

Summary
In Vibrio cholerae, virulence gene expression is regulated by a transmembrane‐localized transcription factor complex designated as ToxRS. ToxR harbours two cysteines in the periplasmic domain that can form inter‐ and intramolecular disulfide bonds. In this study, we investigated the σE‐dependent inner membrane proteolysis of ToxR, which occurs via the periplasmic‐localized proteases DegS and DegP. Both proteases respond to the redox state of the two cysteine thiol groups of ToxR. Interestingly, in the presence of sodium deoxycholate, ToxR proteolysis is blocked independently of ToxS, whereas ToxR activation by bile salts requires ToxS function. From these data, we identified at least two levels of control for ToxR activation by sodiumdeoxycholate. First, bile inhibits ToxR degradation under starvation and alkaline pH or under conditions in which DegPS responds to the reduced disulfide bonds of ToxR. The second level links bile to ToxRS complex formation and further activation of its transcription factor activity. Overall, our data suggest a comprehensive bile sensory function for the ToxRS complex during host colonization.
Introduction
Studies on Vibrio cholerae pathogenesis have revealed that the production of cholera toxin (CT) and toxin‐coregulated pili (TCP) is coordinated by a regulatory network that has been historically referred to as the ToxR regulon (Matson et al., 2007). This system is comprised of several transcriptional factors, which include AphAB, TcpPH, ToxRS and ToxT (Miller and Mekalanos, 1984; Miller et al., 1989; DiRita et al., 1991; Hase and Mekalanos, 1998; Skorupski and Taylor, 1999). The AphAB complex, in which aphA is under the transcriptional control of the LuxOP quorum‐sensing system (Rutherford et al., 2011), is active under anaerobic conditions (Kovacikova et al., 2010), regulates tcpPH (Skorupski and Taylor, 1999) and enhances toxRS transcription (Xu et al., 2010). TcpPH is a labile complex that is activated via bile salts, which cause rearrangements of TcpP‐intramolecular disulfide bonds to promote an intermolecular disulfide bond (Yang et al., 2013). In contrast, under virulence‐promoting conditions, ToxR is constitutively expressed (Kanjilal et al., 2010), and together with TcpP activates the transcription of toxT (Higgins and DiRita, 1994; Hase and Mekalanos, 1998; Bina et al., 2003; Childers and Klose, 2007). ToxT subsequently activates the transcription of the ctx and tcp loci. V. cholerae strains that lack either TcpP, ToxT or ToxR do not produce CT or TCP and are non‐virulent (Champion et al., 1997). ToxR directly regulates the transcription of many genes (Bina et al., 2003), the best characterized of which are ompT and ompU (Miller and Mekalanos, 1988), which encode the major porins of V. cholerae.
Structurally, ToxR is related to the OmpR‐type regulators (Ottemann et al., 1992). The N‐terminus of ToxR is located in the cytoplasm and contains a winged helix‐turn‐helix DNA‐binding motif, followed by a single transmembrane domain (TM) and a periplasmic C‐terminal domain (Miller et al., 1987). Experimentally defined operator binding sites of ToxR are termed ToxR boxes and have been identified 40 to 180 bp upstream of the ctx, ompU/T and toxT promoters (Pfau and Taylor, 1996; Goss et al., 2013). As demonstrated by domain analysis, the ToxR TM segment functions in ToxR activity and may be involved in a bile‐dependent ToxR activation (Dziejman et al., 1999; Crawford et al., 2003; Hung and Mekalanos, 2005). The ToxR periplasmic domain has been proposed to be a sensor for environmental stimuli, containing two cysteine residues at amino acid positions 236 and 293 that can form homodimer or intramolecular disulfide bonds (Ottemann and Mekalanos, 1996). Recently, we showed in greater detail that intramolecular disulfide bond formation in ToxR produces an active conformation of ToxR that is necessary for the proper regulation of the porin genes ompU and ompT but not for the activation of toxT transcription. Other transcriptional regulators also contain cysteine residues, including AphB and TcpP. Interestingly, the reduction of the cysteine thiol groups in some of these regulators has been shown to be important for virulence gene regulation and is dependent on oxygen limitation and the presence of bile salts (Liu et al., 2011; Yang et al., 2013). Additionally, cysteine residues in TcpP and TcpH have been reported to be crucial for protein stability (Morgan et al., 2015), indicating that replacing these cysteine residues can disrupt inter‐ and intramolecular‐disulfide bonds within TcpP and TcpH and alter TcpPH stability.
Similar to tcpH and tcpP, a second gene is co‐transcribed with toxR, termed toxS (Miller et al., 1989b). ToxS is also an inner membrane‐localized protein (DiRita et al., 1991) with only 6‐8 amino acids exposed to the cytoplasm, followed by a single TM domain and a C‐terminal region located in the periplasm. The current model suggests that ToxR and ToxS are interacting partners in the periplasm (DiRita and Mekalanos, 1991) and form heterodimers (Ottemann and Mekalanos, 1996). The intramolecular disulfide form of ToxR monomers was previously shown to be a binding partner for ToxS and the inability to form intramolecular disulfide bonds is associated with the attenuated regulation of OmpT and OmpU porin expression (Fengler et al., 2012). Furthermore, knockout mutations of toxS negatively influence the transcriptional activity of ToxR (Miller et al., 1989), suggesting that either ToxS facilitates the activity of ToxR or that ToxS affects ToxR protein stability (DiRita and Mekalanos, 1991; Pfau and Taylor, 1998). Recently, it was demonstrated that ToxR is subject to proteolysis under starvation and alkaline pH conditions both in a classical and an El Tor O1 isolate (Almagro‐Moreno et al., 2015a; 2015b). ToxS plays a crucial role in this process, since RseP‐ and site‐1‐mediated proteolysis can either be prevented by WT ToxS or promoted by mutant ToxSL33S (Almagro‐Moreno et al., 2015a; 2015b). Furthermore, the interaction between ToxR and ToxS has been described to be influenced by the binding of bile acids, which results in an enhanced active transcriptional complex, influencing OmpU and OmpT expression (Midgett et al., 2017).
The recently published data addressing the ToxRS interaction with bile acids and ToxR proteolysis prompted us to elucidate the molecular relationship between ToxRS activation and stability. In this study, we describe two major routes of ToxR proteolysis. One route involves DegS and DegP, which respond towards the redox state of the ToxR cysteine residues. The other route appears to be independent of the redox state but is sensitive to starvation and alkaline conditions. However, both degradation paths are inhibited if bile salts (sodium deoxycholate) are present in the environment. Since bile salts control proteolysis and influence the activity of the ToxRS transcriptional complex, our data extend the current model of ToxRS and indicate a bile acid‐sensing function of ToxRS.
Results
ToxR protein stability depends on its redox state, its interaction with ToxS and the proteases DegS and DegP
As previously demonstrated, cysteine residues within the periplasmic domain of ToxR contribute to inter‐ and intramolecular disulfide bonds and have implications for ToxR transcription factor activity (Ottemann and Mekalanos, 1996; Fengler et al., 2012). Prompted by the newly described regulated intramembrane proteolysis (RIP) of ToxR (Almagro‐Moreno et al., 2015b), we revisited the cysteine replacement mutant toxRCC, which has a very low ToxR activation phenotype (Fengler et al., 2012) and characterized ToxRCC protein stability. For these experiments, we compared ToxR and ToxRCC in WT, dsbA, FLAGtoxRCC, FLAGtoxRCCdegPS (Fig. 1A), dsbC and degSdsbA V. cholerae strains (Fig. S1). From these cultures, whole cell lysates (WCL) were generated from cells grown in M9 maltose media for up to 32 h, solubilized in Laemmli buffer under nonreducing conditions and used for SDS‐PAGE followed by the immunoblot analysis. Two protein bands were observed, the reduced (ToxRred) and the oxidized (ToxRoxy) form, the latter of which contains an intramolecular disulfide bond (Fig. 1A) (Otteman and Mekalanos, 1996). At time points 6‐32 h, both, ToxRred and ToxRoxy were observed in the WT strain and no degradation was detected. In contrast, the endogenous ToxR in the dsbA strain and the FLAGToxRCC mutant could hardly be detected past the 12 h time point (Fig. 1A), corresponding to an early stationary growth phase (Fig. S2). In the dsbA mutant, ToxRred became the major protein form of monomeric ToxR from time point 6 h onwards. These results support the conclusion that DsbA acts as an oxido‐reductase on ToxR, forming intramolecular disulfide bonds and affecting ToxR stability. A dsbC mutant exhibited a stable ToxR expression profile with less recognizable ToxRred when compared to the WT strain (Fig. S1). To assess DegS and DegP protease activities, we constructed degSdsbA and FLAGtoxRCCdegPS mutant strains and monitored ToxR and FLAGToxRCC respectively. As observed in the degSdsbA mutant (Fig. S1), ToxR exhibited a prolonged and stable expression period, suggesting the participation of DegS during ToxR proteolysis. However, when compared to the WT strain, ToxR degradation was observed after incubating for 21‐24 h, indicating the presence of additional active proteases. As a result of these observations, we monitored a FLAGtoxRCCdegPS strain (Fig. 1A) and showed that ToxRCC indeed stayed stable over 32 h if compared to the FLAGtoxRCC strain. To further elucidate the contribution of the two proteases DegS and DegP, the knockout mutant strain FLAGtoxRCCdegPS was used to perform complementation studies. The expression of either of these proteases in trans led to increased ToxRCC degradation compared to the strains harbouring control plasmids (Fig. 1B). Furthermore, a degS knockout showed impaired σE response with significantly decreased degP transcription (Fig. S3). These data provide strong genetic evidence that the DegS and DegP proteases independently promote ToxR degradation if ToxRred or FLAGToxRCC is encountered.
Figure 1.
