

Abstract
Astroviruses (AstV) are a major cause of diarrhoea in children. Interestingly, some wildlife species, including bats, remain phenotypically asymptomatic after infection. Disease symptoms, however, may only be less visible in bats and enteric viruses may indeed perturb their gut microbial communities. Gut microbiomes represent an important driver of immune defence mechanisms but potential effects of enteric virus-host microbiome interactions are largely unexplored. Using bats as a natural model system, we show that AstV-infections affect the gut microbiome, with the strength of the effect depending on host age. The gut microbial α- and β-diversity and the predicted microbial functional orthologs decreased in young bats but surprisingly increased in adult AstV + bats. The abundance of bacterial taxa characteristic for healthy microbiomes was strongly reduced in young AstV+ bats, possibly attributable to their immature immune system. Regardless of age, pathogen-containing genera exhibited negative interactions with several commensal taxa and increased after AstV-infection, leading to pathobiont-like shifts in the gut microbiome of all infected bats. Thus, in apparently healthy bats, AstV-infections disturb gut bacterial homeostasis, possibly increasing previously suppressed health risks by promoting co-infections. If similar processes are present in humans, the effects of enteric virus infections might have longer-term impacts extending beyond the directly observed symptoms.
Introduction
Astroviruses (AstV) represent a world wide leading cause of infectious diarrhoea in children, in the elderly and in immunocompromised people, mainly transmitted through a faecal-oral route [1]. In humans, HAstV-infections lead to an increased intestinal epithelial cell permeability by disrupting cellular tight junctions and thereby causing the loss of ability of the intestine to reabsorb water and nutrients [2]. However, infections in healthy children, in adults and, especially, in some animal species remain asymptomatic, although little is known about the pathogenesis of the disease [3]. For example, murine-AstV-infected laboratory mice exhibit no diarrhoea, despite an increase in intestinal permeability. This implies that intestinal permeability is independent of diarrhoea [4, 5] and that immune modulation might instead play a role in the defence against AstV-infection. The gut microbiome is an important driver of immune defence mechanisms and shifts in its composition with age influence the susceptibility to enteropathogens [6, 7]. Contrariwise, because of the direct contact of the gut microbiome and its protective role, it is also probably a target of enteric viruses such as AstV. Thus, an investigation into changes in the diversity and bacterial abundance pattern of the gut microbiome might be a missing link in understanding the effects of an AstV-infection and also its potential as a driver of co-infections, an area of research that has been largely neglected so far [3].
Bats represent an excellent model for studying host-pathogen-microbiome interactions, since they serve as an important source of several crucial RNA viruses of humans and livestock, including AstV [8]. As in humans, a remarkably high AstV-prevalence, viral genetic diversity and inferred zoonotic potential has been reported for bats. Contrary to inbred mouse lines, bats exhibit comparable genomic background diversity as humans [1, 9, 10]. Additionally, bat species can live in diverse habitats, including disturbed ones, and thus come into frequent contact with humans and their livestock. To understand the effects of AstV-infections on gut microbiomes in bats, we studied changes in the bacterial diversity and abundance pattern of naturally AstV-infected young and adult Jamaican fruit bats (Artibeus jamaicensis). We identified age-dependent changes in the α- and β-diversity of the gut microbiome in AstV+ bats. All infected bats revealed a higher abundance of pathogen-containing genera. Our results in a natural model system have thus revealed that AstV-infections distort the composition of gut bacterial communities and might provide missing links for a better understanding of the effects of AstV-infections in humans, and also as potential drivers of co-infections, an area of research that has been largely neglected so far.
Materials and Methods
Sample collection and Astrovirus testing
To investigate the influence of AstV on the gut microbiome of bats, we chose the Jamaican fruit bat, Artibeus jamaicensis (Phyllostomidae: Stenodermatinae), a species inhabiting a wide range of habitats and widely distributed in tropical America. It shows high dispersal capability and often a high abundance, even in modified landscapes. It has been reported to carry several RNA and DNA viruses [8] and has been previously used as a model system for studying host-virus interactions [11].
We captured A. jamaicensis by using mist nets (Ecotone, Gdynia, Poland) in Central Panama in 2013 (Brändel et al., in review). Individuals were sexed and aged based on the ossification of the epiphyses of the fingers [12]. Young individuals (<1-year) have gaps between the epiphyses of the fingers and are sexually not active (termed hereafter as ‘young’). All sexually mature individuals (>1-year-old) have closed epiphyses (termed hereafter as ‘adults’). Immediately after capture, bats were individually placed in clean cloth bags. Faecal samples were collected either from the bags or obtained by using Nylon swabs in the rectal area and preserved in Eppendorf vials filled with 500 µl RNAlater (Life Technologies).
Viral RNA was extracted from the faeces by using the MagNAPure 96 DNA and the Viral NA Small Volume Kit (Roche) according to the manufacturer’s instructions. AstV prevalence was determined by the broadly reactive nested reverse transcription-PCR (RT-PCR) assay as described previously [13]. We included all AstV positive samples (NAstV+ = 82 (36 young, 46 adults; AstV-prevalence in overall data set: 11.8%) and 169 randomly chosen AstV-negative samples (68 young, 101 adults) in the microbiome analysis.
Bacterial DNA extraction and 16S rRNA gene amplicon sequencing
Following homogenization of faecal samples (SpeedMill PLUS Homogenizer, Analytik Jena, Germany), bacterial genomic DNA was extracted by using NucleoSpin 96 Soil kit (Macherey-Nagel, Germany) according to the manufacturer’s instructions. We amplified the hypervariable V4 region of the 16 S rRNA gene (291 bp) with the primer pair 515 F (5-GTGCCAGCMGCCGCGGTAA-3) and 806 R (5-GGACTACHVGGGTWTCTAAT-3)[14]. We followed the approach of the Fluidigm System (Access Array™ System for Illumina Sequencing Systems, © Fluidigm Corporation) for primer tagging. The PCR (15ul of volume) was performed as described previously [15]. After purification (NucleoMag bead based size selection, Macherey-Nagel, Germany) and quantification (DropSense, Trinean, US) of barcoded samples, the pooled sample library was paired-end sequenced in a single run on an Illumina® MiSeq platform.
The initial processing and merging of forward and reverse reads was performed as previously described [16]. We followed the workflow pipeline of the QIIME 1.9.1 software package [17] for quality filtering (q = 25) and further analysis. Potential chimeras were removed by using USEARCH 6.1 (Greengenes database, version 13.8, http://greengenes.lbl.gov). Operational Taxonomic Units (OTUs) were picked by using the open-reference OTU picking approach at a 97% similarity threshold with UCLUST as the default. Taxonomy was assigned by using the Ribosomal Database Project (RDP) classifier. Singletons and OTUs belonging to chloroplast, mitochondria, archaea and eukaryota were further removed from the data set. The 16 S rRNA gene sequences are available in the NCBI sequence read archive under BioProject PRJNA473909 with the accession number SRP149464.
Alpha and beta diversity analysis
To investigate the effect of AstV-infection on gut microbial diversity, we calculated alpha diversity measurements for each sample by using three different diversity indices (number of observed species (OTUs), Chao1 and phylogenetic diversity (PD)) after rarefying the data to 11,000 sequences per sample. All further analyses were performed in R version 3.3.2 ([18]). To analyse the effect of AstV-infection on these alpha diversity metrics, we performed General Linear Modelling (GLM) after log-transforming the data by using the lme4 package in R [19]. We included AstV-infection (NAstV- = 169, NAstV+ = 82), age (young = 104, adult = 147), interaction between these two (AstV*age) and sex (female = 150, male = 101) in the model as explanatory variables. Furthermore, we calculated effect sizes (Cohen’s d) with 95%-confidence intervals for each significant comparison by using the effsize package in R [20].
To investigate the effect of the AstV-infection on gut microbial beta diversity, we calculated unweighted and weighted UniFrac metrics after rarefying the data to 11,000 sequences per sample by using the phyloseq package in R [21]. The PERMANOVA approach (adonis in R package vegan) was used to test the significance of the differences in the community composition with 999 permutations. For both beta diversity metrics, we included, as before, AstV-infection, age, interaction between these two (AstV*age) and sex in the models as explanatory variables. Furthermore, in order to visualize the pattern of separation between the various categories of samples, principal coordinates analyses (PCoA) were performed based on the UniFrac metrics.
Relative abundance of major phyla and identification of OTUs associated with AstV-infections
Using GLMs, we compared the relative abundance of the five major phyla in relation to AstV-infection, age, interaction between these two (AstV*age) and sex as explanatory variables as performed for alpha and beta diversity. In order to identify the OTUs accountable for differences in the microbiome of AstV− / AstV+ young and adult bats, we employed two different approaches after the removal of spurious OTUs (i.e., OTUs present in less than three samples for each category, OTUs not classified at phylum level). We used G-tests (goodness of fit) in QIIME and a negative binomial-model-based approach (Exact binomial test generalized for overdispersed counts) available in the edgeR package in R [22] to identify OTUs that significantly differed (p ≤ 0.001) in their relative abundance after correction for multiple testing by using the Benjamini–Hochberg procedure and reported jointly identified OTUs by both approaches.
Microbial network analyses
To infer the relationships among OTUs differing in their relative abundance in AstV− and AstV+ samples of young and adult bats, we prepared microbial correlation networks by using CoNet plugin for Cytoscape [23]. We restricted these analyses to those OTUs jointly identified by both statistical approaches (G test, Exact test). To calculate all pairwise taxa (OTU) scores, we applied an ensemble approach by using five similarity measures (‘Pearson correlation’, ‘Spearman correlation’, ‘Bray–Curtis dissimilarity’, ‘Kullback-Leibler dissimilarity’ and ‘mutual information’). This approach is more precise than the use of a single correlation detection strategy, especially for sparse data [24]. We followed the network-building parameters as suggested by Faust and Raes [23].
Microbiome functionality analyses
To anticipate functional differences of the gut microbiome due to AstV-Infection based on 16 S rRNA gene sequencing, we used PICRUSt [25]. After removing all de novo OTUs from the data set and normalization for copy number variation (as implemented in PICRUSt [25]), the metagenome prediction was carried out by using the KEGG Orthology (KOs) classification. To assess the accuracy of the prediction, weighted Nearest Sequenced Taxon Index (weighted NSTI) scores were calculated. The NSTI score represents the sum of phylogenetic distances of each organism in a sample to its nearest relative in a sequenced bacterial genome [25]. The NSTI value for A. jamaicensis (mean NSTI = 0.034 ± 0.02 s.d.) was close to humans (mean NSTI = 0.03 ± 0.02 s.d.) [25] and was sufficiently low to allow the accurate prediction of the metagenomes. To investigate the effect of the AstV-infection on the KEGG composition, we calculated ‘gower’ distances after rarefying the data (using the phyloseq package in R [21]). ‘Gower’ distances consider presence-absence as well as abundance of taxa and perform especially well for detecting differences between clusters. We used the PERMANOVA approach to test the significance of the differences in the KEGG composition with 999 permutations. We included AstV-infection, age, the interaction (AstV*age) and sex of the individual in the model as explanatory variables to explain differences in the gower metrics. PCoA plots were used to visualize the pattern of separation between young and adult individuals. We categorized KOs to major functional pathways by applying the KEGG classification at the hierarchy level 3. To identify the pathways, which show differential abundance in AstV + and AstV− young and adult individuals, we used Exact tests implemented in the edgeR package in R [22] and only pathways that remained significant (p ≤ 0.05) after Benjamini–Hochberg correction were reported.
Results
AstV-infection perturbs bacterial alpha and beta diversity differently in young and adult bats
We characterized changes in the gut microbiome in relation to AstV-infection in 251 Jamaican fruit bats by performing high-throughput sequencing of the V4 region of the bacterial 16 S ribosomal RNA gene. We recovered on average 33,249 high quality reads per sample after taxonomic assignments.
We used General Linear Modelling (GLM) to test whether inter-individual differences within three alpha diversity estimates (number of observed species (OTUs), Chao1 and phylogenetic diversity (PD)) can be explained by AstV-infection, host age, sex or an AstV*age interaction. We detected a strong influence of AstV*age interactions on inter-individual differences in all three alpha diversity metrics (OTUs: p = 0.013, Chao1: p = 0.008, and PD: p = 0.004, all other predictors p > 0.05) (Supplementary Table 1, Fig. 1). In young bats, all three alpha diversity metrics were lower in AstV + than AstV− individuals (number of observed species: Cohen’s d = 0.53, Chao1: Cohen’s d = 0.55, and PD: Cohen’s d = 0.54), whereas in adults, the alpha diversity values were higher (number of observed species: Cohen’s d = 0.30, Chao1: Cohen’s d = 0.32, and PD: Cohen’s d = 0.30) in infected individuals (Fig. 1).
Fig. 1.

Differential effect of AstV-infection on bacterial alpha diversity of young and adult bats. Shown is the effect of AstV-infection (AstV−, AstV+) on (a) number of observed species (p = 0.011), b Chao1 (p = 0.006) and (c) PD (p = 0.005) in young (orange) and adult (grey) individuals
The use of PERMANOVA models based on unweighted and weighted UniFrac metrics revealed a significant effect of host age (unweighted: R2 = 0.009, p = 0.001, weighted: R2 = 0.022, p = 0.001), sex (unweighted: R2 = 0.006, p = 0.008, weighted: R2 = 0.009, p = 0.028) and AstV*age interaction (unweighted: R2 = 0.008, p = 0.001, weighted: R2 = 0.015, p = 0.002) on gut microbial beta diversity (Supplementary Table 2). An AstV-infection showed no effect on the composition of the microbiome itself (unweighted: p = 0.246) but on the abundance pattern (weighted: R2 = 0.014, p = 0.004) (Supplementary Table 2). The differential effect of AstV-infections on the bacterial community composition of young and adult bats were visualised by PCoA. AstV-infections explained 51.2 and 45.7% of the variance in weighted UniFrac distances of young and adult bats, respectively (Fig. 2).
Fig. 2.

Differential effect of AstV-infections on the bacterial community composition of young and adult bats. Principal-coordinate plots of unweighted and weighted UniFrac distances in young (a, b) (unweighted R2 = 0.016, p = 0.003, weighted R2 = 0.042, p = 0.001) and adult (c, d) (unweighted R2 = 0.008, p = 0.072, weighted R2 = 0.017, p = 0.019) bats. Dots and surrounding dashed ellipses (95% confidence level) represent gut bacterial communities of AstV− (blue) and AstV + (red) individuals
AstV-infection induces shifts in relative abundance of major phyla and OTUs according to bat age
The gut bacterial community of A. jamaicensis is dominated by the phyla Proteobacteria (59.98%) and Firmicutes (25.45%), followed by Tenericutes (3.9%), Actinobacteria (2.7%) and Bacteroidetes (0.9%) (Supplementary Figure 1). AstV-infection caused significant changes in the relative abundance of the phyla Proteobacteria (p = 0.04) and Firmicutes (p = 0.006) and showed a tendency in Tenericutes (p = 0.055) and Actinobacteria (p = 0.054) in relation to host age (Supplementary Figure 1).
Using two different approaches, we jointly identified 39 OTUs in young bats and 41 OTUs in adult bats, which differed significantly (p ≤ 0.001) in their mean abundance between AstV− and Ast+ individuals (Supplementary Table 3, Supplementary Table 4, Supplementary Figure 2). In infected young, the abundance of 22 OTUs (56.4%) increased in total 7.9-fold but the remaining 17 OTUs decreased 30.1-fold. In infected adults, the abundance of the majority of OTUs (26, 63.4%) increased in total 6.7-fold but only 15 OTUs revealed a decreased abundance (10.5-fold).
Whereas in young AstV+ bats, six OTUs reported in previous studies as essential members of healthy microbiomes [26, 27] greatly decreased (617.5-fold), one such identified OTU showed a 5.1-fold increase in infected adults. In both age groups, OTUs belonging to reportedly pathogen-containing taxa increased after AstV-infection (young: 13 OTUs, 6.9-fold increase; adults: 15 OTUs, 5.1-fold increase) (Supplementary Table 3, Supplementary Table 4).
Picked OTUs that showed a lower abundance in AstV+ young bats mainly belonged to the families Lachnospiraceae and Ruminococcaceae and to the genera Oscillospira and Bacteroides. OTUs showing a higher abundance in young AstV+ individuals mainly belonged to the families Clostridiaceae, Peptostreptococcaceae and Enterobacteriaceae and to several genera, such as Aggregatibacter, Clostridium, Acinetobacter and Pseudomonas. In addition, one OTU was identified to the species level as Haemophilus influenzae (Supplementary Table 3, Fig. 3a).
Fig. 3.

Differential abundance of OTUs in relation to AstV-infection of young and adult bats. Shown are OTUs that were jointly picked by both the G test and negative binomial Exact test and that differ in their mean abundance in relation to the infection status of (a) young and (b) adult (39 and 41 OTUs, respectively; Supplementary Figure 2) bats. The values indicate a log10-fold (logFC) decrease (blue) or increase (red) after AstV-infection. OTUs are arranged according to increasing values of log-fold change. The highest possible taxonomic assignment (maximal to the genus level) is shown for each OTU. OTU-IDs are coloured according to previous studies (see Supplementary Table 3 and Supplementary Table 4 for details and references) as an essential member of healthy microbiomes (green) or as pathogen-containing taxa (red); *includes unclassified OTUs at genus level
In adult bats, OTUs that showed a lower abundance in AstV + individuals were mainly from the families Xanthomonadaceae and Enterobacteriaceae and from the genera Candidatus Portiera and Veillonella, whereas OTUs that showed higher abundances belonged to families Peptostreptococcaceae and Enterobacteriaceae. Moreover, Mycoplasma, Streptococcus and other pathogen-containing genera were also detected (Supplementary Table 4, Fig. 3b). Most OTUs changing in abundance after AstV-infection in both young and adult bats belonged to the families of Peptostreptococcaceae and Enterobacteriaceae. OTUs of the Peptostreptococcaceae primarily increased in abundance in both AstV+ young and adult individuals whereas the abundance of OTUs assigned to Enterobacteriaceae mainly increased (n = 5 OTU, except for OTU537290) in young AstV+ bats but decreased (n = 5 OTU, except for OTU537290) in adult AstV + animals.
Microbial networks differ between AstV− and AstV+bats
The microbial co-occurrence network based on OTUs that differed in abundance between AstV− and AstV+ bats (Fig. 3) revealed that decreasing OTUs often showed a negative relationship with certain other OTUs that increased after infection (Fig. 4). For example, in the network of young bats, the OTU belonging to Haemophilus influenzae (OTU316852), which was the most abundant among all 39 OTUs, showed a negative relationship with Lachnospiraceae_OTU4155396 (Fig. 4a, Supplementary Table 3). Similarly, Peptostreptococcaceae (OTU351976) showed negative relationships with both Lachnospiraceae (OTU4155396) and Ruminococcaceae (OTU11308) (Fig. 4a, Supplementary Table 3). Likewise, in the OTU network of adult bats, by far the most abundant OTU (OTU1141646) belonging to Streptococcus and three OTUs of Peptostreptococcaceae (OTU308309, OTU351976, OTU531374), which showed an increased abundance in AstV+ bats, had negative relationships with various other OTUs with decreased abundance after infection (Fig. 4b, Supplementary Table 4).
Fig. 4.

Microbial networks of OTUs differing in their relative abundance in relation to AstV-infection. Blue nodes mark decreasing OTUs and red nodes increasing OTUs in (a) young and (b) adult bats; sizes are according to the relative proportion of the OTUs. Red edges indicate negative relationships and grey edges positive relationships between the interacting OTUs. The highest possible taxonomic assignment (maximal to the genus level) is shown for each OTU. OTU-IDs are coloured according to previous studies (see Supplementary Table 3 and Supplementary Table 4 for details and references) as an essential member of healthy microbiomes (green) or as pathogen-containing taxa (red). *includes unclassified OTUs at genus level
Predicted metagenomes and higher functional pathways differ between young and adult bats after AstV-infections
We applied a PERMANOVA-based model approach to investigate whether differences in the predicted KEGG orthologs (KOs) occur in relation to AstV-infection, age, sex or AstV*age interaction. In the model, AstV-infection (R2 = 0.012, p = 0.013) and the interaction AstV*age (R2 = 0.012, p = 0.02) had a significant effect (Supplementary Table 5, Supplementary Figure 3), but neither age (p = 0.057) nor sex (p = 0.117). The AstV-infection explained 56.4 and 62.4% of the variance of the two PCoA-axes in young and adult bats, respectively (Supplementary Figure 3).
In adult bats, 16 predicted higher functional pathways (KEGG) were identified which differed in their abundance in AstV− compared to AstV+ individuals (negative binomial based Exact tests, p ≤ 0.05) but not significantly different KEGG pathway were detected in young bats (all p > 0.05). Identified pathways that occurred in a lower abundance in AstV+ adults were related to metabolism (“metabolism”, “carbohydrate metabolism”, “metabolism of other amino acids”), “membrane transport” and “cellular processes and signalling”, whereas the abundance of other metabolic pathways (“amino acid metabolism”, “glycan biosynthesis and metabolism”, “energy metabolism”) increased in AstV+ individuals (Fig. 5). Also, the relative abundance of pathways related to “cell growth and death”, “transcription”, “translation”, “cell motility”, “replication and repair” was higher in AstV+ compare to AstV− adult individuals (Fig. 5).
Fig. 5.
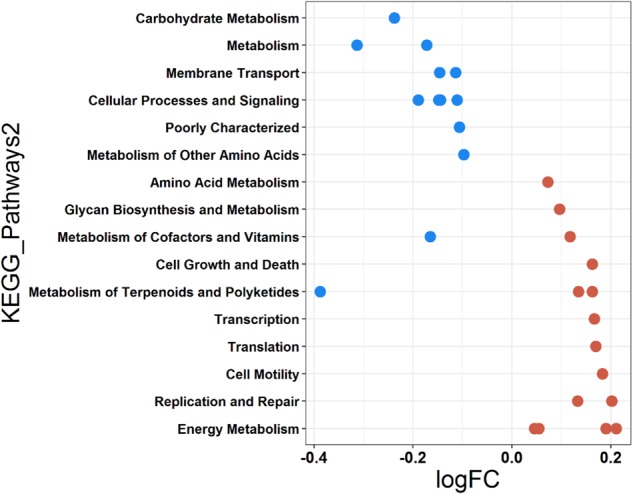
Differential abundance of predicted major functional pathways in relation to AstV infection in adult bats. Differences in the mean abundance of major functional pathways (identified by PICRUSt prediction by using KEGG classification) identified by Exact tests (p < 0.05). The values indicate a log10-fold (logFC) decrease and increase in AstV− (blue) and AstV+ (red) adults, respectively. Functional pathways are arranged according to increasing values of logFC
Discussion
Evidence has accumulated that virus infections may induce shifts in gut microbial communities [28, 29]. Astroviruses are a major cause of diarrhoea in children [1]. Interestingly, - and in contrast to strong health effects they have in humans—some wildlife species, including bats, remain phenotypically asymptomatic after infection [3]. However, disease symptoms after AstV-infection may only be less visible in bats. Enteric viruses may cause disturbances of the gut microbial communities but the potential link between AstV-infections and gut microbiomes have not been explored yet. Understanding the changes in the diversity and bacterial abundance pattern of the bat gut microbiome after AstV-infection may contribute to a better understanding of their effects in humans, and also for their potential as drivers of co-infections, an area of research that has been largely neglected so far.
Using bats as a natural model system, we have been able to explore, for the first time, the effects of AstV-infection on gut microbiome diversity and pathobiont-like-shifts in a naturally AstV-infected mammal. Our study has revealed that AstV-infection perturbs the gut microbiome of Jamaican fruit bats at both alpha and beta diversity levels and, even more importantly, that this disturbance in gut microbial community mainly depends on the interaction of infection and host age. AstV-infections were associated with a reduction in alpha and beta bacterial diversity in young individuals but, surprisingly, the opposite was observed in adult individuals in which both diversity metrics increased after AstV-infection. We did not observe differences in the body condition (i.e. reduced body mass) of young and adult AstV+ bats compared to uninfected individuals. Nevertheless, the higher AstV-induced effect (measured by Cohen’s d value) on the gut microbiome of young compared to adult bats suggests an increased disease severity in young infected bats.
Given the fact that AstV has such a high prevalence in the study population, one could question the cause-and-effect relationship and argue that the observed changes were primarily due to invasions of harmful bacteria in the gut that caused an increased host’s susceptibility towards AstV-infections. In other words, AstV-infections could be considered as a secondary effect of pathogenic bacteria infections [30]. According to this scenario, one would expect a higher AstV prevalence in adults. However, in our study we observed a higher number of potential pathogenic taxa with increased abundance in adult AstV-infected bats compared to young individuals. The AstV*age interaction was significant in the models. In Jamaican fruit bats as well as in other studies on bats [31] and humans [3] the AstV prevalence was found to be higher in young individuals. More research including experimental AstV-infection of individuals with healthy and dysbiotic microbiomes can help to proof such cause-and-effect relationships.
Because of the important role of the microbiome in maintaining gut homeostasis and in providing protection against gut-invading pathogens, shifts in its diversity beyond the normal range leading to dysbiosis might cause either diarrhoea directly (as reported in humans) or indirectly by increasing the gut’s susceptibility to enteric bacterial pathogens, which in turn may cause diseases [32, 33]. By comparing our results with previous studies on microbial dysbiosis, we noticed two broadly contrasting patterns. The reduction in gut microbial diversity in young AstV+ bats is in accordance with the observation in a recent meta-analysis, where a decrease in microbial diversity was consistently observed during diarrhoea and Inflammatory Bowel Disease (IBD) [34]. The increase in gut microbial diversity in adult bats is similar to reported effects of virus infections that directly interfere with the host immune system and indirectly affect the host gut microbiome, such as during Human Immunodeficiency Virus (HIV) infection in humans [35], Simian Immunodeficiency Virus (SIV) infection in chimpanzees [28] or Canine Distemper Virus (CDV) infection in giant panda [29]. In adult bats, the increase in bacterial diversity might be the result of a better immune-mediated counter-response against AstV-infection, either simply because of the more effective immune system in adulthood or because of an acquired immunity against repeated AstV-infections [36]. This might be the reason that AstV-infections in children cause more pathological symptoms and higher disease severity than in adults [3]. If this is the case, one would expect that not all bacterial taxa are similarly affected by AstV-infection but react in a different manner depending on their function. This leads to questions as to which bacterial taxa are affected following AstV-infection in young and adult bats and whether shifts in abundance hit commensal and potential pathogenic bacteria alike?
At the phylum level, contrary to dysbiotic states in humans [37], the abundance of Proteobacteria in bats decreases after AstV-infection. Firmicutes and Actinobacteria decrease in young bats but increase in adult AstV-infected bats. Both bacterial phyla are involved in immune-mediated colonization resistance against enteric pathogens with a decreased abundance during several diseases [38]. Tenericutes abundance, however, is only elevated in infected young, corresponding to observations in human patients with colorectal adenomas [39].
At the more specific genus and family level, we have detected, in young AstV+ individuals, a decrease in many essential bacteria of core microbiomes (i.e., groups of microbes commonly found within a host’s microbiome with critical functions [40]) such as OTUs from Bacteroides, Oscillospira, Lachnospiraceae and Ruminococcaceae that reportedly contribute to host health [26, 27]. For example, Bacteroides plays a critical role in maintaining gut homeostasis [41]. The colonization of germ-free mice with Bacteroides can rectify several immune system defects [42]. Lachnospiraceae and Ruminococcaceae are two further commensal families specialized in the degradation of complex plant material in the human gut. Other than metabolism, they also provide protection against enteric infections [43]. Oscillospira, Ruminococcaceae (clostridial cluster IV) and Lachnospiraceae (cluster XIVa) are also butyrate producers, an important source of energy for gut epithelial cells, and they maintain epithelial barrier integrity, the loss of which can result in diarrhoea [44, 45]. All of these taxa are reduced in abundance during various diseases such as in Crohn’s disease [26, 46].
At the same time, an increase of OTUs from taxa that harbour potential pathogens, such as Haemophilus, Enterobacteriaceae, and Peptostreptococcaceae, have been noted in AstV+ young bats. Haemophilus is a well-known bacterial genus that harbours many pathogenic species such as H. influenzae, which shows an increased presence in gut diseases (e.g. Crohn’s disease; [46]) or after influenza virus infection [47]. The family of Enterobacteriaceae harbour many pathogenic and commensal species (e.g. Escherichia coli) [32]. Enterobacteriaceae are usually present at low densities in the gut; however, their abundance has been shown to increase when the microbiome is in dysbiosis, e.g. in Crohn’s diseases [46], other IBDs [48] and in pathogen-induced gut infections [49] such as HIV infection [50]. Similarly, members of the Peptostreptococcaceae have been shown to increase in various dysbiosis conditions, e.g., those attributable to colorectal cancer [51] and SIV infection [28].
Interestingly, in adult AstV+ bats, we did not observe a decrease in OTUs belonging to the core microbiome as in young bats. Instead, one member of the core microbiome, Lactococcus, even increased in infected individuals. This could be attributable to a more active and competent immune system in adults and an immune-mediated control of bacterial taxa [52, 53]. Indeed, we observed a lower number of OTUs that decreased in abundance (e.g. Candidatus Portiera, Veillonella, Xanthomonadaceae and Enterobacteriaceae) than OTUs that increased in abundance. As in young bats, the increasing OTUs mainly belonged to taxa that harbour potential pathogens such as Mycoplasma, Streptococcus, Peptostreptococcaceae and Enterobacteriaceae. Pathogenic Mycoplasma and Streptococcus species have been reported to increase during various diseases such as in Crohn’s disease [46, 54] and others [47, 55]. However, OTUs of some Enterobacteriaceae mainly increased in abundance in young bats (n = 5 OTU) but decreased in adult bats (n = 5 OTU) after AstV-infection (but see OTU537290). This might again be explained by the better immune system of adult bats. We further noticed an increase in abundance of two Peptostreptococcaceae OTUs (OTU308309, OTU351976) in both AstV+ young and adult individuals, suggesting that, here, the increase in abundance of these OTUs is regardless of bat age and is instead a common feature after AstV-infection.
The microbial networks confirmed negative interactions between essential bacteria constituting the core microbiome and potentially pathogenic taxa. For example, in young individuals, one OTU from Lachnospiraceae (OTU4155396) showed a negative relationship with Haemophilus influenzae. Analogously, both Lachnospiraceae (OTU4155396) and Ruminococcaceae (OTU11308) showed negative relationships with Peptostreptococcaceae (OTU351976). This is in accordance with previous findings that members from both Lachnospiraceae and Ruminococcaceae exhibit negative interactions with invading pathogens [56], are important components of gut health and their loss can result in diarrhoea [44]. Similarly, in adult bats, we observed a negative interaction of a potential pathogenic member of Streptococcus (OTU1141646) with Xanthomonadaceae (OTU548694, OTU3055791) and Enterobacteriaceae (OTU746679) and, likewise, of Peptostreptococcaceae (OTU308309) with Enterobacteriaceae (OTU211). Overall, we observed pathobiont-like shifts (an increase in potential pathogens, e.g., Haemophilus, Streptococcus, Mycoplasma) in both young and adult bats after AstV-infection and negative interactions between potential pathogens and commensals. Such a pathobiont-like shift on its own can cause diarrhoea in children [57].
The AstV-infections not only perturbed the alpha and beta diversity levels, also the predicted metagenomes and higher functional pathways were affected in a comparable way. In young bats, the microbial KOs were reduced after AstV-infection whereas they were increased in adult bats. The reduction in microbial KOs might be due to a less efficient gut bacterial community (e.g., reduction in abundance of butyrate producing bacteria) in AstV+ young bats, similar to the observations in IBD [58], whereas the increase in overall KOs in adult AstV + bats could be related to an overall increase in bacterial diversity. Accordingly in adults, several predicted bacterial metabolism pathways changed in abundance after AstV-infection. Especially pathways related to basic cellular functions such as “cell growth and death”, “transcription”, “translation”, “cell motility”, “replication and repair” increased in AstV+ compared to AstV− individuals. This might be due to active and competent immune system in adults, which crosstalk with the gut microbiome in a diseased state [53]. However, one methodical caveat is that PICRUSt is based on a closed reference OTU picking approach, which does not include de novo OTUs and thus, the function of de novo OTUs could not be predicted in our analysis. Future research requires carefully planned experimental AstV-infection studies to observe potential changes in the immune system and gut microbiome while controlling for host age to disentangle the respective roles of each component of this tripartite relationship.
In conclusion, using bats as a natural model system, we show that the enteric AstV indeed affects the gut microbiome with the strength of the effect depending on host age. Interestingly, the gut microbial alpha and beta diversities and predicted microbial functions (KOs) decreased in young bats but increased in adult AstV+ bats. We observed a reduction in the bacteria community characteristic for a healthy microbiome in young bats, possibly attributable to their immature immune system. However, regardless of age, pathogen-containing genera increased after AstV-infection leading to pathobiont-like shifts in the gut microbiome. We have thus demonstrated that in apparently healthy bats that AstV-infections disturb the gut bacterial homeostasis, possibly increasing previously suppressed health risks by promoting co-infections.
Electronic supplementary material
Acknowledgements
We thank the Smithsonian Tropical Research Institute in Panamá for providing the essential infrastructure and, especially, Oris Acevedo and Belkys Jimenéz for their constant help during our fieldwork. We are grateful to all field assistants and Mark AF Gillingham for statistical advice. We thank Kerstin Wilhelm, Ulrike Stehle, Lara Jeworowski, Tobias Bleicker, Monika Eschbach-Bludau and Sebastian Brünink for excellent technical assistance. We are also grateful to Theresa Jones for language editing. This research was funded by the German Science Foundation (DFG) and is part of the DFG Priority Program SPP 1596/2 Ecology and species barriers in emerging infectious diseases (SO 428/ 9-1, 9-2; TS 81/7-1, 7-2; DR 772/8-1).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41396-018-0239-1) contains supplementary material, which is available to authorized users.
References
- 1.Donato C, Vijaykrishna D. The broad host range and genetic diversity of mammalian and avian Astroviruses. Viruses. 2017;9:102. doi: 10.3390/v9050102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moser LA, Carter M, Schultz-Cherry S. Astrovirus increases epithelial barrier permeability independently of viral replication. J Virol. 2007;81:11937–45. doi: 10.1128/JVI.00942-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson C, Hargest V, Cortez V, Meliopoulos V, Schultz-Cherry S. Astrovirus pathogenesis. Viruses. 2017;9:22. doi: 10.3390/v9010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng TFF, Kondov NO, Hayashimoto N, Uchida R, Cha Y, Beyer AI, et al. Identification of an Astrovirus commonly infecting laboratory mice in theUS and Japan Khudyakov YE (ed) PLoS ONE. 2013;8:e66937. doi: 10.1371/journal.pone.0066937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yokoyama CC, Loh J, Zhao G, Stappenbeck TS, Wang D, Huang HV, et al. Adaptive immunity restricts replication of novel murine Astroviruses. J Virol. 2012;86:12262–70. doi: 10.1128/JVI.02018-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–30. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeng MY, Cisalpino D, Varadarajan S, Hellman J, Warren HS, Cascalho M, et al. Gut microbiota-induced immunoglobulin g controls systemic infection by symbiotic bacteria and pathogens. Immunity. 2016;44:647–58. doi: 10.1016/j.immuni.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:531–45. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Benedictis P, Schultz-Cherry S, Burnham A, Cattoli G. Astrovirus infections in humans and animals; Molecular biology, genetic diversity, and interspecies transmissions. Infect Genet Evol. 2011;11:1529–44. doi: 10.1016/j.meegid.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer K, Pinho dos Reis V, Balkema-Buschmann A. Bat Astroviruses: towards understanding the transmission dynamics of a neglected virus family. Viruses. 2017;9:34. doi: 10.3390/v9020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munster VJ, Adney DR, van Doremalen N, Brown VR, Miazgowicz KL, Milne-Price S, et al. Replication and shedding of MERS-CoV in Jamaican fruit bats (Artibeus jamaicensis) Sci Rep. 2016;6:21878. doi: 10.1038/srep21878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Handley CO Jr, Wilson DE, Gardner AL. Demography and Natural History of the Common Fruit Bat, Artibeus jamaicensis, on Barro Colorado Island, Panama. Smithson Contrib to Zool. Report No. 511. Washington, DC: Smithsonian Institution; 1991.
- 13.Chu DKW, Poon LLM, Guan Y, Peiris JSM. Novel Astroviruses in insectivorous bats. J Virol. 2008;82:9107–14. doi: 10.1128/JVI.00857-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caporaso JG, Lauber CL, Walters Wa, Berg-Lyons D, Huntley J, Fierer N, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012;6:1621–4. doi: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Menke S, Wasimuddin, Meier M, Melzheimer J, Mfune JKE, Heinrich S, et al. Oligotyping reveals differences between gut microbiomes of free-ranging sympatric Namibian carnivores (Acinonyx jubatus, Canis mesomelas) on a bacterial species-like level. Front Microbiol. 2014;5:526. doi: 10.3389/fmicb.2014.00526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wasimuddin S, Menke S, Melzheimer J, Thalwitzer S, Heinrich S, Wachter B, et al. Gut microbiomes of free-ranging and captive Namibian cheetahs: Diversity, putative functions and occurrence of potential pathogens. Mol Ecol. 2017;26:5515–27. doi: 10.1111/mec.14278. [DOI] [PubMed] [Google Scholar]
- 17.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high- throughput community sequencing data. Nat Publ Gr. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2011. [Google Scholar]
- 19.Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67:1–48. doi: 10.18637/jss.v067.i01. [DOI] [Google Scholar]
- 20.Torchiano M. Efficient effect size computation [R package effsize version 0.7.1]. https://cran.r-project.org/web/packages/effsize/. Accessed 11 Oct 2017.
- 21.McMurdie PJ, Holmes S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data Watson M (ed) PLoS ONE. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faust K, Raes J. CoNet app: inference of biological association networks using Cytoscape. F1000Research. 2016;5:1519. doi: 10.12688/f1000research.9050.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiss S, Van Treuren W, Lozupone C, Faust K, Friedman J, Deng Y, et al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 2016;10:1669–81. doi: 10.1038/ismej.2015.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–21. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Konikoff T, Gophna U. Oscillospira: a central, enigmatic component of the human gut microbiota. Trends Microbiol. 2016;24:523–4. doi: 10.1016/j.tim.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Rajilić-Stojanović M, de Vos WM. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol Rev. 2014;38:996–1047. doi: 10.1111/1574-6976.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moeller AH, Shilts M, Li Y, Rudicell RS, Lonsdorf EV, Pusey AE, et al. SIV-induced instability of the chimpanzee gut microbiome. Cell Host Microbe. 2013;14:340–5. doi: 10.1016/j.chom.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao N, Li M, Luo J, Wang S, Liu S, Wang S, et al. Impacts of canine distemper virus infection on the giant panda population from the perspective of gut microbiota. Sci Rep. 2017;7:39954. doi: 10.1038/srep39954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gulraiz F, Bellinghausen C, Bruggeman CA, Stassen FR. Haemophilus influenzae increases the susceptibility and inflammatory response of airway epithelial cells to viral infections. FASEB J. 2015;29:849–58. doi: 10.1096/fj.14-254359. [DOI] [PubMed] [Google Scholar]
- 31.Mendenhall IH, Skiles MM, Neves ES, Borthwick SA, Low DHW, Liang B, et al. Influence of age and body condition on Astrovirus infection of bats in Singapore: An evolutionary and epidemiological analysis. One Health. 2017;4:27–33. doi: 10.1016/j.onehlt.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bäumler AJ, Sperandio V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature. 2016;535:85–93. doi: 10.1038/nature18849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh P, Teal TK, Marsh TL, Tiedje JM, Mosci R, Jernigan K, et al. Intestinal microbial communities associated with acute enteric infections and disease recovery. Microbiome. 2015;3:45. doi: 10.1186/s40168-015-0109-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duvallet C, Gibbons SM, Gurry T, Irizarry RA, Alm EJ. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun. 2017;8:1784. doi: 10.1038/s41467-017-01973-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lozupone CA, Li M, Campbell TB, Flores SC, Linderman D, Gebert MJ, et al. Alterations in the gut microbiota associated with HIV-1 infection. Cell Host Microbe. 2013;14:329–39. doi: 10.1016/j.chom.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marvin SA. The immune response to Astrovirus infection. Viruses. 2016;9:1. [DOI] [PMC free article] [PubMed]
- 37.Shin NR, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015;33:496–503. doi: 10.1016/j.tibtech.2015.06.011. [DOI] [PubMed] [Google Scholar]
- 38.Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol. 2013;13:790–801. doi: 10.1038/nri3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu Y, Chen J, Zheng J, Hu G, Wang J, Huang C, et al. Mucosal adherent bacterial dysbiosis in patients with colorectal adenomas. Sci Rep. 2016;6:26337. doi: 10.1038/srep26337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hernandez-Agreda A, Gates RD, Ainsworth TD. Defining the core microbiome in Corals’ microbial soup. Trends Microbiol. 2017;25:125–40. doi: 10.1016/j.tim.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 41.Hong SN, Rhee PL. Unraveling the ties between irritable bowel syndrome and intestinal microbiota. World J Gastroenterol. 2014;20:2470–81. doi: 10.3748/wjg.v20.i10.2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ivanov II, Frutos R de L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–49. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wlodarska M, Willing BP, Bravo DM, Finlay BB. Phytonutrient diet supplementation promotes beneficial Clostridia species and intestinal mucus secretion resulting in protection against enteric infection. Sci Rep. 2015;5:9253. doi: 10.1038/srep09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Antharam VC, Li EC, Ishmael A, Sharma A, Mai V, Rand KH, et al. Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J Clin Microbiol. 2013;51:2884–92. doi: 10.1128/JCM.00845-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2007;27:104–19. doi: 10.1111/j.1365-2036.2007.03562.x. [DOI] [PubMed] [Google Scholar]
- 46.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–92. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ganz HH, Doroud L, Firl AJ, Hird SM, Eisen JA, Boyce WM. Community-level differences in the microbiome of healthy wild mallards and those infected by influenza A viruses. mSystems. 2017;2:e00188–16. doi: 10.1128/mSystems.00188-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of enterobacteriaceae. Cell Host Microbe. 2007;2:119–29. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 50.Vujkovic-Cvijin I, Dunham RM, Iwai S, Maher MC, Albright RG, Broadhurst MJ, et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med. 2013;5:193ra91. doi: 10.1126/scitranslmed.3006438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, et al. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst. 2013;105:1907–11. doi: 10.1093/jnci/djt300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kimoto H, Mizumachi K, Okamoto T, Kurisaki JI. New Lactococcus strain with immunomodulatory activity: enhancement of Th1-type immune response. Microbiol Immunol. 2004;48:75–82. doi: 10.1111/j.1348-0421.2004.tb03490.x. [DOI] [PubMed] [Google Scholar]
- 53.Burcelin R. Gut microbiota and immune crosstalk in metabolic disease. Mol Metab. 2016;5:771–81. doi: 10.1016/j.molmet.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roediger WEW, Macfarlane GT. A role for intestinal mycoplasmas in the aetiology of Crohn’s disease? J Appl Microbiol. 2002;92:377–81. doi: 10.1046/j.1365-2672.2002.01531.x. [DOI] [PubMed] [Google Scholar]
- 55.Wang H, Dai W, Qiu C, Li S, Wang W, Xu J, et al. Mycoplasma pneumoniae and Streptococcus pneumoniae caused different microbial structure and correlation network in lung microbiota. J Thorac Dis. 2016;8:1316–22. doi: 10.21037/jtd.2016.04.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Borton MA, Sabag-Daigle A, Wu J, Solden LM, O’Banion BS, Daly RA, et al. Chemical and pathogen-induced inflammation disrupt the murine intestinal microbiome. Microbiome. 2017;5:47. doi: 10.1186/s40168-017-0264-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pop M, Walker AW, Paulson J, Lindsay B, Antonio M, Hossain M, et al. Diarrhea in young children from low-income countries leads to large-scale alterations in intestinal microbiota composition. Genome Biol. 2014;15:R76. doi: 10.1186/gb-2014-15-6-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gong D, Gong X, Wang L, Yu X, Dong Q. Involvement of reduced microbial diversity in Inflammatory Bowel Disease. Gastroenterol Res Pract. 2016;2016:6951091. doi: 10.1155/2016/6951091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.