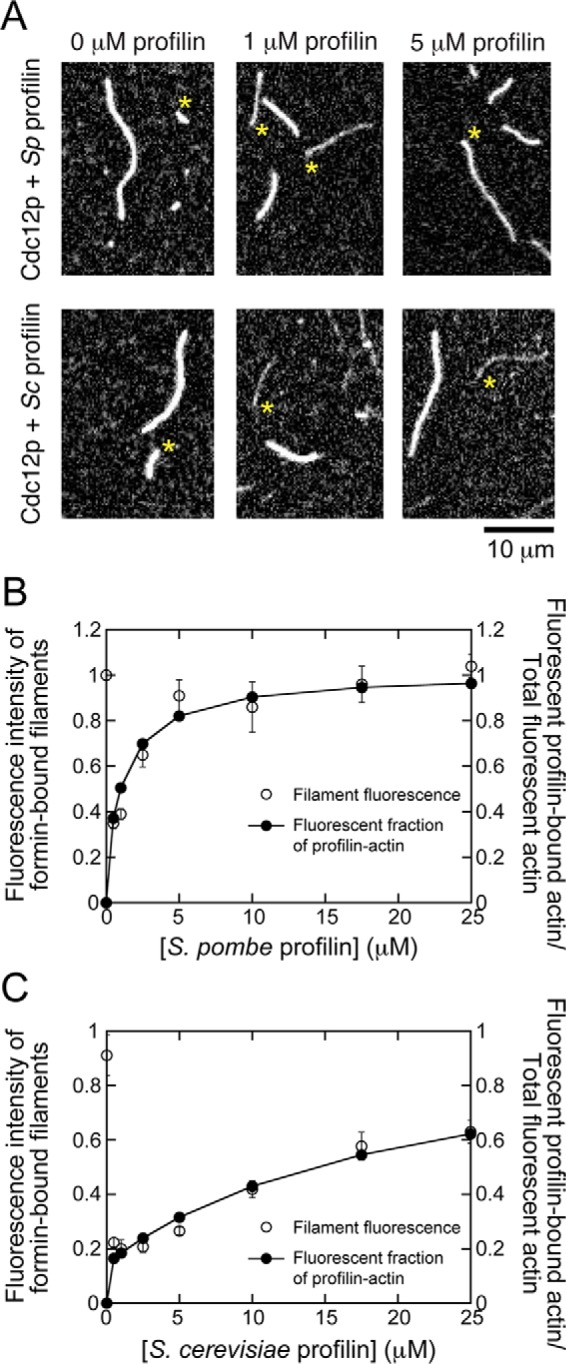
Cdc12p polymerizes filaments exclusively via the FH1 pathway in the presence of profilin. The conditions were as follows: 0.75 μm actin (33% Oregon Green-labeled) in microscopy buffer with varying concentrations of profilin. The data were collected by TIRF microscopy. A, representative fluorescence micrographs of actin filaments elongating in the presence of Cdc12p and a range of concentrations of S. pombe and S. cerevisiae profilin. Asterisks indicate the barbed ends of filaments bound by formin. Filaments lacking an asterisk are not formin-bound and serve as internal controls for measuring filament elongation rates and fluorescence intensity. B and C, dependences of the fluorescence intensity of formin-bound filaments (open circles) and the fluorescent fraction of profilin-bound actin (filled circles) on the concentration of S. pombe profilin (B) and S. cerevisiae profilin (C). Fluorescence intensities of formin-bound filaments were normalized to the intensities of filaments that are not formin-bound. The fluorescent fraction of profilin-bound actin was calculated using published binding affinities (32, 33) and normalized to the fraction of fluorescent actin included in each reaction (i.e. 0.33). Error bars are standard errors of the mean fluorescence intensity of at least 10 filaments.
The dependence of the fluorescence of Cdc12p-polymerized filaments on the profilin concentration closely mirrored the profilin dependence of the fraction of fluorescent profilin-bound actin, which we calculated using the published binding affinity (Kd = 0.2 μm (33)) (Fig. 2B). This agreement is consistent with our prediction for a formin that incorporates actin subunits into elongating filaments exclusively via the FH1 pathway in the presence of profilin.
To further establish the relationship between filament fluorescence and the fraction of profilin-bound, fluorescent actin monomers, we next performed similar polymerization assays using Cdc12p and S. cerevisiae profilin, which binds actin monomers more weakly than does S. pombe profilin (Fig. 1C; Kd = 2.9 μm for S. cerevisiae profilin (32, 33)). Because of this difference in affinity, a smaller fraction of profilin-bound actin is fluorescent in reactions containing identical concentrations of S. cerevisiae and S. pombe profilin (Fig. 1C). Correspondingly, we found that filaments polymerized by Cdc12p in the presence of S. cerevisiae profilin were dimmer than those polymerized with S. pombe profilin (Fig. 2, A and B). At each concentration of profilin, the average fluorescence intensity of Cdc12p-bound filaments matched the fraction of fluorescent profilin-bound actin, consistent with incorporation of actin subunits into filaments exclusively via the FH1 pathway in the presence of profilin (Fig. 2C). These results confirm that filament fluorescence is directly correlated with the fraction of labeled, profilin-bound actin monomers, which varies depending on the profilin isoform.
Bni1p incorporates actin subunits through both the FH1 and FH2 pathways
To determine whether formin-mediated elongation can occur through the FH1 and FH2 pathways simultaneously, we next measured the fluorescence intensities and elongation rates of filaments polymerized by the S. cerevisiae formin Bni1p in the absence and presence of S. cerevisiae profilin. In the absence of profilin, Bni1p-bound filaments elongate at approximately half the rate observed for filaments with free barbed ends, corresponding to a gating factor of 0.5 (19, 22). It is therefore likely that the FH2 pathway would contribute to polymerization even in the presence of profilin. Inclusion of profilin speeds filament elongation rates up to 4-fold, indicating that subunit addition through the FH1 pathway of Bni1p is also robust (Fig. 3D, black circles) (19, 22).
Figure 3.

