

SUMMARY
Innate lymphoid cells (ILCs) are important for mucosal immunity. The intestine harbors all ILC subsets, but how these cells are balanced to achieve immune homeostasis and mount appropriate responses during infection remains elusive. Here, we show that aryl hydrocarbon receptor (Ahr) expression in the gut regulated ILC balance. Among ILCs, Ahr was most highly expressed by gut ILC2s, and controlled chromatin accessibility at the Ahr gene locus via positive feedback. Ahr signaling suppressed Gfi1 transcription factor-mediated expression of the interleukin 33 receptor ST2 in ILC2s and expression of ILC2 effector molecules IL-5, IL-13 and amphiregulin in a cell-intrinsic manner. Ablation of Ahr enhanced anti-helminth immunity in the gut, while genetic or pharmacological activation of Ahr suppressed ILC2 function but enhanced ILC3 maintenance to protect the host from Citrobacter rodentium infection. Thus, the host regulates the gut ILC2-ILC3 balance by engaging the Ahr pathway to mount appropriate immunity against various pathogens.
In Brief
The aryl hydrocarbon receptor (Ahr) promotes ILC3 maintenance and function. Li et al. report that Ahr restricts intestinal ILC2 function in a cell-intrinsic manner, suggesting a central role for gut adaptation of Ahr expression in regulating the ILC2-ILC3 balance.
Graphical Abstract

Introduction
Innate lymphoid cells (ILCs) represent an emerging family of cell types, including natural killer (NK) cells that express both transcription factors T-bet and Eomes, group 1 ILCs (ILC1s) that express T-bet but not Eomes, group 2 ILCs (ILC2s) that express transcription factor GATA3, and group 3 ILCs (ILC3s) that express transcription factor RORγt (Artis and Spits, 2015). ILCs play important roles in tissue remodeling and innate immunity, particularly at barrier surfaces (Ebbo et al., 2017; Klose and Artis, 2016). Various tissues contain distinct ILC subsets. For instance, the liver is enriched with ILC1s; the lung contains ILC2s in mice under the steady state despite the existence of other ILCs in human; in contrast, the mouse gut harbors all three major ILC subsets (Li et al., 2017; Simoni and Newell, 2018). It is unknown how various ILCs are kept in balance for gut immune homeostasis and how the host orchestrates a diverse population of ILCs to control infection and inflammation.
The aryl hydrocarbon receptor (Ahr) is a ligand-dependent environmental sensor and its transcriptional activity requires ligand-induced nuclear translocation and dimerization with its partner, the aryl hydrocarbon receptor nuclear translocator (ARNT) (Cella and Colonna, 2015). Best known for sensing environmental toxins [e.g., 2,3,7,8-tetrachloro-dibenzo-p-dioxin (TCDD), Ahr can also respond to endogenous ligands generated from host cells, diet, and microbiota (Zhou, 2016). Ahr is expressed in barrier tissues (e.g., the gut and the skin) by immune cells such as lymphocytes and by tissue structural cells such as epithelial and stromal cells (Stockinger et al., 2014). The presence of Ahr-expressing cells in those locations may represent a mechanism of tissue adaptation by allowing host to readily respond to external environmental changes. Consistent with this notion, regulatory T (Treg) cells in the gut expressed the highest amounts of Ahr compared to those in other lymphoid or non-lymphoid tissues (Ye et al., 2017).
Recent studies show that Ahr plays important roles in regulating ILCs. For example, Ahr is required for liver-resident ILC1s maintenance (Zhang et al., 2016). Ahr is also essential for ILC3s maintenance and function (Kiss et al., 2011; Lee et al., 2011; Qiu et al., 2012). Ahr-expressing ILC3s regulate intestinal T helper 17 (Th17) cells and microbiota, and thus gut inflammation (Hepworth et al., 2013; Qiu et al., 2013). Mechanistically, Ahr works cooperatively with transcription factors RORγt and STAT3 to induce interleukin-22 (IL-22) for anti-bacterial immunity (Guo et al., 2014; Qiu et al., 2012). Ahr activity in ILC3s can be inhibited by a zinc finger transcription factor, Ikaros, via a mechanism involving disruption of Ahr-ARNT complex (Li et al., 2016). Despite the progress of our understanding of Ahr in ILCs, how Ahr expression is regulated in ILCs, and its function in other ILCs (e.g., ILC2s) are unknown. In this study, we showed that among various ILCs in the gut, Ahr was expressed at the highest levels by ILC2s. Specific chromatin events marked a tissue adaptation of Ahr expression in gut ILC2s. In contrast to its function in promoting ILC3s maintenance, Ahr suppressed ILC2 function, thus representing a critical node of regulation of the ILC balance for gut immunity against various pathogenic infections.
