

Abstract
The CTX-M β-lactamases have emerged as the most widespread extended-spectrum β-lactamases (ESBLs) in Gram-negative bacteria. These enzymes rapidly hydrolyze cefotaxime, but not the related cephalosporin, ceftazidime. ESBL variants have evolved, however, that provide enhanced ceftazidime resistance. We show here that a natural variant at a nonactive site, i.e. second-shell residue N106S, enhances enzyme stability but reduces catalytic efficiency for cefotaxime and ceftazidime and decreases resistance levels. However, when the N106S variant was combined with an active-site variant, D240G, that enhances enzyme catalytic efficiency, but decreases stability, the resultant double mutant exhibited higher resistance levels than predicted on the basis of the phenotypes of each variant. We found that this epistasis is due to compensatory effects, whereby increased stability provided by N106S overrides its cost of decreased catalytic activity. X-ray structures of the variant enzymes in complex with cefotaxime revealed conformational changes in the active-site loop spanning residues 103–106 that were caused by the N106S substitution and relieve steric strain to stabilize the enzyme, but also alter contacts with cefotaxime and thereby reduce catalytic activity. We noted that the 103–106 loop conformation in the N106S-containing variants is different from that of WT CTX-M but nearly identical to that of the non-ESBL, TEM-1 β-lactamase, having a serine at the 106 position. Therefore, residue 106 may serve as a “switch” that toggles the conformations of the 103–106 loop. When it is serine, the loop is in the non-ESBL, TEM-like conformation, and when it is asparagine, the loop is in a CTX-M–like, cefotaximase-favorable conformation.
Keywords: drug resistance, enzyme kinetics, enzyme catalysis, protein evolution, protein structure, protein stability, protein drug interaction, conformational change, antibiotic resistance, β-lactamase
Introduction
Increasing bacterial resistance to antibiotics is decreasing antibiotic treatment options and creating a threat to effective therapy. A common mechanism of resistance is the production of an enzyme that modifies the antibiotic such that it no longer binds its target. Examples of antibiotics subject to this mechanism of resistance include β-lactams, aminoglycosides, and chloramphenicol. The level of resistance conferred depends on in vivo bacterial expression levels as well as the catalytic activity and specificity of the enzymes. Because of this, the evolution of increased resistance can proceed via multiple pathways including increased gene dosage, promoter mutations, increased enzyme stability and solubility, altered catalytic properties, or a combination of these mechanisms. Here, we describe the mechanism by which evolved variants of CTX-M β-lactamase provide increased resistance to oxyimino-cephalosporin antibiotics by acquiring mutations that balance increased enzyme stability and expression levels with altered catalytic activity and substrate specificity.
Worldwide, β-lactams represent ∼65% of antibiotic usage and are a key component of antimicrobial therapy (1). The most common mechanism of resistance to these drugs in Gram-negative bacteria is the production of β-lactamase enzymes that catalyze the hydrolysis of the amide bond present in the β-lactam ring to create a product that no longer binds the transpeptidase target enzymes involved in cell wall biosynthesis (2). CTX-M β-lactamases are grouped into four classes (A–D) based on primary amino acid sequence homology. Class A β-lactamases are all serine hydrolases (3), but they differ with respect to substrate specificity. The TEM-type enzymes, including TEM-1 and SHV-1, efficiently hydrolyze only penicillins and early generation cephalosporins. After the introduction of third-generation oxyimino-cephalosporins, such as cefotaxime and ceftazidime in the 1980s, the CTX-M family of extended spectrum β-lactamases (ESBLs) 3 emerged to confer antibiotic resistance (4, 5). CTX-M β-lactamases, which are ∼40% identical to TEM-1 (6), are ESBLs that are divided into five subgroups based on amino acid sequence homology, including CTX-M-1, CTX-M-2, CTX-M-8, CTX-M-9, and CTX-M-25; the names are based on the prominent member of each subgroup. The enzymes exhibit ≥10% sequence differences across subgroups, and within the subgroups the variants differ by one or a few amino acid substitutions (<5% sequence difference) (7). In total, there are over 160 unique CTX-M variants known (7, 8).
Although CTX-M enzymes efficiently hydrolyze cefotaxime (∼1500-fold higher kcat/Km than TEM-1), they hydrolyze the related oxyimino-cephalosporin ceftazidime poorly (4, 6). Variants with amino acid substitutions that result in increased ceftazidime hydrolysis, however, have emerged (6–8). For example, the P167S and D240G substitutions occur in multiple CTX-M subgroups and are associated with 10-fold increased hydrolysis of ceftazidime (9–12). The P167S and D240G substitutions are localized in the active site of CTX-M enzymes and have been proposed to increase the active-site volume or flexibility to accommodate the bulky R group of ceftazidime (Fig. 1) (9, 13). However, these substitutions also destabilize the enzyme and decrease expression levels in Escherichia coli (9, 12). A few other substitutions, including A77V and N106S, have also been found in multiple CTX-M groups and are often associated with the P167S or D240G substitutions (7). In contrast to P167S and D240G, these substitutions are not located in the active site and, when present as single substitutions in CTX-M enzymes, do not increase hydrolysis of oxyimino-cephalosporins (12, 14). We have previously shown the A77V substitution has little effect on the kinetics of oxyimino-cephalosporin hydrolysis, but it stabilizes the CTX-M enzyme and increases in vivo expression levels, thereby compensating for the loss in stability resulting from the P167S and D240G mutations (12). The role of the N106S substitution, however, has not been investigated.
Figure 1.

Structures of CTX-M-14 β-lactamase and oxyimino-cephalosporins. A, ribbon diagram of CTX-M-14 β-lactamase. The Asn-106 and Asp-240 residues are highlighted in blue. The catalytic Ser-70 residue is highlighted in red. B, schematic illustration of cefotaxime. C, schematic illustration of ceftazidime.
In this study, CTX-M-14, an intensively studied enzyme from the CTX-M-9 family, was used to examine the mechanism of action of the N106S substitution (Fig. 1). Alone or in combination with D240G, the N106S substitution increases CTX-M enzyme stability and steady-state expression levels in bacteria. However, N106S negatively affects both cefotaxime and ceftazidime hydrolysis rates. Nevertheless, on balance, the increase in enzyme stability and expression levels outweighs the negative catalytic effects, resulting in the double mutant enzyme providing higher resistance levels to both cefotaxime and ceftazidime.
X-ray crystallography shows that although Asn-106 is not located in the active site, the N106S substitution results in a conformational change of the residue 103–106 loop that alters the position of active-site residues Asn-104 and Tyr-105 leading to decreased interactions with cefotaxime, consistent with the observed reduction in catalytic activity. Interestingly, the structural shift caused by the N106S mutation creates a 103–106 loop that closely matches the conformation that is seen in TEM-1 β-lactamase, which has a serine at position 106. Therefore, residue 106, although not located in the active site, acts to switch the 103–106 loop between a cefotaximase-favorable CTX-M conformation and the non-ESBL TEM-1 conformation. Furthermore, examination of ϕ, ψ, and ω dihedral angles reveals the 103–106 loop in CTX-M is in a strained conformation, and the N106S-induced shift to a TEM conformation reduces strain, consistent with the enhanced enzyme stability associated with N106S. Thus, the CTX-M enzymes that maintain the WT Asn at position 106 pay a price in stability to maintain the 103–106 loop in a conformation that is favorable for cefotaxime catalysis.
Results
Oxyimino-cephalosporin resistance levels conferred by N106S, D240G, and N106S/D240G reveal epistasis between mutations
The levels of antibiotic resistance provided to E. coli by the WT CTX-M-14 and N106S mutant were assessed by determining minimum inhibitory concentrations (MICs) for cefotaxime and ceftazidime (Table 1). The introduction of the N106S substitution resulted in a 16-fold reduction in cefotaxime MIC while not affecting the ceftazidime MIC compared with WT CTX-M-14. Therefore, in the absence of other substitutions, N106S does not provide a resistance advantage for ceftazidime or cefotaxime. This is not what would be expected for a mutation that is associated with increased resistance to oxyimino-cephalosporins.
Table 1.
Minimum inhibitory concentrations for E. coli containing empty vector control, wild-type, and mutant enzymes
| Plasmid | MIC (μg/ml) |
|
|---|---|---|
| CTXa | CAZ | |
| pTP123 | <0.05 | 0.19 |
| CTX-M-14 | 4.0 | 0.38 |
| N106S | 0.25 | 0.38 |
| D240G | 1.5 | 0.5 |
| N106S/D240G | 6.0 | 0.75 |
a CTX is cefotaxime; CAZ is ceftazidime.
In natural isolates, N106S is found in combination with other substitutions, such as D240G, and is thought to be a modifier of D240G function (7, 14, 15). Therefore, the N106S substitution was introduced into a CTX-M-14 D240G variant. CTX-M-14 containing only the D240G substitution exhibited a 3-fold reduction in cefotaxime resistance levels (MIC) compared with WT and a modest 1.3-fold increase in ceftazidime MIC, similar to previous observations (12). Combining N106S and D240G into a double mutant increased the MIC for cefotaxime relative to either WT (1.5-fold) or the N106S (24-fold) and D240G (4-fold) single mutants (Table 1). For ceftazidime, the N106S/D240G double mutant has a 2-fold higher MIC than WT, a 2-fold higher MIC than N106S alone, and a 1.5-fold higher MIC compared with D240G. Therefore, the combination of mutations that each decrease or have a neutral effect on resistance levels relative to WT results in an enzyme (N106S/D240G) that confers more resistance than WT. The nonadditive (epistasis) interactions between the N106S and D240G substitutions with reference to resistance levels are consistent with a proposed role of N106S as a modifier of the D240G phenotype.
Enzyme kinetic parameters alone do not explain antibiotic resistance levels
The effects of the N106S, D240G, and N106S/D240G mutations on the kinetics of cefotaxime and ceftazidime hydrolysis were determined using the purified enzymes. The N106S substitution results in a large increase in the Km value for cefotaxime hydrolysis such that it could not be accurately determined (Table 2). The kcat/Km value was therefore determined using the equation v = kcat/Km [E][S], where [S] ≪ Km (see under “Experimental procedures”). The results indicate the catalytic efficiency of N106S for cefotaxime was reduced 5-fold relative to WT CTX-M-14. The kcat/Km value for ceftazidime hydrolysis by N106S indicates a modest decrease in catalytic efficiency (1.6-fold). This is similar to the effects on resistance levels (MIC) (Table 1).
