

Abstract
Antitumor T-cell responses have the potential to be curative in cancer patients, but the induction of potent T-cell immunity through vaccination remains a largely unmet goal of immunotherapy. We previously reported that the immunogenicity of peptide vaccines could be increased by maximizing delivery to lymph nodes (LNs), where T-cell responses are generated. This was achieved by conjugating the peptide to 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-PEG (DSPE-PEG) to promote albumin binding, which resulted in enhanced lymphatic drainage and improved T-cell responses. Here, we expanded upon these findings and mechanistically dissected the properties that contribute to the potency of this amphiphile-vaccine (amph-vaccine). We found that multiple linkage chemistries could be used to link peptides with DSPE-PEG, and further, that multiple albumin-binding moieties conjugated to peptide antigens enhanced LN accumulation and subsequent T-cell priming. In addition to enhancing lymphatic trafficking, DSPE-PEG conjugation increased the stability of peptides in serum. DSPE-PEG-peptides trafficked beyond immediate draining LNs to reach distal nodes, with antigen presented for at least a week in vivo, whereas soluble peptide presentation quickly decayed. Responses to amph-vaccines were not altered in mice deficient in the albumin-binding neonatal Fc receptor (FcRn), but required Batf3-dependent dendritic cells (DCs). Amph-peptides were processed by human DCs equivalently to unmodified peptides. These data define design criteria for enhancing the immunogenicity of molecular vaccines to guide the design of next-generation peptide vaccines.
Keywords: vaccine, T cell, lymph node
Introduction
The capacity of antitumor T-cell responses to be curative in cancer patients is known from clinical studies of adoptive cell therapy, but the induction of potent endogenous T-cell responses through therapeutic vaccination remains a challenge (1,2). Trials where patients received personalized vaccines targeting predicted neoantigens have generated significant enthusiasm in the field because they demonstrate the feasibility of immunizing against antigens that should not be restrained by tolerance (3,4). Yet, the immunogenicity of the vaccines reported in these trials remain modest. For example, Ott et al. employed “long” neoantigen-containing peptides as immunogens, which are attractive due to their defined nature, relative ease of manufacturing, favorable safety profile, and requirement of professional antigen presenting cells (APCs) for presentation (5). However, as observed in many other peptide vaccine trials, CD8+ T-cell responses were only detectable after extended peptide stimulation ex vivo (4). Much room remains for improvement in the immunogenicity of these vaccines.
To generate more potent peptide vaccines, a variety of approaches based on optimized formulation and immunization regimens have been demonstrated. Vaccine responses have been boosted by the utilization of altered peptide ligands, or high-affinity, non-self epitopes that prime cross-reactive T-cell responses against tumor antigens (6). Significant effort has been put into developing adjuvants to enhance T-cell responses following peptide vaccination, including the use of TLR agonists (7), type I interferons (8), and Treg–disrupting monoclonal antibodies (7,9). Vaccine formulation can also contribute to the persistence and quality of antitumor immunity. One of the most common emulsion formulations, incomplete Freund’s adjuvant, has been found to depot peptide antigens at the injection site and sequester T cells distal from the tumor, which may harm antitumor immunity, whereas formulations that are non-persisting at the site of vaccination may prove to be more effective (10).
The poor lymphatic drainage and short in vivo half-life of linear peptides likely contribute to the low potency of peptide vaccines (11–14). We previously reported that enhanced lymph node (LN)-targeting of peptides can be achieved by covalently modifying peptides and molecular adjuvants with diacyl lipids, with the peptides linked through a PEG spacer, and this trafficking enhancement results in a corresponding increase in T-cell priming (14). The mechanism of action is thought to involve binding of the lipid tails to endogenous albumin, allowing peptides and molecular adjuvants to “hitchhike” on albumin to the draining LN, similar to the mechanism of action of sentinel LN mapping dyes in clinical use (15,16).
Here, we explored in detail mechanisms of action of these molecular vaccines and evaluated the role of distinct components of the amphiphile (amph)-vaccine platform. We tested the relative efficacy of several different albumin-binding moieties, the stability of amph-vaccine conjugates in presence of serum proteases, the effect of employing different adjuvants with amph-peptides, and the importance of the neonatal Fc receptor and cross-presenting dendritic cells (DCs) in amph-vaccine immunogenicity. We also assessed the duration and location of antigen presentation following amph-vaccine administration in vivo. Together, our data support the use of albumin-binding as a general design criterion for improving peptide vaccines by improving stability and lymphatic distribution, resulting in enhanced and prolonged antigen presentation in the LNs for superior CD8+ T-cell priming.
Materials and Methods
Oligo conjugate synthesis
Solid phase DNA synthesis, purification, and characterization of lipo-CpG (ODN 1826) were carried out as previously described (14). Synthesis of the Vitamin E-CpG conjugate was performed as described in (14) using α-Tocopherol-TEG Phosphoramidite (Glen Research) with the following changes: after conjugation in solid phage, cleavage and ultra-mild deprotection was performed using 3:1 tertbutylamine in water for 6 hours at 65°C and the 5’ dimethoxytrityl (DMT) group was removed using 80% acetic acid at room temperature for 2 hours, followed by an ethanol precipitation. Following cleavage and deprotection, oligos were purified via RP-HPLC and quantified using UV-VIS. For synthesis of TAMRA-labeled Vit. E-CpG, a 3’ TAMRA bead was utilized (TAMRA lcaa CPG 500Å, ChemGenes), followed by the ultra-mild cleavage and deprotection conditions described above. Synthesis of Vitamin B3 CpG was performed using 5’-Niacin-CE Phosphoramidite (Link Technologies). All CpG oligonucleotide conjugates used an ODN class B sequence 1826 with two guanine spacers.
Synthesis of DSPE-PEG-EGP constructs
Reaction partners and conjugate structures are shown in Supplementary Fig. S1. The peptide antigen was first synthesized using 9-fluorenyl methoxycarbonyl (Fmoc) solid-phase peptide synthesis with pre-loaded Wang resin (EMD Millipore). Synthesis was performed on a CEM Liberty Blue automated microwave peptide synthesizer. Coupling reactions were performed using 4 eq. of Fmoc-protected amino acid, 4 eq. of N,N’-diisopropylcarbodiimide (DIC)) and 8 eq. of ethyl(hydroxyimino)cyanoacetate (Oxyma pure) and removal of Fmoc groups on resin-attached amino acids was achieved with 20% 4-methylpiperidine in DMF. After synthesis, peptides were cleaved off the resin in a 95:2.5:2.5 trifluoroacetic acid (TFA)/triisopropylsilane (TIPS)/H2O mixture for 3 hours and washed with dichloromethane. Rotary evaporation and precipitation in cold diethyl ether yielded the crude product. Crude peptides were purified by HPLC (Waters Prep 150) on a C18 Phenomenex Kinetex column in a water-acetonitrile gradient containing 0.1% v/v TFA. Pure fractions were collected and identified using ESI-MS. The combined fractions were subjected to rotary evaporation to remove acetonitrile, frozen in liquid nitrogen, and lyophilized to dryness. Purity was verified to be >95% using LC-MS on an Agilent QTOF 6520 LC-MS. Example chromatograms and ESI-MS spectra are shown in Supplementary Fig. S2.
For the DSPE-PEG2000-AVGALEGPRNQDWLGVPRQL-COOH, purified H2N-AVGALEGPRNQDWLGVPRQL-COOH was dissolved in DMF along with excess diisoproplyethylamine (DIEA). This mixture was then added to approximately 2 molar equivalents of DSPE-PEG2000-NHS (Nanosoft Polymers) also dissolved in DMF. Reaction progress was monitored by ESI-MS until the peptide peak was consumed (approximately 18 hours) and only the polydisperse product peak, and some DSPE-PEG2000 starting material were seen. The reaction mix was then diluted with water, and product was purified by HPLC using a Phenomenex Jupiter C12 column. After lyophilization to dryness, LCMS confirmed purity to be >95% and the product mass was a polydisperse mixture centered around 5162 m/z.
To synthesize DSPE-PEG2000-CAVGALEGPRNQDWLGVPRQL-COOH, purified H2N-CAVGALEGPRNQDWLGVPRQL-COOH was dissolved in DMF and added to approximately 1.25 molar equivalents of DSPE-PEG2000-maleimide (Laysan Bio) also dissolved in DMF. Reaction progress was monitored by ESI-MS until the peptide peak was consumed (approximately 24 hours), and only the polydisperse product peak and some DSPE-PEG2000-maleimide were seen. The reaction mix was then diluted with water and purified as above. After lyophilization to dryness, LCMS confirmed purity to be >95% and the product mass was a polydisperse mixture centered around 5349 m/z.
For synthesis of DSPE-PEG2000-(click)-AVGALEGPRNQDWLGVPRQL-COOH, with peptide H2N-AVGALEGPRNQDWLGVPRQL still on resin, 2 molar equivalents of 5-azidopentanoic acid, 1.9 molar equivalents of HBTU and 8 molar equivalents of DIEA in DMF were added. This mixture was allowed to react overnight before standard cleavage from resin and purification as described above. The purified peptide (Az)-AVGALEGPRNQDWLGVPRQL-COOH was then dissolved in DMSO along with DSPE-PEG2000-DBCO (Avanti) in approximately a 1:1 molar ratio. The reaction was monitored until the peptide peak was nearly consumed and the product was then purified as above. After lyophilization to dryness, LCMS confirmed purity to be >95% and the product mass was a polydisperse mixture centered around 5405 m/z.