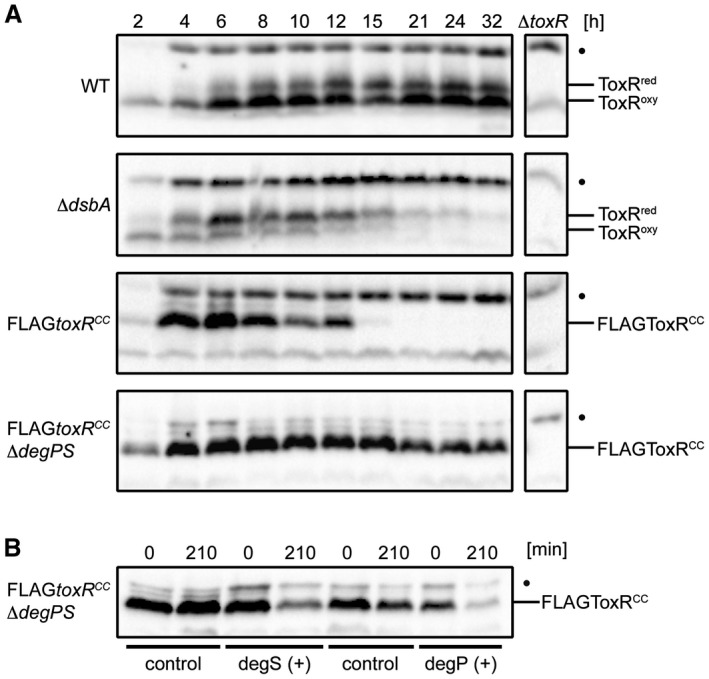
The cysteine oxidation and reduction state of ToxR affects protein stability in a DegPS‐dependent way. A. Shown are ToxR immunoblots of V. cholerae WT, dsbA, FLAGtoxRCC and FLAGtoxRCCdegPS grown in M9 maltose. B. Complementation studies of V. cholerae FLAGtoxRCCdegPS are demonstrated by ToxR immunoblots. Cells harboured pBAD18‐KandegS, pMMB67EHdegP or the corresponding plasmid controls. Protein biosynthesis was inhibited by the addition of Cm. Immunoblots were performed under standard nonreducing Laemmli buffer conditions utilizing α‐ToxR antiserum. The migration patterns of ToxRred/oxy are indicated. (•): Represents a nonspecific cross‐reacting background band.
To further characterize the instability of ToxRred and the involvement of ToxS, temporal expression levels of chromosomally produced ToxR protein were examined in the WT and toxS mutant strains (Fig. 2A and B). To prevent de novo protein biosynthesis, chloramphenicol was added to monitor the fate of the ToxR protein. In this assay, cells were incubated with or without sublethal concentrations of the reducing agent DTT to promote the conversion of ToxRoxy into ToxRred. Without the DTT treatment, the amount of ToxRoxy was increased over ToxRred, while the opposite effect was detected in the presence of DTT in the WT and toxS strains. In these assays, ToxR was observed to remain stable in WT cells regardless of the presence or absence of DTT, whereas in the toxS mutant, increased ToxR degradation was detected in DTT‐treated cells (Fig. 2A). Using densitometric analysis, we determined that this result was significant (Fig. 2B, Fig. S4). Intriguingly, proteolysis was observed for ToxRCC or for ToxR in a dsbA strain starting at time point 15 h (Fig. 1A). Therefore, we tested the WT and toxS knockout mutant strains grown to 15 h for ToxR stability (Fig. S5). After incubation, the cultures were divided and further incubated with or without DTT for an additional 6 h. Similar to the results presented in Fig. 2A, but without the addition of chloramphenicol, the greatly increased proteolysis of ToxR was only visible in the toxS knockout mutant in the presence of DTT (Fig. S5). Since DTT‐treated cells predominantly express ToxRred, we concluded that ToxR proteolysis correlates with a cysteine‐reduced state that is enhanced in the absence of ToxS. Importantly, we have no evidence for the selective degradation of a specific form of ToxRred/oxy. In addition, we obtained no evidence that DTT increases the proteolysis of ToxRCC (Fig. S6); therefore, we conclude that DTT does not activate DegS and DegP. Furthermore, the involvement of DegS was confirmed in this assay, showing that in a toxSdegS double mutant, no ToxR degradation was observed, with or without DTT treatment (Fig. S7).
Figure 2.
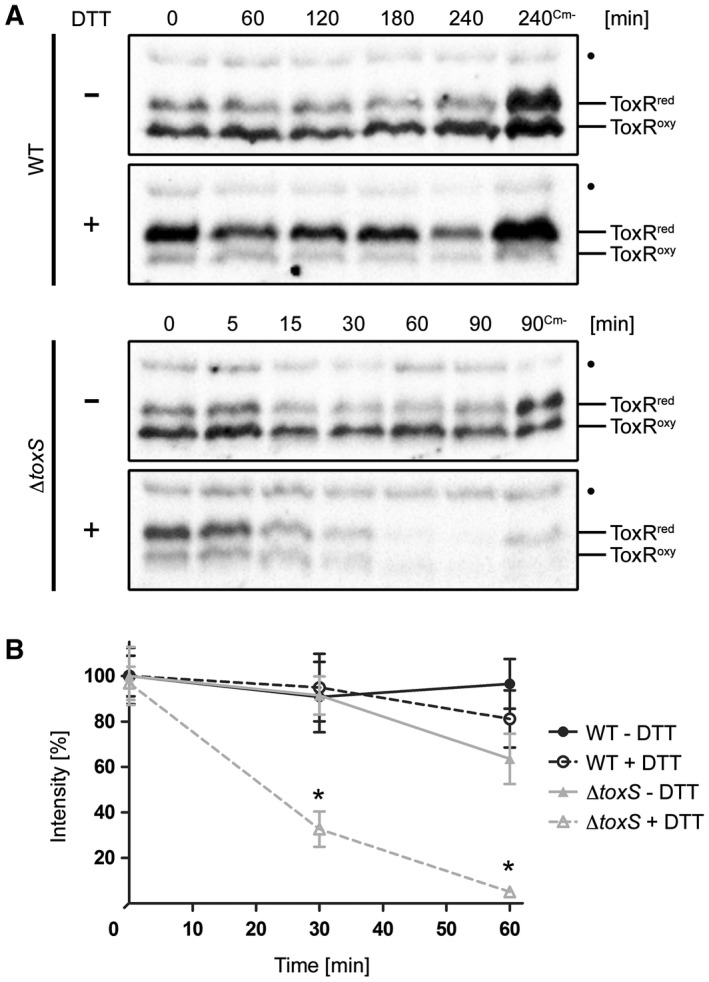
Effects of DTT treatment on the redox state and protein stability of ToxR in V. cholerae WT and toxS mutants. ToxR temporal stability levels were measured by the immunoblot analysis in WT and toxS mutant strains grown in M9 maltose with or without DTT (+/–). Protein biosynthesis was inhibited by the addition of Cm. Samples without chloramphenicol (Cm‐) served as negative controls. A. Immunoblots of WT and toxS WCL were performed under standard nonreducing Laemmli buffer conditions utilizing α‐ToxR antiserum. The migration patterns of ToxRred/oxy are indicated. (•): Represents a nonspecific cross‐reacting background band. B. Graphs show band intensities (%) of WCL samples treated under reducing Laemmli buffer conditions defined by densitometry of similar blots (see one set of representative immunoblots Fig. S4). For each time point, the sample number was n ≥ 6 and the mean values with standard deviation are shown. Two‐way ANOVA with Bonferroni post hoc analysis indicates significant differences between toxS strains without (grey filled triangle, solid line) and toxS cells with DTT (grey open triangle, dotted line) with P < 0.001 at time points 30 and 60 min. No significant differences were seen between WT without DTT (black filled circle, solid line) and WT incubated with DTT (black open circle, dotted line).
Characterization of the DegS‐dependent ToxR and RseA proteolysis
DegS is a serine site‐1 protease and a stress‐sensor protein that activates the σE response pathway by recognizing unfolded OMPs in the periplasm of Escherichia coli (Lima et al., 2013). Several studies of the σE pathway in V. cholerae have been published (Kovacikova and Skorupski, 2002; Ding et al., 2004; Mathur et al., 2007; Davis and Waldor, 2009) and RseP, DegS and DegP were recently shown to participate in ToxR proteolysis (Almagro‐Moreno et al., 2015b). However, the release of σE via the activity of DegS was not well studied in these investigations. To evaluate DegS activity against its known substrate, the anti‐sigma factor RseA homologue of V. cholerae was monitored in a proteolysis assay (Fig. 3A). We observed that FLAGRseA was a substrate for the DegS protease under the conditions tested, as was FLAGToxRCC monitored in degS+/– strains (Fig. 3B). For ToxRCC several residual degradation products at approximately 31 and 26 kDa were observed, as proteolysis may have been incomplete due to its overexpression from a plasmid. Of note, the WT ToxR protein remained stable, even when overexpressed from a plasmid. Furthermore, as shown in Fig. 3C, the degradation of FLAGToxRCC was accelerated in the presence of stressor molecules, which was accomplished via the over‐expression of a synthetic C‐terminal OmpU fragment derived from pTrc99ApelBYYF (for a detailed description see Experimental Procedures). This assay was performed because it has been shown that the C‐terminal peptides derived from stress‐induced, unfolded porins act as binding partners for the PDZ domain of DegS, which in turn leads to σE pathway activation (Walsh et al., 2003; Wilken et al., 2004; Chaba et al., 2007). In contrast, no activation of proteolysis was observed for the WT ToxR, neither in the absence nor in the presence of ToxS. Taken together, these results confirm that DegS acts proteolytically on the V. cholerae homologue of RseA, as shown in E. coli. In agreement with these observations, our data indicate that the activation of DegS triggered by C‐terminal YYF oligopeptide exposure also increases FLAGToxRCC proteolysis.
Figure 3.