Actin elongation mediated by Bni1p proceeds at a constant rate through the FH2 pathway. The conditions were as follows: 0.75 μm actin (33% Oregon Green) in microscopy buffer with varying concentrations of profilin. The data were collected by TIRF microscopy. A, representative fluorescence micrographs of actin filaments elongating in the presence of Bni1(FH1FH2)p or Bni1(FH2)p and a range of concentrations of S. cerevisiae profilin. Asterisks indicate the barbed ends of filaments bound by formin. Filaments lacking an asterisk are not formin-bound and serve as internal controls for measuring filament elongation rates and fluorescence intensity. B, the dependence of the fluorescence intensities of Bni1(FH1FH2)p- and Bni1(FH2)p-bound filaments (open circles and squares) and the predicted fluorescent fraction of profilin-bound actin (filled circles) on the concentration of profilin. Fluorescence intensity was normalized to the intensity of filaments that are not formin-bound. The fluorescent fraction of profilin-bound actin was normalized to the fraction of fluorescent actin included in each reaction (i.e. 0.33). Error bars are standard errors of the mean fluorescence intensity of at least 10 filaments. C, fraction of actin subunits incorporated into filaments by Bni1(FH1FH2)p via the FH1 (filled circles) or FH2 (open circles) pathway. At each profilin concentration, the contribution of each pathway to the overall rate of elongation was calculated using the average filament fluorescence intensity (see Fig. 3B) and the fluorescent fraction of profilin-bound actin. The data are plotted as a function of the concentration of both profilin (top axis) and profilin–actin (bottom axis). Error bars are standard errors of the mean fraction measured for at least 10 filaments. D, dependence of the overall barbed end elongation rate mediated by Bni1(FH1FH2)p (black circles), and the rates mediated via the FH1 and FH2 pathways (purple and orange circles, respectively) on the profilin–actin (bottom axis) and profilin (top axis) concentrations. Elongation rates mediated by Bni1(FH2)p (open squares) are also included for comparison. Error bars are standard errors of the mean elongation rate of at least 10 filaments.
Consistent with our measurements of Cdc12p-bound filaments, filaments polymerized by an FH1FH2 construct of Bni1p in the absence of profilin (i.e. exclusively through the FH2 pathway) were as bright as control filaments (Fig. 3, A and B; Bni1(FH1FH2)p). In the presence of profilin, Bni1p-bound filaments were dimmer than control filaments. Thus, Bni1p polymerizes filaments at least partially through its FH1 pathway in these conditions. At low (0.5–5 μm) profilin concentrations, the fluorescence of Bni1p-bound filaments exceeded the predicted intensity corresponding to the fraction of fluorescent profilin–actin (Fig. 3B). The difference between the fluorescence intensity of Bni1-bound filaments and the intensity corresponding to the fraction of fluorescent profilin–actin was maximal at 0.5 μm profilin and decreased as the profilin concentration increased, until these values became approximately equal at profilin concentrations exceeding 5 μm.
To rule out the possibility that changes in filament fluorescence intensity might arise from profilin-sensitive subunit incorporation via the FH2 pathway, we performed similar fluorescence measurements of filaments assembled by the FH2 domain of Bni1p (Fig. 3A; Bni1(FH2)p). Filaments assembled by Bni1(FH2)p were as bright as control filaments in all profilin conditions (Fig. 3B), suggesting that free and profilin-bound actin monomers are incorporated into filaments via the FH2 pathway with similar efficiency. Thus, differences between the fluorescence of formin-bound and control filaments must arise from the selective incorporation of unlabeled profilin–actin monomers via the FH1 pathway. Consistent with published results (19), the elongation rates of both control and Bni1(FH2)p-bound filaments decreased approximately linearly over the range of profilin concentrations used in our reactions, such that rates measured in the presence of 25 μm profilin were ∼40% slower than rates measured in the absence of profilin (Fig. 3D). Despite the decrease in overall elongation rates, these data indicate that the gating factor of Bni1p is insensitive to profilin.
We calculated the fraction of actin subunits polymerized by Bni1p through each pathway and found that although Bni1p mediates filament elongation exclusively via its FH2 pathway in the absence of profilin, polymerization proceeds via a combination of both the FH1 and FH2 pathways in the presence of profilin (Fig. 3C). As the profilin concentration increases, the concentration of profilin–actin also increases (Fig. 3C, top and bottom axes). As a result, the relative contribution of the FH1 pathway increases from 0 in the absence of profilin to ∼100% in the presence of 10 μm profilin. This change in flux through the FH1 pathway is coupled with a decrease in the contribution of the FH2 pathway from 100% in the absence of profilin to ∼0% at concentrations equal to or exceeding 10 μm profilin.
To determine how the flux of actin subunits through each pathway contributes to the overall rate of polymerization mediated by Bni1p, we multiplied our observed barbed end elongation rates by the fraction of polymerized actin that was incorporated via either the FH1 or the FH2 pathway at each profilin concentration (Fig. 3D). This revealed that although Bni1p assembles filaments solely through its FH2 pathway in the absence of profilin, the contribution of the FH2 pathway to the overall rate of elongation decreases steadily as the concentration of available profilin–actin increases. The rates of subunit addition mediated via the FH2 pathway in the presence of profilin–actin are slower than the rates of elongation mediated by Bni1(FH2)p (Fig. 3D). A Student's t test confirmed that the differences between rates mediated via the FH2 pathway and those mediated by Bni1(FH2)p are significant (p < 0.0001) in the presence of 2.5 μm and higher concentrations of profilin. This suggests that the two pathways constitute competitive modes of delivery of actin to the barbed end.
Subunit incorporation via the FH1 and FH2 pathways is insensitive to fluorescent labeling of actin
To test the robustness of our approach to measuring the flux of actin subunits polymerized through the FH1 and FH2 pathways, we varied the fraction of labeled actin in our reactions. The rate of actin elongation mediated by Bni1p slowed as the fraction of labeled actin monomers increased, consistent with published studies of filaments with free barbed ends (26) and with kinetic models of formin-mediated elongation, which propose that the efficiency of FH1-mediated subunit incorporation depends on the concentration of profilin–actin (19, 21). The rate of elongation is more sensitive to the fraction of labeled actin monomers in reactions containing 1 μm profilin than in the presence of higher amounts of profilin. This suggests that free profilin can attenuate the dependence of the rate of elongation on the profilin–actin concentration via competition for binding to the polyproline tracts in the FH1 domain. (Fig. 4A). Despite this effect on the rate of filament elongation, the fraction of actin subunits incorporated into filaments through each pathway was insensitive to the amount of labeled actin present in our reactions (Fig. 4B). Thus, our approach for measuring the relative contributions of the FH1 and FH2 pathways to elongation is not influenced by the ratio of labeled and unlabeled actin monomers included in each reaction.
Figure 4.