Table 2.
Kinetic parameters for wild type and mutant CTX-M enzymes
| β-Lactamase | Parameter | Antibiotica |
|
|---|---|---|---|
| CTX | CAZ | ||
| CTX-M-14 | kcat (s−1) | 203 ± 13.4 | ND |
| Km (μm) | 72 ± 6.7 | >500 | |
| kcat/Km (s−1 m−1) | 2.8 ± 0.3 | 0.001 ± 0.0007 | |
| N106S | kcat (s−1) | ND | ND |
| Km (μm) | >500 | >500 | |
| kcat/Km (s−1 m−1) | 0.6 ± 0.02 | 0.0006 ± 4.5E-05 | |
| D240G | kcat (s−1) | 321 ± 46.2 | ND |
| Km (μm) | 52 ± 8.6 | >500 | |
| kcat/Km (s−1 m−1) | 6.2 ± 1.4 | 0.013 ± 0.0007 | |
| N106S/D240G | kcat (s−1) | 475 ± 59 | ND |
| Km (μm) | 310 ± 10 | >500 | |
| kcat/m (s−1 m−1) | 1.5 ± 0.2 | 0.003 ± 2E-0.5 | |
| N104A | kcat (s−1) | ND | ND |
| Km (μm) | >500 | >500 | |
| kcat/Km (s−1 m−1) | 0.2 ± 0.01 | 0.0001 ± 1.4E-06 | |
a CTX is cefotaxime; CAZ is ceftazidime; ND is not determined.
The D240G substitution results in a >2-fold increase in catalytic efficiency for cefotaxime hydrolysis relative to WT due to increased kcat and decreased Km values (Table 2). This finding is surprising given the observed 3-fold reduction in cefotaxime resistance levels for E. coli containing the D240G mutant would predict lower kcat/Km values if catalytic activity were the sole contributor to resistance levels. The D240G substitution results in a 10-fold increase in kcat/Km for ceftazidime hydrolysis, consistent with previous reports (9, 12). The D240G mutant, however, provides E. coli with a modest 1.3-fold increase in ceftazidime resistance, which is less than might be expected from a 10-fold increase in catalytic efficiency. The inconsistency between the effects of D240G on kcat/Km versus MIC for cefotaxime and ceftazidime can be explained by the previous finding that the substitution destabilizes the enzyme and leads to lower protein expression levels in E. coli and lower resistance (12).
The N106S/D240G double mutant enzyme, in contrast to the N106S single mutant, has a Km value in the measurable range and shows a 2.5-fold increase in catalytic efficiency for cefotaxime hydrolysis compared with the N106S mutant (Table 2). Therefore, the addition of the D240G substitution partially compensates for the apparent loss in cefotaxime affinity due to the N106S substitution. However, the kcat/Km value for cefotaxime hydrolysis by the double mutant is still nearly 2-fold lower than the value for WT CTX-M-14 (Table 2). Nevertheless, the N106S/D240G double mutant displays a higher cefotaxime MIC than WT. In addition, the N106S/D240G double mutant enzyme exhibits lower catalytic efficiency for ceftazidime hydrolysis than the D240G enzyme and yet provides higher ceftazidime resistance than the D240G mutant, consistent with the idea that bacterial resistance is influenced by factors beyond catalytic activity.
Enzyme stability and in vivo protein expression levels contribute to resistance levels
Because catalytic activity does not fully correlate with cefotaxime and ceftazidime resistance levels, the effect of the mutations on enzyme stability and protein expression levels was determined. The thermal stabilities of the WT CTX-M-14, N106S, D240G, and N106S/D240G enzymes were determined using temperature-controlled CD spectroscopy, which reports the melting temperature (Tm) of the purified CTX-M β-lactamases (Table 3 and Fig. 2) (9, 16). The Tm of the WT enzyme was determined to be 52.8 °C. The N106S single substitution resulted in a large increase in stability relative to WT (+5.2 °C). In contrast, the D240G single substitution results in a decrease in stability (−3.2 °C) when compared with the WT enzyme. The decrease in stability of D240G is consistent with previous studies by Chen et al. (9) that determined a 1 °C decrease relative to CTX-M-14. When the N106S and D240G substitutions are combined in the double mutant, the N106S mutation rescues the protein stability that is lost due to the D240G mutation, increasing the Tm of the enzyme by 4.6 °C above that observed for the WT enzyme.
Table 3.
Thermal stabilities of the wild type and mutant CTX-M enzymes determined by CD
| Variant | Tm | ΔTm | ΔΔG | ΔH |
|---|---|---|---|---|
| °C | °C | kcal/mol | kcal/mol | |
| CTX-M-14 | 52.8 ± 0.01 | 221 ± 2 | ||
| N106S | 58.0 ± 0.04 | +5.2 | 3.20 ± 0.13 | 212 ± 9 |
| D240G | 49.6 ± 0.81 | −3.2 | −1.59 ± 0.37 | 152 ± 7 |
| N106S/D240G | 57.4 ± 0.23 | +4.6 | 2.49 ± 0.04 | 187 ± 8 |
Figure 2.
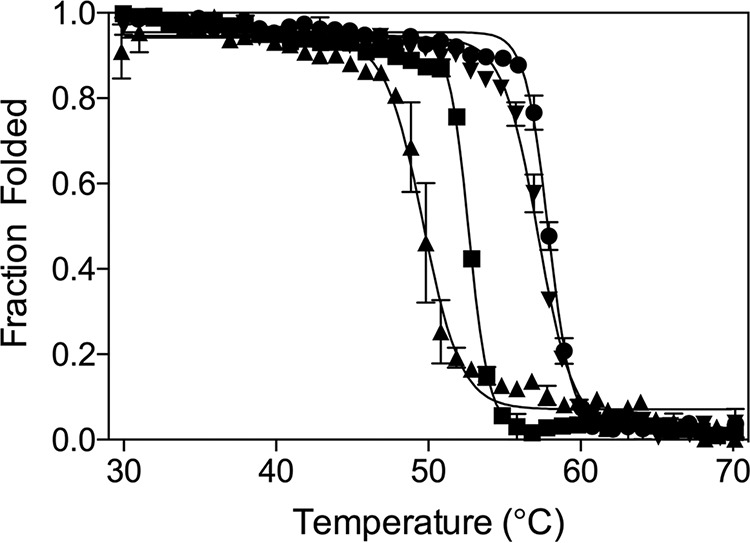
Thermal denaturation curves for CTX-M-14 (WT) and mutant enzymes. The melting curves are shown for the WT CTX-M-14 (square symbol), D240G (upward triangle), N106S (circle), and N106S/D240G (downward triangle) mutant enzymes. The Tm values (Table 3) were determined by fitting a sigmoidal function to the transition from the folded to unfolded state (see “Experimental procedures”).
Protein expression levels often correlate with protein stability and resistance to antibiotics (16–18). Stable β-lactamase enzymes remain folded and escape degradation by periplasmic proteases leading to higher in vivo protein expression levels and higher antibiotic resistance in E. coli (16, 18). In contrast, less stable variants unfold and are proteolyzed more rapidly, decreasing steady-state protein levels, which, in turn, decreases resistance to antibiotics (17, 19). Steady-state levels of the enzymes were examined by Western blot analysis with anti-CTX-M-14 polyclonal antibody on the periplasmic fraction of E. coli expressing the WT and mutant CTX-M enzymes (Fig. 3). The in vivo protein expression level of the N106S mutant enzyme was increased relative to the CTX-M-14 WT enzyme, whereas the expression level of the D240G mutant enzyme was decreased relative to WT, as predicted based on the stability measurements. The addition of the N106S substitution to the D240G enzyme restored the in vivo protein expression levels of the double mutant to above WT levels. Therefore, the strong stabilizing effect of the N106S substitution correlates with increased enzyme expression levels in the periplasm of E. coli for both the CTX-M N106S single mutant and N106S/D240G double mutant. Although it is possible that the nucleotide changes associated with the N106S and D240G substitutions could affect mRNA levels and thereby alter protein expression levels, the results are consistent with increased enzyme stability leading to higher expression.
Figure 3.
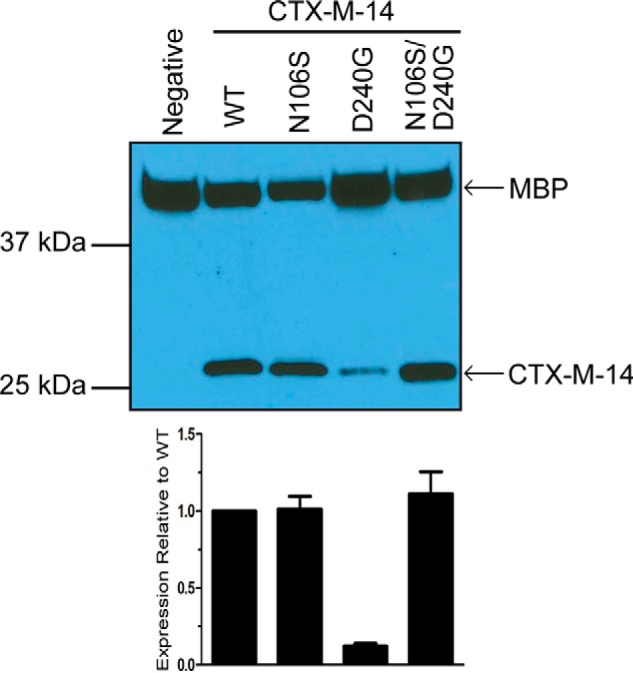
Steady-state protein levels of WT CTX-M-14 and the mutant β-lactamases. Western blot analysis with an anti-CTX-M-14 polyclonal antibody shows protein expression levels in the periplasmic fraction of E. coli encoding WT CTX-M-14 and the N106S, D240G, and N106S/D240G mutant enzymes. Polyclonal antibody to the periplasmic protein MBP was used as a loading control. The signal was visualized by chemiluminescence and quantified by densitometry. The signal for CTX-M-14 β-lactamase was normalized to that for MBP in the same sample. The lower panel shows protein levels relative to the WT enzyme. The error bars represent the standard deviation of protein levels based on three experiments.