Results
Ahr is expressed specifically in gut-resident ILC2s
To gain insight of the role of Ahr in ILCs, we asked how Ahr is expressed in various ILC lineages. Compared to ILC progenitors (CHILP and ILC2P) and other mature ILCs (including ILC3), gut ILC2s expressed the highest amounts of Ahr protein and mRNA (Figures 1A, 1B, S1A and S1B). We have recently developed an Ahr knock-in mouse model, AhrdCAIR/+, which expresses a constitutively active form of Ahr (CA-Ahr) and IRES-GFP from the endogenous Ahr gene locus (Figure S1C) (Ye et al., 2017). The IRES-GFP is controlled by the endogenous promoter/enhancer of Ahr locus in AhrdCAIR/+ reporter mice, thus reflecting the transcription of Ahr gene. However, the constitutively active Ahr also expressed in the same cells may affect endogenous Ahr expression. Thus, we compared endogenous Ahr protein expression in wildtype mice and GFP in AhrdCAIR/+ reporter mice. Data showed that gut ILC2s highly expressed Ahr, in contrast to other tissue-resident or circulating ILC2s in the lung, fat, mesenteric lymph node, skin, or blood, supporting a hypothesis of gut-restricted induction and/or maintenance of Ahr expression (Figures 1C to 1E and S1D). Ahr expression in ILC2s was not significantly changed between germ-free mice (GF) and specific pathogen free mice (SPF) (Figure S1E), suggesting a dispensable role for microbiota in Ahr expression by gut ILC2s. Despite expression of Ahr, ILC2s did not make IL-22 (data not shown), presumably due to lack of RORγt and/or STAT3 that work cooperatively with Ahr and are essential for Il22 transcriptional induction in ILC3s (Guo et al., 2014; Qiu et al., 2012).
Figure 1. Ahr is highly expressed by gut ILC2s with unique chromatin features at the Ahr gene locus.

(A) Intracellular staining of Ahr in NK cells (Lin− T-bet+EOMES+RORγt−), ILC1s (Lin−T-bet+EOMES−RORγt−), ILC2s (Lin− GATA3+RORγt−) and T-bet+ or T-bet− ILC3s (Lin−RORγt+GATA3−) isolated from the GATA3+RORγt−) and T-bet+ or T-bet− ILC3s (Lin−RORγt+GATA3−) isolated from the lamina propria of small intestine (SI) or large intestine (LI). Data are representative of three independent experiments. (B) ∆MFI (Mean Fluorescence Intensity) of Ahr expression was calculated as follows: MFI of indicated Ahr+/+ cell populations – MFI of indicated Ahr−/− cell populations. Data are shown as mean ± SEM (n=6). ANOVA followed by Bonferroni’s test for ILC2s versus other ILC subsets in SI or LI. (C) Intracellular staining of Ahr in ILC2s from indicated organs. Data are representative of three independent experiments. (D) Flow cytometry analysis of GFP expression by ILC2s from the various tissues of indicated mice. Data are representative of five independent experiments. (E) Percentages of GFP+ proportion in ILC2s in various tissues of AhrdCAIR/+ mice. Data are shown as mean ± SEM (n=5 for SI, n=6 for LI, lung, and fat). ANOVA followed by Bonferroni’s test for the SI or LI versus other tissues. (F) Representative ATAC-seq tracks at the Ahr locus in ILC2s sorted from SI or LI of Ahr+/+Rag1−/− or Ahr−/− Rag1−/− littermate mice (upper). Recruitment of Gfi1 or GATA3 to the Ahr locus in ILC2s by analyzing published ChIP-seq data (middle). Representative RNA-seq tracks at the Ahr locus in ILC2s sorted from SI or LI of Ahr+/+Rag1−/− or Ahr−/−Rag1−/− littermate mice (lower). (G to I) ILC2s (Lin−KLRG1+CD90+) sorted from LI of Ahr+/+ or Ahr−/− littermate mice were expanded in vitro and subjected to ChIP assay. Enrichment of Ahr (G), Gfi1 (H), or GATA3 (I) at the sites 54 kb (+54 kb) or 14 kb (+14 kb) downstream of the transcription start site was determined by real-time PCR. Data are representative of two independent experiments, and shown as mean ± SEM (n=3). Rabbit IgG isotype antibody was used as a negative control for ChIP of Gfi1 or GATA3. (J) Fragments Per Kilobase of transcript per Million mapped reads (FPKM) of Ahr in RNA-seq of sorted ILC2s from SI or LI of Ahr+/+Rag1−/− or Ahr−/−Rag1−/− littermate mice. The q-value was generated by Cufflinks analysis as described in STAR Methods. ***q<0.001. See also Figures S1 and S2.