For the ABP-antigen constructs, following synthesis of the antigen component, the PEG2000 linker (Creative PEGworks) was coupled manually onto peptidyl resin containing the antigen peptide; the resin was then transferred back to the synthesizer to complete the construct. The crude peptides were cleaved overnight at room temperature (by volume, 82.5% trifluoroacetic acid, 5% water, 5% thioanisole, 5% phenol, and 2.5% ethane dithiol), triturated with −80 °C diethyl ether, and purified via RP-HPLC. The ABP moieties were cyclized by incubating the purified constructs (0.1 mg/mL) in 0.1 M NaHCO3 (Sigma), pH 8.0 for 24h to facilitate disulfide formation. The cyclized constructs were desalted via solid phase extraction, lyophilized, and analyzed via LC-MS to verify identity and purity. An example chromatogram and corresponding mass spectrum is shown in Supplementary Fig. S3A-B. Conditions for cyclization were verified on a monodisperse construct with G4 spacer (Supplementary Fig. S3C-D).
For the labeled constructs, EGP long and ABP-PEG2000-EGP were synthesized as described above, incorporating a C-terminal propargylglycine to serve as a chemical handle. Prior to disulfide cyclization, purified EGP long and ABP-PEG2000-EGP long peptides were coupled to TAMRA-azide (tetramethylrhodamine, 5-isomer, Lumiprobe) via Cu(I)-catalyzed click chemistry. Briefly, ~0.5 μmol of each peptide was dissolved in 720 μL of 50:50 b.v. solution of t-butanol (Sigma) and water. The following reagents were then added (final concentration in parentheses): TAMRA-azide (500 μM), Tris (pH 8.0, final concentration 50 mM; Roche), ascorbic acid (100 mM, Sigma), TCEP (1 mM, Hampton Research), TBTA (100 μM; Alfa Aeser), and copper sulfate (5 mM; Sigma) to a final volume of 1.00 mL. The reactions were mixed thoroughly and incubated at room temperature for one hour. The reactions were quenched in 20 mL 95:5 water: acetonitrile with 0.1% TFA additive, purified via RP-HPLC, and lyophilized. The TAMRA-labeled constructs were cyclized and desalted as described above.
Peptide antigen sequences used are as follows: gp100 EGP long, EGP20 (gp10020–39): AVGALEGPRNQDWLGVPRQL (18); gp100 EGP short, EGP9: EGPRNQDWL; E7 from HPV: RAHYNIVTF; SIV-gag AL11: AAVKNWMTQTL; the MHC II-restricted neoantigen MUT30: VDWENVSPELNSTDQ (3); OVA251–270: GLEQLESIINFEKLTEWTSS; and HIV gag p1764–93: LQPSLQTGSEELRSLYNTVATLYCVHQRIE. The ABP sequence chosen was the minimal core sequence identified in a phage display library of albumin binders: DICLPRWGCLW (19).
Mice and immunizations
All animal studies were carried out under an institute-approved IACUC protocol following federal, state, and local guidelines for the care and use of animals. C57BL6/J mice were procured from Jackson Laboratory. Neonatal Fc receptor knockout mice (FcRn–/–, B6.129X1-Fcgrttm1Dcr/DcrJ), Batf3–/– mice (B6.129S(C)-Batf3tm1Kmm/J), and pmel-1 mice (B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J) were obtained from Jackson Laboratory and bred in-house. 6-to 12-week old female mice were used for these studies.
Immunizations comprised of peptides or peptide-conjugates mixed with adjuvant were administered subcutaneously at the base of the tail in 100 μL total, 50 μL on each side, with sterile saline diluent. The same vaccine preparation and injection technique was used for both prime (day 0) and boost (day 14). Whole protein OVA (LowEndo™, Worthington Biochemical) was dosed at 10 μg. Peptides were dosed at 10 μg and conjugates were dosed at the molar equivalent. Adjuvants c-di-GMP (InvivoGen) and polyI:C (HMW, InvivoGen) were dosed at 25 μg, lipo-CpG and vitamin E-CpG were dosed at 1.24 nmol (equivalent to 7.9 µg CpG), and polyI:C was used at 25 μg. For LN trafficking studies using the Spectrum In Vivo Imaging System (IVIS, PerkinElmer), the CpG dose was 3.3 nmol, and the peptide dose was 15 nmol.
For trafficking studies and immunization studies using IFA (Montanide ISA 51 VG, SEPPIC), aqueous peptide solutions were formulated at a 1:1 ratio with IFA and emulsified using a syringe adapter emulsifier provided by the manufacturer until a stable emulsion was formed. In this experiment only, mice were boosted twice, on days 14 and 28, and intracellular cytokine staining was performed on day 35.
Adoptive transfer proliferation studies
Pmel-1 T cells were isolated from spleens of pmel-1 mice with an EasySep CD8+ T-cell Isolation Kit (Miltenyi), according to the manufacturer’s protocol. Purified pmel-1 T cells were labeled by incubating for 20 minutes at 37°C in 5 μM CFSE or CellTrace Violet (Thermo Fisher Scientific) in PBS plus 0.1% BSA, quenched with RPMI plus 20% FBS, and washed. T cells were resuspended in sterile saline, and 5 × 105 pmel-1 T cells were transferred retro-orbitally into recipient wild-type (w.t.) mice. Inguinal, axillary, brachial, lumbar, cervical, and mesenteric LNs were collected, mechanically dissociated, stained, and analyzed by flow cytometry on a BD LSR Fortessa with the antibodies indicated in the “Evaluation of murine immune responses and flow cytometry” section below. Proliferation index (PI) was quantified using PI = log(FInd/MFIall)/log(2) where MFIall = median fluorescence intensity of live Thy1.1+ pmel-1 T cells and FInd=peak fluorescence intensity of viable non-divided pmel-1 T cells.
In vitro peptide stability assay
To assess serum stability of peptides, four EGP20 constructs (EGP20, amph-EGP20 (N), amph-EGP20 (C), and PEG2000-EGP20) were subjected to overnight incubation at 37°C in RPMI with 10% fresh mouse serum over concentrations ranging from 10 ng/mL to 2.4 µg/mL. Serum-treated or control constructs were then incubated with pooled splenocytes from 3 previously DSPE-PEG-EGP20 immunized mice (50,000 splenocytes per condition) for 24 hours at 37°C (in RPMI with 10% heat inactivated FBS), with brefeldin A added at 18 hours and stained and run on the flow cytometer as described below.
Evaluation of murine immune responses and flow cytometry
Antibodies to IFNγ (clone XMG1.2), TNFα (clone MP6-XT22), CD69 (clone H1.2F3), PD-1 (clone 29F.1A12), Tim-3 (clone B8.2C12), CD44 (clone IM7), CD62L (clone MEL-14), IL-4 (clone 11B11), and CD8α (clone 53–6.7) were purchased from BioLegend, and CD4 (clone RM4–5) and IL-17A (clone eBio17B7) were purchased from eBioscience. Gp100 tetramer (iTAg Tetramer/PE H-2Db gp100), E7 (iTAg Tetramer/PE H-2Db E7), and OVA tetramer (iTAg Tetramer/PE H-2Kb OVA) were purchased from MBL. For all experiments where murine blood was used, blood was collected retro-orbitally using VWR® Microhematocrit Capillary Tubes into MiniCollect tubes (0.5mL K2E K2EDTA, Greiner Bio-One GmbH) 6 days following boost unless otherwise noted. Antibody staining was performed at a dilution of 1:100 for 25 minutes at 4°C in the presence of mouse Fc block (Mouse BD Fc Block™) in PBS with 1% BSA and 5mM EDTA unless otherwise indicated. Tetramer staining was performed in buffer containing 50 nM dasatinib (Sigma-Aldrich) at room temperature for 30 minutes, followed by surface staining at 4°C for 15 minutes. Viability was assessed by LIVE/DEAD Fixable Aqua (Life Technologies) or DAPI. Cells were analyzed using BD LSR Fortessa and BD FACSCanto flow cytometers. Data was analyzed using FlowJo v10. Intracellular cytokine staining (ICS) was performed as previously described (14), with fixation and permeabilization using BD Cytofix/Cytoperm according to the manufacturer’s protocol. Peptides for restimulation were used at a concentration of 10 µg/mL with sequences as follows - for gp100 immunizations: gp10025–33 native, EGSRNQDWL; SIV gag AL11: AAVKNWMTQTL; OVA: SIINFEKL; HIV gag p17: RSLYNTVAT.
Primary human monocyte-derived DC presentation assay
The human HLA-B57-restricted KF11-specific CD8+ T-cell clone (recognizing KAFSPEVIPMF (20)) was maintained in the presence of IL2 (50 U/mL, R&D Systems), using CD3-specific monoclonal antibody 12F6 (0.1 µg/mL, Enzo Life Sciences), and irradiated allogeneic feeder PBMCs.
Human peripheral blood mononuclear cells (PBMCs) were freshly isolated from HLA-B57+ blood donors with Ficoll-Hypaque (Sigma-Aldrich). PBMCs were isolated from buffy coats collected from HLA-typed blood donors after written informed consent and approval by the Partners Human Research Committee (protocol #2005P001218). Monocytes were enriched using CD14+ magnetic isolation, according to the manufacturer’s instructions (EasySep™ Human Monocyte Isolation Kit, StemCell). DCs were differentiated during a 6-day culture at 106 cells/mL in AIM-V medium (Thermo Fisher), supplemented with 1% HEPES, 1% human AB serum, IL4 (20 ng/mL; CellGenix), and GM-CSF (10 ng/mL; CellGenix). On days 2 and 4, fresh IL4 and GM-CSF were added. DCs were harvested on day 6 and incubated with free HIV p2428–60 peptide (VEEKAFSPEVIPMFSALSEGATPQDLNTMLNTV) or DSPE-PEG-p2428–60 for 4 hours at 37°C and then washed. The DCs were then plated with the KF11 CD8+ T-cell clone in a 2:1 effector-to-target ratio in 96-well plates (Millipore) coated with anti-IFNγ (clone 1-D1K, Mabtech, 2 µg/mL). DCs pulsed with equivalent molar concentrations of the optimal KF11 peptide were used as controls. Cells then were cocultured overnight wrapped in foil at 37°C in AIM V medium supplemented with 1% HEPES and 10% human AB serum on human IFNγ ELISPOT plates (EMD Millipore). The ELISPOT plate was developed according to the manufacturer’s protocol. Plates were scanned using a CTL-ImmunoSpot Plate Reader, and data were analyzed using CTL ImmunoSpot Software. The magnitude of the epitope-specific response was reported as the number of SFU per 104 T cells.