X-ray structures of CTX-M-14 N106S and N106S/D240G reveal a conformational change in an active-site loop
The enzyme kinetics results described above indicate the N106S substitution results in a large increase in Km and a decrease in kcat/Km values for cefotaxime hydrolysis compared with WT and increases the enzyme's stability. To understand the molecular basis for these effects, the structure of the N106S enzyme was determined at 1.7 Å resolution (Fig. 4 and Table 4). The N106S structure reveals how a substitution outside of the active site can communicate a change in conformation to an active-site residue. Asn-106 is conserved in CTX-M enzymes and is part of a structure called the 103VNYN106 loop (Fig. 4) (20). As seen in Fig. 5A, in the previously published S70G/CTX (CTX = cefotaxime) structure this loop contains Asn-104 and Tyr-105 that are both in the active site and make direct interactions with substrate. In the S70G/CTX structure, the Asn-104 side chain is pointed into the active site and makes a hydrogen bond with the side chain NH of Asn-132 and with the carbonyl oxygen of the acyl-amide group of cefotaxime in CTX-M-14 (Fig. 5A) (21). Tyr-105 forms a wall of the active site and makes van der Waals interactions with bound substrate (Fig. 5A) (21). The Asn-106 residue is at the base of the loop and stabilizes its structure by hydrogen-bonding to the main chain NH and carbonyl oxygen of Val-103 as observed in both the WT CTX-M-14 apoenzyme and the S70G/CTX structures (9, 21). Importantly, the peptide bond between Asn-104 and Tyr-105 is oriented such that the carbonyl oxygen is pointed away from the active site (Figs. 4B and 5A). The N106S substitution causes changes in the conformation of the loop that leads to the side chain of Asn-104 rotating out of the active site in the N106S structure (Fig. 4C). The conformational change is due to the shorter Ser-106 side chain not forming hydrogen bonds with the main chain NH of Val-103 but rather with the main chain NH and side chain OH of Thr-133 and the main chain O of Val-103 in an altered position (Fig. 4C). This causes a flip in the orientation of the Asn-104–Tyr-105 peptide bond so that the carbonyl oxygen is pointed into the active site where it forms a hydrogen bond to Asn-132, and the Asn-104 side chain is pointed out of the active site (Fig. 4, C and D). In addition, Tyr-105 assumes two conformations in the N106S structure with one conformation similar to that observed in WT and the second with the hydroxyphenyl group angled along the active site (Fig. 4, C and D).
Figure 4.
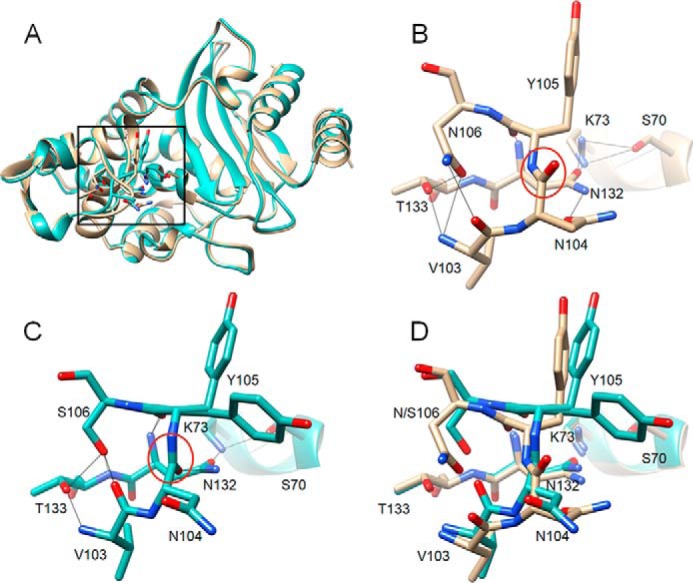
Alignment of structures of WT CTX-M-14 and N106S mutant. A, ribbon diagram of alignment of structures of WT CTX-M-14 (PDB code 1YLT) (9) (tan) and N106S-chain B (green). Side chains for residues 103–106, 132–133, and 70, 73 are shown as sticks. The box indicates the region of the structure shown in B–D. B, WT CTX-M-14 residues 103–106, 132–133, and 70, 73 are shown as sticks and labeled. Hydrogen bonds are indicated by black lines. The carbonyl oxygen of the peptide bond between positions Asn-104 and Tyr-105 is pointed upwards away from Asn-132 and circled in red. C, N106S structure with residues 103–106, 132–133, and 70, 73 shown as sticks and labeled. Hydrogen bonds are indicated by black lines. The carbonyl oxygen of the peptide bond between positions Asn-104 and Tyr-105 is circled in red and is pointed downwards forming a hydrogen bond with the side chain of residue Asn-132. The side chain of residue Asn-104 is rotated away from Asn-132 and out of the active site. The Tyr-105 side chain exists in two conformations in the structure. D, alignment of 103–106, 132–133, and 70, 73 from the WT CTX-M-14 (tan) and N106S (green) structures. In all panels, oxygen and nitrogen atoms are colored red and blue, respectively.
Table 4.
X-ray crystallography data collection and refinement statistics for CTX-M-14 mutant enzymes
| N106S | S70G/N106S/CTX | N106S/D240G | S70G/N106S/D240G/CTX | |
|---|---|---|---|---|
| PDB code | 6CYK | 6CYQ | 6CYN | 6CYU |
| Data collection | ||||
| Space group | P 21 | P 32 | P 32 | P 32 2 1 |
| a, b, c (Å) | 45.2, 107.2, 47.8 | 83.4, 83.4, 232.1 | 83.2, 83.2, 232.0 | 41.4, 41.4, 230.6 |
| α, β, γ (°) | 90.0, 101.7, 90.0 | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 |
| Resolution range (Å) | 23.40–1.70 | 35.0–1.70 | 35.0–1.60 | 50.0–1.82 |
| (1.73–1.70) | (1.73–1.70) | (1.63–1.60) | (1.85–1.82) | |
| R-merge (%) | 9.1 (51.6) | 12.6 (77.0) | 11.9 (62.0) | 5.5 (10.6) |
| I/σ | 8.8 (2.0) | 12.2 (2.5) | 18.9 (4.8) | 25.6 (15.0) |
| Multiplicity | 3.6 (3.5) | 4.3 (4.8) | 6.3 (6.3) | 4.7 (4.3) |
| Completeness (%) | 99.6 | 98.3 | 99.4 | 99.8 |
| Wilson B-factor (Å2) | 11.9 | 12.8 | 11.8 | 17.8 |
| Refinement | ||||
| Molecules per asymmetric unit | 2 | 8 | 8 | 1 |
| No. of unique reflections | 48,725 (4787) | 195,781 (9881) | 236,763 (11761) | 21,800 (1050) |
| R-work/R-free (%) | 15.1/18.5 | 17.0/20.0 | 16.8/20.4 | 16.0/20.0 |
| No. of protein residues | 523 | 2072 | 2087 | 261 |
| Ramachandran | ||||
| Favored (%) | 98 | 99 | 99 | 99 |
| Outliers (%) | 0 | 0 | 0 | 0 |
| Average B-factor (Å2) | 15.7 | 19.3 | 18.7 | 25.0 |
| Protein | 13.2 | 17.8 | 16.0 | 23.5 |
| Ligand | 27.9 | 23.1 | – | 32.3 |
| Solvent | 28.2 | 30.3 | 30.7 | 33.3 |
| Root mean square deviations | ||||
| Bond length (Å) | 0.006 | 0.006 | 0.006 | 0.006 |
| Bond angles (°) | 0.853 | 0.864 | 0.810 | 0.819 |
Figure 5.

Comparison of the CTX-M-14 S70G structure in complex with cefotaxime (PDB code 4PM5) (21) with that of the S70G/N106S enzyme in complex with CTX. A, S70G/CTX structure (tan) showing residues 103–106, 132–133, and 70, 73 as sticks and labeled. Hydrogen bonds are indicated by black lines. Note that the carbonyl oxygen of the peptide bond between positions Asn-104 and Tyr-105 is pointed upwards away from Asn-132, whereas the Asn-104 side chain makes hydrogen bonds to Asn-132 and the acyl-amide group of cefotaxime. B, alignment of residues 103–106, 132–133, and 70, 73 and cefotaxime from the eight molecules in the asymmetric unit of the S70G/N106S/CTX (light blue) structure. Cefotaxime is colored dark gray. The peptide bond between Asn-104 and Tyr-105 has a flipped orientation compared with A for all eight molecules. C, S70G/N106S/CTX-chain A structure showing residues 103–106, 132–133, and 70, 73 as sticks and labeled. Note that the carbonyl oxygen of the peptide bond between positions Asn-104 and Tyr-105 is pointed down and forms a hydrogen bond with Asn-132, whereas the Asn-104 side chain is rotated out of the active site, and no longer forms a hydrogen bond with the acyl-amide group of cefotaxime. D, alignment of residues 103–106, 132–133, and 70, 73 and cefotaxime from S70G/CTX (tan) and S70G/N106S/CTX-chain A (light blue) structures. Cefotaxime from the S70G structure is colored white and that from S70G/N106S is dark gray. E, S70G/N106S/CTX-chain C (light blue) structure showing residues 103–106, 132–133, and 70, 73 as sticks and labeled. Note the altered conformations of the Tyr-105 and Asn-104 side chains compared with chain A shown in C and the dual conformations of the oxime moiety and aminothiazole ring of cefotaxime. F, alignment of residues 103–106, 132–133, and 70, 73 and cefotaxime from S70G/CTX (tan) and S70G/N106S/CTX-chain C (light blue) structures. Cefotaxime from the S70G structure is colored white and that from S70G/N106S is dark gray.
The N106S structure suggests the mutation results in a loss of a hydrogen bond between Asn-104 and cefotaxime as well as altered interactions between Tyr-105 and substrate. However, the positioning of residues could change upon substrate binding. Therefore, the structure of CTX-M-14 S70G/N106S in complex with cefotaxime was determined (Table 4). The S70G substitution prevents substrate hydrolysis to allow the structure of the enzyme with intact cefotaxime to be determined (21, 22), although, as discussed below, the structures determined in this study with the S70G mutation had hydrolyzed cefotaxime in the active site, likely due to the long soaking time with cefotaxime in the preparation of the crystals. The structure in complex with cefotaxime was superimposable with the N106S apoenzyme, with the peptide bond between residues 104 and 105 flipped relative to WT, and the Asn-104 side chain pointed away from the bound cefotaxime (Fig. 5). The S70G/N106S/CTX structure has eight molecules in the asymmetric unit, and cefotaxime is present in each. A comparison of the eight molecules with each other reveals close similarity throughout the structure but variation in the position of the Asn-104 and Tyr-105 side chains (Fig. 5B and Fig. S1). The differences in the conformation of residues 104 and 105 among the different chains suggest flexibility in side-chain positions and orientation of residues 104 and 105. In contrast, cefotaxime is bound in a nearly identical position for all eight molecules (Fig. 5B and Fig. S1). Cefotaxime is found in two conformations in chains B–F in nearly equal occupancy with the oxime moiety and aminothiazole ring flipped relative to one another (Fig. 5E and Fig. S1). In all cases, the hydrogen bond between Asn-104 and the acyl-amide group of cefotaxime is lost. The loss of this hydrogen bond, the conformational heterogeneity of the Tyr-105 side chain in the complex, and the altered positions of the oxime and aminothiazole groups may contribute to the large increase in Km for cefotaxime hydrolysis associated with the N106S substitution.