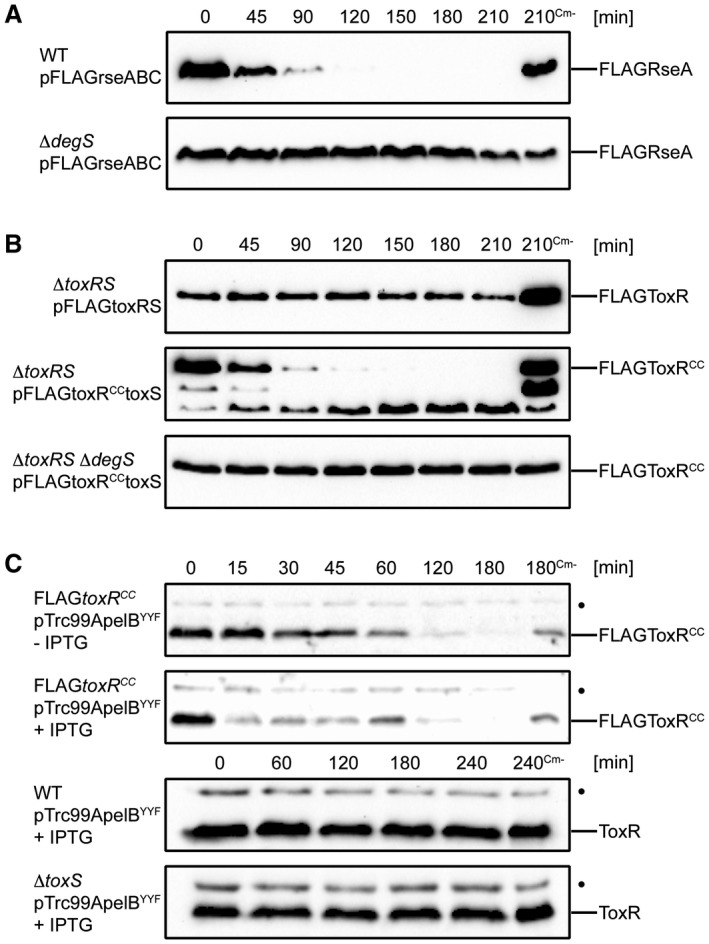
FLAGToxRCC undergoes DegS‐regulated proteolysis, which can be enhanced upon the overexpression of a synthetic C‐terminal OmpU fragment (YYF). Degradation assay was conducted with plasmid‐carrying cells grown in M9 maltose to mid‐log growth phase. Cultures were induced with IPTG for 1 h followed by inhibition of protein translation by Cm. Samples without chloramphenicol (Cm‐) served as negative controls. A. Proteolysis of anti‐sigma factor RseA is controlled by site‐1 protease DegS. Immunoblots utilizing anti‐FLAG antibodies show temporal stability levels of FLAGRseA in WT and degS background. B. Regulated proteolysis of FLAGToxRCC is controlled by DegS. Immunoblots utilizing anti‐FLAG antibodies are showing temporal stability levels of FLAGToxRCC in toxRS and toxRSdegS background. C. DegS‐PDZ activation by a synthetic C‐terminal OmpU fragment (YYF) induces an acceleration of FLAGToxRCC degradation. Immunoblots utilizing α‐ToxR antiserum show chromosomal expressed levels of ToxR or FLAGToxRCC. (•): Represents a nonspecific cross‐reacting background band.
Bile salts interfere with ToxR proteolysis
A well‐known effect of bile salts on V. cholerae is the activation of the ToxR transcriptional activity, which leads to high levels of ompU transcription and ompT repression (Provenzano et al., 2000; Midgett et al., 2017). Bile salts do not activate toxRS transcription per se (Mey et al., 2015), but this surfactant may influence ToxRS interaction strength, as described recently (Midgett et al., 2017). Therefore, we focused on the effects of bile salts on FLAGToxRCC stability, with the results showing that DC, but not TC, could inhibit FLAGToxRCC proteolysis (Fig. 4) in a ToxS‐independent manner. To determine whether DC inhibits DegS activity directly, a FLAGRseA degradation assay was performed. The results did not reveal an inhibition of DegS‐dependent proteolysis of FLAGRseA (Fig. S8), indicating that the DC‐mediated inhibition is ToxR specific but does not target the inhibition of the protease DegS. Interestingly, adjusted WCLs analysed by SDS‐PAGE and Kang staining revealed DC, but not TC, ToxR‐induced OmpU expression (Fig. S9 and S10).
Figure 4.
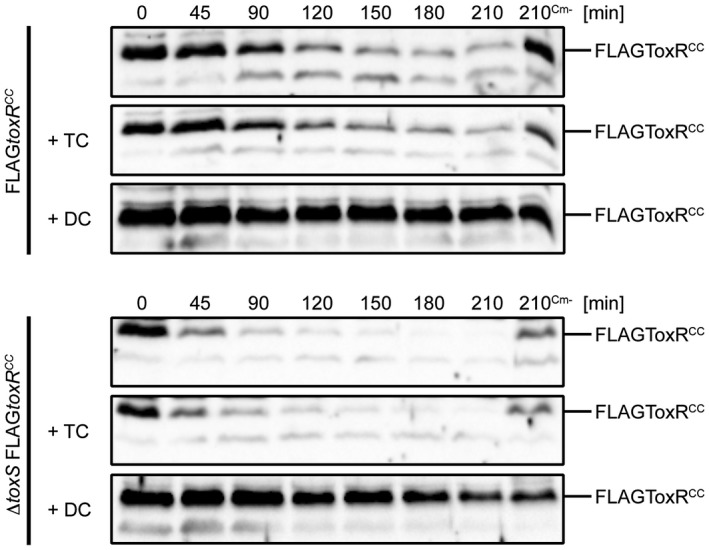
Sodium deoxycholate (DC) protects FLAGToxRCC degradation in WT and in toxS mutant strains. Degradation assays of V. cholerae FLAGtoxRCC and toxS FLAGtoxRCC strains are subjected to immunoblotting using α‐ToxR antiserum. Cells were grown in M9 maltose in the absence or presence of 0.1% sodium taurocholate or sodium deoxycholate. Protein biosynthesis was inhibited by the addition of Cm, whereas samples without chloramphenicol (Cm‐) served as negative controls. Shown in Fig. S9 and S10 are Kang‐stained gels, which represent loading and quality controls to monitor influences of bile salts on protein expression patterns.
As reported previously (Almagro‐Moreno et al., 2015b), ToxR proteolysis was identified under conditions of starvation and alkaline pH incubation (PBS buffer pH 8.1). We detected ToxR in the WT strain and observed that DC also inhibits ToxR degradation under starvation and alkaline incubation conditions (Fig. 5). We also monitored ToxR stability in a toxS knockout mutant and monitored enhanced ToxR instability, which was partially stabilized by added DC. Additionally, we detected decreased ToxR levels under alkaline PBS conditions in the toxSdegPS triple mutant, indicating that other proteases recognize ToxR as a substrate.
Figure 5.
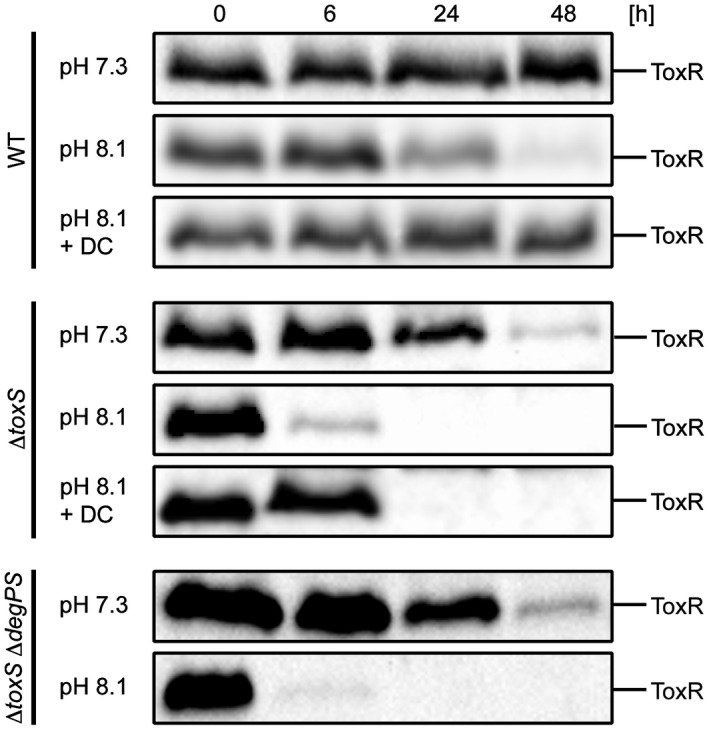
ToxR proteolysis under starvation and alkaline pH conditions is inhibited by DC. Shown are immunoblots of WCL samples of ToxR in WT, toxS and toxSdegPS strains treated under reducing Laemmli buffer conditions. Cells were grown in LB medium ON and then shifted into PBS (pH 7.3), alkaline PBS (pH 8.1) and alkaline PBS (pH 8.1) supplemented with DC (0.01%). Immunoblots were performed utilizing α‐ToxR antiserum.
As the interaction between bile salts and purified ToxR was published recently (Midgett et al., 2017), we hypothesized that bile salts directly adhere to ToxR and shield it from protease activity, thereby interfering with ToxR degradation. Midgett et al. also indicated that the binding of bile acids facilitates the ToxRS interaction, transducing bile acid signalling into ToxR transcription activity. To address this issue, we monitored PhoA activity of strains with chromosomal ompT::phoA and ompU::phoA fusions and ToxR levels in the corresponding strains. We observed that the addition of DC activated ompU and repressed ompT transcription in WT cells but did not alter the ToxR protein levels in the WT or toxS mutant strains (Fig. 6A and B). DC‐dependent activation of ToxR transcriptional activity was significantly decreased in the toxS background, supporting the view of Midgett et al. (2017) that ToxRS‐bile acids are the relevant activator complex for ToxR. To answer the question of whether the disulfide bond formation or protein stabilization correlates with the DC activation of the ToxR transcriptional activity, the toxRCC mutant was monitored for ompU and ompT transcription. The results showed that the toxRCC strain with the DC treatment could still be transcriptionally activated and showed enhanced ToxRCC protein expression, indicating increased protein stability (Fig. 6A and B). As expected, the toxR mutant control showed no activated ompU transcription or ompT repression with or without DC. To summarize, these results suggest that DC but not TC mediates the protection of ToxR degradation under conditions in which the DegS protease is active. Moreover, we can conclude that ToxS is required for the activation of bile acid‐dependent ToxR transcriptional activity and this does not correlate with a change in the ToxR protein levels.
Figure 6.