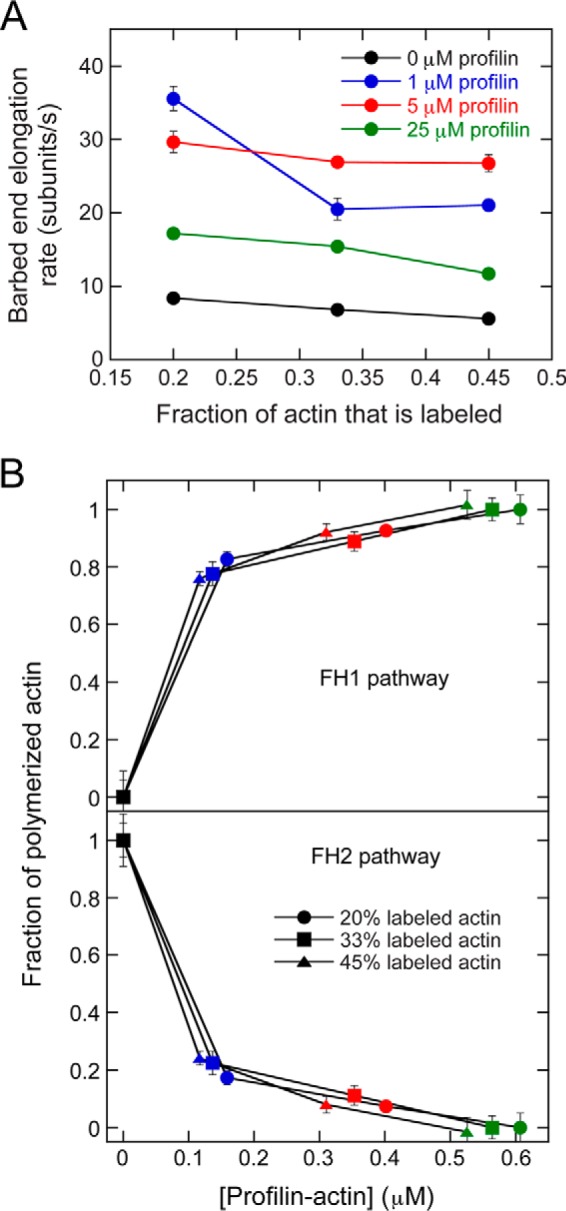
The contribution of the FH2 pathway to formin-mediated filament elongation is independent of the fraction of fluorescently labeled actin. The data were collected by TIRF microscopy. A, the dependence of the barbed end elongation rate mediated by Bni1(FH1FH2)p on the fraction of fluorescently labeled actin. The rates were measured in the absence (black) or presence of 1 μm (blue), 5 μm (red), or 25 μm (green) S. cerevisiae profilin. Error bars are standard errors of the mean elongation rate of at least 10 filaments. B, fraction of actin subunits incorporated into filaments via the FH1 (top panel) and FH2 (bottom panel) pathways in the presence of a range of fractions of fluorescently labeled actin, plotted against the concentration of profilin–actin. Elongation rates were measured in the presence of 20% (circles), 33% (squares), or 45% (triangles) fluorescently labeled actin and a range of profilin concentrations (colors are the same as in A). Error bars are standard errors of the mean fraction measured for of at least 10 filaments.
An increase in the flux through the FH2 pathway compensates for a reduction in the efficiency of the FH1 pathway
We next sought to test whether changes in the polymerization efficiency of Bni1p would alter the fractions of actin subunits incorporated through the FH1 and FH2 pathways. We did so by altering the reaction landscape in two ways.
Global changes in polymerization efficiency
First, we investigated the effects of altering the overall rate of actin polymerization mediated by Bni1p by varying the concentration of actin monomers in our reactions (Fig. 5A). The rate of filament elongation is proportional to the concentration of available monomers, so we expected that a decrease in the actin concentration would slow the elongation of Bni1p-bound filaments. Consistent with previous studies, we found that FH2 gating was insensitive to actin concentration (19, 22), so Bni1p-bound filaments elongated at approximately half the rate of filaments with free barbed ends independent of the actin concentration. Inclusion of profilin sped Bni1p-mediated elongation in a concentration-dependent manner. Notably, the concentration of profilin that produced peak elongation rates was actin concentration-dependent, with lower actin concentrations requiring less profilin to produce optimal rates (Fig. 5A). Because of profilin's inhibitory effect on filament nucleation, we were unable to visualize sufficient numbers of actin filaments for analysis in reactions containing 0.25 μm actin and 25 μm profilin. Therefore, we analyzed filament elongation in the presence of up to 17.5 μm profilin in reactions containing 0.25 μm actin.
Figure 5.

Competition for polyproline tract binding by profilin and profilin–actin decreases the relative contribution of the FH1 pathway to elongation. The data were collected by TIRF microscopy. A, dependence of the barbed end elongation rate mediated by Bni1(FH1FH2)p on the concentration of profilin. The data were collected in the presence of 0.75 μm (black), 0.5 μm (blue), and 0.25 μm (red) actin. Error bars are standard errors of the mean elongation rate of at least 10 filaments. B, dependence of the fluorescence intensities of formin-bound filaments on the concentration of profilin–actin. Fluorescence intensities were measured for filaments polymerized in the presence of 0.25 (red), 0.5 μm (blue), or 0.75 μm (black) actin and were normalized to the intensity of filaments that were not formin-bound. Error bars are standard errors of the mean fluorescence intensity of at least 10 filaments. C, fraction of actin subunits incorporated into filaments via the FH1 (top panel) or FH2 (bottom panel) pathways of Bni1(FH1FH2)p in the presence of 0.25 μm (red), 0.5 μm (blue), or 0.75 μm (black) actin, plotted against the concentration of profilin–actin. Error bars are standard errors of the mean fraction measured for at least 10 filaments. D and E, elongation rates mediated by Bni1(FH1FH2)p (black circles), and rates mediated via the FH1 (purple circles) or FH2 (orange circles) pathways, in the presence of 0.25 μm (D) or 0.5 μm (E) actin monomers, plotted against the concentration of profilin–actin. Error bars are standard errors of the mean elongation rate measured for at least 10 filaments.
We quantified the fluorescence of Bni1p-assembled filaments in each actin concentration condition (Fig. 5B) and determined the fraction of filamentous actin polymerized by Bni1p via its FH1 or FH2 pathway (Fig. 5C). To highlight differences among the data sets, we plotted both the filament fluorescence and the fraction of polymerized actin incorporated via each pathway as functions of the concentration of profilin–actin.
Consistent with the shift in the profilin concentration required to produce peak elongation rates, lower profilin–actin concentrations were required to produce dimly fluorescent Bni1p-bound filaments (Fig. 5B) by stimulating efficient actin subunit incorporation through the FH1 pathway in the presence of low actin concentrations (Fig. 5, C–E). The FH1 pathway became a less efficient mode of elongation at higher profilin–actin concentrations, likely because of the vast excess of free profilin that competed with profilin–actin for binding to FH1's polyproline tracts in these reactions. These results suggest that the relative efficiencies of the FH1 and FH2 pathways depend not only on the concentration of profilin–actin but also on the availability of free actin monomers, which can only be incorporated via the FH2 pathway, and on the concentration of free profilin, which inhibits the FH1 pathway.