Transforming growth factor β (TGFβ) promotes Ahr expression in CD4+ T cells (Kimura et al., 2008; Quintana et al., 2008; Veldhoen et al., 2008), and is shown to regulate the differentiation and function of tissue-resident ILCs (Cortez et al., 2016; Denney et al., 2015). As TGFβ is enriched in the gut, we sought to examine if TGFβ is responsible for the high Ahr expression in the gut ILC2s. Sorted gut ILC2s gradually lost Ahr expression in vitro with or without TGFβ (Figures S1F and S1G). Treatment of TGFβ did not induce Ahr expression in ILC2s that had lost Ahr after culturing in vitro (Figures S1H and S1I). Together, these data suggest that in vivo gut milieu supports Ahr expression in ILC2s, and despite its abundance in the gut, TGFβ itself was not sufficient to sustain and/or induce the expression of Ahr in ILC2s in the in vitro culture system.
Ahr locus in gut ILC2s has unique chromatin structure
To determine the potential mechanism underlying the gut-specific expression of Ahr in ILCs, the chromatin accessibility at the Ahr locus was analyzed using Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) data generated from various ILC lineages (Shih et al., 2016), and several chromatin features at the Ahr locus emerged. Despite various levels of expression of Ahr in ILC subsets and Treg cells (Figures 1A, 1B and S2A), the chromatin conformation was generally open at the transcription start site of Ahr (Figure S2B). At least six regions (boxed in red) were identified at the Ahr locus that showed unique gut ILC2-specific ATAC peaks, i.e., Peak-8, Peak+7.6, Peak+14, Peak+24, Peak+41, and Peak+54, indicating open chromatin (Figure S2B). These peaks were neither evident in other ILCs (NK cells, ILC1s, or ILC3s), nor in lung ILC2s (Figure S2B). Gut Treg cells also express higher amounts of Ahr compared to Treg cells in the lymphoid organs (e.g., thymus, spleen, and lymph nodes) or other tissue-resident Treg cells in the lung, fat, skin, or liver (Ye et al., 2017). However, these gut ILC2-specific open chromatin events at the Ahr locus were mostly absent in gut Treg cells (Figure S2B). Together, these data suggest that these chromatin accessible regions may function as transcriptional enhancers and are responsible for tissue-specific and/or high expression of Ahr by ILC2s in the gut.
Ahr regulates gut-specific chromatin events at the Ahr locus
We next sought to determine whether deletion of Ahr could impact chromatin accessibility at the Ahr locus. To exclude the confounding impact on the ILC2 phenotypes by the adaptive immune system (Halim et al., 2016; Oliphant et al., 2014), gut ILC2s (Lin−KLRG1+CD90+) (Figure S2C) (Pelly et al., 2016) were sorted from Ahr−/− Rag1−/− or littermate Rag1−/− mice. ATAC-seq conducted with the sorted intestinal ILC2 revealed that without prominent changes of other gut ILC2-specific peaks, certain peaks including Peak+54 and Peak+14 were markedly decreased in the absence of Ahr, suggesting that Ahr regulates its own locus chromatin accessibility (Figure 1F). Consistent with unchanged Ahr expression (Figure S1E), the gut ILC2-specific chromatin events at the Ahr locus (e.g., Peak+54) were minimally affected in GF mice without statistical significance compared to SPF mice as revealed by ATAC-seq analysis (Figure S2D).