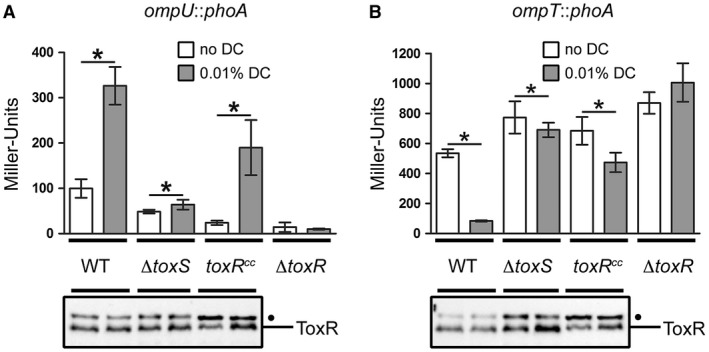
ToxR transcriptional control of ompT and ompU is dependent on ToxS under DC activating conditions. Shown are reporter gene activities of alkaline phosphatase PhoA (Miller Units) linked as operon fusions to either ompU (A) or ompT (B) in V. cholerae WT, toxS, FLAGtoxRCC and toxR strains. Simultaneously, immunoblot analyses were performed utilizing α‐ToxR antiserum. Strains were grown in M9 maltose in the presence (dark bars) or absence (open bars) of 0.01% DC until OD600 of 0.8‐1. Data shown are mean and standard deviation of six independent samples for each condition. The asterisks indicate a statistically significant difference between bile salt‐treated and non‐treated cells, with P‐values by paired t‐test (*): P < 0.05. (•): Represents a nonspecific cross‐reacting background band.
Discussion
Recently published data showed that V. cholerae cells switch into a persistent state under defined laboratory growth conditions, including an alkaline pH and starvation (Almagro‐Moreno et al., 2015a). Such culture conditions exposed the RIP mechanism of ToxR in a RseP (site‐2 protease)‐dependent manner, and further studies indicated the involvement of numerous site‐1 proteases (Almagro‐Moreno et al., 2015b). Our data confirmed this ToxR proteolysis and extended the current model. For example, our analyses revealed that the reduction of ToxR cysteines targets it for site‐1‐mediated proteolysis by DegS and DegP. The impact of the disulfide bond formation has recently attracted increased attention because of its involvement in bacterial virulence and survival (Landeta et al., 2018). In previous observations (Fengler et al., 2012), we demonstrated that toxR mutants lacking cysteine residues and dsbA strains exhibited decreased ToxR transcriptional activity. This was best demonstrated by the deactivation and the derepression of ompU and ompT transcription respectively. However, such a phenotype may also correlate well with ToxR proteolysis, prompting us to further investigate ToxR protein stability. To test the fate of ToxR, WT cells were compared with dsbA, dsbC, dsbAdegS, FLAGtoxRCC and FLAGtoxRCCdegPS mutant strains over a 32 h time course. For the WT strain, monomeric ToxRred/oxy molecules underwent a shift towards the reduced form during entry into the stationary growth phase, and ToxR remained stable at these late time points. In contrast, strong degradation of ToxR/FLAGToxRCC in the dsbA and FLAGtoxRCC strains was visible past the 12 h time point. For FLAGToxRCC, no degradation was observed in a degPS strain over the entire time course of the experiment. For the degSdsbA mutant, a delay in ToxR proteolysis was monitored, indicating the activities of additional protease activities at later time points, e.g. DegP. Based on the results of the degPS knockout and complementation studies, it appears that the sensitive ToxRCC form is recognized by both proteases independently (Fig. 7). However, this result does not allow for a detailed interpretation since overexpression of proteases may cause artificial effects. Therefore, we cannot currently make inferences on the importance of synergy, additive or sequential mechanisms by which the two proteases act upon ToxR, although these issues will be addressed in future studies. Furthermore, it could be observed that in a dsbA strain, the primary fraction of ToxR became ToxRred prior to proteolysis. Since DsbAB activity depends on the respiratory electron chain activity, a change in ToxRred/oxy may correlate with growth status. Complex scenarios may contribute to this activity, for example: (i) a change in the expression of more abundant periplasmic proteins that are substrates for the DsbAB system; (ii) a higher oxidized state of components of the respiratory chain (e.g. ubiquinones) that could act as a better sink for electrons derived from DsbB; or (iii) the existence of a differentially active DsbAB system, either through changes in expression levels or activity and half‐life time. We did not observe ToxR degradation in the dsbC mutant but did observe less pronounced ToxRred levels, indicating some change in ToxRred maintenance, a circumstance that requires future study.
Figure 7.
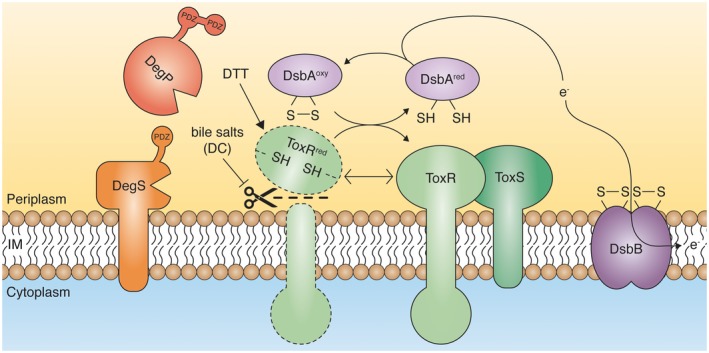
Proteolysis of ToxR is controlled by cysteine‐thiol redox state and bile salts. During de novo synthesis, ToxR molecules are inserted into the inner membrane, exposing thiol groups on the periplasmic located domain. Such thiol groups are then oxidized by the DsbAB system to form intramolecular disulfide bonds. ToxRoxy together with ToxS represents a robust ToxRS transcription complex. Any modifications to the disulfide bond formation of ToxR (e.g. DTT, loss of DsbA activity or exchange of the cysteine residues to serine) lead to DegS‐ and DegP‐mediated proteolysis. Evidence presented here indicates that ToxR molecules, in complex with ToxS, are protected from proteolysis, even under reducing conditions. Standard growth conditions with physiological relevant amounts of bile salts (DC) favour ToxR stability in a ToxS‐ and DegPS‐independent way. [Colour figure can be viewed at https://wileyonlinelibrary.com]
To promote cysteine reduction in ToxR, V. cholerae was incubated with sub‐lethal concentrations of DTT, after which reduced ToxR monomers became the major detectable form. The level of ToxR undergoing proteolysis was significantly elevated in a toxS mutant, which was DegS‐ and DegP‐dependent. Thus, these data confirm that the proteolysis‐sensitive form is the reduced ToxR molecule, which then becomes a substrate for proteolysis, as outlined in the model (Fig. 7). Furthermore, these results are in agreement with previously published data, showing that ToxS is needed to stabilize ToxR (Almagro‐Moreno et al., 2015b; Midgett et al., 2017). As a possible scenario, we assume that the interaction between ToxR and ToxS during de novo synthesis is weakened if ToxRred is dominant, as proteases may act on ToxR. The following observations would support this hypothesis: ToxR mutants that constantly produced or changed the ToxRred/oxy equilibrium to ToxRred, such as dsbA and toxRCC, were degraded instantly. Comparatively, treatment with DTT only resulted in ToxR degradation in the absence of ToxS. Therefore, it may be tempting to speculate that if ToxRred is primarily present during de novo synthesis, ToxR und ToxS may not interact strongly. In contrast, if de novo synthesis in the WT strain is not affected in the presence of reducing conditions such that ToxR/ToxS can interact, challenging ToxR with DTT will lead to the production of ToxRred, but it could still interact with ToxS. Thus, ToxRred would remain protected from degradation. Future studies are needed to determine which of these scenarios occurs for de novo ToxRS complex assembly.
We also characterized DegS in greater detail. DegS and RseP are part of the σE stress‐sensor complex, which exclusively cleaves the RseA substrate in E. coli (Lima et al., 2013). To the best of our knowledge, no other DegS substrate is known. In this study, as was previously observed (Almagro‐Moreno et al., 2015a), DegS recognizes ToxR in V. cholerae. Thus, we were interested in evaluating DegS activity with respect to RseA in V. cholerae. As expected, the RseA homologue in V. cholerae was confirmed to be a substrate of DegS. Furthermore, the overproduction of a synthetically designed and secreted C‐terminal OmpU oligopeptide, containing a DegS responsive tripeptide signature (YYF) (Walsh et al., 2003; Chaba et al., 2007), also resulted in the stimulation of ToxRCC degradation. Therefore, DegS has similar activation mechanisms in V. cholerae as are observed in E. coli, but for RseA and ToxR it recognizes two substrates. It would be interesting to determine if more DegS substrates are present in V. cholerae, although none have been identified. Additionally, we identified a transcriptional feedback regulatory mechanism between the σE response and DegP, showing that a degS knockout mutant exhibits decreased degP transcription. Thus, a degS knockout phenotype is also associated with a decrease in DegP levels. Since both proteases are involved in the RIP of ToxR (Fig. 7), it will be interesting to characterize the interaction between the σE pathway and the ToxR regulon and its physiological importance. A significant indication of a physiologically relevant association between the two systems came from the study by Almagro‐Moreno and colleagues. They showed that σE pathway‐deficient mutants rpoE or rseP exhibit no ToxR RIP after being incubated under alkaline and starvation conditions (Almagro‐Moreno et al., 2015b), which interfered with a dormant survival program (Almagro‐Moreno et al., 2015a). In this study, we characterized two key players of the σE pathway, DegP and DegS, shown to be involved in the control of ToxR proteolysis. We observed that the redox state of ToxR cysteines regulates its proteolysis. Furthermore, we showed that a double degPS knockout does not prevent WT ToxR proteolysis under conditions associated with the initiation of a dormant stage. This may simply indicate the existence of unknown environmental conditions that influence the RIP of ToxR in a DegPS and ToxR redox state‐dependent manner.
Previous studies have indicated that bile acids play a major role in stimulating the transcriptional activity of ToxRS (Provenzano and Klose, 2000) and TcpPH (Yang et al., 2013). Furthermore, Midgett and co‐workers reported an enhanced interaction between ToxR and ToxS due to the binding of chenodeoxycholate or cholate (Midgett et al., 2017). To revisit bile acid‐dependent activation, we characterized two primary substituents of bile salts, the sodium salts TC and DC. Interestingly, we demonstrated that incubation with DC but not TC completely blocked FLAGToxRCC degradation in cells and generally enhanced ToxR‐dependent transcriptional regulation of the porin genes ompU and ompT. DC‐dependent inhibition of RIP on ToxR was also observed under alkaline and starvation conditions, indicating that DC rescues ToxR from becoming a substrate for RIP independently of the proteases. Additionally, we obtained strong evidence that in the presence of bile salts, proteolysis control is ToxS independent, whereas ToxR transcriptional activity is highly accelerated in the presence of ToxS. For ToxRCC, a correlation between increased levels of ToxRCC protein and transcriptional activity of porin expression occurs in the presence of DC. Thus, it seems that bile salts activate ToxR independently of disulfide bond formation and protein stability in the WT strain. These analyses also indicated that without notable changes in the WT ToxR levels, the ToxR transcription factor activity was highly stimulated by DC. Similarly, ToxR activity was linked to a different stimulus. Deletions in RND efflux systems were previously observed to lead to the high expression of the leuO gene, which is under the control of ToxR (Bina et al., 2018). The accumulation of secreted substances, such as L‐malate, has been shown to lead to binding of activated ToxR, which could constitute a similar mechanism as observed with bile salts (Midgett et al., 2017). However, based on recently published studies, it has become evident that ToxR acts as a receptor for not only bile salts but also perhaps an array of effectors that have yet to be identified.