Targeted changes in FH1 efficiency
We next sought to test the limits of the competition for subunit addition between the FH1 and FH2 pathways by introducing mutations into the FH1 domain of Bni1p that decrease its interactions with profilin. We analyzed actin filaments assembled by two variants of Bni1p, in which we replaced either two or three of the four native polyproline tracts in the FH1 domain with polyglycine/serine chains that cannot bind profilin (Fig. 6A). Bni1(PCPDFH2)p retains the two polyproline tracts located closest to the FH2 domain, whereas Bni1(PDFH2)p retains only the most C-terminal tract. Published studies have shown that although mutations in the FH1 domain of Bni1p do not impact FH2-mediated gating, variants containing fewer tracts elongate filaments more slowly than does the WT protein (19). These variants therefore provide a means to test whether the effects of FH1 domain mutations on the rate of polymerization arise solely from changes in the flux through the FH1 pathway or whether mutations in the FH1 domain also affect polymerization via the FH2 pathway.
Figure 6.

Reducing the efficiency of the FH1 domain promotes elongation through the FH2 pathway. The data were collected by TIRF microscopy. A, schematic representation of the FH1 and FH2 domains of Bni1p and variants thereof. The FH1 domain of Bni1p contains four polyproline tracts (purple ovals), which are sites for profilin binding. The variants were constructed by replacing multiple polyproline tracts with polyglycine/serine sequences (white ovals with X). The Bni1(PCPDFH2)p variant retains the two tracts located closest to the FH2 domain, whereas the Bni1(PDFH2)p variant retains only the C-terminal tract. B, representative fluorescence micrographs of actin filaments elongating in the presence of Bni1(FH1FH2)p, Bni1(PCPDFH2)p, or Bni1(PDFH2)p and 1 μm S. cerevisiae profilin. Asterisks indicate the barbed ends of filaments bound by a formin. Filaments lacking an asterisk are not formin-bound. C, dependence of the fluorescence intensities of filaments polymerized by Bni1(PCPDFH2)p (blue triangles) or Bni1(PDFH2)p (purple squares) on the concentration of profilin–actin. The data for WT Bni1(FH1FH2)p are also shown for comparison (black circles). Fluorescence intensities were normalized to the intensities of filaments that were not formin-bound. Error bars are standard errors of the mean fluorescence intensity of at least 10 filaments. D, fraction of actin subunits incorporated into filaments via the FH1 (top panel) or FH2 (bottom panel) pathways of Bni1(PCPDFH2)p (blue triangles) or Bni1(PDFH2)p (purple squares). Data for WT Bni1(FH1FH2)p are also included for comparison (black circles). Error bars are standard errors of the mean fraction measured for at least 10 filaments. E and F, elongation rates (black circles) mediated by Bni1(PDFH2)p (E) or Bni1(PCPDFH2)p (F) and rates mediated via the FH1 (purple circles) or FH2 (orange circles) pathways, plotted against the concentration of profilin–actin. Error bars are standard errors of the mean elongation rate measured for at least 10 filaments.
We confirmed that FH2 gating is unaffected in both Bni1p variants and that these variants mediate slower rates of elongation than does WT Bni1p in the presence of profilin (Fig. S1A). Filaments assembled by the variants were brighter than those polymerized by the WT protein (Fig. 6, B and C). Bni1(PDFH2)p-associated filaments were also brighter than Bni1(PCPDFH2)p-associated filaments. Thus, at each profilin concentration, Bni1(PDFH2)p incorporated a larger fraction of fluorescently labeled actin subunits than did Bni1(PCPDFH2)p, which in turn incorporated more fluorescent actin than did WT Bni1p.
Compared with WT Bni1p, the FH2 pathway played a larger role in polymerization mediated by the variants at all profilin and profilin–actin concentrations. The fraction of actin subunits polymerized by Bni1(PDFH2)p via its FH2 pathway decreased from 1, as measured in the absence of profilin, to ∼0.3, measured in the presence of 2.5–17.5 μm profilin (Fig. 6, D and E, and Fig. S1B). The fraction polymerized via the FH2 pathway of Bni1(PCPDFH2)p was smaller at each profilin concentration and reached a minimum of ∼0.05 in the presence of 17.5 μm profilin (Fig. 6, D and F, and Fig. S1B). This suggests that the FH2 pathway plays a more significant role in filament elongation mediated by formins with inefficient FH1 domains.
In the presence of 25 μm profilin, the fraction of actin subunits incorporated via the FH2 pathway increased sharply for both variants, consistent with a reduction in the efficiency of the FH1 pathway arising from competition between free profilin and profilin–actin for binding to the polyproline tracts. This increase was not observed for the WT protein, suggesting that the extent of competition scales with the number of available profilin-binding sites (Fig. 6, D–F).
Taken together, these two sets of experiments reveal that the fractions of actin subunits incorporated through the FH1 and FH2 pathways depend not only on the concentration of profilin–actin, but also on the concentrations of free actin and free profilin, as well as the number of binding sites for profilin–actin in the FH1 domain. This relationship among reaction parameters ensures that an increase in the flux of actin subunits through the FH2 pathway accompanies and partially compensates for a decrease in the efficiency of profilin–actin transfer from the FH1 domain, which may arise via a decrease in the available profilin–actin concentration, an increase in the free profilin concentration, or a mutation in the FH1 domain itself.
Discussion
Formins mediate actin filament elongation by regulating the rate at which incoming subunits successfully bind barbed ends. This regulation occurs via two major pathways. The FH2 pathway enables direct addition of actin monomers from the bulk solution via conformational changes in the dimeric, barbed end-bound FH2 domains. The FH1 pathway requires the formation of profilin–actin complexes, which bind and are transferred to the barbed end from polyproline tracts located within the flexible FH1 domains (Fig. 1A). We measured the flux of actin subunit addition that proceeds through these two pathways during filament elongation mediated by two formins: Cdc12p, which has an extremely low gating factor and is predicted to elongate filaments solely via the FH1 pathway in the presence of profilin, and Bni1p, which has an intermediate gating factor and could possibly elongate filaments through a combination of both pathways. We found that the fraction of filament polymerization that occurs via each pathway is directly dependent on the efficiency of the other pathway, indicating that these pathways are competitive mechanisms for subunit addition by formins. This competition allows formins to compensate for changes in the efficiency of one pathway by adjusting the frequency of subunit addition via the other.