Transcription factors can auto-regulate their transcription by binding to their own locus (e.g., the promoter and/or enhancers). To test the potential self-regulation of Ahr, chromatin immunoprecipitation (ChIP) of Ahr was conducted with ILC2s sorted from Ahr−/− or Ahr+/+ mice. Among two Ahr-dependent ATAC-seq peaks, Peak+54, located around 54 kb downstream of the transcription start site showed enrichment of Ahr (Figure 1G), suggesting a differential requirement for Ahr binding in chromatin accessibility. Together, these data show that Ahr regulates chromatin remodeling at its own locus in gut ILC2s.
Cooperative action of Ahr and Gfi1 promotes Ahr transcription in ILC2s
Through an in silico motif analysis of Peak+54 region, the evolutionarily conserved consensus sequences for Gfi1 and Gata3, transcription factors that play important roles in ILC2 development and function, were identified (data not shown). Recruitment of Gfi1 and GATA3 to Peak+54 was further confirmed by analysis of ILC2s ChIP-seq that was previously published (Spooner et al., 2013; Zhong et al., 2016) (Figure 1F). These data prompted us to assess the potential synergy between Ahr and other key ILC2 regulators, which may potentiate the Ahr transcription. To this end, we performed Gfi1- and GATA3-ChIP experiments with sorted large intestinal ILC2s from Ahr−/− or Ahr+/+ littermate mice. Strikingly, Ahr deficiency in ILC2s significantly reduced the recruitment of Gfi1 and GATA3 to Peak+54 region (Figures 1H and 1I), suggesting that binding of Gfi1 and GATA3 to the Ahr locus is dependent on Ahr, presumably facilitated by Ahr-directed permissive chromatin changes. In accordance to these binding events at the Ahr locus, Ahr-deficient ILC2s and Gfi1-deficient ILC2s showed reduced Ahr transcription (Figures 1F, 1J and S2E) (Spooner et al., 2013), while deletion of GATA3 has minimal effect on Ahr expression in ILC2s (Yagi et al., 2014) (Figure S2F). In addition, Ahr-deficient Treg cells showed no detectable reduction of Ahr mRNA (Figure S2G). Together, these data suggest that Ahr may cooperate with Gfi1 to promote its own transcription in ILC2s.
Ahr deficiency alters ILC2 transcriptional program
To gain more insight into the regulation of ILC2 transcriptional program by Ahr, we performed genome-wide analysis of mRNA expression with RNA-seq data of gut ILC2s. Gene expression analysis identified 424 genes that were significantly changed (q-value ≤ 0.05, fold change ≥ 1.5) out of 11286 expressed genes (FPKM ≥ 1) in the Ahr-deficient ILC2s, among which 278 genes were upregulated and 146 genes were downregulated in the small intestine of Ahr−/−Rag1−/− mice (Figure 2A). Large intestinal ILC2s that lacked Ahr showed 168 genes significantly changed out of 11458 expressed genes, among which 109 genes increased and 59 genes decreased (Figure 2A). Despite an overlapping set of differentially-expressed genes, unique genes were found regulated by Ahr in the small or large intestinal ILC2s, consistent with the distinct features of these two organs (Figure 2B). When cross-referencing to the previously published 32 ILC2-characteristic protein-coding genes whose expressions are at least 4-fold higher than those in other ILC subsets in the small intestines (Robinette et al., 2015), we found differential regulation of these genes (16 out of 32 for the small intestine, and 12 out of 32 for the large intestine) by Ahr (Figures 2C and 2D). Together, these data suggest that Ahr regulates a broader set of gene transcription in ILC2s that is not limited to ILC2 signature program.
Figure 2. Ahr deficiency alters ILC2 transcriptional program, and affects chromatin landscape in ILCs.