In summary, our data demonstrated that cysteine oxidation of ToxR is most likely important for proper protein function and stability. Interchangeable dynamics between reduced versus oxidized forms were observable during the transition from early to late stationary growth phases. Furthermore, we showed that ToxR degradation responds to interventions during the disulfide bond formation in a DegS‐ and DegP‐dependent manner. We therefore propose that an increase in thiol group formation and the disruption of the ToxR and ToxS interaction will subsequently lead to ToxR proteolysis, as outlined in the model (Fig. 7).
Several questions remain: What could influence such a thiol redox state switch? Is there an intrinsic half‐life of stable disulfide bonds for ToxR? A good example of regulated disulfide bond formation comes from studies of TcpP homodimer conformation in V. cholerae (Yang et al., 2013). There, it was reported that intramolecular disulfide bonds in TcpP are resolved by the presence of TC to form intermolecular disulfide bonds between two TcpP molecules, leading to a homodimer and a transcriptionally active conformation for the toxT promoter. Moreover, DsbA and other oxido‐reductases were reported to exhibit activity after binding to TC (Xue et al., 2016). By monitoring the ToxR thiol redox status, we did not observe evidence of a change in the ratio of ToxRred/oxy, neither when cells were incubated in alkaline PBS nor in bile salts. Of note, the bile salt concentrations used in this study are physiologically relevant, as was determined in the small intestine of humans (Stadler et al., 1988). The ToxR response to bile acids leads to an inverse regulation of porin expression, associated with cell survival (Provenzano and Klose, 2000). Under these conditions, ToxR may be stabilized to facilitate proper porin regulation. Furthermore, considering that the activation of TcpP depends on bile salts (Yang et al., 2013), it seems that V. cholerae has adapted to different bile acid species accordingly and has integrated that in a complex virulence regulatory network. Based on our data, we suggest specific conditions that may lead to ToxR‐regulated RIP, e.g. participation of the σE pathway leads to an upregulation of DegP, and probably other proteases, due to periplasmic protein folding stress. Varying environments may change the ToxRred/oxy equilibrium due to changes in growth status. Finally, bile acids, which are present during infection cycles of V. cholerae in humans or other vertebrates (Hofmann et al., 2010), unexpectedly interfere with ToxR stability. Based on our hypothesis, all three conditions described above may occur in separate parts in the gut and help determine the genetic program of virulence expression due to ToxR control. This complex interaction requires further characterization. In summary, extending the original finding that ToxR proteolysis is involved in environmental persistence (Almagro‐Moreno et al., 2015a), our finding that bile acid regulates the control of ToxR proteolysis may also bolster the concept of ToxR undergoing RIP during the infection cycle.
Experimental procedures
Bacteria, plasmids and growth conditions
All bacterial strains and plasmids used in this study are listed in Table 1. V. cholerae P27459‐S, a spontaneous streptomycin‐resistant mutant of the clinical isolate P27459 (O1 El Tor Inaba), was used as the wild‐type (WT) strain (Pearson et al., 1993). The Escherichia coli strains XL1‐Blue, AB1157, LE392, DH5α λpir and SM10 λpir were used for genetic manipulations, with the latter strain being used for introducing plasmids into V. cholerae by conjugation. Bacteria were grown in Luria‐Bertani (LB) broth or on LB agar plates with aeration at 37°C. V. cholerae was typically inoculated to a starting OD600 of 0.1 and grown to mid‐log phase (OD600 of 0.4‐0.5), late‐log phase (OD600 of 0.8‐1) or for the indicated time in M9 maltose minimal medium with aeration at 37°C. For starvation and alkaline pH conditions, cells were grown in LB overnight and were resuspended in either PBS or PBS pH 8.1 buffer adjusted with sodium hydrogen carbonate (0.3%). When required, supplements were used at the following final concentrations: streptomycin (Sm; 100 μg ml–1), ampicillin (Ap; 50 or 100 μg ml–1), chloramphenicol (Cm; 2, 5 or 100 μg ml–1), kanamycin (Km; 50 μg ml–1), isopropyl ß‐D‐1‐thiogalactopyranoside (IPTG; 0.05 or 1 mM), L‐arabinose (0.05%), sucrose (10%), sodium‐deoxycholate (DC; 0.01 or 0.1%), sodium‐taurocholate (TC; 0.1%) and dithiothreitol (DTT; 6 mM).
Table 1.
Strains and plasmids used in this study.
| Strains/Plasmids | Descriptions | References |
|---|---|---|
| E. coli strains | ||
| DH5αλpir | F ‐ Δ(lacZYA‐argF)U169 recA1 endA1 hsdR17 supE44 thi‐1 gyrA96 relA1 λ::pir | Hanahan (1983) |
| SM10λpir | thi thr leu tonA lacY supE recA::RPA‐2‐Te::Mu λpirR6K, Kmr | Miller and Mekalanos, (1988) |
| XL1‐Blue | F ‐ ::Tn10 proA + B + lac q Δ( lacZ )M151 recA1 endA1 gyrA46 (Nal r ) thi hsdR17 (r K − m K + ) supE44 relA1 lac | Bullock et al. (1987) |
| AB1157 | thr‐1 ara‐14 leuB6 Δ(gpt‐proA)62 lacY1 tsx‐33 supE44 amber galK2 hisG4 rfbD1 mgl‐51 rpsL31 kdgK51 xyl‐5 mtl‐1 argE3 thi‐1 | Bachmann (1987) |
| LE392 | F ‐ supF supE hsdR galK trpR metB lacY tonA | Silhavy et al. (1984) |
| V. cholerae strains | ||
| WT | P27459‐S , O1 Inaba, El Tor, clinical isolate, Bangladesh 1976, spontaneous Smr | Pearson et al. (1993) |
| ΔdsbA | P27459‐S ΔdsbA::km, dsbA replaced by km cassette, Smr, Kmr | Fengler et al. (2012) |
| dsbC::pGP | P27459‐S dsbC::pGP704 insertion, Smr, Apr | Fengler et al. (2012) |
| ΔtoxR | P27459‐S with deletion in toxR, Smr | Fengler et al. (2012) |
| ΔtoxS | P27459‐S with deletion in toxS, Smr | This study |
| ΔtoxRS | P27459‐S with deletion in toxR and toxS, Smr | Fengler et al. (2012) |
| FLAGtoxR CC | P27459‐S ΔtoxR::FLAGtoxR C236SC293S , toxR replaced by FLAGtoxR C236SC293S , Smr | Fengler et al. (2012) |
| ΔtoxS FLAGtoxR CC | P27459‐S ΔtoxS ΔtoxR::FLAGtoxR CC , toxR replaced by FLAGtoxR C236SC293S , Smr | This study |
| ΔdegS | P27459‐S ΔdegS::cat, degS replaced by cat cassette, Smr , Cmr | This study |
| ΔdegS ΔdsbA | P27459‐S ΔdegS ΔdsbA::km, dsbA replaced by km cassette, Smr , Cmr, Kmr | This study |
| ΔtoxRS ΔdegS | P27459‐S ΔtoxRS ΔdegS::cat, deletion in degS replaced by cat cassette, Smr , Cmr | This study |
| ΔtoxS ΔdegS | P27459‐S ΔtoxS ΔdegS::cat, deletion in degS replaced by cat cassette, Smr , Cmr | This study |
| ΔtoxS ΔdegPS | P27459‐S ΔtoxS ΔdegPS::cat, deletion in degPS replaced by cat cassette, Smr, Cmr | This study |
| FLAGtoxR CC ΔdegPS | P27459‐S FLAGtoxR CC ΔdegPS::cat, deletion in degPS replaced by cat cassette, Smr, Cmr | This study |
| WT ompU::phoA | Insertion of pGP704phoA downstream of ompU in P27459‐S, Smr, Apr | This study |
| ΔtoxS ompU::phoA | P27459‐S ΔtoxS with insertion of pGP704phoA downstream of ompU, Smr, Apr | This study |
| FLAGtoxR CC ompU::phoA | P27459‐S FLAGtoxR CC with insertion of pGP704phoA downstream of ompU, Smr, Apr | This study |
| ΔtoxR ompU::phoA | P27459‐S ΔtoxR with insertion of pGP704phoA downstream of ompU, Smr, Apr | This study |
| WT ompT::phoA | Insertion of pGP704phoA downstream of ompT in P27459‐S, Smr, Apr | This study |
| ΔtoxS ompT::phoA | P27459‐S ΔtoxS with insertion of pGP704phoA downstream of ompT, Smr, Apr | This study |
| FLAGtoxR CC ompT::phoA | P27459‐S FLAGtoxR CC with insertion of pGP704phoA downstream of ompU, Smr, Apr | This study |
| ΔtoxR ompT::phoA | P27459‐S ΔtoxR with insertion of pGP704phoA downstream of ompT, Smr, Apr | This study |
| WT degP::phoA | Insertion of pGP704phoA downstream of degP, Smr, Apr | This study |
| ΔdegS degP::phoA | P27459‐S ΔdegS with insertion of pGP704phoA downstream of degP, Smr, Apr | This study |
| Plasmids | ||