A robust method for accurate determination of the flux through the FH1 and FH2 pathways
To determine the relative contributions of the FH1 and FH2 pathways to overall rates of elongation mediated by formins, we measured the fluorescence intensities of actin filaments assembled from a mixture of unlabeled actin monomers and monomers labeled with Oregon Green 488 at cysteine 374. By comparing the fluorescence of filaments polymerized by formins to that of control filaments that assembled spontaneously, we quantified the biased incorporation of unlabeled actin via the profilin-dependent FH1 pathway. The relative fraction of unlabeled monomers that were incorporated into filaments depended on both the profilin concentration and the affinity of the profilin–actin interaction (Fig. 2).
We first validated our approach by performing experiments with Cdc12p. When normalized to the fluorescence of spontaneously assembled filaments, the average fluorescence intensity of filaments polymerized by this formin was equal to the fraction of profilin-bound actin that was fluorescent in each reaction (Fig. 2, B and C). This relationship held true when we repeated the measurements using S. cerevisiae profilin, which has a much weaker affinity for actin than does S. pombe profilin. Consistent with the very low gating factor for Cdc12p, which permits only extremely slow FH2-mediated binding of subunits to barbed ends, our results support a model in which Cdc12p mediates filament elongation solely via its FH1 pathway in the presence of profilin.
We further tested the robustness of our fluorescence-based approach by varying the fraction of labeled subunits in polymerization reactions that included the S. cerevisiae formin Bni1p. We found that the fraction of labeled actin present in the reactions does not influence our measurements of the contributions of the FH1 and FH2 pathways to polymerization (Fig. 4B). We therefore propose that our method provides robust and accurate measurements of the flux through the two major pathways for subunit incorporation by formins.
A balance among reaction parameters controls the relative flux of actin subunits through each pathway
Unlike Cdc12p, which elongates actin filaments exclusively via its FH1 pathway in the presence of profilin, we found that Bni1p can mediate polymerization via both its FH1 and FH2 pathways under certain conditions. The magnitude of the flux through the FH1 pathway depends on the concentration of profilin–actin (Figs. 3C and 7A). In reactions containing 0.75 μm actin and at least 10 μm profilin, the FH1 pathway contributes all of the subunits incorporated via Bni1p during elongation. Thus, under certain conditions, the extent of competition for actin subunit incorporation via the FH1 and FH2 pathways can result in the full attenuation of one pathway in favor of the other.
Figure 7.

A balance among reaction parameters controls the relative flux through the FH1 and FH2 pathways. A and B, heat maps depicting the flux of subunit addition mediated by Bni1p via the FH1 and FH2 pathways in a range of profilin–actin and free profilin concentrations (A) and profilin concentrations and numbers of polyproline tracts present in the FH1 domain (B). Both plots were constructed using experimental data and smoothed by interpolation. Dark purple and yellow data correspond to polymerization mediated solely through the FH1 or FH2 pathway, respectively (see color bar on right).
Perturbation of the polymerization conditions revealed that the relative fluxes through the FH1 and FH2 pathways depend on the specific combination of parameters (Fig. 7). For example, the concentration of profilin–actin complexes depends on the concentrations of both actin and profilin; a change in the concentration of any of these species in turn modulates polymerization via the FH1 pathway (Fig. 7A). Similarly, deletion of profilin-binding sites decreases the relative efficiency of the FH1 domain (Fig. 7B). In all cases, when the FH1 pathway becomes a less efficient mode for polymerization, the FH2 pathway becomes relatively more efficient. In this manner, alleviation of the competition between the two pathways ultimately tempers the effect of a reduction in FH1 efficiency on the overall rate of elongation.
The role of gating in regulating the flux through the FH1 and FH2 pathways for elongation
Our experiments with Cdc12p and Bni1p suggest a role for gating in regulating the extent of competition for actin subunit addition among the FH1 and FH2 pathways. First, we found that any amount of profilin–actin can cause elongation to occur exclusively via the FH1 pathway when the gating factor is extremely small, as is the case for Cdc12p. In contrast, larger concentrations of profilin–actin were necessary to fully inhibit subunit addition through the FH2 pathway of Bni1p. This suggests a positive correlation between a formin's gating factor and the fraction of subunit addition that occurs via the FH2 pathway in the presence of profilin. Formins with larger gating factors might therefore require higher concentrations of profilin–actin to promote sufficient actin subunit incorporation via the FH1 pathway to compete with the FH2 pathway. On the other hand, the efficiency of the FH1 pathway also increases with the number of polyproline tracts present in the FH1 domain (Fig. 6C), suggesting that the role played by the FH2 pathway during polymerization might depend on the FH1 domain sequence in addition to the gating factor (Fig. 7B).
The relationship between the FH1 and FH2 pathways is likely more complex for formins whose gating factors are sensitive to profilin concentration, such as mDia1 (22). In these cases, we would expect the rate of actin subunit incorporation via the FH2 pathway to more strongly depend on the concentrations of profilin–actin and excess profilin than we observed for Bni1p. In contrast, chimeric formin constructs consisting of FH1 and FH2 domains from different formins have been demonstrated to polymerize actin less efficiently than their WT counterparts in the presence of profilin (35). As a result, we would expect chimeric formins to incorporate a larger fraction of actin subunits through the FH2 pathway in the presence of profilin than would be observed for more efficient, WT formins.
Biological implications of the competition among the FH1 and FH2 pathways during filament elongation mediated by formins
The competition between the two pathways for filament elongation imposes both lower and upper limits to polymerization rates mediated by formins. The lower limit for elongation is the rate mediated by the FH2 pathway and is dictated by the gating factor of the formin. This rate is observed in conditions where the FH1 pathway is turned off, such as in the absence of profilin or in the presence of very high concentrations of free profilin (Fig. 7A). As such, this lower limit for elongation ensures a nonzero rate of polymerization even in conditions that are unfavorable for FH1-mediated subunit addition. Our experiments with Cdc12p and Bni1p suggest that the upper limit for elongation is likely to be the rate mediated by the FH1 pathway when it is “maximally efficient” and can therefore fully inhibit the FH2 pathway. This rate increases in proportion to the available profilin–actin concentration and is inversely proportional to the concentration of free profilin and free actin, which inhibit the FH1 pathway and activate the FH2 pathway, respectively. This upper limit establishes a narrower range of formin-mediated polymerization rates than would be observed if the FH1 and FH2 pathways contributed to elongation in an additive manner. The competitive mechanism for subunit incorporation might therefore provide a polymerization “buffer” that minimizes the effects of mutations in the FH1 domain, changes in the local concentration of profilin—actin, or the application of linear force, all of which could reduce the flux through the FH1 pathway on the overall rate of formin-mediated elongation.