(A) Scatterplot of gene expression of log2 (FPKM) comparing ILC2s sorted from littermate Ahr+/+Rag1−/− (WT) or Ahr−/−Rag1−/− (KO) mice. Differentially-expressed genes (q-value ≤ 0.05 and fold change ≥ 1.5) are highlighted in blue (decreased) or red (increased). (B) Venn diagrams of upregulated and downregulated genes identified in (A) indicating the overlap of differentially-expressed genes regulated by Ahr in the ILC2s between the small intestine (SI) and large intestine (LI). (C and D) Heatmap of ILC2-characteristic genes in the SI (C) or LI (D) of littermate Ahr+/+Rag1−/− (WT) or Ahr−/−Rag1−/− (KO) mice. (E) FPKM of ILC2s key transcriptional regulator genes in ILC2s from SI or LI of littermate Ahr+/+Rag1−/− or Ahr−/−Rag1−/− mice. The q-value was generated by Cufflinks analysis as described in STAR Methods. *q<0.05, **q<0.01, ***q<0.001. (F) Scatterplot comparing the Log2 fold change (KO/WT) of ATAC-seq signals versus Log2 fold change (KO/WT) of RNA-seq signals. The r-value represents r-squared goodness of fit value for linear regression. (G) Global annotation of ATAC-seq peak locations in ILC2s and ILC3s from SI and LI of littermate Ahr+/+Rag1−/−(WT) or Ahr−/−Rag1−/− (KO) mice. (H) Annotation of differentially-expressed ATAC-seq peak locations in ILC2s and ILC3s from SI and LI of littermate Ahr+/+Rag1−/− (WT) or Ahr−/−Rag1−/− (KO) mice. See also Figures S3 and S4.
We further determined the impact of Ahr on the expression of genes that have been implicated in the literature to play an important role in transcriptional control of ILC2s (Li et al., 2017; Sonnenberg and Artis, 2015). In the small intestine, Gfi1,Tcf7, Nfil3, Id2, Ets1, and Tox expression were increased in Ahr-deficient ILC2s (q-value ≤ 0.05), while Gata3, Rorα, Bcl11b, and Ehmt2 (G9a) expression were not significantly changed (Figure 2E). In the large intestine, Gfi1 was upregulated (q-value ≤ 0.05) in the absence of Ahr, while the other key regulators mostly remained statistically unchanged or decreased (e.g., Tox) (Figure 2E). Together, these data suggest that Ahr selectively regulates a subset of ILC2-related genes in the gut.
Genes that were increased in the small intestines of Ahr−/−Rag1−/− mice showed enrichment for pathways involved in cytokine-cytokine receptor interaction, chemokine signaling pathway, and T cell receptor signaling pathway (Figure S3A). Downregulated genes show enrichment for pathways involved in adherens junction, Jak-STAT signaling, tight junction, lysosome and lysine degradation signaling pathways (Figure S3A). Cytokine-cytokine receptor interaction pathway also showed downregulation in Ahr-deficient ILC2s (Figure S3A), most likely from the downregulation of IL-2 in Ahr-deficient ILC2s (data not shown). Together, these data suggest that Ahr targets various signaling pathways to regulate ILC2 gene transcription.
Ahr regulates chromatin accessibility at select gene loci in ILCs
Globally, ATAC-seq peaks did not change between Ahr-deficient and Ahr-sufficient ILC2s or ILC3s sorted from Ahr−/−Rag1−/−and Ahr+/+Rag1−/− littermate mice (Figure S3B to S3E); however, loss of Ahr did affect a number of specific peaks in ILC2s (around 1.5% of total peaks for the small intestine, and 1.4% of total peaks for the large intestine), as well as in ILC3s (around 7.4% of total peaks for the small intestine, and 6.6% of total peaks for the large intestine) (Figure S3F), indicating that Ahr does not function as a general chromatin remodeler in ILCs but instead regulates selective chromatin events in the genome. The differentially-expressed genes in ILCs correlated with the changes in ATAC-seq peak signals (e.g., Il1rl1 for ILC2s and Il22 for ILC3s) (Figures 2F and S3F). Chromatin changes observed at the Ahr and Il1rl1 loci in Ahr-deficient ILC2s on Rag1−/− background were similar to those on C57BL/6 background (Figures S4A and S4B). The location of ATAC-seq peaks in the genome were mostly unchanged between Ahr−/− and Ahr+/+ ILC2s or ILC3s, with majority of peaks located in the promoters (18–23%), introns (39–41%), and intergenic regions (29–37%) (Figure 2G). However, the differentially-regulated peaks were preferentially located in the introns (48–51%) or intergenic regions (41–44%) but not the promoters (3–6%) of ILCs (Figure 2H), suggesting a critical role for Ahr in regulation of ILC distal regulatory gene elements (e.g., enhancers).