Mass spectrometry analysis of peptide degradation products
Mass spectrometry (MS) analysis of peptide fragments generated after incubation in cytosolic, endosomal, or lysosomal degradation conditions was carried out as previously described (21). Briefly, 2 nmol of p2428–60 peptide or DSPE-PEG-p2428–60 were incubated with cell extracts from each cellular compartment that were isolated from antigen presenting cells derived from primary human monocytes at 37°C in 50μL of degradation buffer (50 mM Tris-HCl, 1 mM MgCl2, 1 mM ATP, and 137 mM potassium acetate at pH 7.4, pH 5.5, or pH 4.0). The reaction was stopped with formic acid, and peptides were purified via trichloroacetic acid precipitation. The degradation mix was injected into a Nano-HPLC (Eksigent) at a 400 nL/minute flow rate. A Nano cHiPLC trap column (200 µm x 0.5 mm ChromXP c18-CL 5 µm 120Å; Eksigent) was used to remove salts in the sample buffer. Peptides were separated in a Nano cHiPLC column (75 µm x 15 cm ChromXP c18-CL 5 µm 300Å; Eksigent) with 0.1% formic acid in water and 0.1% formic acid in acetonitrile. Mass spectra were recorded in the range of 370 to 2000 daltons (Da) on a LTQ Orbitrap Discovery (Thermo). In tandem MS mode, the eight most intense ions were selected with a 1 Da window for fragmentation in a collision cell using Helium with 35 V as collision voltage. MS fragments were searched against source peptide databases with Protein Discoverer 1.4 (Thermo Scientific). For semi-quantification the integrated area under a peak generated by a given peptide is proportional to the abundance of that peptide. Each degradation time point was run on the MS at least twice. The MHC class I and class II epitopes from gag p24 were identified using the from HIV Los Alamos Immunology database (http://www.hiv.lanl.gov/).
Statistical analysis
Statistical methods were not used to determine sample size, but sample numbers were chosen based on estimates from pilot studies and published results, such that appropriate statistical tests may yield statistically significant results. Unless otherwise stated, analysis of tetramer and ICS results were analyzed by using one-way ANOVA with a Tukey post-test using GraphPad Prism software. Where ANOVA was used, variance between groups was found to be similar by Bartlett’s test. Where comparisons were predetermined, as indicated in figure legends, ANOVA with Bonferroni correction was used. Pmel-1 proliferation data was analyzed using student’s t-test within a given LN to compare EGP20 peptide and DSPE-PEG-EGP20. No samples were excluded from analysis.
Results
Albumin-binding moieties enhance lymphatic drainage and immunogenicity of vaccines
We previously reported that peptide antigens conjugated to a DSPE phospholipid tail through a poly(ethylene glycol) spacer (Fig. 1A) exhibit increased lymphatic trafficking following immunization, and, thereby, a 20–30-fold increased immunogenicity in mice (14). This enhanced lymphatic trafficking was associated with the ability of the conjugates to bind serum albumin, suggesting that albumin serves as an in situ molecular chaperone delivering amph-vaccines to LNs. If this proposed mechanism of action is correct, other albumin-binding moieties might be equally well-suited to target peptide vaccines to lymphoid tissues. To test this idea, we prepared cyclized albumin-binding peptides linked via a PEG2000 spacer to the melanoma gp100 antigen EGP20 (ABP-PEG-EGP20; Fig. 1B, Supplementary Fig. S3)(19). When mice were immunized with TAMRA-labeled EGP20 peptide or ABP-PEG-EGP20, the ABP-conjugated peptide resulted in 13.6-fold and 18.2-fold higher fluorescence in inguinal and axillary LNs 24 hours later, respectively, compared to unmodified EGP20 (Fig. 1C-D). Vaccination with ABP-PEG-EGP20 elicited a 5-fold increase in antigen-specific T cells compared to unmodified peptide (Fig. 1E).
Figure 1: Albumin-binding peptide conjugation enhances LN targeting and immunogenicity of peptide antigens.
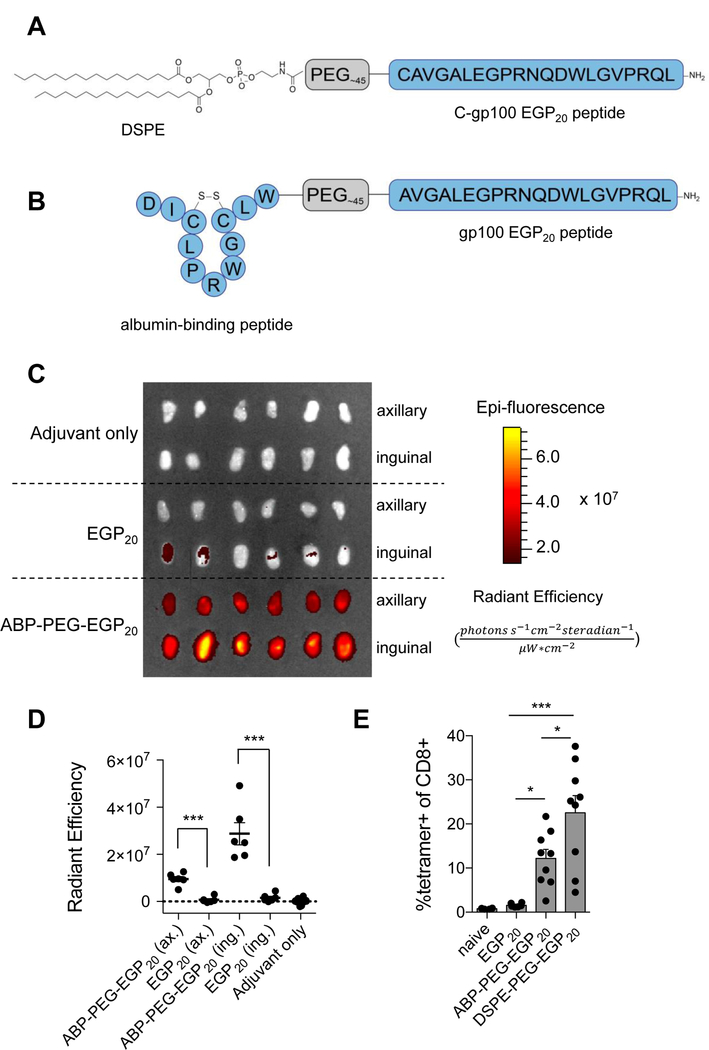
A, Schematic of DSPE conjugated to EGP20 via a PEG spacer. B, Schematic of cyclized albumin-binding peptide conjugated to EGP20 peptide via a PEG spacer. C-D, C57BL6/J mice were injected s.c. at the tail base with 15 nmol TAMRA labeled EGP20 peptide or ABP-PEG-EGP20 and 25 µg c-di-GMP adjuvant. C, Axillary (ax) and inguinal (ing) LNs were resected 24 hours later and imaged using IVIS. Shown are resected nodes. D, Quantification of background-subtracted radiant efficiency of images shown in C ((photons*s−1*cm−2*steradian−1)/(µW*cm2)). n = 6/LN type; student’s t-test; representative of 2 independent experiments. E, C57BL6/J mice were primed on day 0 and boosted on day 14 with 5 nmol EGP20 peptide or EGP20-conjugate and 25 µg c-di-GMP. Tetramer staining was performed on day 21. Mean tetramer responses quantified as % tetramer+ of CD8+. n = 6–9/group; ANOVA with Bonferroni correction for shown preplanned comparisons; representative of 2 independent experiments. *P<0.05, **P<0.01, ***P<0.001.
As a second test of the generality of albumin binding as a mechanism for targeting antigen/adjuvants to LNs, we also evaluated vaccine conjugates of the most biologically active form of vitamin E, α-tocopherol, which has been shown to bind to serum albumin with an affinity of KD ~7 μM (22). Constructs utilizing vitamin E as the albumin-binding domain for EGP20 peptide or the TLR9 agonist CpG ODN 1826 were synthesized (Fig. 2A-B). Fluorescence imaging of excised LNs after immunization with fluorophore-tagged Vit. E-CpG showed 2.9-fold and 3.1-fold higher CpG levels in the draining inguinal and axillary nodes, respectively, compared to unmodified oligonucleotides (Fig. 2C). Consistent with this enhanced lymphatic delivery, mice immunized with whole protein ovalbumin (OVA) adjuvanted with Vit. E-CpG showed 10-fold higher frequencies of OVA-specific CD8+ T cells compared to vaccination with unmodified CpG or CpG conjugated with Vitamin B3, which is not known to bind to serum albumin (Fig. 2D-E). Vit. E-CpG performed comparably to the diacyl lipid-CpG conjugate lipo-CpG, which was previously demonstrated to exhibit enhanced adjuvanticity by virtue of its improved LN trafficking (14). To see if α-tocopherol offered similar enhancements to peptide antigens, α-tocopherol-PEG-maleimide was used to conjugate vitamin E-PEG to the gp100 antigen EGP20 long via an N-terminal cysteine (Vit. E-PEG-EGP20; Fig. 2B). Mice immunized with Vit. E-PEG-EGP20 showed T-cell responses approximately 10-fold higher than unmodified peptides (Fig. 2F-G). Vitamin E conjugation, thus, improves the adjuvant activity of CpG and immunogenicity of peptide antigens, and together with the ABP conjugate data, suggests that directing molecular vaccines to bind albumin may be a general design principle to increase LN accumulation and immunogenicity.
Figure 2: Vitamin E conjugation enhances lymphatic drainage and immunogenicity of molecular vaccines.