Experimental procedures
Protein purification
Constructs encoding the FH1 and FH2 domains of Cdc12p (residues 882–1375) and Bni1p (residues 1227–1776 and variants thereof) were cloned into pGEX-4T-3 plasmids (GE Healthcare Life Sciences) and expressed in 1-liter cultures of BL21 DE3 RP Codon Plus cells (Agilent Technologies). Bni1p constructs also encoded an N-terminal TEV protease recognition sequence and a C-terminal His6 tag in addition to the N-terminal glutathione S-transferase tag that is included in pGEX vectors. Unless otherwise specified, chemicals were purchased from Millipore Sigma.
For purification of Bni1p constructs, the cells were lysed by sonication in 50 mm Tris (pH 8.0), 500 mm NaCl, 1 mm DTT, and centrifuged at 16,000 rpm for 40 min to remove insoluble material. Each supernatant was incubated with 2 ml of glutathione–Sepharose resin (Gold Biotechnology) with rotation for 1 h and then transferred into an empty glass column. The resin was washed with 20 ml of lysis buffer, followed by 20 ml of low-salt wash buffer (50 mm Tris, pH 8.0, 100 mm NaCl, 1 mm DTT). Each protein was then eluted with 6 ml of 100 mm GSH (pH 8.0) in low-salt wash buffer, and the eluted protein was incubated with ∼2–5 μm TEV protease overnight at 4 °C. Bni1p constructs were purified from cleaved glutathione S-transferase and TEV protease via nickel affinity chromatography, concentrated using 30,000 molecular-weight cutoff spin columns (Millipore), dialyzed into KMEI (50 mm KCl, 1 mm MgCl2, 1 mm EGTA, 10 mm imidazole, pH 7.0) with 1 mm DTT, flash-frozen in 50- and 500-μl aliquots, and stored at −80 °C.
The cells expressing Cdc12p were lysed by sonication in 20 mm HEPES (pH 7.4), 200 mm NaCl, 10% glycerol, 5 mm DTT and centrifuged at 16,000 rpm for 40 min to clarify the lysate. The supernatant was incubated with glutathione–Sepharose resin for 1 h with rotation at 4 °C, transferred to an empty column, and washed with lysis buffer. Fifty units of thrombin were added to the resin followed by an overnight incubation at 4 °C. Cleaved Cdc12p was collected and dialyzed into 20 mm Tris (pH 8.5), 25 mm NaCl, 5% glycerol, 1 mm DTT. The protein was then applied to 20 ml of DE52 anion exchange resin and eluted with a linear gradient of 25–500 mm NaCl. Fractions containing the Cdc12p were pooled; dialyzed into 20 mm HEPES (pH 7.4), 200 mm NaCl, 5% glycerol, 1 mm DTT; flash-frozen; and stored at −80 °C. We used ProtParam (http://web.expasy.org/protparam (36)) to calculate extinction coefficients for Bni1p and Cdc12p constructs.
S. cerevisiae and S. pombe profilin were expressed from pMW172 vectors in BL21 DE3 pLysS cells (19, 24). The cells were lysed by sonication in 20 mm Tris (pH 7.5), 150 mm KCl, 0.2 mm DTT and centrifuged for 30 min at 16,000 rpm to clarify the lysate. Supernatants were applied to 10 ml of poly-l-proline resin, washed with lysis buffer followed by lysis buffer with 2 m urea, and profilin was eluted with lysis buffer with 7 m urea. Eluted profilin was dialyzed into KMEI + 1 mm DTT and stored at 4 °C. We used extinction coefficients of 19,060 m−1 cm−1 and 19,940 m−1 cm−1 at λ = 280 nm for S. cerevisiae and S. pombe profilin, respectively.
Skeletal muscle actin was purified from an acetone powder prepared from frozen chicken breasts (Trader Joe's) by one cycle of polymerization and depolymerization (37). Monomers were gel-filtered on S-300 resin in G-buffer (2 mm Tris, pH 8.0, 0.2 mm ATP, 0.5 mm DTT, 0.1 mm CaCl2). Actin was labeled on cysteine 374 with Oregon Green 488 iodoacetamide (Thermo Fisher Scientific) (26). Labeled and unlabeled actin monomers were stored at 4 °C. We used an extinction coefficient of 26,000 m−1 cm−1 at λ = 290 nm for unlabeled actin. For labeled actin, we used an extinction coefficient of 78,000 m−1 cm−1 at λ = 491 nm to measure the concentration of Oregon Green and the following relation to calculate the concentration of actin in each fraction: [total actin] (μm) = [A290 − (A491 × 0.171)]/26,000 m−1 cm−1.
Flow-chamber preparation
Glass coverslips (22 mm × 50 mm; Fisher Scientific) and slides were sonicated for 45 min in 2% Hellmanex III, rinsed extensively with water, and sonicated in ethanol. Chambers were prepared as described previously (19). Briefly, two thin strips of parafilm were applied longitudinally across a coverslip, creating a 4-mm channel. A slide was positioned perpendicularly to the coverslip, and pressure was applied to create a seal. Chambers were flamed, stored at room temperature, and used within 1 week.
Prior to each polymerization experiment, the chamber was incubated with a solution of 0.5% Tween 20 in High-Salt TBS (HS-TBS; 50 mm Tris, pH 7.5, 600 mm NaCl) and ∼1 μm N-ethylmaleimide–inactivated chicken skeletal muscle myosin (26) in HS-TBS and 100 mg/ml BSA in HS-TBS. The chamber was washed with HS-TBS between each incubation step and with KMEI immediately prior to introduction of a polymerization reaction.
To initiate polymerization, mixtures of unlabeled and labeled Ca2+-ATP actin were converted to Mg2+-ATP actin by addition of 0.25 mm MgCl2, 1 mm EGTA to exchange Ca2+ ions for Mg2+. After a 5-min incubation, polymerization was initiated by addition of 2× microscopy buffer (1× microscopy buffer: 10 mm imidazole, pH 7.0, 50 mm KCl, 1 mm MgCl2, 1 mm EGTA, 50 mm DTT, 0.2 mm ATP, 15 mm glucose, 20 μg/ml catalase, 100 μg/ml glucose oxidase, 0.5% (w/v) methylcellulose (4,000 cP at 2%)) with formin and profilin. Samples were immediately introduced into the chamber upon initiation of polymerization.