Ahr suppresses ILC2 function
In contrast to its positive role in promoting ILC3s (Kiss et al., 2011; Lee et al., 2011; Qiu et al., 2012), Ahr inhibited the ILC2 compartment, as revealed by the increased percentage and absolute number of ILC2s (Lin−KLRG1+GATA3+) in the gut but not the lung of Ahr−/− mice (Figures 3A, 3B, S5A and S5B). Of note, the ILC2 progenitor (ILC2P) in the bone marrow of Ahr−/− mice was nevertheless decreased (Figure S5C), despite minimal expression of Ahr in ILC2P (Figure S1A), suggesting a cell-extrinsic mechanism of regulation.
Figure 3. ILC2 function is enhanced in the absence of Ahr.
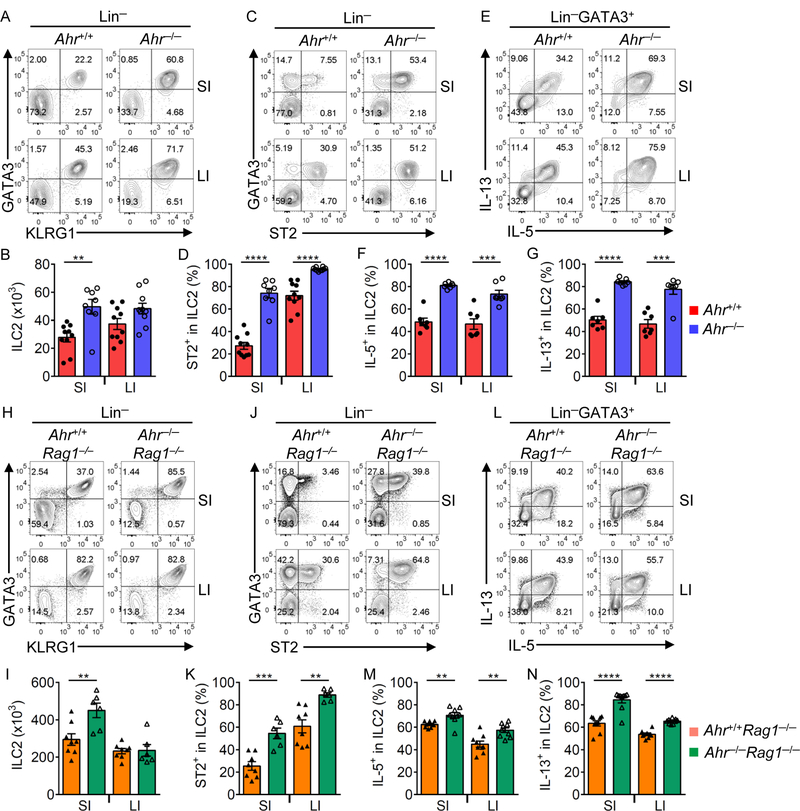
(A to G) FACS analyses of KLRG1 and GATA3 expression (A) and ST2 and GATA3 expression (C) after gating on Lin− cells, and IL-5 and IL-13 expression (E) after gating on Lin−GATA3+ lamina propria lymphocytes (LPLs) in the small intestine (SI) and large intestine (LI) of Ahr+/+ or Ahr−/− littermate mice. Data are representative of five independent experiments. Absolute numbers of KLRG1-ILC2s (Lin−GATA3+KLRG1+) (B) and percentages of ST2+ (D), IL-5+ (F), and IL-13+ (G) cells in ILC2s (Lin−GATA3+) in the SI and LI of Ahr+/+ or Ahr−/− littermate mice. Data are shown as mean ± SEM (n=7–10 per group). (H to N) FACS analyses of KLRG1 and GATA3 expression (H) and ST2 and GATA3 expression (J) after gating on Lin− cells, and IL-5 and IL-13 expression (L) after gating on Lin−GATA3+ LPLs in the SI and LI of Ahr+/+Rag1−/− or Ahr−/−Rag1−/− littermate mice. Data are representative of three independent experiments. Absolute numbers of KLRG1+ILC2s (Lin− GATA3+KLRG1+) (I) and percentages of ST2+ (K), IL-5+ (M), and IL-13+ (N) cells in ILC2s (Lin−GATA3+) in the SI and LI of Ahr+/+Rag1−/− or Ahr−/−Rag1−/− littermate mice. Data are shown as mean ± SEM (n=6–8 per group). See also Figure S5.