The structures of the N106S apoenzyme and the S70G/N106S/CTX complex suggest that the movement of the Asn-104 side chain out of the active site and consequent loss of the hydrogen bond with cefotaxime contributes to the large increase in Km and decrease in kcat/Km for cefotaxime hydrolysis associated with the N106S substitution. This, in turn, predicts that a N104A substitution in CTX-M-14 would lead to an increase in Km and a decrease in kcat/Km for cefotaxime hydrolysis. To test this hypothesis, kinetic parameters for cefotaxime hydrolysis were determined for the N104A mutant enzyme (Table 2). Similar to the N106S enzyme, the N104A mutant exhibits a large increase in Km to a level that cannot be accurately measured and a 14-fold decrease in kcat/Km for cefotaxime hydrolysis. These results show that residue Asn-104 is an important determinant of cefotaxime hydrolysis by WT CTX-M-14 and support the hypothesis that the reduced cefotaxime hydrolysis observed for the N106S mutant is due to the rotation of the Asn-104 side chain out of the active site and loss of a hydrogen bond with the substrate.
The kinetic results indicate that addition of the D240G substitution to N106S lowers the Km and increases the kcat/Km values for cefotaxime hydrolysis relative to the N106S enzyme. To address the basis for the compensatory effect of D240G on catalysis, the structure of the N106S/D240G enzyme was determined (Fig. 6). Eight molecules are present in the asymmetric unit for the N106S/D240G apoenzyme structure, and a comparison of the molecules indicates they are superimposable, including the Val-103–Ser-106 region (Fig. 6B). Next, the structure of the S70G/N106S/D240G enzyme in complex with cefotaxime was determined and had one molecule in the asymmetric unit. The β-lactamase portion of the S70G/N106S/D240G structure in complex with cefotaxime is superimposable with the N106S/D240G apoenzyme indicating there are no large changes in residue positions upon cefotaxime binding (Fig. 6A). In addition, the conformations of the residues in the Val-103–Ser-106 region are very similar to those in the N106S/D240G apoenzyme with only slight movement of the Tyr-105 side chain (Fig. 6, C and D). The S70G/N106S/CTX structure has eight molecules in the asymmetric unit with multiple conformations of residues Asn-104 and Tyr-105. In contrast, the eight molecules in the asymmetric unit of the N106S/D240G structure and the one molecule in the S70G/N106S/D240G/CTX structure assume one conformation, which shows close similarity to chains A and B and E–H in the asymmetric unit of the S70G/N106S/CTX crystal structure (Fig. 6, E and F, and Fig. S1). In addition, the cefotaxime molecule is in a similar position in the S70G/N106S/CTX and S70G/N106S/D240G/CTX structures (Fig. 6, E and F).
Figure 6.

Alignments of structures of CTX-M-14 S70G/N106S/CTX, N106S/D240G, and S70G/N106S/D240G/CTX. A, ribbon diagram of alignment of structures of CTX-M-14 N106S/D240G-chain A (pink), S70G/N106S/CTX-chain C (light blue), and S70G/N106S/D240G/CTX (light green). Cefotaxime structures from S70G/N106S/CTX and S70G/N106S/D240G/CTX are colored white and dark gray, respectively. Side chains for residues 103–106, 132–133, and 70, 73 are shown as sticks. The box indicates the region of the structure shown in B–F. B, structural alignment of residues Val-103–Ser-106, Asn-132–Thr-133, Ser-70, and Lys-73 for the eight molecules in the asymmetric unit of the N106S/D240G structure. C, stick representation of residues Val-103–Ser-106, Asn-132–Thr-133, Ser-70, and Lys-73 for the single molecule in the asymmetric unit of the N106S/D240G/CTX structure shown in light green. The cefotaxime molecule is shown in dark gray. Two conformations of the cefotaxime side chain are observed at nearly equal occupancy. Hydrogen bonds are shown as black lines. D, alignment of residues Val-103–Ser-106, Asn-132–Thr-133, Ser-70, and Lys-73 for chain A of the N106S/D240 structure (pink) versus the S70G/N106S/D240G/CTX structure (light green). The cefotaxime molecule associated with the S70G/N106S/D240G/CTX structure is shown in dark gray. E, alignment of residues Val-103–Ser-106, Asn-132–Thr-133, Ser-70, and Lys-73 for chain A of the S70G/N106S/CTX structure (light blue) versus the S70G/N106S/D240G/CTX structure (light green). The cefotaxime molecule associated with the S70G/N106S/CTX structure is shown in white and that associated with S70G/N106S/D240G/CTX is shown in dark gray. F, alignment of residues Val-103–Ser-106, Asn-132–Thr-133, Ser-70, and Lys-73 for chains A–H of the S70G/N106S/CTX structure (light blue) versus the S70G/N106S/D240G/CTX structure (light green). The cefotaxime molecule associated with the S70G/N106S/CTX structure is shown in white and that associated with S70G/N106S/D240G/CTX is shown in dark gray. In all panels, oxygen and nitrogen atoms are colored red and blue, respectively.
The addition of the D240G mutation to the N106S enzyme, as reflected in the S70G/N106S/D240G/CTX structure, results in a wider entrance to the active site due to removal of the Asp-240 side chain, which is consistent with a higher binding affinity for cefotaxime for the N106S/D240G double mutant and thus a lower Km value (Fig. 7). In addition, the eight molecules in the asymmetric unit of the N106S/D240G apoenzyme and one molecule in the S70G/N106S/D240G/CTX structure all exhibit the same conformation of Asn-104 and Tyr-105, which is in contrast to the multiple conformations observed in the S70G/N106S/CTX structure. Although there are too few molecules to be definitive, it is possible that the D240G substitution reduces the conformational heterogeneity of the enzyme, leading to increased activity, as has been suggested for ESBL mutations that lead to increased cefotaxime hydrolysis by the TEM-1 β-lactamase (23, 24).
Figure 7.

Surface representations of CTX-M-14 β-lactamase mutants in complex with cefotaxime. A, structure of pseudo-WT S70G/cefotaxime (CTX). Gly-70 is in red, Asn-104 and Asn-106 in cyan, and Tyr-105 and Asp-240 in yellow. Cefotaxime is shown as sticks with nitrogen colored blue, oxygen colored red, and sulfur colored yellow. B, structure of S70G/N106S/CTX, chain A. C, structure of S70G/N106S/D240G/CTX.
X-ray structures of CTX-M-14 S70G/N106S and S70G/N106S/D240G in complex with cefotaxime reveal hydrolyzed product
As described above, the S70G substitution was used to avoid substrate turnover and trap cefotaxime in the active site for structural determination. This approach has been used to capture intact cefotaxime for a number of different structures, including CTX-M-9 S70G, CTX-M-14 S70G, CTX-M-14 S70G/S237A, CTX-M-14 S70G/R276A, and CTX-M-14 S70G/S237A/R276A (12, 21, 22). In all of these cases, high-resolution structures were obtained with intact cefotaxime bound in the active site. In contrast, examination of cefotaxime in all eight molecules of the S70G/N106S/CTX structure and the single molecule of the S70G/N106S/D240G/CTX structure indicates that cefotaxime is hydrolyzed with a carboxyl group present at the C-8 position (Fig. S2). This reaction presumably occurs via direct attack of a water molecule on the carbonyl carbon of the amide.
Serine active-site β-lactamases proceed through the attack of the serine oxygen on the C-8 carbonyl carbon of the substrate with protonation of the leaving group N-5 nitrogen (Fig. S2). For some cephalosporin substrates, rather than protonation of the nitrogen, tautomerization can occur with the double bond in the dihydrothiazine ring shifting from positions 3–4 to 4–5 and formation of a double bond at position 3–3′ with consequent elimination of the R2 group (Fig. S2D) (25, 26). The structures of a deacylation-deficient mutant (E166A) of Toho-1 (CTX-M-44) β-lactamase as well as an E166A/R274N/R276N variant of Toho-1 in complex with acylated cefotaxime show the R2 group is eliminated, and a double bond is present at position 3–3′ (27, 28). Similarly, in our structures the R2 group is eliminated, and the bond angles about C-3 are 120°, suggesting sp2 hybridization and consistent with a double bond at position 3–3′.
Discussion
CTX-M β-lactamases have become an important source of bacterial resistance to β-lactam antibiotics, particularly oxyimino-cephalosporins (7). These enzymes readily hydrolyze cefotaxime but not the related cephalosporin, ceftazidime. Nevertheless, in response to the introduction of these extended-spectrum antibiotics, variants have evolved with amino acid substitutions that provide enhanced ceftazidime resistance. We show a complex interplay of effects between substitutions determines the ultimate impact on oxyimino-cephalosporin resistance levels in bacteria. The N106S substitution enhances enzyme stability but at the cost of catalytic efficiency, whereas the D240G substitution enhances enzyme catalytic efficiency at the cost of stability. This interplay between stability and catalytic efficiency leads to epistasis whereby the double mutant exhibits higher resistance levels than predicted based on the phenotypes of the single mutants.
There are a number of examples of enzymes where mutations increase catalytic activity but are associated with a decrease in stability, i.e. they trade off stability for increased activity (29–32). Here, we show that drug-resistance mutations in CTX-M β-lactamase observed in natural isolates exhibit trade-offs in two directions. The D240G substitution exhibits the commonly observed trade-off of increased catalytic activity but decreased stability. The N106S substitution, however, trades off in the opposite direction with decreased catalytic efficiency but increased stability. The ultimate effect on bacterial fitness conferred by the double mutant in the presence of cephalosporins depends on the relative magnitude of the trade-offs. In this case, the positive stability effect outweighs the negative catalytic effect to result in increased enzyme expression and enhanced resistance for E. coli containing the N106S/D240G double mutant relative to that containing WT.