| pKEK229 | OriR6K, mobRP4, sacB, Apr | Correa et al. (2000) |
| pCVD442 | OriR6K, mobRP4, sacB, Apr | Donnenberg and Kaper (1991) |
| pGP704 | OriR6K, mobRP4, Apr | Miller and Mekalanos (1988) |
| pBAD18‐Kan | Expression vector, oriColE1, arabinose Inducible, Kmr | Guzman et al. (1995) |
| pMMB67EH | Expression vector, oriColE1, IPTG inducible, Apr | Morales et al. (1991) |
| pACYC184 | Cloning vector, orip15A, Tetr, Cmr | Rose (1988) |
| pFLAG‐MACTM | Expression vector with N‐terminal FLAG‐Tag, IPTG inducible, Apr | Sigma‐Aldrich |
| pTrc99A | Expression vector oriColE1, lacI q, IPTG inducible, Apr | Amann et al. (1988) |
| pKEK229degS::cat | pKEK229 carrying up and down fragments of degS flanking a cat cassette, Apr, Cmr | This study |
| pKEK229dsbA::km | pKEK229 carrying up and down fragments of dsbA flanking a km cassette, Apr, Kmr | Fengler et al. (2012) |
| pCVD442degPS::cat | pCVD442 carrying up and down fragment of degP and degS flanking a cat cassette, Apr, Cmr | This study |
| pCVD442toxS | pCVD442 carrying up and down fragments of toxS, Apr | This study |
| pCVD442toxRS | pCVD442 carrying up fragment of toxR and down fragment of toxS, Apr | Fengler et al. (2012) |
| pCVD442FLAGtoxRCC | pCVD442 carrying up and down fragments of FLAGtoxR C236SC293S, Apr | Fengler et al. (2012) |
| pGP704phoA | pGP704 with promoterless phoA of SM10λpir, Apr | Berg et al. (2007) |
| pGP704phoAdegP | pGP704phoA with degP gene fragment, Apr | This study |
| pGP704phoAompU | pGP704phoA with ompU gene fragment, Apr | This study |
| pGP704phoAompT | pGP704phoA with ompT gene fragment, Apr | This study |
| pBAD18‐KandegS | degS of P27495‐S in pBAD18‐Kan, Kmr | This study |
| pMMB67EHdegP | degP of P27495‐S in pMMB67EH, Apr | This study |
| pFLAGrseABC | rseABC of P27495‐S in pFLAG‐MACTM, Apr | This study |
| pFLAGtoxRS | toxR and toxS of P27495‐S in pFLAG‐MACTM, Apr | Fengler et al. (2012) |
| pFLAGtoxRCCtoxS | toxR C236SC293S point mutant and toxS of P27495‐S in pFLAG‐MACTM, Apr | Fengler et al. (2012) |
| pTrc99ApelBYYF | C‐terminal 50 residues of OmpU (ending in YYF) fused to a N‐terminal pelB leader sequence in pTrc99A, Apr | This study |
Strain and plasmid constructions
Start‐to‐stop deletions of toxS, degS and degPS in V. cholerae P27459‐S were generated via PCR or SOE‐PCR (splicing by overlap extension) (Horton et al., 1989), cloning two approximate DNA fragments (300‐1,000 bp) upstream and downstream of the region of interest into the suicide plasmids pKEK229 or pCVD442 (Donnenberg and Kaper, 1991). To improve the selection of degS or degPS deletion mutants, a cat cassette was obtained from pACYC184 and ligated between the degS or the degPS upstream and downstream DNA fragments. Corresponding primers for the amplification of the degPS‐cat, degS‐cat and toxS fragments, as well as sequencing primers for validation are listed in Table 2. Synthesized oligonucleotides (Thermo Fisher Scientific) harbouring recognition sites for restriction endonucleases were used to facilitate directed cloning. The resulting constructs were electroporated into E. coli DH5α λpir, isolated, transformed into SM10 λpir and subsequently conjugated into V. cholerae derivatives (Donnenberg and Kaper, 1991). Double‐crossover recombinant deletion mutants were selected using 10% sucrose agar plates. The ompU::phoA, ompT::phoA and degP::phoA transcriptional fusion strains were constructed by cloning internal fragments (500‐800 bp) of the respective gene into the plasmid pGP704phoA (Berg et al., 2007), which contains a promoterless phoA reporter. After the gene of interest was PCR amplified from the template DNA derived from the WT strain SP27459 and was digested with the corresponding restriction endonucleases, the fragments were ligated into similarly digested pGP704phoA. The resulting constructs were introduced into V. cholerae derivatives by conjugation (Miller and Mekalanos, 1988) and were selected for ampicillin resistance.
Table 2.
Oligonucleotidesa (5′‐3′) used in this study.
| XbaI_dtoxS_fwd | TTTTCTAGATGGATTATTCTAAGTCTGCAT | |
| SacI_dtoxS _rev | ATTGAGCTCCCATGAAACTATTTTTTGTCTC | |
| SOE_dtoxS_rev | TCAGTCAGGAGCAAGATCCTACTCACACACTTTGAT | |
| SOE_dtoxS_fwd | GATCTTGCTCCTGACTGAGCGTAGAATAGGACATAA | |
| HindIII_toxRCC_dtoxS_rev | TTTAAGCTTAGCAAGATCCTACTCAGACA | |
| HindIII_dtoxS_fwd | TATAAGCTTCCTGACTGAGCGTAGAATA | |
| NcoI_cat_fwd | TTTACCGGTCAACCAGGCGTTTAAGGG | |
| NcoI_cat_rev | TTTACCGGTTTCCAACTTTCACCATAATGA | |
| SacI_ddegS _fwd | AAAGAGCTCAAAAATCCCCAAACTTGCA | |
| NcoI_ddegS_rev | TAAACCGGTTATAAATATTGACGACGGCA | |
| NcoI_ddegS_fwd | TTTACCGGTCGCCAGAATGTCACAGATA | |
| XbaI_ddegS_rev | ATATCTAGAGAAGCTGGCTAAGAAGTAATGCT | |
| SacI_ompU_fwd | TTTGAGCTCCTTTAATAGTCTATCGAGTTCTT | |
| SthI_ompU_rev | TTTTGGTACCGAAGTACCTTTCGCGCT | |
| SacI_ompT_fwd | ATGAGCTCGGTGATTTGGCTGTGCACG | |
| SthI_ompT_rev | TAGGTACCTTACCAGTAGATACGAGCACC | |
| XhoI_rseA_fwd | TAACTCGAGACTATGGTGAATAGAATGGC | |
| KpnI_rseC_rev | ATTGGTACCTTTCGTTAGGGTTCGCATCA | |
| template for pelBYYF | GCCGACCGCTGCTGCTGGTCTGCTGCTCCTCGCTGCCCAGCCGGCGATGGCCCACCACCACCACCACCACTCAGCAGATAATTTTGCTATCGACGCAACTTACTACTTCAAGCCAAACTTCCGCTCTTACATCTCTTACCAGTTCAATCTGCTAGATTCAGACAAAGTTGGTAAAGTAGCATCAGAAGACGAACTGGCTA | |
| NcoI_tripep_fwd | ATTCCATGGGAAAATACCTGCTGCCGACCGCTGCTGCTGGTCT | |
| Pst_YYF_tripep_rev | AATCTGCAGTTAGAAGTAGTAACGTAGACCGA TAGCCAGTTCGTCTTCTGATG | |
| bla_inv_rev | CCGTAAGATGCTTTTCTGTGACTGGT | |
| ompU_seq_fwd | CCAACAAACATTAAAATCATTTAA | |
| pFLAGMAC_fwd | AACGGTTCTGGCAAATATTC | |
| pTricHis_rev | CTTCTGCGTTCTGATTTAATCTG | |
| rseB_seq_fwd | GACTCGTGATTCGGTGGA | |
| M13rev‐48_fwd | AGCGGATAACAATTTCAC | |
| SphI_degP_fwd | TTAGCATGCAGGTCGTACCACTGGCT | |
| HindIII_degP_rev | AAAAAGCTTAACGTCCGGTTGAAGT | |
| SacI_depP_phoA‐fusion_fwd | TTAGAGCTCCGGTGGCAACGTCGGTAT | |
| KpnI_degP_phoA‐fusion_rev | TTAGGTACCTTAACGAACAACCAAGTAAAGCG | |
| EcoRI_VC0566_compl_fwd | TTTGAATTCTTTGTTGAGGAGCTTATGATG | |
| BamHI_VC0566_compl_rev | TATGGATCCTTAACGAACAACCAAGTAAAGC | |
| BamHI_degS_fwd | TTTGGATCCTCGCCAGAATGTCACAGAT | |
| SmaI_degS_rev | TTTCCCGGGTGACTGACGAACAGGAAC | |
| SacI_degS_fwd | ATAGAGCTCAAAAATCCCCAAACTTGC | |
| XbaI_degS_rev | ATATCTAGAAGCTGGCTAAGAAGTAAT | |
| HindIII_cat_fwd | ATAAAGCTT AGCACCTCAAAAACACCATC | |
| BamHI_cat_rev | TTAGGATCC CACCAGGCGTTTAAGGGCA | |
| M13_pUC_rev | AGCGGATAACAATTTCACACAGG | |
| pGP704_CVD_rv|15 | GATGTAACGCACTGAGAAG | |
Restriction sites are underlined
The expression plasmids pBAD18‐KandegS, pMMB67EHdegP, pFLAGrseABC and pTrc99ApelBYYF were constructed by PCR of the respective coding regions derived from the template DNA of the WT strain SP27459. The obtained amplicons were digested with the corresponding restriction endonucleases indicated in the oligonucleotide name (Table 2). For pTrc99ApelBYYF, a synthetic oligonucleotide (template for pelBYYF, Table 2) comprised of the pelB leader sequence and the 3′ region of ompU (C’‐terminal 50 amino acid residues) was synthesized (IDT‐Integrated DNA Technologies) and served as template DNA. For the amplification of this fragment, the oligonucleotide primers NcoI_tripep_fwd and Pst_YYF_tripep_rev were used, with the latter containing a point mutation within the DNA sequence that changed the amino acid sequence YDF to YYF for optimized DegS PDZ recognition according to Walsh et al. (2003). PCR fragments were ligated into similarly digested pBAD18‐Kan, pMMB67EH, pFLAG‐MACTM and pTrc99A expression plasmids, which harbour arabinose or IPTG inducible promoters. Subsequently, the expression plasmids were electroporated into DH5α λpir and monitored for kanamycin or ampicillin resistance before being introduced into V. cholerae derivatives. All deletion mutants, reporter strains and plasmids described in this study were confirmed by PCR and sequencing (data not shown).
