

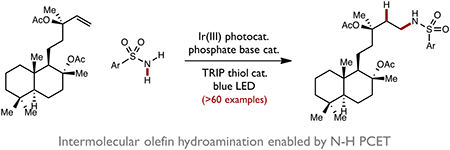
Abstract
Here we report a catalytic method for the intermolecular anti-Markovnikov hydroamination of unactivated alkenes using primary and secondary sulfonamides. These reactions occur at room temperature under visible light irradiation and are jointly catalyzed by an iridium(III) photocatalyst, a dialkyl phosphate base, and a thiol hydrogen atom donor. Reaction outcomes are consistent with the intermediacy of a N-centered sulfonamidyl radical generated via proton-coupled electron transfer (PCET) activation of the sulfonamide N–H bond. Studies outlining the synthetic scope (>60 examples) and mechanistic features of the reaction are presented.
Graphical Abstract
Introduction
Olefin hydroamination is an ideal method for the synthesis of aliphatic amines, combining alkenes and simple N–H functional groups in a direct and atom-economical fashion.1 While appealing in principle, these transformations are often challenging in practice: general methods for the intermolecular hydroamination of unactivated alkenes are rare2 and even fewer provide access to the anti-Markovnikov series of addition products.3 New olefin amination technologies that address these limitations have the potential to create a significant synthetic benefit.4
To this end, our group recently reported a photo-driven olefin amination method based on the proton-coupled electron transfer (PCET) activation of anilide N–H bonds.5 In this work, an excited state redox catalyst and a weak phosphate base jointly mediate the concerted homolytic activation of the strong N–H bonds of N-aryl amide derivatives under visible light irradiation to afford a transient amidyl radical. While cyclizations of these reactive N-centered radicals onto pendant alkenes were highly efficient, efforts to extend this protocol to intermolecular C–N bond formation proved unsuccessful (Figure 1a). This lack of reactivity likely stems from the comparatively high stability of N-aryl amidyls, which enables charge recombination between the reduced photocatalyst and the N-radical to occur at rates faster than bimolecular olefin addition. To overcome this limitation, we sought to develop PCET activations of alternative N–H functional groups where the resulting N-radical intermediate would undergo intermolecular olefin addition at rates competitive with back electron transfer. In particular, we focused on the PCET activation of sulfonamide N–H bonds (Figure 1b). In a recent report describing a directed C–H alkylation method,6 we demonstrated that a N-radical species derived from the PCET activation of a 2° sulfonamide could activate distal aliphatic C–H bonds via 1,5–hydrogen atom transfer (HAT). As sulfonamide-derived radicals are known to undergo olefin addition with high levels of anti-Markovnikov regioselectivity,7 we reasoned that this approach might serve as the basis for a new regioselective hydroamination method.
Figure 1.

(a) PCET-mediated intramolecular hydroamidation with N-aryl amides (b) PCET-mediated intermolecular hydroamination with sulfonamides
To put this approach in context, it is important to note that several intermolecular sulfonamide-based aminations of unactivated olefins have been reported in recent years involving transition metal complexes and Brønsted acids (Figure 2).8 However, these methods typically exhibit high levels of Markovnikov regioselectivity in the C–N bond-forming step. With respect to intermolecular anti-Markovnikov couplings, Nicewicz has reported the lone example - a novel photocatalytic method for the intermolecular hydroamination of electron-rich alkenes with sulfonamides and a variety of azoles.3b In these reactions, a highly oxidizing excited state organic photocatalyst converts an alkene substrate to its corresponding radical cation. These electrophilic intermediates can then react with nucleophilic amine donors in the key C–N bond-forming event.9 While powerful, these methods are limited by the thermodynamic challenges associated with one-electron oxidation of terminal and disubstituted olefins, and intermolecular variants of these aminations are currently limited to the reactions of styrenes and trisubstituted alkyl olefins. With respect to other examples of anti-Markovnikov hydroamination catalysis, Hull has recently reported an elegant method for the directed addition aliphatic of amines to simple alkenes.3c,10 Also, numerous anti-Markovnikov aminations of styrenes have been reported, including seminal early reports from Beller and Hartwig.1d,11,12
Figure 2.

Prior work in intermolecular olefin hydroamination with sulfonamides.
Results and Discussion
We envisioned a prospective catalytic cycle for a PCET-based olefin amination as shown in Figure 3. In analogy to our previous work,5 we anticipated that the phosphate base would first form a hydrogen bonded complex with the sulfonamide N–H bond. The resulting non-covalent adduct would participate in a concerted PCET event with the excited state of the iridium photocatalyst [Ir(dF(CF3)ppy)2(5,5’-dCF3bpy)]PF6 (A), resulting in formal homolysis of the N–H bond and formation of the key sulfonamidyl radical intermediate. When considered as a formal hydrogen atom acceptor, the oxidant/base catalyst pair has an effective bond strength of 103 kcal/mol.6, 13 As such, the homolytic activation of the strong N–H bonds in both primary and secondary sulfonamides (N–H BDFEs ~105 & ~97 kcal/mol, respectively)14 are feasible on thermochemical grounds. Next, in accord with literature precedent, we expected the N-radical species to undergo anti-Markovnikov addition to an olefin partner to furnish a new C–N bond and a vicinal carbon-centered radical. This alkyl radical can then be reduced by the thiol co-catalyst via HAT, and the resulting thiyl radical can undergo single-electron reduction (E[ArS•/ArS−] = −0.22 V vs. Fc+/Fc)15 to the corresponding thiolate by the reduced Ir(II) state of the photocatalyst (E[IrIII/IrII] = −1.07 V vs. Fc+/Fc). Favorable proton transfer between the thiolate (pKa[PhSH] ≈ 21 in MeCN)16 and the phosphoric acid (pKa ≈ 12 in MeCN)17 should follow, returning the active forms of all three catalysts.
Figure 3.

Prospective catalytic cycle
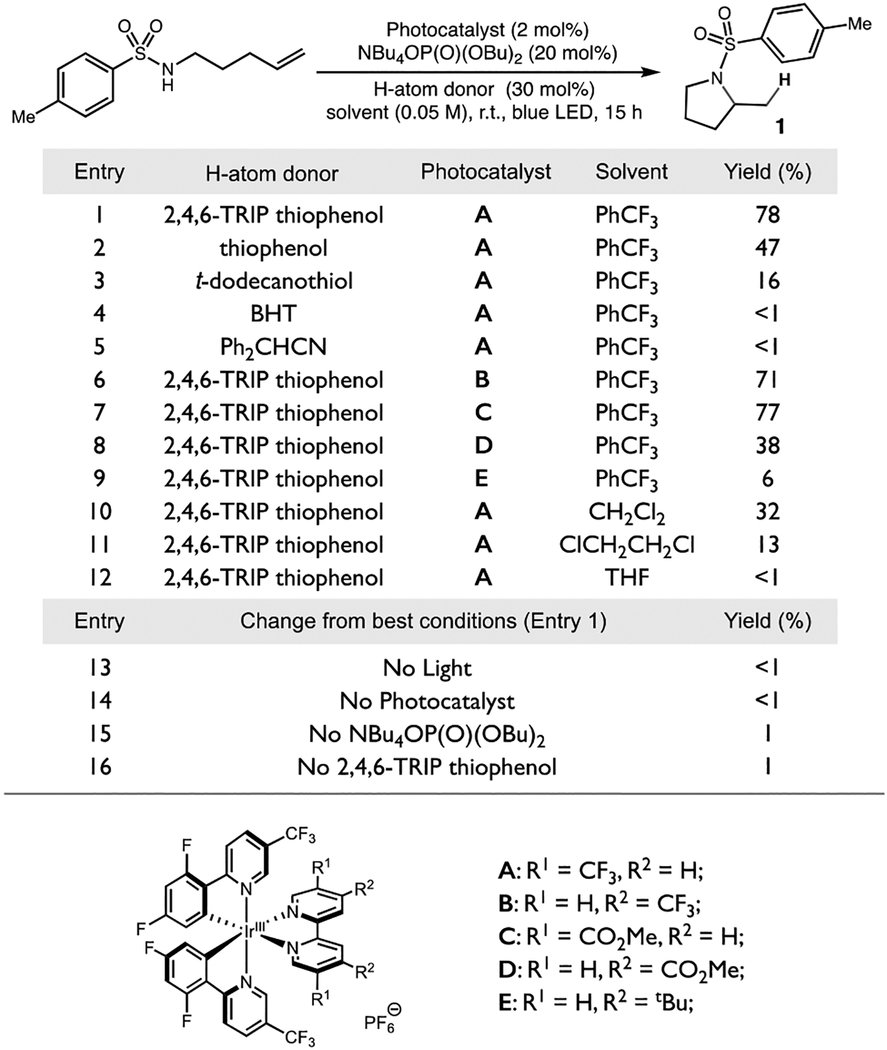
Notably, success in the proposed scheme requires that PCET activation of the substrate N–H bond occurs in the presence of the aryl thiol - a known substrate class for multi-site PCET18 that exhibits a much weaker S–H bond (S–H BDFE ~ 79 kcal/mol).19, 18b Moreover, the thiol H-atom donor must not preemptively reduce the highly reactive and electrophilic sulfonamidyl radical intermediates, which are known to be potent hydrogen atom abstractors.20 While seemingly problematic, recent PCET methods for olefin amination5a and alcohol β-scission21 have demonstrated these surprising selectivities and implied the feasibility of the proposed transformation. Accordingly, we were pleased to find that under conditions similar to our C–H abstraction work,6 treatment of model terminal olefin substrate 4-methyl–N-(pent-4-en-1-yl) benzenesulfonamide with 2 mol % of Ir photocatalyst A, 20 mol % of tetrabutyl ammonium dibutylphosphate base, and 30 mol % of 2,4,6-triisopropyl-thiophenol (TRIP thiol) provided the desired hydroamination product 1 in 78% GC yield following irradiation with blue LEDs in trifluorotoluene at room temperature (Table 1, entry 1). We next explored the sensitivity of these reactions to various changes in the standard reaction conditions. Other hydrogen atom donors were moderately successful, but uniformly less effective than TRIP thiol (entries 2–5). Similarly, a number of structurally similar iridium photocatalysts to A were also effective in these reactions (entries 6, 7), but the reaction yields diminished as the potential of the excited state species decreased (entries 8, 9). Also, the reaction is moderately successful in dichloromethane (entry 10), but considerably less so in other solvents (entries 11, 12). Notably, control reactions of 1 lacking the phosphate base, photocatalyst A, light, or TRIP thiol were uniformly unsuccessful (entries 13–16).
Table 1.
Reaction sensitivity screen. Reactions were run on 0.05 mmol scale and yields are determined by GC analysis relative to an internal standard.
|
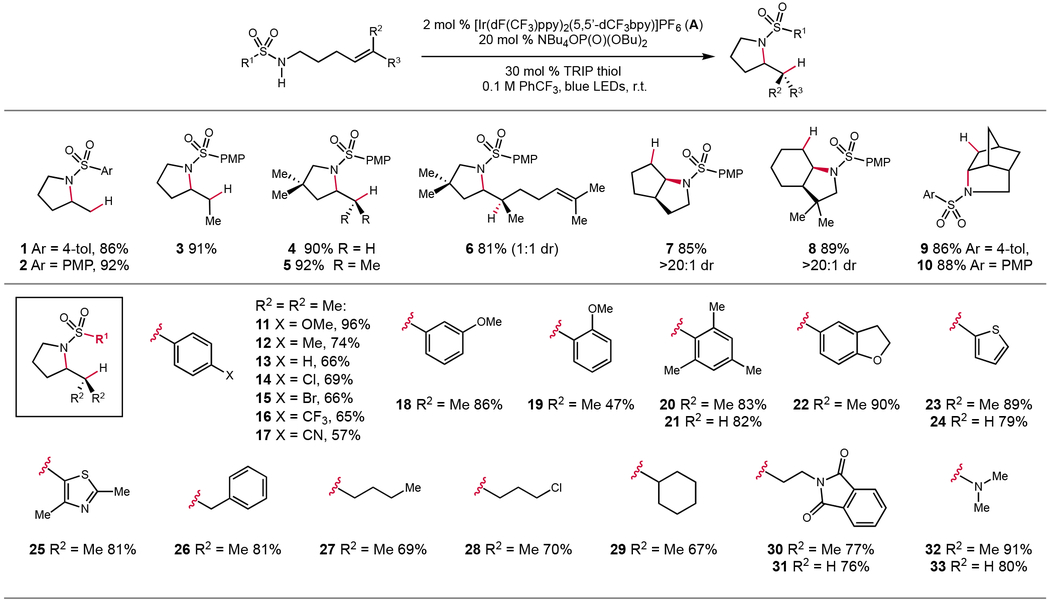
In the intramolecular version of this reaction we found that a number of olefin substitution patterns were accommodated, including terminal (1, 2 and 4), 1,2-disubstituted (3), and trisubstituted olefins (5) (Table 2). For a substrate containing two electronically similar alkenes, only the double bond proximal to the nitrogen was aminated (6). Various bicyclic and bridged ring systems can also be accessed using this method with high levels of diastereoselectivity (7–10). We also found these conditions were successful for aminations with a wide range of aryl and alkyl substituted sulfonamides. A para-methoxy phenyl substituted sulfonamide was found to cyclize in 96% yield (11). Similarly, tosyl- and phenylsulfonamide bearing substrates afforded the desired amination products 12 and 13 in 74% and 66% yield, respectively. Halogenated and more electron-deficient sulfonamides also afford the corresponding products 14–17 in more moderate yields. Aminations with substrates bearing meta- (18) and ortho- (19) substituted methoxyphenyl groups also proceed smoothly under the standard conditions, as did cyclization of a hindered mesityl derivative (20, 21). Heterocyclic sulfonamides were also tolerated in this reaction, furnishing products 22–25. Alkyl-substituted sulfonamides were also successful substrates, affording the desired cyclization products 26–31 in good yields. We also observed that both a primary alkyl chloride (28) and phthalimide (30, 31) were tolerated under these photoredox conditions. In addition, two different dimethylaminosulfamate substrates delivered hydroamination products 32 and 33 in 91% and 80% yield, respectively. Notably, in this series of substrates, several sterically and electronically distinct sulfonamides were shown to react comparably well with both the trisubstituted model olefin and a much less reactive terminal alkene (21, 24, 31, and 33), suggesting that more reactive olefins are not required for efficient intramolecular cyclizations.
Table 2.
Scope of the intramolecular amination. Yields are for isolated material following chromatography on silica gel and are the average of two experiments. Reactions conducted on 0.5 mmol scale.
|
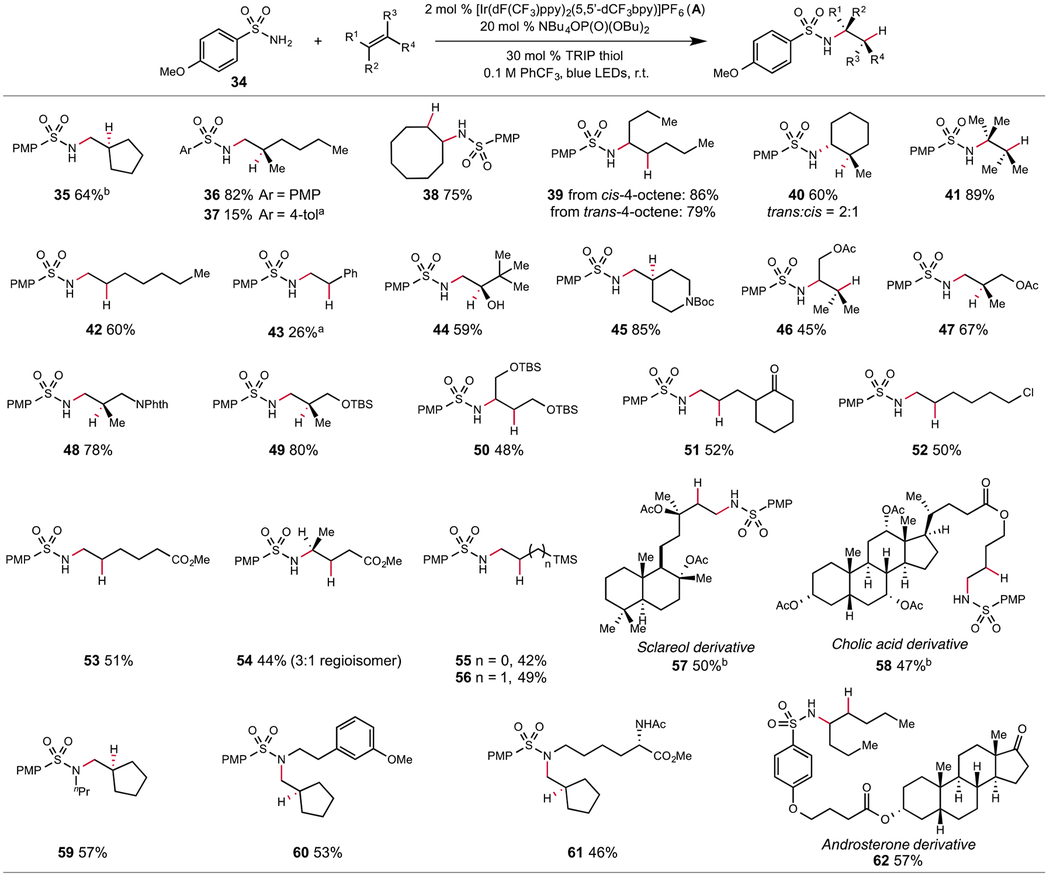
Next, we examined the viability of more difficult intermolecular alkene aminations (Table 3). We were pleased to find that numerous alkenes are effective reaction partners for the N-centered radical generated from sulfonamide 34. A wide range of olefin substitution patterns are tolerated, including 1,1-disubstituted (35–37), 1,2-disubstituted (38, 39), tri- and tetrasubstituted (40, 41), and even terminal alkenes (42). Both cyclic and acyclic alkene partners can be employed, and the reaction is not sensitive to alkene configuration, as both cis- and trans-4-octene afford the same product with similar efficiency (39). As sulfonamidyl radicals are electrophilic, they are expected to react most readily with nucleophilic olefin partners. Accordingly, we found that a silyl enol ether can be efficiently aminated to afford vicinal amino alcohol product 44 following desilylation. In terms of functional group tolerance, carbamates (45), acetates (46, 47), phthalimides (48), silyl ethers (49, 50), ketones (51), primary alkyl chlorides (52), and methyl esters (53) are all tolerated under the standard conditions. In addition, an unsymmetrical but electronically biased 1,2 disubstituted alkene could be hydroaminated to provide 54 as a 3:1 mixture of regioisomers. The major isomer exhibits connectivity consistent with attack of the electrophilic N-radical on the more electron rich carbon of the alkene. We also found that both vinyl and allyl silanes could hydroaminated, though with modest reaction efficiencies (55, 56). Several more complex substrates, including natural product and pharmaceutical derivatives, were also aminated successfully (57, 58). In this study, we generally found that the 2° sulfonamide products resulting from hydroamination with 34 do not participate in a second olefin amination step to furnish 3° sulfonamide products. This selectivity for mono-alkylation likely results from the increased stability of the 2° N-radical species formed upon PCET activation, which should decrease the rate of olefin addition relative to that of charge recombination. However, we observed that with more nucleophilic olefin partners, such as methylenecyclopentane, secondary N-alkyl sulfonamides could also be aminated effectively (59, 60). We also observed that a lysine-based amino acid derivative (61) and a steroid conjugate (62) could be successfully alkylated. Notably, in 61 the secondary sulfonamide was selectively alkylated in the presence of a secondary amide N–H bond, a potentially competitive site for PCET. With respect to limitations, the intermolecular reactions are generally less efficient with sulfonamide donors other than 34 (37) and styrene was found to be a poor substrate (43) in this reaction. Efforts to address these constraints are the focus of future work.
Table 3.
Scope of the intermolecular hydroamination. Yields are for isolated material following chromatography on silica gel and are the average of two experiments. Reactions conducted on 0.5 mmol scale with 3 equivalents of olefin unless otherwise noted. a yields determined by 1H–NMR analysis of the crude reaction mixture; b 2 equivalents of olefin were used.
|
Lastly, we attempted to highlight the synthetic versatility of sulfonamide PCET activation in the context of a tandem olefin amination/directed C–H bond alkylation sequence (Scheme 1). First, the terminal alkenes 63 and 66 were subjected to the intermolecular anti-Markovnikov hydroamination protocol described above to afford the alkylated sulfonamides in 75% and 74% yield, respectively. The newly installed secondary sulfonamide was then activated in a second oxidative PCET event using the same Ir/phosphate pair, leading to site-selective abstraction of the δ-C–H bond to afford a carbon-centered radical that can be trapped by an electron-deficient olefin (64, 65, 67). These reactions underscore the synthetic benefits of PCET-based methods for homolytic bond activation, generating versatile radical intermediates from common organic functional groups under mild catalytic conditions.22
Scheme 1.

Tandem amination/C–H alkylation. Yields are for isolated material following chromatography on silica gel and are the average of two experiments. Reactions conducted on 0.5 mmol scale with 5.0 equivalents of 57 and 60 in the first step and 2.0 equivalents of alkene were used in the second step.
In addition to these synthetic studies, we also sought to understand the elementary steps leading to N-radical generation. Using cyclic voltammetry, we found the oxidation of N-propyl-4-methoxybenzenesulfonamide occurs at +1.8 V vs. Fc+/Fc. As such, direct electron transfer between this sulfonamide and the excited state of photocatalyst A (E[*Ir (III)/(II)] = +1.30 V vs. Fc+/Fc) is endergonic by nearly 500 mV. Accordingly, we found that solutions of sulfonamide alone do not quench the luminescence of A. Other sulfonamides that do not bear electron rich aromatic groups have even higher oxidation potentials (Ep/2(N-propyltosylamide) = +2.2 V vs. Fc+/Fc, Ep/2 (N-propyl-4-cyanobenzensulfonamide) > +3.0 V vs. Fc+/Fc). These values indicate that a stepwise ET-PT mechanism for sulfonamidyl radical formation is unlikely to be operative. Similarly, the pKa of benzenesulfonamide is approximately 27 in MeCN23 - roughly 15 pKa units less acidic (ΔG° = +20.6 kcal/mol) than the conjugate acid of the dibutylphosphate base. This highly unfavorable equilibrium discounts mechanisms involving an initial proton transfer step between the sulfonamide and the phosphate base to form a more easily oxidized sulfonamide anion. Lastly, the potentials required for oxidation of terminal and disubstituted alkenes24 are more than 600 mV endergonic relative to the excited state potential of A, inconsistent with the viability of alkene radical cation formation under these conditions.
Based on these outcomes, we reasoned that concerted PCET might be the operative mechanism of N-radical generation. Consistent with this view, we observed quenching of *A in CH2Cl2 solutions containing varied concentrations of N-propyl-4-methoxybenzenesulfonamide in the presence of a constant concentration of dibutyl phosphate. While the degree of quenching was modest (Ksv = 3.0 M−1), the decrease in luminescence intensity was linearly correlated to the sulfonamide concentration, consistent with a first-order kinetic dependence (Figure 4, top panel). As described in detail in prior work from our group, the dialkyl phosphate base alone also quenches the luminescence of A in CH2Cl2 solution at rt.6 However, the concentration dependence of the quenching process is complex and exhibits saturation behavior – a feature that may reflect the formation of a favorable iridium-phosphate ionic hydrogen-bonded adduct under these reaction conditions. However we note that phosphate oxidation occurs at potentials significantly more positive than those of *A (ΔG° > +1.0 V), arguing against the possible role of phosphoryl radicals in the N–H activation step. The thermodynamic driving force for the proposed excited state PCET process is favorable (ΔG° = −6.0 kcal/mol) based on a N–H BDFE of N-ethyl-benzenesulfonamide of 97 kcal/mol and an effective BDFE of 103 kcal/mol for the Ir/phosphate pair.
Figure 4.

(top) Stern-Volmer luminescence quenching of *A in the presence of varying concentrations of N-propyl-4-methoxybenzenesulfonamide. (middle) Cyclic voltammograms of 4-methoxybenzenesulfonamide (2.5mM) in CH2Cl2 containing 0.1M NBu4PF6 and varying concentrations of NBu4OP(O)(OBu)2. Ag reference electrode, glassy carbon working electrode, Pt mesh counter electrode were used. Scan rate: 0.1 V/s. (bottom) Cyclic voltammograms of 4-methoxybenzenesulfonamide (2.5mM) and of NBu4OP(O)(OBu)2 (5mM) in CH2Cl2 containing 0.1M NBu4PF6. Ag reference electrode, glassy carbon working electrode, Pt mesh counter electrode. Scan rate: 0.1 V/s.
We further evaluated the role of PCET in sulfonamide activation using electrochemical techniques.25 Specifically, we carried out cyclic voltammetry studies on 4-methoxybenzenesulfonamide in CH2Cl2 containing 0.1 M NBu4PF6 in the presence of varying concentrations of monobasic dibutyl phosphate (Figure 4, middle panel). While all of the voltammograms were irreversible, we observed that the onset potentials were shifted to less positive potentials and current response increases with increasing concentrations of phosphate. Control experiments revealed that analogous scans of sulfonamide or phosphate alone do not give rise to these current features (Figure 4, bottom panel). Qualitatively, these outcomes are also consistent with the proposed PCET process.
Conclusions
We have developed a catalytic anti-Markovnikov hydroamination of unactivated alkenes with simple sulfonamides enabled by a PCET activation of the sulfonamide N–H bond. This work further demonstrates the potential of PCET for the direct homolytic activation of strong heteroatom–hydrogen bonds found in many common organic functional groups, and utilization of the resulting radical intermediates in synthetically useful transformations.26
Supplementary Material
ACKNOWLEDGMENT
Financial support was provided by NIH R01 GM113105.
Footnotes
Supporting Information.
This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
SUPPORTING INFORMATION
Experimental details; characterization data and NMR spectra; voltammetric, thermochemical and luminescence quenching data
REFERENCES
- (1).(a) Huang L; Arndt M; Gooβen K; Heydt H; Gooβen LJ Chem. Rev 2015, 115, 596. [DOI] [PubMed] [Google Scholar]; (b) Müller TE; Hultzsch KC; Yus M; Foubelo F; Tada M Chem. Rev 2008, 108, 3795. [DOI] [PubMed] [Google Scholar]; (c) Hong S; Marks TJ Acc. Chem. Res 2004, 37, 673. [DOI] [PubMed] [Google Scholar]; (d) Müller TE; Beller M Chem. Rev 1998, 98, 675. [DOI] [PubMed] [Google Scholar]
- (2).For representative intermolecular N–H addition to unfunctionalized olefins.Ryu J; Li GY; Marks TJ J. Am. Chem. Soc 2003, 125, 12584.Brunet JJ; Chu NC; Diallo O Organometallics 2005, 24, 3104.Zhang Z; Lee SD; Widenhoefer RA J. Am. Chem. Soc 2009, 131, 5372.Reznichenko AL; Nguyen HN; Hultzsch KC Angew. Chem., Int. Ed 2010, 49, 8984.Sevov CS; Zhou J; Hartwig JF J. Am. Chem. Soc 2012, 134, 11960.Sevov CS; Zhou J; Hartwig JF J. Am. Chem. Soc 2014, 136, 3200.
- (3).For anti-Markovnikov hydroamination:Musacchio AJ; Lainhart BC; Zhang X; Naguib SG; Sherwood TC; Knowles RR Science, 2017, 355, 727.Nguyen TM; Manohar N; Nicewicz DA Angew. Chem. Int. Ed 2014, 53, 6198.Ensign SC; Vanable EP; Kortman GD; Weir LJ; Hull KL J. Am. Chem. Soc 2015, 137, 13748.
- (4).For recent representative advances in olefin amination chemistry, see:Zhu L; Xiong P; Mao Z; Wang Y; Yan X; Lu X; Xu H Angew. Chem. Int. Ed 2016, 55, 4930.Gui J; Pan C; Jin Y; Qin T; Lo JC; Lee BJ; Spergel SH; Mertzman ME; Pitts WJ; Cruz TEL; Schmidt MA; Darvatkar N; Natarajan SW; Baran PS Science 2015, 348, 886.Zhu S; Buchwald SL; J. Am. Chem. Soc 2014, 136, 15913.Bernoud E; Oulié P; Guillot R; Mellah M; Hannedouche J Angew. Chem. Int. Ed 2014, 53, 4930.Shigehisa H; Koseki N; Shimizu N; Fujisawa M; Niistu M; Hiroya KJ Am. Chem. Soc 2014, 136, 13534.Rucker RP; Whittaker AM; Dang H; Lalic GJ Am. Chem. Soc 2012, 134, 6571.Pronin SV; Tabor MG; Jansen DJ; Shenvi RA J. Am. Chem. Soc 2012, 134, 2012.Guin J; Mück-Lichtenfeld C; Grimme S; Studer AJ Am. Chem. Soc 2007, 129, 4498.
- (5).(a) Miller DC; Choi GJ; Orbe HS; Knowles RR J. Am. Chem. Soc 2015, 137, 13492. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nguyen LQ; Knowles RR ACS Catal. 2016, 6, 2894. [Google Scholar]; (c) Choi GC; Knowles RR J. Am. Chem. Soc 2015, 137, 9226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Nature 2016, 539, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Kharasch MS; Priestley HM J. Am. Chem. Soc 1939, 61, 3425. [Google Scholar]; (b) Ohashi T; Sugie M; Okahara M; Komori S Tetrahedron Lett. 1968, 9, 4195. [Google Scholar]; (c) Lu H; Chen Q; Li CJ Org. Chem 2007, 72, 2564. [DOI] [PubMed] [Google Scholar]; (d) Kitagawa O; Yamada Y; Fujiwara H; Taguchi T Angew. Chem., Int. Ed 2001, 40, 3865. [DOI] [PubMed] [Google Scholar]; (e) Kitagawa O; Miyaji S; Yamada Y; Fujiwara H; Taguchi TJ Org. Chem 2003, 68, 3184. [DOI] [PubMed] [Google Scholar]; (f) Heuger G; Göttlich R Beilstein J. Org. Chem 2015, 11, 1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).For intermolecular hydroaminations of unactivated alkenes with sulfonamides, see:Fei J; Wang Z; Cai Z; Sun H; Cheng X Adv. Synth. Catal 2015, 357, 4063.Yang C; Fan W; Liu G; Duan L; Li L; Li Y RSC adv. 2015, 5, 61081.Zhang J; Yang C; He CJ Am. Chem. Soc 2006, 128, 1798.Karshtedt D; Bell AT; Tilley TD J. Am. Chem. Soc 2005, 127, 12640.Rosenfeld DC; Shekhar S; Tekemiya A; Utsunomiya M; Hartwig JF Org. Lett 2006, 8, 4179.Intramolecular hydroamination:Sherman ES; Fuller PH; Kasi D; Chemler SR J. Org. Chem 2007, 72, 3896.Komeyama K; Morimoto T; Takaki K Angew. Chem. Int. Ed 2006, 45, 2938.Schlummer B; Hartwig JF Org. Lett 2002, 4, 1471.
- (9).Nguyen TM; Nicewicz DA J. Am. Chem. Soc 2013, 135, 9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ickes AR; Ensign SC; Gupta AK; Hull KL J. Am. Chem. Soc 2014, 136, 11256. [DOI] [PubMed] [Google Scholar]
- (11).Beller M; Trauthwein H; Eichberger M; Breindl C; Herwig J; Muller TE; Thiel OR Chem. - Eur. J 1999, 5, 1306. [Google Scholar]
- (12).(a) Utsunomiya M; Kuwano R; Kawatsura M; Hartwig JF J. Am. Chem. Soc 2003, 125, 5608. [DOI] [PubMed] [Google Scholar]; (b) Takemiya A; Hartwig JF J. Am. Chem. Soc 2006, 128, 6042. [DOI] [PubMed] [Google Scholar]
- (13).(a) Warren JJ; Tronic TA; Mayer JM Chem. Rev 2010, 110, 6961. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weinberg DR; Gagliardi CJ; Hull JF; Murphy CF; Kent CA; Westlake BC; Paul A; Ess DH; McCafferty DG; Meyer TJ Chem. Rev 2012, 112, 4016. [DOI] [PubMed] [Google Scholar]; (c) Waidmann CR; Miller AJM; Ng CA; Scheuermann ML; Porter TR; Tronic TA; Mayer JA Energy Environ. Sci 2012, 5, 7771. [Google Scholar]; (d) Tarantino KT; Liu P; Knowles RR J. Am. Chem. Soc. 2013, 135, 10022. [DOI] [PubMed] [Google Scholar]
- (14).Zhao Y; Bordwell FG; Cheng J; Wang DJ Am. Chem. Soc. 1997, 119, 9125.Also see DFT calculation in supporting information.
- (15).Romero N; Nicewicz DA J. Am. Chem. Soc. 2014, 136, 17024.Also see S–H BDFE = 79 kcal/mol from:Dénès F; Pichowicz M; Povie G; Renaud P Chem. Rev 2014, 114, 2587.
- (16).pKa(PhSH) = 10.3 in DMSO, see:Bordwell FG; Hughes DL J. Org. Chem 1982, 47, 3224.pKa difference between in MeCN and in DMSO is typically ~11 pKa units. See:Chantooni MK; Kolthoff IM J. Phys. Chem, 1976, 80, 1306.
- (17).Rueping M; Nachtsheim BJ; Ieawsuwan W; Atodiresei I Angew. Chem., Int. Ed 2011, 50, 6706. [DOI] [PubMed] [Google Scholar]
- (18).(a) Gagliardi CJ; Murphy CF; Binstead RA; Thorp HH; Meyer TJ J. Phys. Chem. C 2015, 119, 7028. [Google Scholar]; (b) Medina-Ramos J; Oyesanya O; Alvarez JC J. Phys. Chem. C 2013, 117, 902. [Google Scholar]; (c) Kuss-Petermann M; Wenger OS J. Phys. Chem. Lett 2013, 4, 2535. [DOI] [PubMed] [Google Scholar]
- (19).Bordwell FG; Cheng J-P; Ji G-Z; Satish AV; Zhang XJ Am. Chem. Soc. 1991, 113, 9790. [Google Scholar]
- (20).Nikishin GI; Troyanski EI; Lazarea MI Tetrahedron Lett. 1985, 26, 1877. [Google Scholar]
- (21).Yayla HG; Wang H; Tarantino KT; Orbe HS; Knowles RR J. Am. Chem. Soc. 2016, 138, 10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).For deprotection of the PMP sulfonamides, see:Ankner T; Hilmersson G Org. Lett 2009, 11, 503.So CM; Kume S; Hayashi TJ Am. Chem. Soc. 2013, 135, 10990.
- (23).Estimated from Bordwell pKa table: pKa (PhSO2NH2) = 16 in DMSO.
- (24).Roth HG; Romero NA; Nicewicz DA Synlett, 2016, 27, 714. [Google Scholar]
- (25).Costentin C Chem. Rev 2008, 108, 2145. [DOI] [PubMed] [Google Scholar]
- (26).(a) Yayla HG; Knowles RR Synlett. 2014, 20, 2819. [Google Scholar]; (b) Gentry EC; Knowles RR Acc. Chem. Res 2016, 49, 1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.