The N106S substitution stabilizes the CTX-M enzyme but also alters catalysis by greatly increasing the Km and decreasing the kcat/Km values for cefotaxime hydrolysis (Table 2). The structure of the N106S mutant provides an explanation for the effects on catalysis in that the substitution changes the hydrogen bonding pattern in the Val-103–Ser-106 loop region that results in flipping the orientation of the peptide bond between Asn-104 and Tyr-105 which, in turn, results in rotation of the Asn-104 side chain out of the active site. The structural evidence suggests that this loss of a hydrogen bond interaction with cefotaxime is responsible for the observed increase in the Km and the decrease in the kcat/Km values. This idea was confirmed by the finding that an N104A mutant enzyme, which cannot hydrogen-bond to substrate regardless of its conformation, also displays increased Km and decreased kcat/Km values for cefotaxime hydrolysis. In addition, the structure of the S70G/N106S enzyme in complex with cefotaxime revealed conformational heterogeneity of Asn-104 and Tyr-105 side chains. All of the Asn-104 side-chain conformations were pointed away from cefotaxime, consistent with this residue no longer contributing to substrate binding and catalysis. Tyr-105 assumed a range of side chain conformations, none of which matched that observed in the WT apoenzyme. The heterogeneity of Tyr-105 side-chain conformations also suggests a smaller contribution of this residue to catalysis in the N106S mutant.
The addition of the D240G substitution to the N106S enzyme lowered the Km and increased the kcat/Km values for cefotaxime hydrolysis (Table 2). The conformation of residues Asn-104 and Tyr-105 in the S70G/N106S/D240G/CTX structure matched the dominant conformation of these residues in the S70G/N106S/CTX structure (chains A and B and E–H) (Fig. 6 and Fig. S1). In addition, the cefotaxime molecules were in similar positions in both structures (Fig. 6). However, the removal of the Asp-240 side chain increases access of substrate to the active site, particularly in the context of Tyr-105 assuming an altered conformation where it is angled down the active site, which could improve affinity for oxyimino-cephalosporins (Fig. 7). In addition, it is possible that reduced conformational heterogeneity of Asn-104 and Tyr-105, as observed for the N106S/D240G apoenzyme and the S70G/N106S/D240G/CTX structures, could increase activity toward cefotaxime by reducing nonproductive conformations. Several enzymes show a cost to catalysis by diluting a catalytically optimal conformation among many nonproductive conformations (23, 33). Furthermore, detailed characterization of the mutations that accumulate during directed evolution experiments reveals that many mutations act to reduce conformational heterogeneity and the number of nonproductive sub-states (34, 35). Structural, computational, and experimental results on TEM-1 ESBLs suggest that stabilizing an active conformation and restricting the sampling of nonfunctional conformations leads to increased hydrolysis of cefotaxime by TEM-1 ESBL variants (23, 24).
The hydrogen-bonding interaction of Asn-104 with cefotaxime is unique to the CTX-M enzymes and clearly contributes to cefotaxime hydrolysis, as indicated by the increased Km and the decreased kcat/Km values of the N104A mutant (Table 2). Among class A enzymes that do not hydrolyze cefotaxime such as TEM-1 and SHV-1, the side chain of residue 104 is oriented out of the active site, similar to its conformation in the CTX-M N106S mutant. Also, in TEM-1 and similar class A enzymes, residue 106 is naturally a serine. A comparison of the 103–106 loop region of WT CTX-M-14, CTX-M-14 N106S, and TEM-1 shows that the loop has different conformations in CTX-M-14 and TEM-1 with the peptide bond between residues 104 and 105 flipped for CTX-M versus TEM-1 (Fig. 8). In contrast, the loop region of the N106S mutant closely resembles that of TEM-1 with the 104–105 peptide bond in matching orientation and the side chain of residue 104 pointed away from the active site (Fig. 8). Therefore, residue 106 may be a primary determinant of the conformation of the 103–106 loop. When it is serine, the loop is in the non-ESBL, TEM-like conformation, and when it is asparagine, the loop is in a CTX-M-like, cefotaximase-favorable conformation with the 104 side chain directed into the active site.
Figure 8.
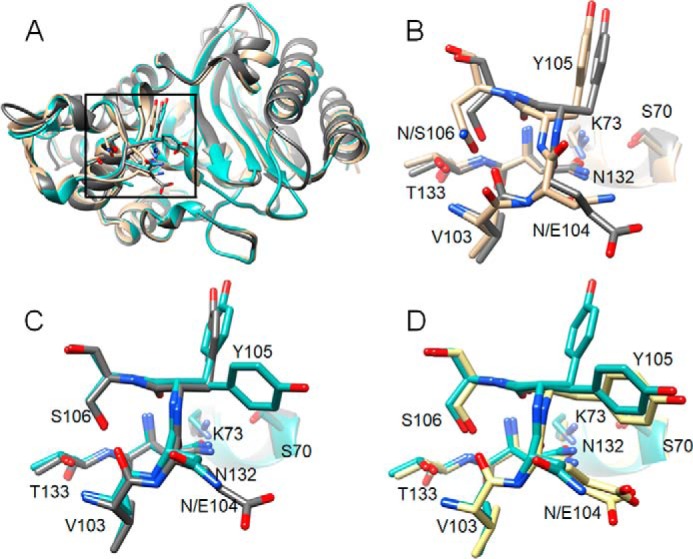
Structural alignments of CTX-M-14, CTX-M-14 N106S, and TEM-1 β-lactamases. A, ribbon diagram of alignment of structures of WT CTX-M-14 (PDB code 1YLT) (9) (tan), N106S (blue-green), and TEM-1 (PDB code 1XPB) (dark gray). Side chains for residues 103–106, 132–133, and 70, 73 are shown as sticks. Box indicates the region of the structure shown in B–D. B, alignment of residues 103–106, 132–133, and 70, 73 for WT CTX-M-14 (tan) and TEM-1 (dark gray). C, alignment of residues 103–106, 132–133, and 70, 73 for CTX-M-14 N106S (blue-green) and TEM-1 (dark gray). D, alignment of residues 103–106, 132–133, and 70, 73 for CTX-M-14 N106S (blue-green) and TEM-1 V216AcrF mutant (PDB code 4ZJ1) (39) (yellow).
The switch in conformation of the 103–106 loop in the CTX-M N106S enzyme also affects the side-chain conformation of Tyr-105. Altered positioning of Tyr-105 has not been observed previously in CTX-M structures but has been seen in TEM-1 structures, including TEM-1 in complex with the protein-based inhibitors BLIP and BLIP-II as well as a structure with a bound boronic acid inhibitor, and in a TEM-1 mutant containing a non-natural amino acid at residue 216 (Fig. 8) (36–39). Doucet and Pelletier (40) have noted that the observed Tyr-105 conformations in TEM-1 correspond to the t (trans, t80°) and m (minus, m-85°, m-30°) rotamer orientations allowed for tyrosine residues, with the t rotamer being the conformation observed in WT TEM-1 (and CTX-M-14) and the m rotamer being the altered conformation with the Tyr-105 side chain angled down the active site. These authors also noted that significant electron density exists for the m rotamer in the WT 1BTL (41) and PDB code 1XPB (42) TEM-1 structures. They used a molecular dynamics simulated annealing approach to show that Tyr-105 is dynamic in TEM-1 and alternates between the t and m rotamers in solution (40). The results presented here suggest the CTX-M N106S substitution and the resulting change in the 103–106 loop to a TEM-like conformation also leads to dynamic behavior of Tyr-105.
The CTX-M N106S substitution is also associated with a large increase in enzyme stability relative to WT CTX-M-14 (Fig. 3). As noted above, the N106S substitution changes the hydrogen-bonding pattern in the 103–106 loop and the orientation of the 104–105 peptide bond. There are also differences in the ϕ, ψ, and ω dihedral angles for residues 103–105 in the loop for the N106S enzyme versus WT CTX-M-14, and they closely resemble those in TEM-1 (Fig. S3). In addition, the dihedral angles of the 103–105 residues in the S70G/N106S/CTX and S70G/N106S/D240G/CTX structures are similar to those of N106S and TEM-1 and different from those of WT CTX-M-14 (Fig. S3). The ϕ, ψ angles for residues 103–105 for the CTX-M enzymes containing the N106S substitution are in favorable regions of the Ramachandran plot with the largest change from WT being the angles for Val-103 moving to a favorable region (Fig. S3) (43). More strikingly, the ω angles for residues 103 and 105 in the N106S-containing enzymes are more favorable compared with WT (Table S1). The conformational angle ω of an ideal trans-peptide is 180° (44). The ω angles for Val-103 and Tyr-105 in the WT CTX-M-14 are 166.9° and 168.1°, whereas in the N106S enzyme structure they are 172.8° and 174.1°, representing a shift toward the ideal value of 180° (Table S1). Furthermore, an assessment of the ω angles for Val-103 and Tyr-105 for available CTX-M-14 structures that contain the WT Asn-106 (seven chains) reveals an average of 167.6° ± 1.63 for Val-103 and 166.2° ± 1.27 for Tyr-105. In contrast, structures of enzymes containing Ser-106 (19 chains) have an average ω angle of 177.5° ± 2.13 for Val-103 and 177.0° ± 2.95 for Tyr-105 (Table S1). This demonstrates a consistent change of the ω angle toward the optimal value in enzymes containing the N106S substitution. Note, however, that the ω angle is under constraint by refinement programs, which could impact the results (45). This issue was addressed by Brereton and Karplus (45), who indicate meaningful ω values varying significantly from 180° can be obtained despite refinement constraints. Thus, favorable ϕ, ψ angles and, particularly, the improved ω values for residues Val-103 and Tyr-105 are consistent with the increased stability of the N106S-containing enzymes versus WT CTX-M-14 due to the relief of the dihedral strain.
Because Asn-106 is conserved in CTX-M enzymes, the loop conformation is strained across the CTX-M family, and these enzymes pay a cost in terms of enzyme stability to maintain the conformation of the 103–106 loop. However, this loop conformation is key for catalysis in that it is required to place Asn-104 in the proper position to hydrogen-bond to the cefotaxime substrate. Therefore, CTX-M enzymes trade off stability for increased catalytic efficiency of cefotaxime hydrolysis. In addition, non-ESBL enzymes such as TEM-1 have a less strained conformation of the 103–106 loop, but this conformation does not place residue 104 in a position to facilitate cefotaxime hydrolysis.