Cell extracts and immunoblot analysis
WCLs were prepared from V. cholerae cultures grown at 37°C in M9 maltose minimal medium (supplemented +/– DTT or bile salts) or incubated under starvation and alkaline conditions (PBS and PBS pH 8.1, respectively) as described above. For time course experiments, cells were harvested at the indicated time points and resuspended in Laemmli buffer (Laemmli, 1970) with or without β‐mercaptoethanol, corresponding to reducing and nonreducing conditions respectively. The protein content was analysed by standard sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) using 15% polyacrylamide gels (Mini‐PROTEAN® Tetra cell, BIO‐RAD) with PageRuler™ Prestained Protein Ladder (10‐180 kDa, Thermo Fisher Scientific) as a molecular mass standard. Prior to the immunoblot analysis, WCLs were normalized to contain similar protein levels by SDS‐PAGE Kang staining (Kang et al., 2002).
Immunoblot analysis was conducted using an AmershamTM ProtranTM 0.45‐µm nitrocellulose membrane (GE Healthcare Life Sciences). After transfer and blocking in Tris‐Buffered Saline (TBS) with 10% skim milk overnight at 4°C, the membranes were incubated for 2 h at RT with the primary antibody (1:1,000 dilution of rabbit anti‐ToxR (Fan et al., 2014), kindly supplied by Jun Zhu, University of Pennsylvania, USA, or 1:1,000 dilution of mouse anti‐FLAG, Sigma‐Aldrich) diluted in TBS with 10% skim milk. After incubation, the membranes were washed as previously described (Fengler et al., 2012) and then incubated for 1 h at RT with the secondary antibody (1:10,000 or 1:7,500 dilution of horseradish peroxidase‐conjugated goat anti‐rabbit or goat anti‐mouse, Dianova GmbH) diluted in TBS with 10% skim milk. Subsequently, the nitrocellulose membranes were washed, and chemiluminescence detection was performed. Each membrane was incubated for 5 min in ECL solution (Clarity™ Western ECL Blotting Substrates, BIO‐RAD) prior to visualization of the reactive protein bands using a Molecular Imager ChemiDocTM XRS System (BIO‐RAD). Band intensities were determined using Image Lab Software (BIO‐RAD). Quantification of ToxR protein bands was performed for each strain as follows: for the 0 min time point, the mean densitomertic count numbers of equally treated WCL samples were set to 100% for each strain and conditions (+/– DTT). WCLs were mixed with Laemmli buffer containing ß‐mercapthoethanol such that only the reduced ToxR form was visible and used for quantification. Next, the count numbers for the 30 and 60 min time points were normalized to the 0 min time point on the same immunoblots. Kang‐stained SDS‐PAGE gels served as loading controls to determine equal protein concentrations, similar as shown in Fig. S9 or Fig. S10. One set of immunoblots used for this analysis is shown in Fig. S4.
Protein stability and degradation assays
To determine ToxRred/oxy, FLAGToxRCC and FLAGRseA protein levels in V. cholerae, WT strain SP27459 and mutant strains carrying various plasmids (see Table 1) or no plasmid were grown overnight in LB and subcultured into fresh M9 maltose minimal medium to a starting OD600 of 0.1 at 37°C. The medium was supplemented with the appropriate antibiotics, and depending on the experiment, with either 0.1% DC or TC (Sigma‐Aldrich). Cultures were grown to mid‐log phase (OD600 of 0.4‐0.5), at which point Cm (100 μg ml–1) and, if necessary, DTT were added to the bacterial cells to inhibit protein translation, as previously described by Studemann et al. (2003). For bacteriaharbouring plasmids, the expression was initially induced by the addition of IPTG (1 mM for pFLAGrseABC and pTrc99ApelBYYF and 0.05 mM for pFLAGtoxRCCtoxS and pFLAGtoxRS) or arabinose (0.05% pBAD18‐KandegS) for 1 h before protein biosynthesis was blocked by the addition of Cm (100 µg ml–1). pMMB67EHdegP was not induced by IPTG since basal expression levels were sufficient to detect DegP activity. Probes without Cm incubation (Cm‐) served as negative controls. WCLs were harvested in Laemmli buffer with or without β‐mercaptoethanol at the indicated time points and analysed by 15% SDS‐PAGE and subsequent immunoblot analysis.
ToxR transcriptional activity determined by ompU and ompT phoA fusions
To determine the enzymatic activities for transcriptional phoA fusions, alkaline phosphatase activities were assayed as described previously (Miller, 1992). V. cholerae strains were grown to late‐log phase (OD600 of 0.8‐1) or ON at 37°C in M9 maltose minimal media supplemented with or without DC (0.01%). To note, higher DC concentrations, such as 0.1%, lead to reduced PhoA activity most likely due to a leakiness of the outer membrane, associated with a loss of the PhoA enzyme from the periplasm. Bacterial cells were permeabilized with SDS and chloroform. Experiments were performed in biological triplicates and technical duplicates respectively. The activities were expressed in Miller‐Units: (A405 × 1,000)/(A600 × ml × min × 0.96).
Supporting information
Acknowledgements
This work was supported by the Austrian Science Fund (FWF) Grants P29404 (JR), P25691 (SS) and National Institutes of Health (NIH/NIAID) R01AI120489 (JZ). For figure design, we thank Gregor Gruber.
References
- Almagro‐Moreno, S. , Kim, T.K. , Skorupski, K. and Taylor, R.K. (2015a) Proteolysis of virulence regulator ToxR is associated with entry of Vibrio cholerae into a dormant state. PLOS Genetics, 11, e1005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almagro‐Moreno, S. , Root, M.Z. and Taylor, R.K. (2015b) Role of ToxS in the proteolytic cascade of virulence regulator ToxR in Vibrio cholerae . Molecular Microbiology, 98, 963–976. [DOI] [PubMed] [Google Scholar]
- Amann, E. , Ochs, B. and Abel, K.J. (1988) Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli . Gene, 69, 301–315. [DOI] [PubMed] [Google Scholar]
- Bachmann, B. (1987) Derivation and genotype of some mutant derivatives of Eschericha coli K‐12, p 1190–1219In: Neidhardt, F.C. et al., (Ed.) Escherichia coli and Salmonella typhimurium. In Cellular and Molecular Biology. Washington, DC: ASM Press. [Google Scholar]
- Berg, T. , Schild, S. and Reidl, J. (2007) Regulation of the chitobiose‐phosphotransferase system in Vibrio cholerae . Archives of Microbiology, 187, 433–439. [DOI] [PubMed] [Google Scholar]
- Bina, J. , Zhu, J. , Dziejman, M. , Faruque, S. , Calderwood, S. and Mekalanos, J.J. (2003) ToxR regulon of Vibrio cholerae and its expression in vibrios shed by cholera patients. Proceedings of the National Academy of Sciences, 100, 2801–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bina, X.R. , Howard, M.F. , Taylor‐Mulneix, D.L. , Ante, V.M. , Kunkle, D.E. and Bina, J.E. (2018) The Vibrio cholerae RND efflux systems impact virulence factor production and adaptive responses via periplasmic sensor proteins. PLoS Pathog, 14, e1006804 1006810.1001371/journal.ppat.1006804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock, W.O. , Fernandez, J.M. and Short, J.M. (1987) XL1‐Blue—a high‐efficiency plasmid transforming recA Escherichia coli strain with β‐galactosidase selection. Biotechniques, 5, 376–379. [Google Scholar]
- Chaba, R. , Grigorova, I.L. , Flynn, J.M. , Baker, T.A. and Gross, C.A. (2007) Design principles of the proteolytic cascade governing the sigma E‐mediated envelope stress response in Escherichia coli: Keys to graded, buffered, and rapid signal transduction. Genes & Development, 21, 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champion, G.A. , Neely, M.N. , Brennan, M.A. and DiRita, V.J. (1997) A branch in the ToxR regulatory cascade of Vibrio cholerae revealed by characterization of toxT mutant strains. Molecular Microbiology, 23, 323–331. [DOI] [PubMed] [Google Scholar]
- Childers, B.M. and Klose, K.E. (2007) Regulation of virulence in Vibrio cholerae: The ToxR regulon. Future Microbiology, 2, 335–344. [DOI] [PubMed] [Google Scholar]
- Correa, N.E. , Lauriano, C.M. , McGee, R. and Klose, K.E. (2000) Phosphorylation of the flagellar regulatory protein FlrC is necessary for Vibrio cholerae motility and enhanced colonization. Molecular Microbiology, 35, 743–755. [DOI] [PubMed] [Google Scholar]
- Crawford, J.A. , Krukonis, E.S. and DiRita, V.J. (2003) Membrane localization of the ToxR winged‐helix domain is required for TcpP‐mediated virulence gene activation in Vibrio cholerae . Molecular Microbiology, 47, 1459–1473. [DOI] [PubMed] [Google Scholar]
- Davis, B.M. and Waldor, M.K. (2009) High‐throughput sequencing reveals suppressors of Vibrio cholerae rpoE mutations: one fewer porin is enough. Nucleic Acids Research, 37, 5757–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, Y. , Davis, B.M. and Waldor, M.K. (2004) Hfq is essential for Vibrio cholerae virulence and down regulates Sigma E expression. Molecular Microbiology, 53, 345–354. [DOI] [PubMed] [Google Scholar]
- DiRita, V.J. and Mekalanos, J.J. (1991) Periplasmic interaction between two membrane regulatory proteins, ToxR and ToxS, results in signal transduction and transcriptional activation. Cell, 64, 29–37. [DOI] [PubMed] [Google Scholar]
- DiRita, V.J. , Parsot, C. , Jander, G. and Mekalanos, J.J. (1991) Regulatory cascade controls virulence in Vibrio cholerae . Proceedings of the National Academy of Sciences, 88, 5403–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnenberg, M.S. and Kaper, J.B. (1991) Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive‐selection suicide vector. Infection and Immunity, 59, 4310–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziejman, M. , Kolmar, H. , Fritz, H.J. and Mekalanos, J.J. (1999) ToxR co‐operative interactions are not modulated by environmental conditions or periplasmic domain conformation. Molecular Microbiology, 31, 305–317. [DOI] [PubMed] [Google Scholar]