Consistent with RNA-seq data, ST2 protein (encoded by Il1rl1 gene), a receptor for IL-33 that is a key cytokine to promote ILC2 responses (Artis and Spits, 2015), as well as Il1rl1 mRNA were upregulated in Ahr−/− ILC2s (Figures 3C, 3D and S5D). Ahr−/− ILC2s produced more IL-5 and IL-13 (Figures 3E to 3G), indicating enhanced ILC2 function in the absence of Ahr. In addition to type 2 cytokines, ILC2s have been shown to secrete amphiregulin (Areg) (Monticelli et al., 2015). Similar to IL-5 and IL-13, lack of Ahr also led to increased overall Areg expression in ILC2s (Figures S5E, S5F, S5H, S5K, and S5M). However, ILC2s that produced Areg only but not IL-5 or IL-13, were unchanged in the absence of Ahr (Figures S5E, S5F, S5G and S5J). Consistent with the Ahr-deficient mice on C57BL/6 background, enhanced ILC2 frequency, number, ST2 expression, and production of IL-5 and IL-13 were observed in the gut of Ahr−/−Rag1−/− mice (Figures 3H to 3N), indicating that Ahr suppresses ILC2s independent of adaptive immune compartment.
Ahr regulates ILC2s in a cell-intrinsic manner
We generated Ahrf/f Vav1-cre mice to selectively ablate Ahr expression in the hematopoietic compartment. Compared to the littermate control mice, Ahrf/f Vav1-cre mice showed a reduction of ILC3s but a slight increase of ILC2s (Figures S6A to S6E), while ST2+ ILC2s and production of IL-5 and IL-13 showed significant upregulation (Figures S6C, S6F to S6I).
To further corroborate this data and determine the role of Ahr in ILCs in a cell-intrinsic manner, we performed a mixed bone marrow transfer experiment. Specifically, bone marrow from Ahr−/− (CD45.2/CD45.2) and wildtype (CD45.1/CD45.1) mice were mixed at 1:1 ratio, and transferred into Rag2−/−Il2rg−/− mice (Figure S6J). ILCs were isolated from the small and large intestines of the bone marrow chimeric mice and stained with congenic markers to distinguish their origins (Figure S6K). Consistent with the reported role of Ahr in ILC3s (Kiss et al., 2011; Lee et al., 2011; Qiu et al., 2012), marked decrease of Ahr−/− (CD45.2/CD45.2) ILC3s were observed in the intestines (Figures S6L and S6M). Despite reduced ILC2P in the bone marrow of Ahr-deficient mice (Figure S5C), comparable mature ILC2s were differentiated in the gut (Figures S6L and S6M). In addition, ILC2s derived from Ahr−/− bone marrow expressed increased ST2 and IL-13 compared to those from wildtype bone marrow (Figures S6N to S6P), suggesting that Ahr suppresses ILC2 function.
Given the opposite effects of Ahr on ILC3s and ILC2s, we wished to determine if ILC3 deficiency could affect ILC2 phenotypes observed in Ahr−/− mice by using Rorcgfp/gfp mice that completely lack ILC3s. Although ILC2 number and percentage increased in Rorcgfp/gfp mice, the expression of ST2, IL-5 and IL-13 remained unchanged or even decreased (Figures S7A to S7F). These data suggest that the increased ILC2 number observed in Ahr-deficient mice (Ahr−/− and Ahr−/−Rag1−/− mice) is likely due to a cell-extrinsic mechanism, at least in part by the lack of ILC3s; however, the upregulation of ST2, IL-5, and IL-13 expression observed in the absence of Ahr is ILC2-intrinsic. Of note, Ahrf/fVav1-cre mice also showed a trend of increased ILC2 frequency and number (Figures S6D and S6E), consistent ILC3 deficiency in these mice.