The CTX-M N106S mutation provides an example of a naturally occurring change at a nonactive site position, which is a second shell residue that alters the catalytic properties of an enzyme. There are a number of examples from directed evolution studies where changes at second shell residues alter enzyme function (34, 46–49). These substitutions can act by altering positions of active-site residues (46, 48), reducing conformational heterogeneity (34), or altering enzyme dynamics (47, 49). The N106S mutation acts by altering the hydrogen bond network of a loop structure that ultimately changes the position and conformational heterogeneity of active-site residues.
The drug resistance mutation N106S causes a complex effect on enzyme structure and evolution. It exhibits epistasis when combined with other drug resistance mutations due to its positive effects on enzyme stability but negative effects on catalysis. Such compromises may be common in enzyme evolution, with the ultimate impact on fitness depending on the relative magnitude of effects on stability and protein expression versus catalytic activity.
Experimental procedures
Bacterial strains and plasmids
The CTX-M-14–pTP123 plasmid was created by inserting the CTX-M-14 gene into the previously constructed pTP123 plasmid, which contains a chloramphenicol resistance marker (21, 50). The CTX-M-14–pTP123 plasmid was transformed into E. coli XL1-Blue [recA1, endA1, gyrA96, thi-1, hsdR17, supE44, relA1, lac, [F9 proAB lacIq lacZΔM15, Tn10 (tetr)]] (Stratagene, Inc., La Jolla CA) for construction of the CTX-M-14 mutants by site-directed mutagenesis (21). The pTP123 plasmids encoding the WT and mutant β-lactamases were transformed into E. coli RB791 (strain W3110 lacIqL8) for MIC determinations, immunoblotting, and protein purification (51).
The pET28a plasmid, which contains a kanamycin resistance marker, was used to express and purify the CTX-M-14 N106S, S70G/N106S, N106S/D240G, and S70G/N106S/D240G mutant enzymes to obtain large amounts of protein for X-ray crystallography. It was also used to express and purify the N104A mutant enzyme because it was unable to be purified effectively when expressed from the pTP123 plasmid. The gene encoding the WT CTX-M-14 or mutant β-lactamase was transferred into the pET28a plasmid using the Gibson Assembly Kit (New England Biolabs, Ipswich, MA) as described previously (12). The pET28a plasmids encoding the WT and mutant β-lactamases were transformed into the E. coli strain BL21 (DE3) (fhuA2[lon]ompTgal(λDE3)[dcm]ΔhsdSλDE3=λsBamHIoΔEcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 Δnin5) for protein expression and purification (52). Proteins expressed from the pET28a plasmid contained an N-terminal His-tag but did not contain a signal sequence. The N-terminal His tag was cleaved with TEV protease before experiments were performed, as described previously (12).
Site-directed mutagenesis
All CTX-M-14 β-lactamase mutants were created in the CTX-M-14–pTP123 or CTX-M-14–pET28a plasmid using Phusion DNA polymerase (New England Biolabs) according to the manufacturer's protocol for site-directed mutagenesis. Primers were obtained from Integrated DNA Technologies (Coralville, IA). The following primers were used to introduce mutations into the CTX-M-14 gene. N106S-F, 5′-CGATCTGGTTAACTACAGCCCGATTGCCGAAAAAC-3′; N106S-R, 5′-GTTTTTCGGCAATCGGGCTGTAGTTAACCAGATCG-3′; D240G-F, 5′-CGGCAGCGGCGGCTACGGCACCAC-3′; D240G-R, 5′-GTGGTGCCGTAGCCGCCGCTGCCGG-3′; N104A-F, 5′-CCTGCCGATCTGGTTGCCTACAATCCGATT-3′. DNA sequencing was performed by Genewiz (South Plainfield, NJ) to verify the sequence of all site-directed mutants
MICs
Etest strips (BioMérieux) were used to determine the MICs for ampicillin, cephalothin, cefotaxime, and ceftazidime. The WT or mutant CTX-M-14 enzyme encoded on the pTP123 plasmid was transformed into E. coli RB791 cells, and the cultures were grown overnight at 37 °C in 10 ml of LB containing 12.5 μg/ml chloramphenicol. The diluted overnight cultures (A600 ∼0.03) were spread onto LB agar plates, and the Etest strip containing the β-lactam antibiotic of interest was placed on the surface of the plate. The plates were incubated at 37 °C overnight, and the concentration of antibiotic at which the zone of inhibition intersected with the Etest strip was reported as the MIC.
Protein expression and purification
The WT CTX-M-14 and the CTX-M-14 D240G mutant enzyme were encoded on the CTX-M-14–pTP123 plasmid and introduced into E. coli RB791 cells for protein expression and purification (50). The cells were grown at 37 °C to a culture density of A600 ∼0.9 in LB medium containing 12.5 μg/ml chloramphenicol. A final concentration of 0.2 mm IPTG was used to induce protein expression, and the cell culture was grown overnight at 23 °C. Bacterial cells were pelleted by centrifuging the culture at 4 °C for 15 min at 4000 rpm. The periplasmic fraction was isolated by resuspending the bacterial cell pellet in 10 mm Tris-HCl, pH 8.0, 20% (w/v) sucrose at a volume of 20 ml/1 g of bacterial cell pellet. The mixture was stirred at 4 °C for 1 h before an equal volume of chilled water was added. The mixture was stirred for an additional hour and centrifuged at 4 °C for 30 min at 9000 rpm. The supernatant at pH 7.0 was collected, and contaminating protein was removed by passing the supernatant through a SP-Sepharose Fast Flow (SPFF) cation-exchange column (GE Healthcare) equilibrated with 10 mm MES, pH 6.0, three times. A positive charge was placed on the CTX-M protein by adjusting the pH of the supernatant to 6.0 using 1 m MES buffer. The supernatant was loaded onto the SPFF column to allow the positively charged CTX-M protein to bind. A NaCl gradient (10 mm MES, 2.5 m NaCl, pH 6.0) was used to elute the protein from the SPFF column. The protein fractions were run on an SDS-polyacrylamide gel, and fractions with >90% purity were collected and concentrated.
The CTX-M-14 N106S, S70G/N106S, N106S/D240G, S70G/N106S/D240G, N104A, N104A/N106S, and N104A/N106S/D240G mutant enzymes were encoded on the CTX-M-14–pET28a plasmid and introduced into E. coli BL21 (DE3) cells for protein expression and purification. The cells were grown in LB medium containing 25 μg/ml kanamycin at 37 °C until the cells reached a culture density of A600 ∼0.9. A final concentration of 0.2 mm IPTG was used to induce protein expression, and the cell culture was grown overnight at 23 °C. Bacterial cells were pelleted by centrifuging the culture at 4 °C for 15 min at 7000 rpm. The bacterial cell pellets were resuspended in 30 ml of lysis buffer (25 mm sodium phosphate, pH 7.2, 40 μm MgCl2, 300 mm NaCl, 10 μg/ml DNase I (Sigma), 1 EDTA-free Protease Inhibitor Mixture Tablet (Roche Applied Science)), and protein was released from the cells using a French press at 1250 p.s.i. The supernatant was isolated by centrifuging the mixture at 10,000 × g for 45 min at 4 °C and passed through a HisTrap FF column (GE Healthcare) equilibrated with 25 mm phosphate buffer, pH 7.2, 300 mm NaCl to allow the His-tagged proteins to bind. An imidazole gradient was used to elute the protein from the column. All β-lactamase enzymes were purified to >90% homogeneity as determined by SDS-PAGE. Finally, 0.5 mg of TEV protease was mixed with 25 mg of CTX-M protein and incubated at 4 °C overnight to remove the N-terminal His tag. Nickel-Sepharose high-performance beads (GE Healthcare) were used to remove TEV protease from the cleaved CTX-M protein. SDS-PAGE was used to confirm cleavage of the His-tag.
Enzyme kinetic analysis
In vitro kinetic parameters for β-lactam antibiotic hydrolysis were determined as described previously (21). Kinetic parameters were determined for the substrates ampicillin, cephalothin, cefotaxime, and ceftazidime with a DU800 spectrophotometer at wavelengths 235, 262, 264, and 260 nm, respectively. All experiments were performed at 30 °C. GraphPad Prism 6 was used to fit initial velocities to the Michaelis-Menten equation, v = Vmax[S]/(Km + [S]), to determine kcat and Km values. The kcat/Km was estimated using the equation v = kcat/Km [E] [S], where [S] ≪ Km, when CTX-M enzymes had a high Km value that prevented determination of Vmax for cefotaxime or ceftazidime hydrolysis. The kinetic parameters for all reactions were determined using initial velocities, which were an average of four measurements. The errors associated with the kinetic parameters were determined as described previously (12).
Thermal stability determinations
For thermal stability analysis, 100 μg of purified protein in 500 μl of 50 mm phosphate buffer, pH 7.0, was measured at 222 nm using a AVIV model 425 circular dichroism spectrometer connected to an Illinois Instruments ZR800 oxygen analyzer. The sample was measured from 30 to 70 °C, with a 6-s equilibration time at each measurement. The temperature was increased at a rate of 1 °C/min. For analysis, the data were fit to a two-state folding mechanism following the Gibbs-Helmholtz equation (53). ΔCP was calculated based on the number of amino acids and solvent-accessible surface area (54). ΔHm was calculated as −1 × (Tm +273.2) × (slope of ΔG/T at Tm). ΔΔG values were calculated at the Tm of WT CTX-M-14 using the equation ΔG(T) = ΔHm (1 − T/Tm) − ΔCP(Tm − T) + T ln(T/Tm).
Immunoblot analysis
The in vivo protein expression levels of the WT and mutant CTX-M β-lactamases were determined using immunoblot analysis as described previously (12, 18). In brief, the CTX-M-14–pTP123 plasmid encoding the WT or mutant CTX-M β-lactamases was transformed into E. coli RB791 cells and grown in LB containing 12.5 μg/ml chloramphenicol at 37 °C to an A600 of 0.6–0.7. The cells were pelleted and resuspended in 0.25 ml (10 mm Tris-HCl, pH 8.0, 20% (w/v) sucrose). After incubation at 4 °C with gentle shaking for 30 min, the same volume of cold water was added and incubated at 4 °C for another 30 min. The cells were then pelleted by centrifugation at 4000 × g for 10 min, and the supernatant containing the periplasmic fraction was collected. 12 μl of the supernatant was mixed with 4 μl of 4× SDS loading buffer, heated at 70 °C for 5 min, and fractionated by running on a 12% SDS-polyacrylamide gel. After being transferred onto a nitrocellulose membrane (GE Healthcare), the expression of CTX-M-14 β-lactamase was detected by probing with a rabbit anti-CTX-M-14 antiserum followed by a donkey anti-rabbit secondary antibody conjugated with horseradish peroxidase (GE Healthcare). In addition, for the purpose of a loading control, the same membrane was also probed with rabbit antiserum raised against maltose-binding protein (MBP) (a gift from Dr. Anna Konovalova, University of Texas Health Science Center at Houston) and the same secondary antibody as used for detection of the periplasmic protein MBP. The SuperSignal West Pico chemiluminescent substrate (ThermoFisher Scientific) was used to develop the immunoblot. The hybridization signal was quantified by densitometry using ImageJ software (National Institutes of Health), and the signal for WT and mutant CTX-M-14 β-lactamase was normalized to that for MBP.
X-ray crystallography
The PEGs, PACT and the JCSG Core I Suites from Qiagen were used to screen conditions for crystal growth using the hanging drop vapor diffusion method. A Mosquito robot (TTP Labtech Ltd., Melbourn, UK) was used to set up crystallization screens by mixing mother liquor and protein in a 1:1 ratio to form a 0.2-μl drop for hanging drop vapor diffusion. The protein was concentrated to 20 mg/ml and stored in 5 mm Tris, pH 7.0, 50 mm NaCl for crystallization. The CTX-M-14 N106S mutant enzyme crystallized in 0.2 m calcium acetate hydrate, 20% (w/v) PEG 3350, pH 7.5. The CTX-M-14 N106S/D240G enzyme crystallized in 25% PEG 1500 (w/v), 0.1 m PCB buffer, pH 6.0. The CTX-M-14 S70G/N106S enzyme crystallized in 0.2 m calcium acetate, 0.1 m MES, pH 6.0, 20% (w/v) PEG 8000. The CTX-M-14 S70G/N106S/D240G enzyme crystallized in 0.2 m MgCl2, 20% (w/v) PEG 3350. The structures of CTX-M-14 S70G/N106S and CTX-M-14 S70G/N106S/D240G mutant enzymes complexed with cefotaxime were obtained by soaking the protein crystals in 50 mm cefotaxime overnight, before collecting data. 20% glycerol was added to the mother liquor and used as the cryoprotectant in all cases. The data for the CTX-M-14 N106S structure were collected at Baylor College of Medicine using a Rigaku FR-E SuperBright High-Brilliance Rotating Anode Generator. The data for the CTX-M-14 N106S/D240G, S70G/N106S/CTX, and S70G/N106S/D240G/CTX structures were collected at the Berkeley Center for Structural Biology at the Advanced Light Source synchrotron beamline. Data were processed using HKL2000, iMosflm (55) in the CCP4i Suite (56). MOLREP was used for molecular replacement using CTX-M-14 (PDB code 1YLT) (9) as the model enzyme for phasing (57). Coot (58) was used to fit the model to the density and the phenix.refine (59), and REFMAC5 (60) programs were used for refinement. All figures were created using the UCSF Chimera program (61).
Author contributions
M. P. P., B. V. V. P., and T. P. conceptualization; M. P. P., L. H., C. J. A., V. S., and B. S. data curation; M. P. P., L. H., B. S., B. V. V. P., and T. P. formal analysis; M. P. P., L. H., Z. S., B. S., and B. V. V. P. validation; M. P. P., L. H., C. A. B., Z. S., C. J. A., V. S., and B. S. investigation; M. P. P., L. H., C. A. B., Z. S., and T. P. visualization; M. P. P., L. H., C. A. B., C. J. A., V. S., and B. S. methodology; M. P. P., B. V. V. P., and T. P. writing-original draft; M. P. P., L. H., C. A. B., C. J. A., V. S., B. S., B. V. V. P., and T. P. writing-review and editing; B. V. V. P. and T. P. supervision; B. V. V. P. and T. P. funding acquisition; B. V. V. P. and T. P. project administration; T. P. resources.
Supplementary Material
Acknowledgments
We thank Hiram Gilbert for discussions and comments on the manuscript. The ALS-ENABLE beamlines are supported in part by NIGMS Grant P30 GM124169-01 from the National Institutes of Health. The Advanced Light Source is a Department of Energy Office of Science User Facility under Contract No. DE-AC02-05CH11231.
This work was supported in part by National Institutes of Health Grant R01 AI32956 (to T. P.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3, Table S1, and supporting Refs. 1–3.
The atomic coordinates and structure factors (codes 6CYK, 6CYQ, 6CYN, and 6CYU) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ESBL
- extended-spectrum β-lactamase
- MIC
- minimum inhibitory concentration
- MBP
- maltose-binding protein
- SPFF
- SP-Sepharose Fast Flow
- PDB
- Protein Data Bank
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- CTX
- cefotaxime
- TEV
- tobacco etch virus.
References
- 1. Livermore D. M. (2006) The β-lactamase threat in Enterobacteriaceae, Pseudomonas and Acinetobacter. Trends Microbiol. 14, 413–420 10.1016/j.tim.2006.07.008 [DOI] [PubMed] [Google Scholar]
- 2. Bonomo R. A. (2017) β-Lactamases: a focus on current challenges. Cold Spring Harb. Perspect. Med. 7, a025239 10.1101/cshperspect.a025239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Strynadka N. C., Adachi H., Jensen S. E., Johns K., Sielecki A., Betzel C., Sutoh K., and James M. N. (1992) Molecular structure of the acyl-enzyme intermediate in b-lactam hydrolysis at 1.7 A resolution. Nature 359, 700–705 10.1038/359700a0 [DOI] [PubMed] [Google Scholar]
- 4. Palzkill T. (2018) Structural and mechanistic basis for extended-spectrum drug-resistance mutations in altering the specificity of TEM, CTX-M, and KPC β-lactamases. Front. Mol. Biosci. 5, 16 10.3389/fmolb.2018.00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Perez F., Endimiani A., Hujer K. M., and Bonomo R. A. (2007) The continuing challenge of ESBLs. Curr. Opin. Pharmacol. 7, 459–469 10.1016/j.coph.2007.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonnet R. (2004) Growing group of extended-spectrum β-lactamases: the CTX-M enzymes. Antimicrob. Agents Chemother. 48, 1–14 10.1128/AAC.48.1.1-14.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. D'Andrea M. M., Arena F., Pallecchi L., and Rossolini G. M. (2013) CTX-M-type β-lactamases: a successful story of antibiotic resistance. Int. J. Med. Microbiol. 303, 305–317 10.1016/j.ijmm.2013.02.008 [DOI] [PubMed] [Google Scholar]
- 8. Cantón R., González-Alba J. M., and Galán J. C. (2012) CTX-M enzymes: origin and diffusion. Front. Microbiol. 3, 110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen Y., Delmas J., Sirot J., Shoichet B., and Bonnet R. (2005) Atomic resolution structures of CTX-M β-lactamases: extended spectrum activities from increased mobility and decreased stability. J. Mol. Biol. 348, 349–362 10.1016/j.jmb.2005.02.010 [DOI] [PubMed] [Google Scholar]
- 10. Bonnet R., Dutour C., Sampaio J. L., Chanal C., Sirot D., Labia R., De Champs C., and Sirot J. (2001) Novel cefotaximase (CTX-M-16) with increased catalytic efficiency due to substitution Asp-240–Gly. Antimicrob. Agents Chemother. 45, 2269–2275 10.1128/AAC.45.8.2269-2275.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kimura S., Ishiguro M., Ishii Y., Alba J., and Yamaguchi K. (2004) Role of a mutation at position 167 of CTX-M-19 in ceftazidime hydrolysis. Antimicrob. Agents Chemother. 48, 1454–1460 10.1128/AAC.48.5.1454-1460.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Patel M. P., Fryszczyn B. G., and Palzkill T. (2015) Characterization of the global stabilizing substitution A77V and its role in the evolution of CTX-M β-lactamases. Antimicrob. Agents Chemother. 59, 6741–6748 10.1128/AAC.00618-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Patel M. P., Hu L., Stojanoski V., Sankaran B., Prasad B. V. V., and Palzkill T. (2017) The drug-resistant variant P167S expands the substrate profile of CTX-M β-lactamases for oxyimino-cephalosporin antibiotics by enlarging the active site upon acylation. Biochemistry 56, 3443–3453 10.1021/acs.biochem.7b00176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Novais A., Cantón R., Coque T. M., Moya A., Baquero F., and Galán J. C. (2008) Mutational events in cefotaximase extended-spectrum β-lactamases of the CTX-M-1 cluster involved in cefazidime resistance. Antimicrob. Agents Chemother. 52, 2377–2382 10.1128/AAC.01658-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Delmas J., Robin F., Carvalho F., Mongaret C., and Bonnet R. (2006) Prediction and evolution of ceftazidime resistance in extended-spectrum β-lactamase CTX-M-9. Antimicrob. Agents Chemother. 50, 731–738 10.1128/AAC.50.2.731-738.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brown N. G., Pennington J. M., Huang W., Ayvaz T., and Palzkill T. (2010) Multiple global suppressors of protein stability defects facilitate the evolution of extended-spectrum TEM β-lactamases. J. Mol. Biol. 404, 832–846 10.1016/j.jmb.2010.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang W., and Palzkill T. (1997) A natural polymorphism in b-lactamase is a global suppressor. Proc. Natl. Acad. Sci. U.S.A. 94, 8801–8806 10.1073/pnas.94.16.8801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marciano D. C., Pennington J. M., Wang X., Wang J., Chen Y., Thomas V. L., Shoichet B. K., and Palzkill T. (2008) Genetic and structural characterization of an L201P global suppressor substitution in TEM-1 β-lactamase. J. Mol. Biol. 384, 151–164 10.1016/j.jmb.2008.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sideraki V., Huang W., Palzkill T., and Gilbert H. F. (2001) A secondary drug resistance mutation of TEM-1 β-lactamase that suppresses misfolding and aggregation. Proc. Natl. Acad. Sci. U.S.A. 98, 283–288 10.1073/pnas.011454198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pérez-Llarena F. J., Kerff F., Abián O., Mallo S., Fernández M. C., Galleni M., Sancho J., and Bou G. (2011) Distant and new mutations in CTX-M-1 β-lactamase affect cefotaxime hydrolysis. Antimicrob. Agents Chemother. 55, 4361–4368 10.1128/AAC.00298-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adamski C. J., Cardenas A. M., Brown N. G., Horton L. B., Sankaran B., Prasad B. V., Gilbert H. F., and Palzkill T. (2015) Molecular basis for the catalytic specificity of the CTX-M extended spectrum β-lactamases. Biochemistry 54, 447–457 10.1021/bi501195g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Delmas J., Leyssene D., Dubois D., Birck C., Vazeille E., Robin F., and Bonnet R. (2010) Structural insights into substrate recognition and product expulsion in CTX-M enzymes. J. Mol. Biol. 400, 108–120 10.1016/j.jmb.2010.04.062 [DOI] [PubMed] [Google Scholar]
- 23. Dellus-Gur E., Elias M., Caselli E., Prati F., Salverda M. L., de Visser J. A., Fraser J. S., and Tawfik D. S. (2015) Negative epistasis and evolvability in TEM-1 β-lactamase–the thin line between an enzyme's conformational freedom and disorder. J. Mol. Biol. 427, 2396–2409 10.1016/j.jmb.2015.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hart K. M., Ho C. M., Dutta S., Gross M. L., and Bowman G. R. (2016) Modelling proteins' hidden conformations to predict antibiotic resistance. Nat. Commun. 7, 12965 10.1038/ncomms12965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mobashery S., and Johnston M. (1986) Reactions of Escherichia coli TEM β-lactamase with cephalothin and with C10-dipeptidyl cephalosporin esters. J. Biol. Chem. 261, 7879–7887 [PubMed] [Google Scholar]
- 26. Pratt R. F., and Faraci W. S. (1986) Direct observation by 1H NMR of cephalosporoate intermediates in aqueous solution during the hydrazinolysis of β-lactamase-catalyzed hydrolysis of cephalosporins with 3′ leaving groups: kinetics and equilibria of the 3′ elimination reaction. J. Am. Chem. Soc. 108, 5328–5333 10.1021/ja00277a044 [DOI] [Google Scholar]
- 27. Shimamura T., Ibuka A., Fushinobu S., Wakagi T., Ishiguro M., Ishii Y., and Matsuzawa H. (2002) Acyl-intermediate structures of the extended-spectrum class A β-lactamase, Toho-1, in complex with cefotaxime, cephalothin, and benzylpenicillin. J. Biol. Chem. 277, 46601–46608 10.1074/jbc.M207884200 [DOI] [PubMed] [Google Scholar]
- 28. Vandavasi V. G., Weiss K. L., Cooper J. B., Erskine P. T., Tomanicek S. J., Ostermann A., Schrader T. E., Ginell S. L., and Coates L. (2016) Exploring the mechanism of b-lactam ring protonation in the class A β-lactamase acylation mechanism using neutron and X-ray crystallography. J. Med. Chem. 59, 474–479 10.1021/acs.jmedchem.5b01215 [DOI] [PubMed] [Google Scholar]
- 29. Wang X., Minasov G., and Shoichet B. K. (2002) Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J. Mol. Biol. 320, 85–95 10.1016/S0022-2836(02)00400-X [DOI] [PubMed] [Google Scholar]
- 30. Mehta S. C., Rice K., and Palzkill T. (2015) Natural variants of the KPC-2 carbapenemase have evolved increased catalytic efficiency for ceftazidime hydrolysis at the cost of enzyme stability. PLoS Pathog. 11, e1004949 10.1371/journal.ppat.1004949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tokuriki N., Stricher F., Serrano L., and Tawfik D. S. (2008) How protein stability and new functions trade off. PLoS Comp. Biol. 4, e1000002 10.1371/journal.pcbi.1000002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Studer R. A., Christin P.-A., Williams M. A., and Orengo C. A. (2014) Stability-activity tradeoffs constrain the adaptive evolution of RubisCO. Proc. Natl. Acad. Sci. U.S.A. 111, 2223–2228 10.1073/pnas.1310811111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bar-Even A., Milo R., Noor E., and Tawfik D. S. (2015) The moderately efficient enzyme: futile encounters and enzyme floppiness. Biochemistry 54, 4969–4977 10.1021/acs.biochem.5b00621 [DOI] [PubMed] [Google Scholar]
- 34. Jiménez-Osés G., Osuna S., Gao X., Sawaya M. R., Gilson L., Collier S. J., Huisman G. W., Yeates T. O., Tang Y., and Houk K. N. (2014) The role of distant mutations and allosteric regulation on LovD active site dynamics. Nat. Chem. Biol. 10, 431–436 10.1038/nchembio.1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Campbell E., Kaltenbach M., Correy G. J., Carr P. D., Porebski B. T., Livingstone E. K., Afriat-Jurnou L., Buckle A. M., Weik M., Hollfelder F., Tokuriki N., and Jackson C. J. (2016) The role of protein dynamics in the evolution of new enzyme function. Nat. Chem. Biol. 12, 944–950 10.1038/nchembio.2175 [DOI] [PubMed] [Google Scholar]
- 36. Strynadka N. C., Jensen S. E., Alzari P. M., and James M. N. (1996) A potent new mode of β-lactamase inhibition revealed by the 1.7Å X-ray crystallographic structure of the TEM-1-BLIP complex. Nat. Struct. Biol. 3, 290–297 10.1038/nsb0396-290 [DOI] [PubMed] [Google Scholar]
- 37. Strynadka N. C., Martin R., Jensen S. E., Gold M., and Jones J. B. (1996) Structure-based design of a potent transition state analogue for TEM-1 β-lactamase. Nat. Struct. Biol. 3, 688–695 10.1038/nsb0896-688 [DOI] [PubMed] [Google Scholar]
- 38. Lim D., Park H. U., De Castro L., Kang S. G., Lee H. S., Jensen S., Lee K. J., and Strynadka N. C. (2001) Crystal structure and kinetic analysis of β-lactamase inhibitor protein-II in complex with TEM-1 β-lactamase. Nat. Struct. Biol. 8, 848–852 10.1038/nsb1001-848 [DOI] [PubMed] [Google Scholar]
- 39. Xiao H., Nasertorabi F., Choi S.-H., Han G. W., Reed S. A., Stevens R. C., and Schultz P. G. (2015) Exploring the potential impact of an expanded genetic code on protein function. Proc. Natl. Acad. Sci. U.S.A. 112, 6961–6966 10.1073/pnas.1507741112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Doucet N., and Pelletier J. N. (2007) Simulated annealing exploration of an active-site tyrosine in TEM-1 β-lactamase suggests the existence of alternate conformations. Proteins 69, 340–348 10.1002/prot.21485 [DOI] [PubMed] [Google Scholar]
- 41. Jelsch C., Mourey L., Masson J. M., and Samama J. P. (1993) Crystal structure of Escherichia coli TEM-1 β-lactamase at 1.8 A resolution. Proteins 16, 364–383 10.1002/prot.340160406 [DOI] [PubMed] [Google Scholar]
- 42. Fonzé E., Charlier P., To'th Y., Vermeire M., Raquet X., Dubus A., and Frère J. M. (1995) TEM-1 β-lactamase structure solved by molecular replacement and refined structure of the S235A mutant. Acta Crystallogr. D Biol. Crystallogr. 51, 682–694 10.1107/S0907444994014496 [DOI] [PubMed] [Google Scholar]
- 43. Ramachandran G. N., and Sasisekharan V. (1968) Conformation of polypeptides and proteins. Adv. Protein Chem. 23, 283–438 10.1016/S0065-3233(08)60402-7 [DOI] [PubMed] [Google Scholar]
- 44. Chellapa G. D., and Rose G. D. (2015) On interpretation of protein X-ray structures: planarity of the peptide unit. Proteins 83, 1687–1692 10.1002/prot.24854 [DOI] [PubMed] [Google Scholar]
- 45. Brereton A. E., and Karplus P. A. (2016) On the reliability of peptide nonplanarity seen in ultra-high resolution crystal structures. Prot. Sci. 25, 926–932 10.1002/pro.2883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Blomberg R., Kries H., Pinkas D. M., Mittl P. R., Grütter M. G., Privett H. K., Mayo S. L., and Hilvert D. (2013) Precision is essential for efficient catalysis in an evolved Kemp eliminase. Nature 503, 418–421 10.1038/nature12623 [DOI] [PubMed] [Google Scholar]
- 47. González M. M., Abriata L. A., Tomatis P. E., and Vila A. J. (2016) Optimization of conformational dynamics in an epistatic evolutionary trajectory. Mol. Biol. Evol. 33, 1768–1776 10.1093/molbev/msw052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang G., Hong N., Baier F., Jackson C. J., and Tokuriki N. (2016) Conformational tinkering drives evolution of a promiscuous activity through indirect mutational effects. Biochemistry 55, 4583–4593 10.1021/acs.biochem.6b00561 [DOI] [PubMed] [Google Scholar]
- 49. Otten R., Liu L., Kenner L. R., Clarkson M. W., Mavor D., Tawfik D. S., Kern D., and Fraser J. S. (2018) Rescue of conformational dynamics in enzyme catalysis by directed evolution. Nat. Commun. 9, 1314 10.1038/s41467-018-03562-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Petrosino J., Rudgers G., Gilbert H., and Palzkill T. (1999) Contributions of aspartate 49 and phenylalanine 142 residues of a tight binding inhibitory protein of β-lactamases. J. Biol. Chem. 274, 2394–2400 10.1074/jbc.274.4.2394 [DOI] [PubMed] [Google Scholar]
- 51. Amann E., Brosius J., and Ptashne M. (1983) Vectors bearing a hybrid trp-lac promoter useful for regulated expression of cloned genes in Escherichia coli. Gene 25, 167–178 10.1016/0378-1119(83)90222-6 [DOI] [PubMed] [Google Scholar]
- 52. Studier F. W., and Moffatt B. A. (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130 10.1016/0022-2836(86)90385-2 [DOI] [PubMed] [Google Scholar]
- 53. Scholtz J. M., and Pace C. N. (1989) in Protein Structure: A Practical Approach (Creighton T. E., ed) IRL Press at Oxford University Press, Oxford [Google Scholar]
- 54. Myers J. K., Pace C. N., and Scholtz J. M. (1995) Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4, 2138–2148 10.1002/pro.5560041020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., and Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 10.1107/S0907444910048675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 10.1107/S0907444909042589 [DOI] [PubMed] [Google Scholar]
- 58. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., and Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 10.1107/S0907444904023510 [DOI] [PubMed] [Google Scholar]
- 61. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.