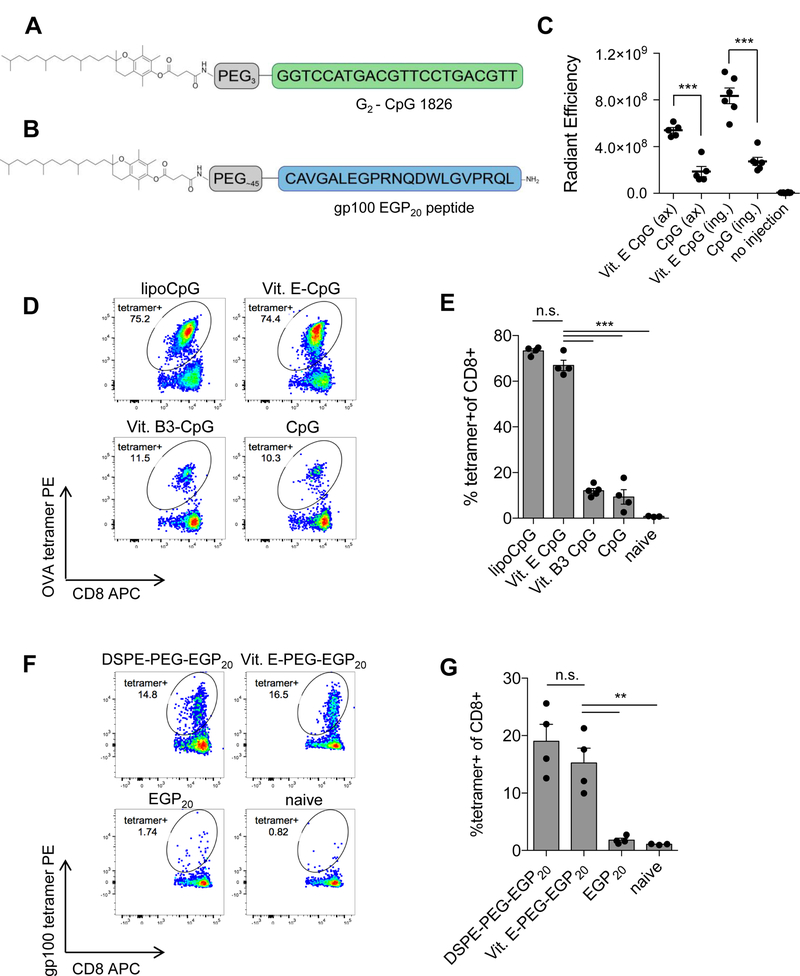
A, Schematic of vitamin E linked to CpG 1826 via triethylene glycol spacer. B, Schematic of EGP20 peptide linked to vitamin E via PEG spacer. C, Mice were injected subcutaneously with 3.3 nmol TAMRA-labeled CpG or lipo-CpG. Axillary (ax) and inguinal (ing) nodes were resected 24 hours later. Uptake was quantified using IVIS. Shown is background-subtracted radiant efficiency. n = 5–6/group; student’s t-test. D-E, Mice were primed on day 0 and boosted on day 14 with 10µg OVA and 1.24 nmol of CpG or CpG conjugate. Tetramer staining was performed on day 21. D, Representative plots gated on live CD8+ T cells. E, Mean tetramer responses quantified as % tetramer+ of CD8+. n = 4/group; ANOVA with Tukey post-test; representative of 2 independent experiments. F-G, C57BL6/J mice were primed on day 0 and boosted on day 14 with 5 nmol EGP20 peptide or EGP20-conjugate and 25 µg c-di-GMP. Tetramer staining was performed 7 days post-boost. F, Representative plots gated on live CD8+ T cells. G, Mean tetramer responses quantified as % tetramer+ of CD8+ n = 4/group; ANOVA with Tukey post-test; representative of 3 independent experiments. *P<0.05, **P<0.01, ***P<0.001.
DSPE-PEG peptides do not rely on FcRn but require Batf3 DCs for optimal responses
We next explored factors controlling the fate of amph-vaccines in LNs. We previously showed that amph-vaccines accumulate in F4/80+ macrophages and CD11c+ DCs (14), but it is unclear how albumin-binding vaccines are acquired by LN APCs. The neonatal Fc receptor (FcRn) is the best characterized receptor for albumin, and studies have implicated FcRn in the cross-presentation of immune complexes (23,24). However, immunization of wild-type (w.t.) or FcRn–/– animals with lipo-CpG and DSPE-PEG-E7 peptides showed no role for FcRn, as FcRn–/– mice generated T-cell responses indistinguishable from w.t. mice (Fig. 3A-B).
Figure 3: Amph-vaccine does not rely on FcRn, and short DSPE-PEG-peptides require Batf3 DCs for optimal responses.
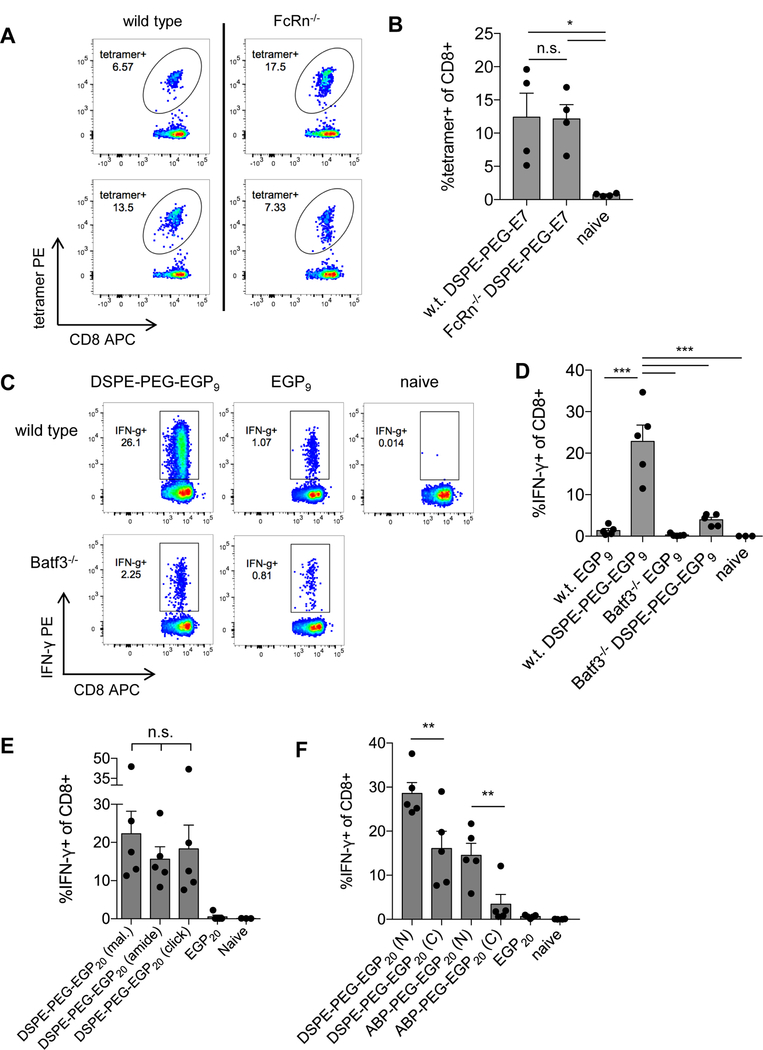
A-B C57BL6/J or FcRn–/– mice were primed on day 0 and boosted on day 14 with 10 µg DSPE-PEG-E7 peptide and 1.24 nmol lipo-CpG. Tetramer staining was performed on day 21. A, Tetramer plots from representative wild-type (w.t.) or FcRn–/– mice gated on live CD8+ T cells. B, Mean tetramer responses quantified as % tetramer+ of CD8+. n = 4/group; ANOVA with Tukey post-test. C-D, C57BL6/J or Batf3–/– mice were primed on day 0 and boosted on day 14 with 5 nmol EGP9 peptide or DSPE-PEG-EGP9 and 1.24 nmol lipo-CpG, and ICS was performed on day 22. C, Shown are representative IFNγ plots gated on live CD8+ T cells. D, Mean T-cell responses quantified as % IFNγ+ of CD8+. n = 5/group; ANOVA with Tukey post-test; representative of 2 independent experiments. E-F, C57BL6/J mice were primed on day 0 and boosted on day 14 with 25 µg c-di-GMP and 5 nmol EGP20 peptide or construct, and ICS was performed 7 days post-boost. Mean T-cell responses quantified as % IFNγ+ of CD8+. n = 5/group; ANOVA with Bonferroni correction for preplanned comparisons. *P<0.05, **P<0.01, ***P<0.001.
Although amph-vaccines were found to accumulate in many APC populations in LNs, we expected that any DSPE-PEG-peptide conjugate (even optimal nonamer epitopes) may require processing to cleave the DSPE-PEG linkage for antigen presentation, and, thus, might require cross-presenting DCs for activity. To test this hypothesis, we immunized Batf3–/– mice, which lack the transcription factor necessary to develop cross-presenting DCs (25). Compared to w.t. mice, priming of T-cell responses to the optimal EGP 9-mer peptide (DSPE-PEG-EGP9) were severely compromised in Batf3–/– mice (Fig. 3C-D), indicating that even “short” DSPE-PEG-peptides may be reliant upon cross-presenting DCs for effective priming.
Immunogenicity of amph-peptide vaccines is insensitive to conjugate linkage chemistry
We previously focused on forming amphiphile conjugates by linking DSPE-PEG-maleimide to the free thiol of an N-terminal cysteine introduced on peptide antigens. However, this conjugation chemistry would be problematic if an antigen sequence of interest had an internal cysteine. To determine whether the immunogenicity of amph-vaccines is sensitive to the linkage chemistry, conjugates were synthesized where DSPE-PEG was either linked using cysteine-maleimide chemistry, copper-free click chemistry, or via amine-succinimidyl ester chemistry to form an amide bond directly to the N-terminus of the peptide antigen (Supplementary Fig. S1). All of these conjugate-peptides demonstrated superior immunogenicity relative to unmodified peptides, and conjugates formed with the different linkages were not statistically different from another (Fig. 3E), suggesting that immunogenicity of peptide constructs is insensitive to the PEG-lipid linkage. When mice were immunized with either N-or C-terminal DSPE-PEG or ABP-PEG antigen conjugates, both showed a trend towards N-terminal conjugation being superior to C-terminal conjugation (Fig. 3F).