- Fan, F. , Liu, Z. , Jabeen, N. , Birdwell, L.D. , Zhu, J. , and Kan, B. (2014) Enhanced interaction of Vibrio cholerae virulence regulators TcpP and ToxR under oxygen limiting conditions. Infection and Immunity, 82, 1676–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fengler, V.H. , Boritsch, E.C. , Tutz, S. , Seper, A. , Ebner, H. , Roier, S. et al (2012) Disulfide bond formation and ToxR activity in Vibrio cholerae . PLoS One, 7, e47756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss, T.J. , Morgan, S.J. , French, E.L. and Krukonis, E.S. (2013) ToxR recognizes a direct repeat element in the toxT, ompU, ompT, and ctxA promoters of Vibrio cholerae to regulate transcription. Infection and Immunity, 81, 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman, L.M. , Belin, D. , Carson, M.J. and Beckwith, J. (1995) Tight regulation, modulation, and high‐level expression by vectors containing the arabinose PBAD promoter. Journal of Bacteriology, 177, 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan, D. (1983) Studies on transformation of Escherichia coli with plasmids. Journal of Molecular Biology, 166, 557–580. [DOI] [PubMed] [Google Scholar]
- Hase, C.C. and Mekalanos, J.J. (1998) TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae . Proceedings of the National Academy of Sciences, 95, 730–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins, D.E. and DiRita, V.J. (1994) Transcriptional control of toxT, a regulatory gene in the ToxR regulon of Vibrio cholerae . Molecular Microbiology, 14, 17–29. [DOI] [PubMed] [Google Scholar]
- Hofmann, A.F. , Hagey, L.R. and Krasowski, M.D. (2010) Bile slats of vertebrates: structural variation and possible evolutionary significance. Journal of Lipid Research, 51, 226–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton, R.M. , Hunt, H.D. , Ho, S.N. , Pullen, J.K. and Pease, L.R. (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene, 77, 61–68. [DOI] [PubMed] [Google Scholar]
- Hung, D.T. and Mekalanos, J.J. (2005) Bile acids induce cholera toxin expression in Vibrio cholerae in a ToxT‐independent manner. Proceedings of the National Academy of Sciences, 22, 3028–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, D. , Gho, Y.S. , Suh, M. and Kang, C. (2002) Highly sensitive and fast protein detection with Coomassie Brilliant Blue in sodium dodecyl sulfate‐polyacrylamide gel electrophoresis. Bulletin of the Korean Chemical Society, 23, 11. [Google Scholar]
- Kanjilal, S. , Citorik, R. , LaRocque, R.C. , Ramoni, M.F. and Calderwood, S.B. (2010) A systems biology approach to modeling Vibrio cholerae gene expression under virulence‐inducing conditions. Journal of Bacteriology, 192, 4300–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacikova, G. and Skorupski, K. (2002) The alternative sigma factor Sigma E plays in important role in intestinal survival and virulence in Vibrio cholerae . Infection and Immunity, 70, 5355–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacikova, G. , Lin, W. and Skorupski, K. (2010) The LysR‐type virulence activator AphB regulates the expression of genes in Vibrio cholerae in response to low pH and anaerobiosis. Journal of Bacteriology, 192, 4181–4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli, U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Landeta, C. , Boyd, D. and Beckwith, J. (2018) Disulfide bond formation in prokaryotes. Nature Microbiology, 3, 210 .1038/s41564-41017-40106-41562 [DOI] [PubMed] [Google Scholar]
- Lima, S. , Guo, M.S. , Chaba, R. , Gross, C.A. and Sauer, R.T. (2013) Dual molecular signals mediate the bacterial response to outer‐membrane stress. Science, 340, 837–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Menghua, M. , Peterfreund, G.L. , Tsou, A.M. , Selamoglu, N. , Daldal, F. et al (2011) Vibrio cholerae anaerobic induction of virulence gene expression is controlled by thiol‐based switches of virulence regulator AphB. Proceedings of the National Academy of Sciences, 108, 810–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur, J. , Davis, B.M. and Waldor, M.K. (2007) Antimicrobial peptides activate the Vibrio cholerae sigma E regulon through an OmpU‐dependent signalling pathway. Molecular Microbiology, 63, 848–858. [DOI] [PubMed] [Google Scholar]
- Matson, J.S. , Withey, J.H. and DiRita, V.J. (2007) Regulatory networks controlling Vibrio cholerae virulence gene expression. Infection and Immunity, 75, 5542–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mey, A.R. , Butz, H.A. and Payne, S.M. (2015) Vibrio cholerae CsrA regulates ToxR levels in response to amino acids and is essential for virulence. MBio, 6, e01064–e01015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgett, C.R. , Almagro‐Moreno, S. , Pellegrini, M. , Taylor, R.K. , Skorupski, K. and Kull, F.J. (2017) Bile salts and alkaline pH reciprocally modulate the interaction between the periplasmic domains of Vibrio cholerae ToxR and ToxS. Molecular Microbiology, 105, 258–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, J.H. (1992) A Short Course in Bacterial Genetics. A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Miller, V. , DiRita, V.J. and Mekalanos, J.J. (1989) Identification of toxS, a regulatory gene whose product enhances ToxR‐mediated activation of the cholera toxin promoter. Journal of Bacteriology, 171, 1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, V.L. and Mekalanos, J.J. (1984) Synthesis of cholera toxin is positively regulated at the transcriptional level by toxR . Proc Nat Acad Sci USA, 81, 3471–3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, V.L. , Taylor, R.K. and Mekalanos, J.J. (1987) Cholera toxin transcriptional activator ToxR is a transmembrane DNA binding protein. Cell, 48, 271–279. [DOI] [PubMed] [Google Scholar]
- Miller, V.L. and Mekalanos, J.J. (1988) A novel suicide vector and its use in construction of insertion mutations: Osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR . Journal of Bacteriology, 170, 2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales, V.M. , Baeckman, A. and Bagdasarian, M. (1991) A series of wide‐host‐range low‐copy‐number vectors that allow direct screening for recombinants. Gene, 97, 39–47. [DOI] [PubMed] [Google Scholar]
- Morgan, S.J. , French, E.L. , Thomson, J.J. , Seaborn, C.P. , Shively, C.A. and Krukonis, E.S. (2015) Formation of an intramolecular periplasmic disulfide bond in TcpP protects TcpP and TcpH from degradation in Vibrio cholerae . Journal of Bacteriology, 81, 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottemann, K.M. , DiRita, V.J. and Mekalanos, J.J. (1992) ToxR proteins with substitutions in residues conserved with OmpR fail to activate transcription from the cholera toxin promoter. Journal of Bacteriology, 174, 6807–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottemann, K.M. and Mekalanos, J.J. (1996) The ToxR protein of Vibrio cholerae forms homodimers and heterodimers. Journal of Bacteriology, 178, 156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, G.D.N. , Woods, A. , Chiang, S.L. and Mekalanos, J.J. (1993) CTX genetic element encodes a site‐specific recombination system and an intestinal colonization factor. Proceedings of the National Academy of Sciences, 90, 3750–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfau, J.D. and Taylor, R.K. (1996) Genetic footprint of the ToxR‐binding site in the promoter for cholera toxin. Molecular Microbiology, 20, 213–222. [DOI] [PubMed] [Google Scholar]
- Pfau, J.D. and Taylor, R.K. (1998) Mutations in toxR and toxS that separate transcriptional activation from DNA binding at the cholera toxin gene promoter. Journal of Bacteriology, 180, 4724–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano, D. and Klose, K.E. (2000) Altered expression of the ToxR‐regulated porins OmpU and OmpT diminishes Vibrio cholerae bile resistance, virulence factor expression, and intestinal colonization. Proceedings of the National Academy of Sciences, 97, 10220–10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano, D. , Schuhmacher, D.A. , Barker, J.L. and Klose, K.E. (2000) The virulence regulatory protein ToxR mediates enhanced bile resistance in Vibrio cholerae and other pathogenic Vibrio species. Infection and Immunity, 68, 1491–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, R.E. (1988) The nucleotide sequence of pACYC184. Nucleic Acids Research, 16, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford, S.T. , van Kessel, J.C. , Shao, Y. and Bassler, B.L. (2011) AphA and LuxR/HapR reciprocally control quorum sensing in vibrios. Genes & Development, 25, 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhavy, T.J. , Berman, M.L. and Enquist, L.W. (1984) Experiments with Gene Fusions. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Skorupski, K. and Taylor, R.K. (1999) A new level in the Vibrio cholerae ToxR virulence cascade: AphA is required for transcriptional activation of the tcpPH operon. Molecular Microbiology, 31, 763–771. [DOI] [PubMed] [Google Scholar]
- Stadler, J. , Stern, H.S. , Yeung, K.S. , McGuire, V. , Furrer, R. , Marcon, N. et al (1988) Effect of high fat consumption on cell proliferation activity of colorectal mucosa and on soluble faecal bile acids. Gut, 29, 1326–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studemann, A. , Noirclerc‐Savoye, M. , Klauck, E. , Becker, G. , Schneider, D. and Hengge, R. (2003) Sequential recognition of two distinct sites in sigma(S) by the proteolytic targeting factor RssB and ClpX. The EMBO Journal, 15, 4111–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, N.P. , Alba, B.M. , Bose, B. , Gross, C.A. and Sauer, R.T. (2003) OMP peptide signals initiate the envelope‐stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell, 113, 61–71. [DOI] [PubMed] [Google Scholar]
- Wilken, C. , Kitzing, K. , Kurzbauer, R. , Ehrmann, M. and Clausen, T. (2004) Crystal structiure of the DegS stress sensor: How a PDZ domain recognizes misfolded protein and activates a protease. Cell, 117, 483–494. [DOI] [PubMed] [Google Scholar]
- Xu, X. , Stern, A.M. , Liu, Z. , Kan, B. and Zhu, J. (2010) Virulence regulator AphB enhances toxR transcription in Vibrio cholerae . BMC Microbiology, 10, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, Y. , Tu, F. , Shi, M. , Wu, C.Q. , Ren, G. , Wang, X. et al (2016) Redox pathway sensing bile salts activates virulence gene expression in Vibrio cholerae . Molecular Microbiology, 102, 909–924. [DOI] [PubMed] [Google Scholar]
- Yang, M. , Liu, Z. , Hughes, C. , Stern, A.M. , Wang, H. , Zhong, Z. et al (2013) Bile salt‐induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proceedings of the National Academy of Sciences, 110, 2348–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials