

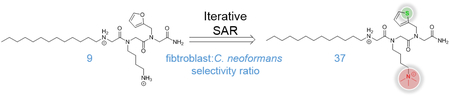
Abstract
As proteolytically stable peptidomimetics, peptoids could serve as antifungal agents to supplement a therapeutic field wrought with toxicity issues. We report the improvement of an antifungal peptoid, AEC5, through an iterative structure-activity relationship study. A sarcosine scan was used to first identify the most pharmacophorically important peptoid building blocks of AEC5, followed by sequential optimization of each building block. The optimized antifungal peptoid from this study, β−5, has improved potency towards Cryptococcus neoformans and decreased toxicity towards mammalian cells. For example, the selectivity ratio for C. neoformans over mammalian fibroblasts was improved from 8 for AEC5 to 37 for β−5.
Keywords: Peptoids, Cryptococcus neoformans, fungal infections, antifungals, structure activity relationship
Graphical Abstract
Cryptococcus neoformans is a common infectious agent among immunocompromised individuals, such as transplant patients and those infected with HIV/AIDS. Initial infection of this yeast-like fungus occurs in the pulmonary system with central nervous system infection following, leading to cryptococcal meningitis.1 The most recent estimates indicate that roughly 220,000 HIV/AIDS patients contract cryptococcal meningitis from C. neoformans each year and that over 181,000 (82%) of these cases will be fatal.2 A second species, C. gattii, which is endemic to parts of the South Pacific and has been observed as an outbreak in the Pacific Northwest regions of Canada and the United States, will infect both immunocompromised and immunocompetent individuals.3,4
Treatment for infections due to pathogenic fungi, such as Cryptococcus spp. vary significantly with the species of the microorganism and the immune status of the patient. First-line treatments, such as flucytosine, and fluconazole, as well as last-line treatments, such as amphotericin B carry their own risks, including high mammalian cytotoxicity that results in gastrointestinal complications, vomiting, QT prolongation, and hepatitis.5–7 Amplifying these side effects is the fact that most current therapeutic regimen include combination or long-term drug treatments due to increasingly observed resistance in many fungal pathogens.6,8 The dearth of suitable treatments necessitates new therapeutic options for patients dealing with fungal infections.
Many organisms utilize antimicrobial peptides (AMPs) as a part of their innate immune response against pathogenic bacteria and fungi.9 AMPs are advantageous given their relatively high specificity for microorganisms over mammalian cells and lack of observed drug resistance, likely stemming from their generally accepted mode of action. Most AMPs used against fungi target the cell membrane, forming pores or causing changes in cell permeability that result in leakage of cytoplasmic components, ultimately resulting in pathogen death.9 These AMPs are able to take advantage of key differences between mammalian and fungal cell membranes to selectively target the pathogen, such as ergosterol, a principle sterol only present in fungal cell membranes.7 Regrettably, peptides are readily recognized and degraded by proteases, thus serving as poor clinical therapeutics for combating in vivo fungal infections.10,11
One solution to the proteolytic instability of AMPs are N-substituted glycines, often referred to as peptoids. In peptides the side chain R group is attached to the α-carbon, whereas in peptoids the R group is attached to the amide nitrogen, making peptoids unrecognizable by proteases. This modification maintains many of the advantageous properties of peptides while significantly reducing the proteolytic susceptibility that results in short peptide half-lives in vivo.12 The antimicrobial utility of peptoids has been demonstrated against both bacteria13–22 and fungi.23–25 By the use of a high-throughput screening assay, we have recently identified a peptoid termed AEC5 with potency against C. neoformans similar to that of first-line clinical antifungals.23 This compound demonstrated relatively minimal toxicity against several mammalian cell types and excellent in vitro proteolytic stability. Herein we report the improvement of AEC5 through sequential optimization of each peptoid submonomer by an iterative structure activity relationship (SAR) process.
AEC5 Sarcosine Scan.
We first sought to determine the pharmacological role of each of AEC5’s three submonomers on C. neoformans potency and mammalian cell cytotoxicity. The structure of AEC5 comprises three unique submonomers, each with a different chemical moiety (Figure 1A). N-terminally is a long hydrophobic tail, followed by a cationic ammonium side chain, and a C-terminal aromatic heterocycle. A sarcosine scan of AEC5 was completed, similar to an alanine scan of peptides, by synthesizing three AEC5 derivatives each with sarcosine in place of one of the submonomers (Figure 1B). All compounds were synthesized on the solid phase using peptoid submonomer26 and traditional Fmoc techniques.27 Following synthesis, compounds were cleaved from the resin using trifluoroacetic acid (TFA) and purified by RP-HPLC. The minimum inhibitory concentration (MIC) against C. neoformans and the cytotoxicity against HepG2 cells was then determined for these compounds, termed AEC5sar1–3, as described previously (Figure 1C).23 The MIC of known antifungal agents fluconazole and amphotericin B was also determined to confirm the susceptibility of this strain of C. neoformans (Figure S3). Substituting sarcosine into any of the three positions had deleterious effects on antifungal potency but mixed effects of mammalian cytotoxicity. The largest change was observed for AEC5sar1 where both potency and toxicity fell above the highest concentrations tested, 400 μg/mL and 800 μg/mL, respectively. This is not surprising given the previously reported importance of the alkyl chain in similar compounds.13,22 Substituting sarcosine in place of the cationic amino group of position two caused a modest 2-fold decrease in antifungal potency compared to AEC5, but concomitantly increased the mammalian cell toxicity (TD50) by 2-fold, from 56.2 μg/mL for AEC5 to 21.1 μg/mL for AEC5sar2. Sarcosine substitution at position two results in a substantial change in the calculated distribution coefficient, cLogD7.4, from −1.18 for AEC5 to 1.44 for AEC5sar2. This increase in hydrophobicity is the likely cause of increased toxicity for AEC5sar2 and stresses the importance of this submonomer in mitigating mammalian toxicity. Interestingly, sarcosine substitution in place of an aromatic heterocycle at position three results in a substantial 8-fold change in antifungal potency with little effect on mammalian toxicity compared to AEC5. The mechanism of action of AEC5 is still being explored, and although we have hypothesized that this compound works in a similar manner to other antifungal peptides and peptoids, through membrane disruption, the importance of this aromatic heterocycle in compound potency suggests that AEC5 may kill C. neoformans through a more complex mechanism of action. These data indicate that the alkyl tail is the most important pharmacophoric moiety in AEC5, and we subsequently sought to optimize this group during round one of our SAR studies.
Figure 1.

(A.) The structure of the antifungal tripeptoid AEC5, showing distinct functionalities at each of the three submonomer positions. (B.) Compound structures for the sarcosine scan of AEC5, replacing the submonomer at each position with sarcosine. Calculated distribution coefficient (cLogD7.4), C. neoformans antifungal potency (MIC), and HepG2 liver cell toxicity (TD50) for AEC5 sarcosine scan compounds. MW = molecular weight; cLogD7.4 = calculated distribution coefficient at pH 7.4; MIC = minimum inhibitory concentration; TD50 = toxic dose 50%; SR = selectivity ratio (TD50/MIC); ND = not determined.
Round 1 SAR.
Several studies have demonstrated the importance of alkyl tail length on potency and toxicity of lipophilic antimicrobial peptoids.13,22 AEC5 derivatives with sixteen, ten, and eight carbon alky tails in position 1, termed α−1, α−2, and α−3, respectively, were synthesized using peptoid submonomer methods (Figure 2A). Given the polyene nature of the potent antifungal amphotericin B, we hypothesized that the inclusion of polyene moieties in this position might improve antifungal potency. The terpene aldehydes farnesal and citral were therefore attached to an N-terminal glycine in position one via reductive amination to afford compounds α−4 and α−5, respectively. The C. neoformans MIC of these compounds was evaluated (Figure 2B), however, significant decreases in potency were observed for compounds α−2 through α−5. This decrease in response to shortening the alkyl tail (compounds α−2 and α−3) was not surprising given literature precedent.13,22 Although increasing the length of the alkyl tail from thirteen to sixteen yielded a compound with a 2-fold improvement in C. neoformans potency, it unsurprisingly resulted in significantly increased toxicity against HepG2 liver cells. Neither polyene tail resulted in improved potency, however, the farnesyl tail did show a large decrease in toxicity (>800 μg/mL) while possessing a similar cLogD7.4, albeit with a 16-fold decrease in antifungal potency. We thought that the significant decrease in toxicity was important and attempted to recover antifungal potency through modification of the aromatic heterocycle in position three during round two of SAR. This resulted in compounds γ−8, γ−9, and γ−10, none of which showed improved potency over α−4 or AEC5 (Figure S4). The use of the farnesyl tail was ultimately abandoned, and we concluded the optimal alkyl tail in position one was the original tridecyl tail from AEC5.
Figure 2.

(A.) Round 1 SAR compounds with varied hydrophobic tails in the first position. (B.) Calculated distribution coefficient (cLogD7.4), C. neoformans antifungal potency (MIC), and HepG2 liver cell toxicity (TD50) for round 1 SAR compounds. MW = molecular weight; cLogD7.4 = calculated distribution coefficient at pH 7.4; MIC = minimum inhibitory concentration; TD50 = toxic dose 50%; SR = selectivity ratio (TD50/MIC); ND = not determined.
Round 2 SAR.
Following the alkyl tail in position 1, the furan in position 3 appeared to have the second largest effect on antifungal potency from the sarcosine scan. AEC5 derivatives containing a variety of aromatic heterocycles in this position were synthesized by peptoid submonomer methods. These included imidazole derivatized compounds γ−1 and γ−3, compound γ−2 containing a thiophene side chain, γ−4 with an indole in this position, and γ−5 displaying a pyridyl ring (Figure 3A). It is important to note that γ−5 required altered synthetic parameters published by Zuckerman et al. to negate the unwanted alkylation reaction at the pyridyl ring.28 This was accomplished using chloroacetic acid in place of bromoacetic acid and extending the length of amine coupling to account for the decreased leaving group ability of chlorine over bromine. Derivatives containing an aromatic phenyl side chain with no heteroatom (γ−6) and a non-aromatic heterocyclic tetrahydrofuran (γ−7) were also synthesized to investigate the role of these individual properties.
Figure 3.

(A.) Round 2 SAR compounds utilizing the tridecyl tail from Round 1 with varied heterocyclic side chains in the third position. (B.) Calculated distribution coefficient (cLogD7.4), C. neoformans antifungal potency (MIC), and HepG2 liver cell toxicity (TD50) for round 2 SAR compounds. MW = molecular weight; cLogD7.4 = calculated distribution coefficient at pH 7.4; MIC = minimum inhibitory concentration; TD50 = toxic dose 50%; SR = selectivity ratio (TD50/MIC).
The C. neoformans potency and HepG2 toxicity of these compounds was determined (Figure 3B). Derivatives γ−1, γ−3, γ−5 all showed 4- to 5-fold decreases in antifungal potency as well as a 2- to 4-fold decrease in mammalian cell toxicity. It is interesting to note that this decrease in potency and toxicity accompanies a decrease in cLogD7.4, supporting our earlier findings that overall lipophilicity of short antimicrobial peptoids is linked to mammalian toxicity.22 The substitution with a non-aromatic heterocycle (γ−7) gave a modest 2-fold decrease in antifungal potency and a 3-fold decrease in mammalian cell toxicity. Interestingly, placement of an aromatic phenyl ring with no heteroatom (γ−6) resulted in a 2-fold improvement in antifungal potency and no significant change in mammalian cell toxicity. A 2-fold improvement in antifungal potency was observed with three of the round 2 derivatives; γ−2 with a thiophene side chain, γ−4 with an indole side chain, and γ−6, as mentioned, with a phenyl side chain. Two of these, γ−4 and γ−6 exhibited increased or unchanged mammalian cell toxicity compared to AEC5. However, γ−2 showed a decrease in HepG2 toxicity with a TD50 of 79.3 μg/mL compared to 56.2 μg/mL for AEC5. With improvement in both potency and toxicity, a selectivity ratio of 25 was calculated for γ−2, compared to 9 for AEC5. A higher selectivity ratio, defined as the TD50 divided by the MIC, is indicative of a compound that is more selective for pathogen over mammalian cells. Data from round 2 of SAR indicated that replacing the furan in position three with a thiophene greatly improved pathogen selectivity by increasing antifungal potency and decreasing mammalian toxicity. Therefore, the thiophene moiety was maintained in this position through round 3 of SAR antifungal optimization.
Round 3 SAR.
The final submonomer to optimize was the amino cation in position 2. Derivatives of γ−2 were synthesized containing various chemical moieties in position two. These include compounds with ammonium groups similar to γ−2 attached via fewer methylene units (β−1 and β−2), or more methylene units (β−3), as well as an arginine mimic (β−4), a trimethylammonium lysine mimic (β−5), and a non-cationic hydroxyl group (β−6) (Figure 4A). This last derivative was synthesized and characterized to evaluate the efficacy of a hydrogen bonding moiety without cationic nature in position two. The MIC of β−6 falls off 4-fold compared to γ−2 while the toxicity against HepG2 cells becomes more than 2-fold worse (Figure 4B). These data, combined with data from AECsar2, highlight the role of the cation in position two in improving C. neoformans potency, and perhaps more importantly, negating compound toxicity against mammalian cells. Bolt et al. recently showed that the chain length between peptoid backbone and amino cation affects mammalian cytotoxicity and antibacterial activity, although the nature of this relationship varied between organisms.29 Modifying the side-chain length of γ−2 had little effect on antifungal activity, with only β−2, possessing the two carbon linker, showing a 2-fold decrease in potency. However, shortening the side-chain length was detrimental to mammalian cytotoxicity with β−1 (3 carbon linker) and β−2 having HepG2 cytotoxicity of 47.3 and 40.5 μg/mL, respectively. Similarly, lengthening the side-chain to six carbons (β−3) increased toxicity moderately to 56.2 μg/mL.
Figure 4.

Synthesis of γ−2 derivatives with unique side chain modifications.
An interesting correlation between side-chain length and cLogD7.4 was observed in the synthesized peptoids which was extrapolated out using distribution coefficients and pKa values calculated in ChemAxon’s MarvinSketch (Figure S5).30 β−3 had the highest distribution coefficient, as expected given the increased number of methylene units. As this side-chain is shortened to five, four (γ−2), or three methylenes (β−1), the distribution coefficient decreases, as anticipated. However, as this linker is shortened further to two methylenes (β−2) or further still to hypothetical compounds containing one or zero methylene units, the distribution coefficient rises sharply, indicating that these compounds are becoming more hydrophobic, even though they possess fewer carbon/hydrogen groups. Given that the calculated distribution coefficient factors in compound ionization, an explanation for this trend can be obtained by observing the calculated pKa values for the amino cation. These values remain relatively constant around a pKa of 10 for compounds possessing side-chain linker lengths of 3–6 carbons. However, as the linker length becomes shorter than three, the calculated pKa begins to drop, meaning that on average this side chain amino group becomes less ionized at neutral pH. Noting our observations regarding increased mammalian cytotoxicity with complete removal of the cation in position 2, we hypothesize that this loss of ionization with shorter side-chain length makes peptoids slightly less potent against C. neoformans and significantly more cytotoxic to mammalian cells. This is further confirmed with the trimethylated γ−2 derivative, β−5, which is permanently ionized. Synthesis of this compound was achieved using standard submonomer methods to install a 4-monomethoxytrityl (Mmt) protected diamine in position 2, followed by Boc protection of the N-terminus, orthogonal Mmt removal with 1% TFA, and methylation using methyl iodide (Figure 5). Cleavage from the resin and removal of the Boc group using 95% TFA yielded only trimethylated β−5 with no mono or dimethylated compound observed. β−5 exhibited no change in MIC but showed further improvement to mammalian cytotoxicity with a TD50 of 91.2 μg/mL.
Figure 5.

(A.) Round 3 SAR compounds utilizing the tridecyl tail from Round 1 and the thiophene aromatic heterocyle from Round 2 with varied cationic moieties in position 2. (B.) Calculated distribution coefficient (cLogD7.4), C. neoformans antifungal potency (MIC), and HepG2 liver cell toxicity (TD50) for round 3 SAR compounds. MW = molecular weight; cLogD7.4 = calculated distribution coefficient at pH 7.4; MIC = minimum inhibitory concentration; TD50 = toxic dose 50%; SR = selectivity ratio (TD50/MIC).
The last derivative explored in this round of SAR possessed the cationic gaunidinium group of an arginine mimic (β−4). This compound was synthesized in a similar manner to β−5, but with guanidinylation using pyrazole carboxamidine after Mmt deprotection instead of methylation (Figure 5). Previous literature has shown that guanidinium containing peptoids have increased mammalian cytotoxicity, as well as improved antibacterial properties,29 however, the effect of guanidinium moieties on antifungal potency has not been explored. We observed that substituting an arginine mimic in place of the lysine mimic in position 2 of γ−2 resulted in increased HepG2 toxicity as expected, but unexpectedly decreased antifungal potency 4-fold. Round 3 ultimately identified β−5 as the most promising antifungal peptoid from this study.
Broad Toxicity Evaluation.
One of the most significant shortcomings of current antifungals is their broad toxicity. We therefore sought to evaluate the broad toxicity of the most promising antifungal peptoids developed through SAR against NIH/3T3 mouse fibroblasts and human erythrocytes, in addition the HepG2 testing already completed (Table 1). As seen with liver cells, both γ−2 and β−5 exhibited decreased toxicity towards NIH/3T3 mouse fibroblasts compared to AEC5, with selectivity ratios improving from 8 for AEC5 to 21 and 37 for γ−2 and β−5, respectively. Interestingly, both γ−2 and β−5 had slightly higher levels of hemolysis compared to AEC5. We note that the HC10 value reported here for AEC5 differs from that published previously. Given that human erythrocytes are primary and originate from different donors, we reevaluated AEC5 along with γ−2 and β−5 to provide the most accurate hemolytic data. Maximum hemolysis at 100 μg/mL (Hmax) further confirmed that the AEC5 derivatives tested here are more hemolytic than the original peptoid. Although slightly increased hemolytic properties are a drawback for γ−2 and β−5, these compounds nonetheless maintain improved selectivity ratios for human erythrocytes compared to AEC5 due to their improved potency against C. neoformans.
Table 1.
| Cell Type | HepG2 | NIH/3T3 | hRBC | hRBC |
|---|---|---|---|---|
| Compound | TD50 (SR) | TD50 (SR) | HC10 (SR) | Hmax (100 μg/mL) |
| AEC5 | 56.2 ± 18.0 (9) | 48.6 ± 3.3 (8) | 56.6 ± 2.4 (9) | 30.4 ± 3.4% |
| γ-2 | 79.3 ± 0.35 (25) | 65.9 ± 3.3 (21) | 37.2 ± 0.83 (12) | 48.6 ± 8.6% |
| β-5 | 91.2 ± 4.7 (29) | 114.6 ± 18.7 (37) | 42.6 ± 1.6 (14) | 46.0 ± 9.4% |
TD50 = toxic dose 50%; HC10 = hemolysis concentration 10%; Hmax = percent hemolysis at 100 μg/mL; SR = selectivity ratio (TD50 or HC10/MIC); all concentrations in μg/mL
In conclusion, we report the iterative improvement of a previously identified antifungal lead compound through sequential optimization of each peptoid submonomer. This work identified two beneficial subtle modifications to AEC5. The first being a thiophene aromatic heterocycle in position 3 in place of a furan, which improved both potency and selectivity through unknown mechanisms. The second being trimethylation of the amino side-chain in position two, locking this nitrogen into a cationic state, and thereby further ameliorating mammalian cell toxicity. Current efforts are focused on elucidating the mechanism of action of these short antifungal peptoids and investigating their in vivo therapeutic properties.
Supplementary Material
BMCL-D-18–00964 Highlights.
An iterative SAR was carried out on the antifungal peptoid AEC5.
Subtle modifications improved both antifungal potency and selectivity.
β−5 shows promise to contribute to the challenging field of antifungals.
Acknowledgments
We thank Dr. Erin McClelland in the Department of Biology at MTSU for her continued assistance in working with C. neoformans. This work was supported by funds provided by MTSU and by the National Institutes of Health AI112861.
Abbreviations
- AMP
antimicrobial peptide
- MIC
minimum inhibitory concentration
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information
Synthetic methods, characterization assay details, NMR analysis of small molecules and key peptoids, as well as supplemental figures, can be found in the supporting information.
The authors declare no competing financial interests.
References
- (1).Movahed E; Munusamy K; Tan GMY; Looi CY; Tay ST; Wong WF PLoS One 2015, 10 (9), e0137457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Rajasingham R; Smith RM; Park BJ; Jarvis JN; Govender NP; Chiller TM; Denning DW; Loyse A; Boulware DR Lancet Infect. Dis 2017, 17 (8), 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Smith RM; Mba-Jonas A; Tourdjman M; Schimek T; DeBess E; Marsden-Haug N; Harris JR PLoS One 2014, 9 (2), e88875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Datta K; Bartlett KH; Baer R; Byrnes E; Galanis E; Heitman J; Hoang L; Leslie MJ; MacDougall L; Magill SS; Morshed MG; Marr KA Emerg. Infect. Dis 2009, 15 (8), 1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pappas PG; Kauffman CA; Andes D; Benjamin DK; Calandra TF; Edwards JE; Filler SG; Fisher JF; Kullberg B-J; Zeichner LO; Reboli AC; Rex JH; Walsh TJ; Sobe JD Clin. Infect. Dis 2009, 48 (5), 503–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Perfect JR; Dismukes WE; Dromer F; Goldman DL; Graybill JR; Hamill RJ; Harrison TS; Larsen RA; Lortholary O; Nguyen M-H; Pappas PG; Powderly WG; Singh N; Sobel JD; Sorrell TC Clin. Infect. Dis 2010, 50 (3), 291–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Walsh TJ; Anaissie EJ; Denning DW; Herbrecht R; Kontoyiannis DP; Marr KA; Morrison VA; Segal BH; Steinbach WJ; Stevens DA; van Burik J-A; Wingard JR; Patterson TF Clin. Infect. Dis 2008, 46 (3), 327–360. [DOI] [PubMed] [Google Scholar]
- (8).Hanson KE; Alexander BD; Perfect J In Antimicrobial Drug Resistance: Clinical and Epidemiological Aspects; Mayers DL, Ed.; Humana Press: Totowa, NJ, 2009; pp 967–985. [Google Scholar]
- (9).Zasloff M Nature 2002, 415 (6870), 389–395. [DOI] [PubMed] [Google Scholar]
- (10).Latham PW Nat. Biotech 1999, 17 (8), 755–757. [DOI] [PubMed] [Google Scholar]
- (11).Zhang L; Falla TJ Expert Opin. Pharmacother 2006, 7 (6), 653–663. [DOI] [PubMed] [Google Scholar]
- (12).Miller SM; Simon RJ; Ng S; Zuckermann RN; Kerr JM; Moos WH Bioorg. Med. Chem. Lett 1994, 4 (22), 2657–2662. [Google Scholar]
- (13).Chongsiriwatana NP; Miller TM; Wetzler M; Vakulenko S; Karlsson AJ; Palecek SP; Mobashery S; Barron AE Antimicrob. Agents Chemother 2011, 55 (1), 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chongsiriwatana NP; Patch JA; Czyzewski AM; Dohm MT; Ivankin A; Gidalevitz D; Zuckermann RN; Barron AE Proc Natl Acad Sci U S A 2008, 105 (8), 2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chongsiriwatana NP; Wetzler M; Barron AE Antimicrob. Agents Chemother 2011, 55 (11), 5399–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kapoor R; Eimerman PR; Hardy JW; Cirillo JD; Contag CH; Barron AE Antimicrob. Agents Chemother 2011, 55 (6), 3058–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kapoor R; Wadman MW; Dohm MT; Czyzewski AM; Spormann AM; Barron AE Antimicrob. Agents Chemother 2011, 55 (6), 3054–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Patch JA; Barron AE J. Am. Chem. Soc 2003, 125 (40), 12092–12093. [DOI] [PubMed] [Google Scholar]
- (19).Lee J; Kang D; Choi J; Huang W; Wadman M; Barron AE; Seo J Bioorg. Med. Chem. Lett 2017, 28 (2), 170–173. [DOI] [PubMed] [Google Scholar]
- (20).Mojsoska B; Zuckermann RN; Jenssen H Antimicrob. Agents Chemother 2015, 59 (7), 4112–4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Fisher KJ; Turkett JA; Corson AE; Bicker KL ACS Comb. Sci 2016, 18 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Turkett JA; Bicker KL ACS Comb. Sci 2017, 19 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Corson AE; Armstrong SA; Wright ME; McClelland EE; Bicker KL ACS Med. Chem. Lett 2016, 7 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Galetti MD; Cirigliano AM; Cabrera GM; Ramírez JA Mol. Divers 2012, 16 (1), 113–119. [DOI] [PubMed] [Google Scholar]
- (25).Luo Y; Bolt HL; Eggimann GA; McAuley DF; McMullan R; Curran T; Zhou M; Jahoda PCAB; Cobb SL; Lundy FT ChemBioChem 2017, 18 (1), 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zuckermann RN; Kerr JM; Kent SBH; Moos WH J. Am. Chem. Soc 1992, 114 (26), 10646–10647. [Google Scholar]
- (27).Amblard M; Fehrentz J-A; Martinez J; Subra G Mol. Biotechnol 2006, 33 (3), 239–254. [DOI] [PubMed] [Google Scholar]
- (28).Burkoth TS; Fafarman AT; Charych DH; Connolly MD; Zuckermann RN J. Am. Chem. Soc 2003, 125 (29), 8841–8845. [DOI] [PubMed] [Google Scholar]
- (29).Bolt HL; Eggimann GA; Jahoda CAB; Zuckermann RN; Sharples GJ; Cobb SL Medchemcomm 2017, 8 (5), 886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).ChemAxon (http://www.chemaxon.com) 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.