Conjugation of peptide antigens with DSPE-PEG enhances serum stability
PEG conjugation has been shown to enhance the serum stability of peptides by protecting from serum proteases (12,26). To test whether amph-peptides show a similar increase in resistance to proteolytic degradation, unmodified EGP20 peptide, DSPE-PEG linked to EGP20 either through an N-terminal or C-terminal cysteine, or PEG-EGP20 without the diacyl lipid (linked N-terminally) were incubated with fresh mouse serum. Serum-treated or fresh peptides were then added to splenocytes from gp100-primed mice, and the IFNγ production of the stimulated cells was quantified by ICS. As shown in Fig. 4A, the percentage of IFNγ+ CD8+ T cells in cultures stimulated by unmodified EGP20 peptide (normalized to the maximal response of the untreated antigen) showed a substantial drop after incubation of the peptide overnight with serum. By contrast, each of the PEG and DSPE-PEG peptide conjugates retained nearly maximal stimulatory activity after the same serum treatment (Fig. 4B-D).
Figure 4: DSPE-PEG conjugation protects peptide integrity; impact of linkage chemistries on priming.
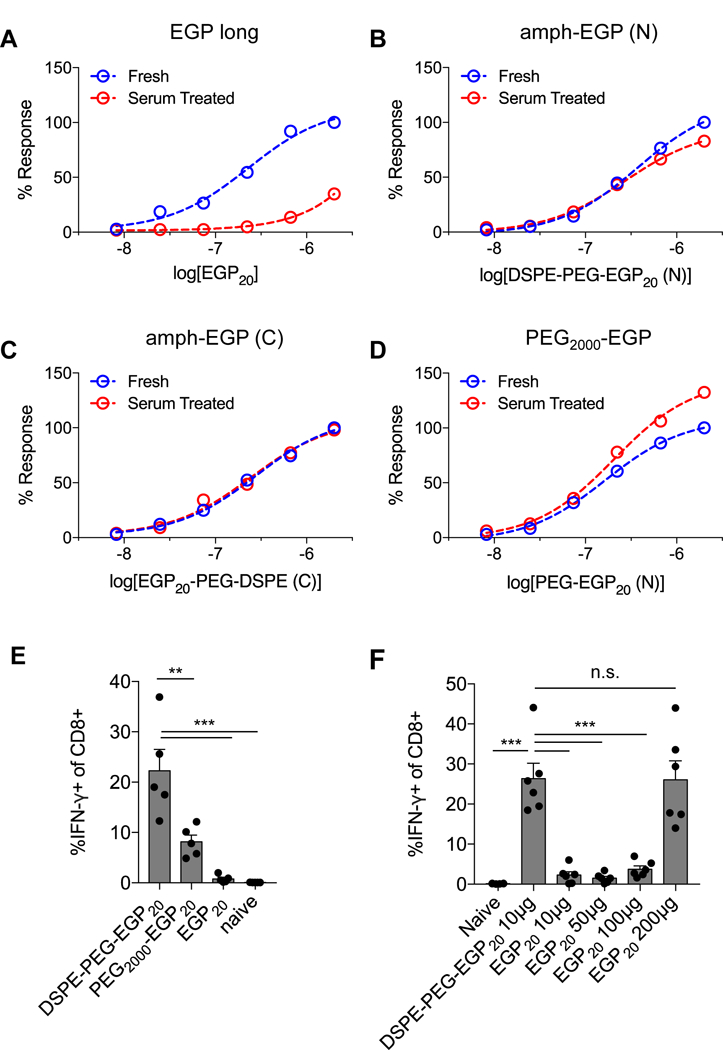
A-D, EGP20 peptide constructs were incubated overnight with 10% fresh mouse serum. Serum-treated or fresh constructs were incubated with pooled splenocytes from previously DSPE-PEG-EGP20 immunized mice for 24 hours, and ICS was performed after 18 hours. A, Normalized IFNγ+ T-cell responses for EGP20 peptide; B, DSPE-PEG-EGP20 with N-terminal DSPE-PEG conjugation; C, DSPE-PEG-EGP20 with C-terminal DSPE-PEG conjugation; and D, PEG-EGP20 with N-terminal PEG conjugation. Representative of 2 independent experiments. E, C57BL6/J mice were primed on day 0 and boosted on day 14 with 25 µg c-di-GMP and 5 nmol EGP20 peptide or peptide construct, and ICS was performed 7 days later. Mean T-cell responses quantified as % IFNγ+ of CD8+. n = 5/group; ANOVA with Tukey post-test. F, Mice were primed on day 0 and boosted on day 14 with 10µg DSPE-PEG-EGP20 or indicated doses of EGP20, and mean T-cell responses quantified as % IFNγ+ of CD8+ 6 days later. n = 5/group; ANOVA with Tukey post-test. *P<0.05, **P<0.01, ***P<0.001.
To evaluate the relative contribution of enhanced serum stability vs. lymphatic trafficking in the immunogenicity of amph-vaccines, mice were vaccinated with DSPE-PEG-EGP20 or the identical construct lacking the diacyl lipid (PEG-EGP20). PEG-EGP20 retained approximately 40% of the activity of the albumin-binding DSPE-PEG-EGP20 (Fig. 4E), indicating that protection from serum proteases is an important but not dominant contributing mechanism in amph-vaccine immunogenicity. Given the observation that albumin-binding conjugates both trafficked to LNs more effectively and showed enhanced stability in vivo, we directly assessed whether any dose of a soluble long peptide vaccine could elicit T-cell responses comparable to the equivalent amph-vaccine. With EGP20 antigen, a 20x higher dose of unmodified antigen was required to elicit comparable priming (Fig. 4F). We also carried out comparative immunizations of free peptides and amphiphile-conjugates of four additional “long” and optimal 9-mer peptide antigens, including both CD4 and CD8 epitopes, to assess the generality of the increased immunogenicity of amph-vaccines, and found consistent increases in T-cell responses elicited by each of these antigens (Supplementary Fig. S4). These data suggest that amph-vaccines promote an increase in vaccine potency that would be difficult to achieve in humans simply by increased free peptide dosing.
DSPE-PEG peptides achieve broad lymphatic distribution and extend in vivo presentation
Enhancement of presentation of an antigen in vivo may result from a number of distinct processes: quantitatively more peptide can be taken up in a given LN, presentation can occur in more nodes throughout the lymphatic chain, or presentation might occur for an extended duration of time. To define more completely the location and longevity of peptide presentation, we utilized pmel-1 TCR-transgenic CD8+ T cells, which recognize EGP9 in the context of H-2Db, as reporters of antigen presentation. Mice were immunized with DSPE-PEG-EGP20 plus adjuvant, EGP20 peptide plus adjuvant, or adjuvant alone, and CFSE-labeled pmel-1 CD8+ T cells were transferred intravenously 24 hours later, followed by transfer of CellTrace Violet-labeled pmel-1 CD8+ T cells an additional 24 hours after that (Fig. 5A-B). Under these conditions, the CFSE-labeled pmel-1 T cells encountering antigen will proliferate in the node in which they encounter antigen and serve as an effective proxy for antigen presence after immunization. The violet labeled pmel-1 T cells (which do not have sufficient time to divide) upregulate CD69 in response to antigen encounter and inform about antigen levels 48 hours after immunization. Substantially higher proliferation of CFSE-labeled reporter cells was seen with DSPE-PEG-EGP20 compared to EGP20 peptide in all nodes examined, except the direct draining inguinal node (Fig. 5C and D). Similarly, CD69 was upregulated among pmel-1 T cells transferred in 48 hours after immunization to a much higher extent in all nodes with DSPE-PEG-EGP20 compared to unconjugated peptide (Fig. 5E, F), and presentation of DSPE-PEG-EGP20 was seen even in the distal mesenteric nodes. Next, mice were immunized and CFSE-labeled pmel-1 T cells were transferred 7 days later. In LNs that were resected 48 hours after transfer, no proliferation in any node was seen with unmodified EGP20 peptide, but DSPE-PEG-EGP20 resulted in proliferation in the inguinal, axillary, and lumbar nodes, indicating that DSPE-PEG-peptide is presented for at least one week in vivo (Fig. 5G-H). Thus, DSPE-PEG-peptides achieved broad lymphatic distribution and extended in vivo presentation.
Figure 5: DSPE-PEG-peptides achieve broad lymphatic distribution and extended presentation.