Microscopy and data analysis
We generated time-lapse images of elongating actin filaments by TIRF microscopy on an Olympus Ti83 motorized microscope equipped with a CellTIRF system that enabled through-objective TIRF microscopy using a 60 × 1.49 N.A. objective and a 488-nm laser. We collected images every 10 s using a Hamamatsu C9100–23B ImagEM X2 EMCCD camera and CellSens Dimension software (Olympus).
Time-lapse movies of growing filaments were processed with ImageJ software (National Institutes of Health (38)). For each reaction, we measured changes in length over time for 10–15 formin-bound and 10 control (i.e. not formin-bound) filaments, typically from at least 15 frames of imaging over a span of at least 200 s. To determine elongation rates, linear fits were applied to plots of filament length over time using Kaleidagraph software (Synergy Software). We measured the fluorescence intensity of formin-bound and control filaments by performing line scans along filaments and neighboring background regions using ImageJ. To avoid any possible influence of photobleaching on our measurements, we performed the line scans along 5–10-μm stretches at filament barbed ends.
Calculations of FH1 pathway contributions to elongation
In each polymerization reaction, the fluorescence intensities of formin-bound filaments were normalized to the fluorescence of control filaments. The fraction of unlabeled actin subunits incorporated via the FH1 pathway (FH1Black) was calculated using the following relation,
| (Eq. 1) |
where FluorescenceFormin-bound is the normalized fluorescence intensity of the formin-bound filaments in the reaction.
The fraction of labeled actin subunits incorporated via the FH1 pathway (FH1Green) was then calculated using the following equation,
| (Eq. 2) |
where Green PA is the fraction of profilin-bound actin that is labeled, and Black PA is the fraction of profilin-bound actin that is unlabeled. Both of these values were calculated using the published affinities of S. pombe or S. cerevisiae profilin for unlabeled actin and actin that is labeled at cysteine 374 (0.2 and 2 μm for S. pombe profilin, and 2.9 and 29 μm for S. cerevisiae profilin (32, 33)).
The total fraction of actin subunits incorporated via the FH1 pathway was then calculated using the following equation.
| (Eq. 3) |
The fraction of actin subunits incorporated via the FH2 pathway (FH2Fraction) was obtained by subtracting FH1Fraction from 1. Polymerization rates mediated through each pathway were obtained by multiplying FH1Fraction and FH2Fraction by the overall elongation rate mediated by the formins in each reaction.
Author contributions
L. A. S., M. E. Z., and N. C. formal analysis; L. A. S., M. E. Z., and N. C. investigation; L. A. S., M. E. Z., and N. C. writing-review and editing; N. C. conceptualization; N. C. supervision; N. C. funding acquisition; N. C. methodology; N. C. writing-original draft; N. C. project administration.
Supplementary Material
Acknowledgment
We thank Melissa Gardner for helpful comments on the manuscript.
This work was supported by National Institutes of Health Grant GM 122787. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
- FH
- formin homology
- TIRF
- total internal reflection fluorescence
- TEV
- tobacco etch virus.
References
- 1. Chang F., Drubin D., and Nurse P. (1997) cdc12p, a protein required for cytokinesis in fission yeast, is a component of the cell division ring and interacts with profilin. J. Cell Biol. 137, 169–182 10.1083/jcb.137.1.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pellegrin S., and Mellor H. (2005) The Rho family GTPase Rif induces filopodia through mDia2. Curr. Biol. 15, 129–133 10.1016/j.cub.2005.01.011 [DOI] [PubMed] [Google Scholar]
- 3. Watanabe N., Kato T., Fujita A., Ishizaki T., and Narumiya S. (1999) Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat. Cell Biol. 1, 136–143 10.1038/11056 [DOI] [PubMed] [Google Scholar]
- 4. Kobielak A., Pasolli H. A., and Fuchs E. (2004) Mammalian formin-1 participates in adherens junctions and polymerization of linear actin cables. Nat. Cell Biol. 6, 21–30 10.1038/ncb1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Feierbach B., and Chang F. (2001) Roles of the fission yeast formin for3p in cell polarity, actin cable formation and symmetric cell division. Curr. Biol. 11, 1656–1665 10.1016/S0960-9822(01)00525-5 [DOI] [PubMed] [Google Scholar]
- 6. Nakano K., Imai J., Arai R., Toh-E A., Matsui Y., and Mabuchi I. (2002) The small GTPase Rho3 and the diaphanous/formin For3 function in polarized cell growth in fission yeast. J. Cell Sci. 115, 4629–4639 10.1242/jcs.00150 [DOI] [PubMed] [Google Scholar]
- 7. Petersen J., Nielsen O., Egel R., and Hagan I. M. (1998) FH3, a domain found in formins, targets the fission yeast formin Fus1 to the projection tip during conjugation. J. Cell Biol. 141, 1217–1228 10.1083/jcb.141.5.1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schönichen A., and Geyer M. (2010) Fifteen formins for an actin filament: a molecular view on the regulation of human formins. Biochim. Biophys. Acta 1803, 152–163 10.1016/j.bbamcr.2010.01.014 [DOI] [PubMed] [Google Scholar]
- 9. Boyer O., Nevo F., Plaisier E., Funalot B., Gribouval O., Benoit G., Huynh Cong E., Arrondel C., Tête M. J., Montjean R., Richard L., Karras A., Pouteil-Noble C., Balafrej L., Bonnardeaux A., et al. (2011) INF2 mutations in Charcot-Marie-Tooth disease with glomerulopathy. N. Engl. J. Med. 365, 2377–2388 10.1056/NEJMoa1109122 [DOI] [PubMed] [Google Scholar]
- 10. Brown E. J., Schlöndorff J. S., Becker D. J., Tsukaguchi H., Tonna S. J., Uscinski A. L., Higgs H. N., Henderson J. M., and Pollak M. R. (2010) Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat. Genet. 42, 72–76 10.1038/ng.505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DeWard A. D., Eisenmann K. M., Matheson S. F., and Alberts A. S. (2010) The role of formins in human disease. Biochim. Biophys. Acta 1803, 226–233 10.1016/j.bbamcr.2009.11.006 [DOI] [PubMed] [Google Scholar]
- 12. Ercan-Sencicek A. G., Jambi S., Franjic D., Nishimura S., Li M., El-Fishawy P., Morgan T. M., Sanders S. J., Bilguvar K., Suri M., Johnson M. H., Gupta A. R., Yuksel Z., Mane S., Grigorenko E., et al. (2015) Homozygous loss of DIAPH1 is a novel cause of microcephaly in humans. Eur. J. Hum. Genet. 23, 165–172 10.1038/ejhg.2014.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pruyne D., Evangelista M., Yang C., Bi E., Zigmond S., Bretscher A., and Boone C. (2002) Role of formins in actin assembly: nucleation and barbed-end association. Science 297, 612–615 10.1126/science.1072309 [DOI] [PubMed] [Google Scholar]
- 14. Moseley J. B., Sagot I., Manning A. L., Xu Y., Eck M. J., Pellman D., and Goode B. L. (2004) A conserved mechanism for Bni1- and mDia1-induced actin assembly and dual regulation of Bni1 by Bud6 and profilin. Mol. Biol. Cell 15, 896–907 10.1091/mbc.e03-08-0621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pring M., Evangelista M., Boone C., Yang C., and Zigmond S. H. (2003) Mechanism of formin-induced nucleation of actin filaments. Biochemistry 42, 486–496 10.1021/bi026520j [DOI] [PubMed] [Google Scholar]
- 16. Michelot A., Guérin C., Huang S., Ingouff M., Richard S., Rodiuc N., Staiger C. J., and Blanchoin L. (2005) The formin homology 1 domain modulates the actin nucleation and bundling activity of Arabidopsis FORMIN1. Plant Cell 17, 2296–2313 10.1105/tpc.105.030908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu Y., Moseley J. B., Sagot I., Poy F., Pellman D., Goode B. L., and Eck M. J. (2004) Crystal structures of a formin homology-2 domain reveal a tethered dimer architecture. Cell 116, 711–723 10.1016/S0092-8674(04)00210-7 [DOI] [PubMed] [Google Scholar]
- 18. Otomo T., Tomchick D. R., Otomo C., Panchal S. C., Machius M., and Rosen M. K. (2005) Structural basis of actin filament nucleation and processive capping by a formin homology 2 domain. Nature 433, 488–494 10.1038/nature03251 [DOI] [PubMed] [Google Scholar]
- 19. Paul A. S., and Pollard T. D. (2008) The role of the FH1 domain and profilin in formin-mediated actin-filament elongation and nucleation. Curr. Biol. 18, 9–19 10.1016/j.cub.2007.11.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paul A. S., and Pollard T. D. (2009) Review of the mechanism of processive actin filament elongation by formins. Cell Motil. Cytoskeleton 66, 606–617 10.1002/cm.20379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vavylonis D., Kovar D. R., O'Shaughnessy B., and Pollard T. D. (2006) Model of formin-associated actin filament elongation. Mol. Cell 21, 455–466 10.1016/j.molcel.2006.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kovar D. R., Harris E. S., Mahaffy R., Higgs H. N., and Pollard T. D. (2006) Control of the assembly of ATP- and ADP-actin by formins and profilin. Cell 124, 423–435 10.1016/j.cell.2005.11.038 [DOI] [PubMed] [Google Scholar]
- 23. Kovar D. R., Kuhn J. R., Tichy A. L., and Pollard T. D. (2003) The fission yeast cytokinesis formin Cdc12p is a barbed end actin filament capping protein gated by profilin. J. Cell Biol. 161, 875–887 10.1083/jcb.200211078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Courtemanche N., and Pollard T. D. (2012) Determinants of formin homology 1 (FH1) domain function in actin filament elongation by formins. J. Biol. Chem. 287, 7812–7820 10.1074/jbc.M111.322958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Higgs H. N. (2005) Formin proteins: a domain-based approach. Trends Biochem. Sci. 30, 342–353 10.1016/j.tibs.2005.04.014 [DOI] [PubMed] [Google Scholar]
- 26. Kuhn J. R., and Pollard T. D. (2005) Real-time measurements of actin filament polymerization by total internal reflection fluorescence microscopy. Biophys. J. 88, 1387–1402 10.1529/biophysj.104.047399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kabsch W., Mannherz H. G., Suck D., Pai E. F., and Holmes K. C. (1990) Atomic structure of the actin:DNase I complex. Nature 347, 37–44 10.1038/347037a0 [DOI] [PubMed] [Google Scholar]
- 28. Oda T., Iwasa M., Aihara T., Maéda Y., and Narita A. (2009) The nature of the globular- to fibrous-actin transition. Nature 457, 441–445 10.1038/nature07685 [DOI] [PubMed] [Google Scholar]
- 29. Schutt C. E., Myslik J. C., Rozycki M. D., Goonesekere N. C., and Lindberg U. (1993) The structure of crystalline profilin-β-actin. Nature 365, 810–816 10.1038/365810a0 [DOI] [PubMed] [Google Scholar]
- 30. Chik J. K., Lindberg U., and Schutt C. E. (1996) The structure of an open state of β-actin at 2.65 A resolution. J. Mol. Biol. 263, 607–623 10.1006/jmbi.1996.0602 [DOI] [PubMed] [Google Scholar]
- 31. Vinson V. K., De La Cruz E. M., Higgs H. N., and Pollard T. D. (1998) Interactions of Acanthamoeba profilin with actin and nucleotides bound to actin. Biochemistry 37, 10871–10880 10.1021/bi980093l [DOI] [PubMed] [Google Scholar]
- 32. Eads J. C., Mahoney N. M., Vorobiev S., Bresnick A. R., Wen K. K., Rubenstein P. A., Haarer B. K., and Almo S. C. (1998) Structure determination and characterization of Saccharomyces cerevisiae profilin. Biochemistry 37, 11171–11181 10.1021/bi9720033 [DOI] [PubMed] [Google Scholar]
- 33. Lu J., and Pollard T. D. (2001) Profilin binding to poly-l-proline and actin monomers along with ability to catalyze actin nucleotide exchange is required for viability of fission yeast. Mol. Biol. Cell 12, 1161–1175 10.1091/mbc.12.4.1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pollard T. D. (1986) Rate constants for the reactions of ATP-actin and ADP-actin with the ends of actin-filaments. J. Cell Biol. 103, 2747–2754 10.1083/jcb.103.6.2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neidt E. M., Scott B. J., and Kovar D. R. (2009) Formin differentially utilizes profilin isoforms to rapidly assemble actin filaments. J. Biol. Chem. 284, 673–684 10.1074/jbc.M804201200 [DOI] [PubMed] [Google Scholar]
- 36. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., and Bairoch A. (2005) Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook (Walker J. M., ed) pp. 571–607, Humana Press, Totowa, NY [Google Scholar]
- 37. Spudich J. A., and Watt S. (1971) The regulation of rabbit skeletal muscle contraction: I. biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 246, 4866–4871 [PubMed] [Google Scholar]
- 38. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.