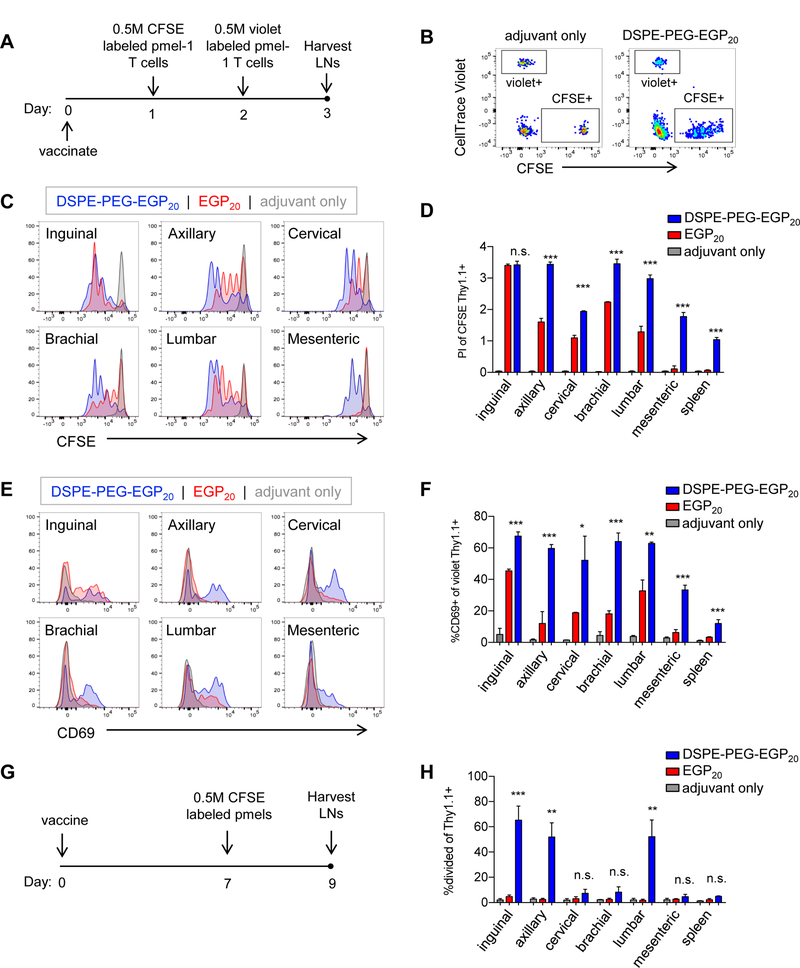
A-F, C57BL6/J mice were immunized with 25 µg c-di-GMP and either 5 nmol EGP20 peptide, DSPE-PEG-EGP20 peptide, or no antigen. 24 hours (h) later, 0.5 million CFSE-labeled pmel-1 CD8+ T cells were transferred intravenously, and 48h after immunization, 0.5 million CellTrace Violet-labeled pmel-1 CD8+ T cells were transferred intravenously. LNs and spleens were isolated from recipient mice 24h later. A, Experimental timeline. B, Gating strategy to identify pmel-1 transferred populations, gated on live CD8+ T cells. C, Representative plots showing CFSE dilution among CD8+Thy1.1+ cells transferred 24h after immunization. D, Mean proliferation index of pmel-1 T cells in indicated LNs or spleen. n = 3/group; student’s t-test between EGP20 and DSPE-PEG-EGP20; representative of 3 independent experiments. E, Representative CD69 expression among CD8+Thy1.1+CellTrace violet+ cells transferred 48h after immunization. F, Mean %CD69+ of CD8+Thy1.1+CellTrace violet+ pmel-1 T cells in indicated LNs or spleen. n = 3/group; student’s t-test between EGP20 and DSPE-PEG-EGP20; representative of 3 independent experiments. G-H, Mice were immunized with 25 µg c-di-GMP and either 5 nmol EGP20 peptide, DSPE-PEG-EGP20 peptide, or no antigen. Seven days later, 0.5 million CFSE-labeled pmel-1 CD8+ T cells were transferred intravenously. LNs were isolated and processed 48h after transfer. F, Experimental timeline. G, % dividing CD8+Thy1.1+ in indicated LNs or spleen. n = 3/group; student’s t-test between EGP20 and DSPE-PEG-EGP20; representative of 2 independent experiments. *P<0.05, **P<0.01, ***P<0.001.
To assess the phenotype of T cells primed by this sustained and systemic antigen presentation, we measured surface markers and cytokine production capabilities of antigen-specific peripheral blood T cells. At 6 days post boost, we observed that approximately 90% of antigen-specific cells primed expressed markers consistent with an effector memory phenotype (CD44highCD62Llow), with <3% PD-1+Tim3+ exhausted cells (Supplementary Fig. S5A-B). We observed that with the adjuvants used here, the CD8+ T-cell response exhibited a Tc1 rather than a Tc2 or Tc17 phenotype, as determined by production of IFNγ but not IL4 or IL17 within tetramer+CD8+ T cells (Supplementary Fig. S5C).
Amph-peptides combine effectively with several different molecular adjuvants
Although DSPE-PEG peptides showed clear superiority over unmodified antigens when administered with molecular adjuvants, peptide vaccines are often clinically administered in combination with Incomplete Freund’s Adjuvant (IFA), a water-in-oil emulsion that induces local inflammation and forms a depot, protecting antigens from degradation and releasing them over time (27). In mouse models, peptides in IFA alone have been shown to create a sink for effector T cells, but addition of TLR agonists to peptides in IFA elicits potent immune responses (10). We, thus, sought to examine the LN trafficking of peptides in IFA vs. DSPE-PEG-conjugates and to compare the immunogenicity of IFA vs. molecular adjuvants for peptides or DSPE-PEG-peptides. First, FAM-labeled DSPE-PEG-EGP20 was formulated with lipo-CpG, c-di-GMP (STING agonist), polyI:C (TLR3 agonist), or IFA, and injected subcutaneously. Drainage to the inguinal and axillary nodes was quantified by IVIS (Fig. 6A-C). Formulation of antigen with lipo-CpG, c-di-GMP, or polyI:C did not substantially alter LN accumulation, but formulation of DSPE-PEG-peptide in IFA prevented efficient lymphatic drainage. To evaluate the impact on vaccine immunogenicity, mice were primed and boosted with EGP20 or DSPE-PEG-EGP20 adjuvanted with c-di-GMP or polyI:C, either injected in saline or formulated in IFA, and responses were quantified with ICS (Fig. 6D). Responses to free peptides adjuvanted with either polyI:C or CDN trended towards improvement with IFA, but responses to DSPE-PEG-peptide were hindered by formulation in IFA, decreasing approximately 50% in magnitude compared to formulation in saline. These data suggest DSPE-PEG-peptides exhibit superior immunogenicity compared to unmodified peptides when combined with a number of molecular adjuvants, including the most potent clinically-employed adjuvant combinations.
Figure 6: DSPE-PEG peptides are immunogenic with multiple adjuvants, and trafficking/priming is diminished by IFA.
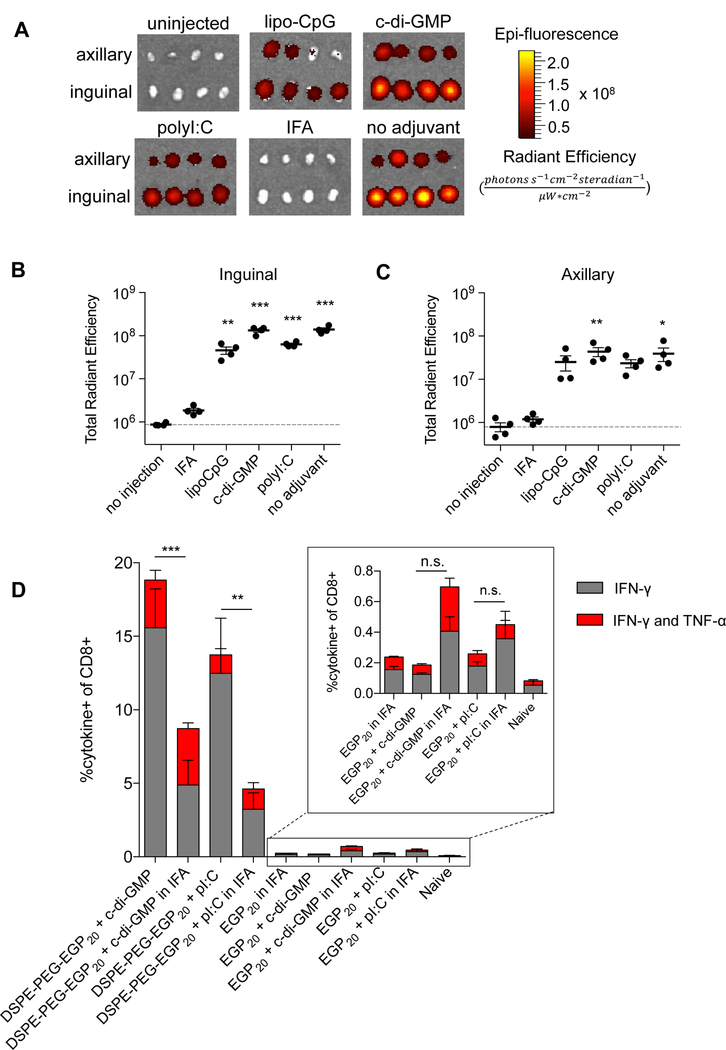
A-C, Mice were immunized with 15 nmol FAM-labeled DSPE-PEG-EGP20 alone, formulated in IFA, or with the indicated adjuvant. LNs were resected 24 hours later. Uptake was quantified using IVIS. A, Images of resected nodes. n=4/group/LN type. B-C, Quantification of background-subtracted radiant efficiency ((photons*s−1*cm−2*steradian−1)/(µW*cm2)) in indicated LNs. n = 4/group; ANOVA with Dunnet’s post-test comparing each group to no injection; representative of 2 independent experiments. D, Mice were primed on day 0 and boosted on days 14 and 28 with 25 µg c-di-GMP and 5 nmol EGP20 peptide construct. ICS was performed on PBMCs on day 35. Mean cytokine responses quantified as % cytokine+ of CD8+. n = 5/group; ANOVA with Bonferroni correction for shown preplanned comparisons. *P<0.05, **P<0.01, ***P<0.001.
Amph-peptide conjugates are processed by human APCs similarly to free peptides
To assess if processing and presentation of amph-peptides is performed efficiently by human antigen presenting cells in vitro, we performed an IFNγ ELISPOT with primary human monocyte-derived DCs. The KF11 CD8+ T-cell clone (recognizing HIV gag p2430–40 in the context of HLA-B57 (20)) was used to probe for presentation in a coculture. DCs generated from an HLA-B57+ donor were pulsed with the HIV-derived gag p2428–60 peptide or DSPE-PEG-p2428–60 at three concentrations and cocultured with KF11-specific CD8+ T cells, and IFNγ spot forming units (SFU) were quantified. As shown in Fig. 7A, similar responses were observed for DSPE-PEG-p24 and p24 peptide, indicating that the functional integration of uptake, processing, and presentation of DSPE-PEG-peptide is equivalent to unmodified peptide in vitro for an immunodominant MHC class I restricted epitope.
Figure 7: DSPE-PEG peptides can be presented by human DCs, with antigen processing similar to unmodified peptides.
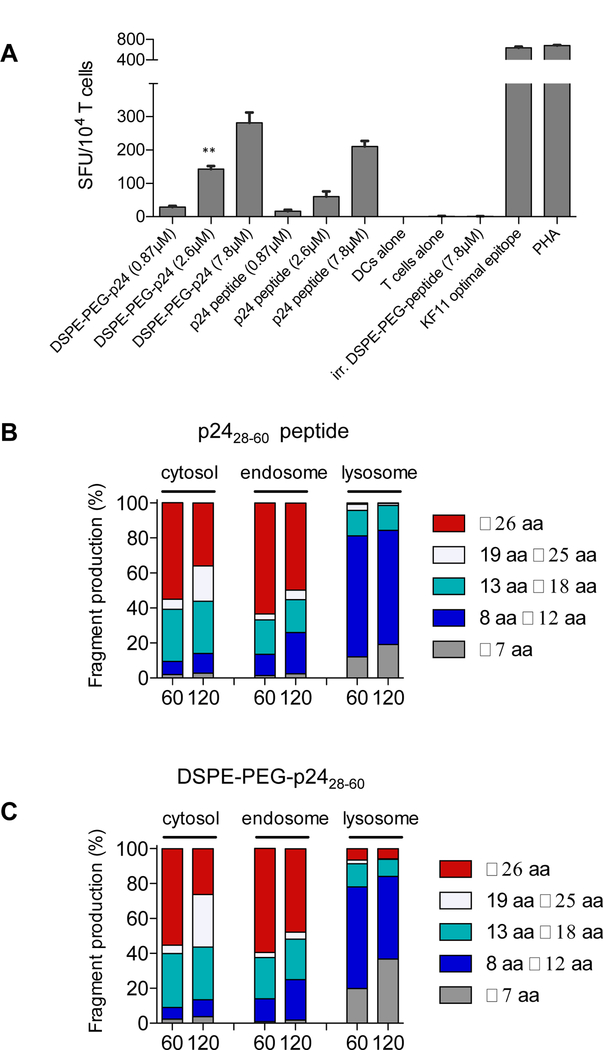
A, Monocyte-derived DCs were generated from HLA-B57+ PBMCs and incubated with free HIV p2428–60 peptide or DSPE-PEG-p2428–60 for 4 hours, washed, and plated overnight with KF11 CD8+ T-cells in a 2:1 effector-to-target ratio. Spot forming units (SFU) per 104 KF11+ T cells quntified. n = 3/group; student’s t-test for indicated comparisons between DSPE-PEG-p2428–60 and p2428–60 peptide for a given concentration. B-C, p2428–60 peptide or DSPE-PEG-p2428–60 (2 nmol) were incubated with cell extracts from endosomal, lysosomal, or cytosolic cellular compartments isolated from human APCs. Resulting peptides were purified and analyzed via mass spectrometry. Shown are the lengths of peptide fragments produced under cytosolic, endosomal, or lysosomal degradation conditions for 60 or 120 minutes. *P<0.05, **P<0.01, ***P<0.001.
The repertoire of peptides generated by antigen processing in APCs is one of many factors that influences T-cell responses (28). To uncover if conjugation of peptide antigens with DSPE-PEG alters the cleavage patterns and resultant peptides generated, HIV gag p2428–60 peptide or DSPE-PEG-p2428–60 were subjected to degradation in cellular extracts from endosomal, lysosomal, or cytosolic compartments derived from human monocyte-derived DCs and analyzed by mass spectrometry as previously described (21). The MHC class I and class II epitopes and the lengths of all fragments generated by degradation of p2428–60 peptide and DSPE-PEG-p2428–60 were virtually identical at both 60 and 120 minutes in each of the cellular compartments analyzed (Fig. 7B-C and Supplementary Table S1), suggesting that conjugation with DSPE-PEG does not interfere with antigen processing machinery by human APCs.
Discussion
We have previously shown that conjugation of CpG oligonucleotide adjuvants or peptide antigens with diacyl lipids (through a PEG spacer in the peptide case) significantly enhances LN accumulation of these vaccine agents, and, thereby, augments T-cell priming (14). Here, we extended those results to show that substitution of the diacyl lipid with α-tocopherol as the albumin-binding moiety similarly enhanced both antigen and adjuvant activity. We also showed that non-lipid modifications, namely fusion of antigens with albumin-binding peptides, also allowed for enhancement of LN delivery and T-cell priming. The former result was noteworthy, given that α-tocopherol has a low affinity for albumin (KD ~7 μM (22)). Effective lymphatic delivery, even with low affinity binders, is likely facilitated by the very high concentrations of albumin in interstitial fluid (29). Our data suggest that amphiphilic peptides utilizing lipid albumin binding domains, DSPE-PEG-EGP20 and Vit. E-PEG-EGP20, are superior to compositions incorporating an ABP. However, we cannot rule out that further optimization of the affinity of these ABPs for albumin or their linkages to the peptide antigens might further increase activity of ABP-PEG-peptide conjugates (19).
Although our prior work provided a reasonable basis for understanding how albumin could mediate vaccine trafficking, the mechanisms underlying antigen capture and processing in the LN remained unclear. The best characterized receptor for albumin, FcRn, does not play a role in the immune response to these albumin-binding conjugates, as FcRn–/– and w.t. mice responded to amph-vaccines similarly. Although other receptors for albumin have been described, including albondin (gp60) and several scavenger receptors that bind conformationally altered albumin to redirect it for degradation, these other receptors are not known to be expressed on antigen presenting cells (30). Albumin is readily internalized by macropinocytosis (31), and this may represent a main mode of uptake of albumin-bound vaccines.
Conjugation of optimal MHC I epitopes with DSPE-PEG has enhanced activity for a number of antigens (14), but it was unclear if this linkage changes the antigen presentation requirements of short peptides. Namely, short soluble peptides may undergo peptide exchange, loading directly onto MHC on the surface of non-professional antigen presenting cells, including T cells and B cells, which may promote tolerance in some circumstances (32–34). By contrast, DSPE-PEG-peptides appear to rely on cross-presenting DCs, even for optimal nonamer antigens, which may allow optimal epitopes to be used in vaccination without concern for presentation by non-professional APCs.
The duration of antigen presentation has been shown to impact the magnitude of T cell effector and memory responses, with longer antigen presentation resulting in more effective CD8+ T cell memory formation compared to brief exposures (35,36), and priming in distal nodes was identified as one of the characteristics of DEC-205–targeted antigens, which promote potent CD8+ T-cell immunity (37). Performing vaccination at multiple injection sites in order to increase the number of LNs utilized for priming has also been shown to enhance the magnitude of T-cell responses (38). We showed here that DSPE-PEG-peptides embodied these important principles, as they are presented in multiple nodes systemically and for at least one week in vivo. We observed that with the adjuvants used here, the T-cell response exhibited a Tc1 phenotype, and we did not see evidence of exhaustion by PD-1 and Tim-3 staining. These results suggest that immunization with DSPE-PEG-peptides recapitulates some traits of systemic viral infection: presentation of antigen in many lymphoid organs for a prolonged duration.
Peptide antigen modification with PEG significantly enhanced T-cell priming, even without including an albumin-binding domain. This was likely mediated through an increase in serum stability and in vivo half-life, as has been demonstrated for PEG modification of erythropoiesis stimulating peptides and anti-viral peptides previously (12,26). This finding indicates that some of the activity of DSPE-PEG-peptides derives from protection of the antigen from serum proteases, although further significant enhancements in immunogenicity are seen with the addition of an albumin-binding domain.
IFA is widely used to enhance immunogenicity of unmodified peptides, but our data suggest that vaccines with lipophilic domains that rely on efficient lymphatic drainage for their mechanism of action may be hindered in activity with formulation in IFA. The mechanism of action of IFA is thought to involve extending in vivo presentation of antigen by forming an antigen depot (27). This may be unnecessary in the case of DSPE-PEG-peptides, which showed an extended in vivo half-life. Although IFA increased priming to free peptides, IFA decreased the T-cell response approximately two-fold for DSPE-PEG-peptides, indicating that IFA should be avoided for translation of DSPE-PEG-peptides clinically. This would have the dual benefit of avoiding T-cell sequestration and induction of apoptosis in IFA depots, as has been observed in mice (10).
Detailed mass spectrometry analysis of fragments produced by degradation in cellular extracts from primary human antigen presenting cells demonstrated that DSPE-PEG conjugation did not alter the repertoire of class I or class II epitopes generated or the overall characteristic lengths of peptide fragments in the pool. Functional antigen presentation assays using HIV gag p24 specific CD8+ T cells confirmed that presentation of the HLA-B57-restricted KF11 epitope was equivalent in vitro. A lack of enhancement of presentation in vitro with DSPE-PEG-peptide over unmodified peptide is consistent with the hypothesized mechanism of action of amph-peptides. Enhancements in trafficking and in vivo serum stability did not play an appreciable role in the in vitro ELISPOT. In summary, simple covalent modification of molecular vaccines with albumin-binding domains was capable of ferrying these molecules throughout the lymphatics, enhancing serum stability and duration of presentation in vivo and increasing T-cell responses by an order of magnitude over unmodified vaccines, and may be of interest for promoting potent antitumor CD8+ T cells clinically.
Supplementary Material
Acknowledgments
DJI is an investigator of the Howard Hughes Medical Institute. We thank the Koch Institute Swanson Biotechnology Center for technical support, specifically the applied therapeutics and whole-animal imaging facility, the histology facility, and the flow cytometry facility. We thank Mark Karver and the Peptide Synthesis Core Facility of the Simpson Querrey Institute at Northwestern University for peptide synthesis. The U.S. Army Research Office, the U.S. Army Medical Research and Materiel Command, and Northwestern University provided funding to develop this core facility and ongoing support is being received from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205). K.D.M. is supported by the Fannie & John Hertz Foundation Fellowship, the NSF Graduate Research Fellowships, and the Siebel Scholarship. D.J.I. is an investigator of the Howard Hughes Medical Institute.
Grant Support
This work was supported in part by the Ragon Institute of MGH, MIT, and Harvard, the Mayo Clinic-Koch Institute Collaboration, the Koch Institute Support (core) grant P30-CA14051 from the National Cancer Institute, the Marble Center for Cancer Nanomedicine, the Bridge Project partnership between the Koch Institute for Integrative Cancer Research and the Dana Farber/Harvard Cancer Center (DF/HCC), the V Foundation, and the NIH (awards EB022433, CA174795, CA206218, and CA172164). The project described was supported by award Number T32GM007753 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Footnotes
Conflict of interest statement: D.J.I. holds equity in Vedantra Pharmaceuticals, which holds a license to the amphiphile–vaccine technology used in these studies.
References
- 1.Rosenberg SA. Decade in review[mdash]cancer immunotherapy: Entering the mainstream of cancer treatment. Nat Rev Clin Oncol 2014;11:630–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Melief CJM, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. The Journal of Clinical Investigation 2015;125:3401–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017;547:222–6 [DOI] [PubMed] [Google Scholar]
- 4.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017;547:217–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melief CJM, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer 2008;8:351–60 [DOI] [PubMed] [Google Scholar]
- 6.Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, et al. gp100/pmel 17 Is a Murine Tumor Rejection Antigen: Induction of “Self”-reactive, Tumoricidal T Cells Using High-affinity, Altered Peptide Ligand. The Journal of Experimental Medicine 1998;188:277–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nava-Parada P, Forni G, Knutson KL, Pease LR, Celis E. Peptide Vaccine Given with a Toll-Like Receptor Agonist Is Effective for the Treatment and Prevention of Spontaneous Breast Tumors. Cancer Research 2007;67:1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sikora AG, Jaffarzad N, Hailemichael Y, Gelbard A, Stonier SW, Schluns KS, et al. IFN-α Enhances Peptide Vaccine-Induced CD8<sup>+</sup> T Cell Numbers, Effector Function, and Antitumor Activity. The Journal of Immunology 2009;182:7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davila E, Kennedy R, Celis E. Generation of Antitumor Immunity by Cytotoxic T Lymphocyte Epitope Peptide Vaccination, CpG-oligodeoxynucleotide Adjuvant, and CTLA-4 Blockade. Cancer Research 2003;63:3281. [PubMed] [Google Scholar]
- 10.Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang X-F, et al. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nat Med 2013;19:465–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathur D, Prakash S, Anand P, Kaur H, Agrawal P, Mehta A, et al. PEPlife: A Repository of the Half-life of Peptides. Scientific Reports 2016;6:36617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan Q, Leuther KK, Holmes CP, Fong K-l, Zhang J, Velkovska S, et al. Preclinical evaluation of Hematide, a novel erythropoiesis stimulating agent, for the treatment of anemia. Experimental Hematology 2006;34:1303–11 [DOI] [PubMed] [Google Scholar]
- 13.Nguyen LT, Chau JK, Perry NA, de Boer L, Zaat SAJ, Vogel HJ. Serum Stabilities of Short Tryptophan-and Arginine-Rich Antimicrobial Peptide Analogs. PLOS ONE 2010;5:e12684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 2014;507:519–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindner V, Heinle H. Binding properties of circulating evans blue in rabbits as determined by disc electrophoresis. Atherosclerosis 1982;43:417–22 [DOI] [PubMed] [Google Scholar]
- 16.Tsopelas C, Sutton R. Why Certain Dyes Are Useful for Localizing the Sentinel Lymph Node. Journal of Nuclear Medicine 2002;43:1377–82 [PubMed] [Google Scholar]
- 17.Mijalis AJ, Thomas Iii DA, Simon MD, Adamo A, Beaumont R, Jensen KF, et al. A fully automated flow-based approach for accelerated peptide synthesis. Nat Chem Biol 2017;13:464–6 [DOI] [PubMed] [Google Scholar]
- 18.van Stipdonk MJB, Badia-Martinez D, Sluijter M, Offringa R, van Hall T, Achour A. Design of Agonistic Altered Peptides for the Robust Induction of CTL Directed towards H-2D<sup>b</sup> in Complex with the Melanoma-Associated Epitope gp100. Cancer Research 2009;69:7784. [DOI] [PubMed] [Google Scholar]
- 19.Dennis MS, Zhang M, Meng YG, Kadkhodayan M, Kirchhofer D, Combs D, et al. Albumin Binding as a General Strategy for Improving the Pharmacokinetics of Proteins. Journal of Biological Chemistry 2002;277:35035–43 [DOI] [PubMed] [Google Scholar]
- 20.Goulder PJ, Tang Y, Pelton S, Walker B.HLA-B57-Restricted Cytotoxic T-Lymphocyte Activity in a Single Infected Subject toward Two Optimal Epitopes, One of Which Is Entirely Contained within the Other. J. Virol 2000; 74(11): 5291–5299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dinter J, Duong E, Lai NY, Berberich MJ, Kourjian G, Bracho-Sanchez E, et al. Variable Processing and Cross-presentation of HIV by Dendritic Cells and Macrophages Shapes CTL Immunodominance and Immune Escape. PLOS Pathogens 2015;11:e1004725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fanali G, Fasano M, Ascenzi P, Zingg J-M, Azzi A. α-Tocopherol binding to human serum albumin. BioFactors 2013;39:294–303 [DOI] [PubMed] [Google Scholar]
- 23.Qiao S-W, Kobayashi K, Johansen F-E, Sollid LM, Andersen JT, Milford E, et al. Dependence of antibody-mediated presentation of antigen on FcRn. Proceedings of the National Academy of Sciences 2008;105:9337–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker K, Qiao S-W, Kuo TT, Aveson VG, Platzer B, Andersen J-T, et al. Neonatal Fc receptor for IgG (FcRn) regulates cross-presentation of IgG immune complexes by CD8−CD11b+ dendritic cells. Proceedings of the National Academy of Sciences 2011;108:9927–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 Deficiency Reveals a Critical Role for CD8α+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008;322:1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Danial M, van Dulmen THH, Aleksandrowicz J, Pötgens AJG, Klok H-A. Site-Specific PEGylation of HR2 Peptides: Effects of PEG Conjugation Position and Chain Length on HIV-1 Membrane Fusion Inhibition and Proteolytic Degradation. Bioconjugate Chemistry 2012;23:1648–60 [DOI] [PubMed] [Google Scholar]
- 27.Aucouturier J et al. Montanide ISA 720 and 51: a new generation of water in oil emulsions as adjuvants for human vaccines. Expert Review of Vaccines 2002;1:111–8 [DOI] [PubMed] [Google Scholar]
- 28.Tenzer S, Wee E, Burgevin A, Stewart-Jones G, Friis L, Lamberth K, et al. Antigen processing influences HIV-specific cytotoxic T lymphocyte immunodominance. Nat Immunol 2009;10:636–46 [DOI] [PubMed] [Google Scholar]
- 29.Lönsmann Poulsen H Interstitial Fluid Concentrations of Albumin and Immunoglobulin G in Normal Men. Scandinavian Journal of Clinical and Laboratory Investigation 1974;34:119–22 [PubMed] [Google Scholar]
- 30.Merlot AM, Kalinowski DS, Richardson DR. Unraveling the mysteries of serum albumin—more than just a serum protein. Frontiers in Physiology 2014;5:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hackstein H, Taner T, Logar AJ, Thomson AW. Rapamycin inhibits macropinocytosis and mannose receptor–mediated endocytosis by bone marrow–derived dendritic cells. Blood 2002;100:1084. [DOI] [PubMed] [Google Scholar]
- 32.Bijker MS, van den Eeden SJF, Franken KL, Melief CJM, van der Burg SH, Offringa R. Superior induction of anti-tumor CTL immunity by extended peptide vaccines involves prolonged, DC-focused antigen presentation. European Journal of Immunology 2008;38:1033–42 [DOI] [PubMed] [Google Scholar]
- 33.Aichele P, Brduscha-Riem K, Zinkernagel RM, Hengartner H, Pircher H. T cell priming versus T cell tolerance induced by synthetic peptides. The Journal of Experimental Medicine 1995;182:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toes RE, Offringa R, Blom RJ, Melief CJ, Kast WM. Peptide vaccination can lead to enhanced tumor growth through specific T-cell tolerance induction. Proceedings of the National Academy of Sciences 1996;93:7855–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johansen P, Storni T, Rettig L, Qiu Z, Der-Sarkissian A, Smith KA, et al. Antigen kinetics determines immune reactivity. Proceedings of the National Academy of Sciences 2008;105:5189–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bachmann MF, Beerli RR, Agnellini P, Wolint P, Schwarz K, Oxenius A. Long-lived memory CD8+ T cells are programmed by prolonged antigen exposure and low levels of cellular activation. European Journal of Immunology 2006;36:842–54 [DOI] [PubMed] [Google Scholar]
- 37.Bonifaz LC, Bonnyay DP, Charalambous A, Darguste DI, Fujii S-I, Soares H, et al. In Vivo Targeting of Antigens to Maturing Dendritic Cells via the DEC-205 Receptor Improves T Cell Vaccination. The Journal of Experimental Medicine 2004;199:815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mould RC, AuYeung AWK, van Vloten JP, Susta L, Mutsaers AJ, Petrik JJ, et al. Enhancing Immune Responses to Cancer Vaccines Using Multi-Site Injections. Scientific Reports 2017;7:8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.