To further determine the regulation of ILC2 function by Ahr, sorted mature ILC2s from the small intestine of wildtype (CD45.1/CD45.1) or Ahr−/− (CD45.2/CD45.2) mice were mixed equally and transferred into Rag2−/−Il2rg−/− mice (Figure 4A). In the presence of wildtype ILC2s, the Ahr-deficient ILC2s still expressed higher level of ST2 and cytokines IL-5 and IL-13, supporting an ILC2-intrinsic role of Ahr (Figures 4A to 4E).
Figure 4. Suppression of ILC2 function by Ahr is cell-intrinsic.

(A to E) Sorted small intestinal (SI) ILC2s (Lin−KLRG1+CD90+) from WT (Ahr+/+) (CD45.1/CD45.1) or KO (Ahr−/−) (CD45.2/CD45.2) age and sex-matched mice were mixed equally (20,000 cells in total) and transferred into Rag2−/−Il2rg−/− littermate recipient mice. Experimental design (A), and FACS analyses of CD45.1 and GATA3 expression (upper), ST2 and GATA3 expression (middle), and IL-5 and IL-13 expression (bottom) after gating on indicated populations (B). Data are representative of two independent experiments. Percentages of ST2+ (C), IL-5+ (D), and IL-13+ (E) cells in recovered WT or KO ILC2s (Lin−GATA3+). Data are shown as mean ± SEM (n=3 per group). (F to K) RFP+ ILC2s (Lin− KLRG1+RFP+) were sorted from Ahrf/f Il5RFP-Cre or littermate Ahr+/+ Il5RFP-Cre mice. FACS analyses of Ahr expression (F), ST2 and GATA3 expression (G), and IL-5 and IL-13 expression (I) after gating on sorted RFP+ ILC2. Data are representative of two independent experiments. Percentages of ST2+ (H), IL-5+ (J), and IL-13+ (K) cells in sorted RFP+ ILC2s (Lin−GATA3+). Data are shown as mean ± SEM (n=3 per group). (L and M) Deletion of Ahr in vitro by retroviral expression of Cre-recombinase in ILC2s. Experimental design (L), and FACS analyses of Thy1.1 and SSC (left) after gating on ILC2s (Lin−GATA3+), and ST2, IL-5, IL-13, and Ahr expression after gating on Thy1.1+ (Cre+) or Thy1.1− (Cre−) ILC2s (M). Data are representative of two independent experiments. See also Figures S6 and S7.
ILC2s are the major producers of IL-5 in the gut and Il5RFP-cre mice have been used to delete genes in ILC2s (Nussbaum et al., 2013). To validate the cell-intrinsic role of Ahr in ILC2s, Ahrf/f Il5RFP-Cre mice were generated. RFP+ ILC2s showed efficient deletion of Ahr (Figure 4F). ST2 expression, and IL-5 and IL-13 production were increased in RFP+ ILC2s of Ahrf/f Il5RFP-Cre mice compared to those of the littermate Ahr+/+ Il5RFP-Cre mice (Figures 4G to 4K). We further determined the role of Ahr in regulation of ILC2 function after development. To this end, Ahr was ablated in vitro by retroviral expression of Cre-recombinase in sorted mature ILC2s from Ahrf/f mice (Figure 4L). Compared to non-transduced cells, transduced ILC2s with the expression of Cre and thus deletion of Ahr had increased expression of ST2 and cytokines IL-5 and IL-13 (Figures 4L and 4M). Collectively, these data demonstrate a cell-intrinsic role for Ahr in regulating ILC2 function.
Suppression of ILC2 function by Ahr is partially dependent on IL-33-ST2 pathway
To determine the mechanism of enhanced ST2 and cytokine production in Ahr-deficient ILC2s, Ahr−/−Il33−/− mice were developed to genetically ablate the IL-33-ST2 pathway. Loss of IL-33 in Ahr−/− mice did not affect ILC2 numbers (Figures 5A and 5B). However, compared to Ahr−/− mice, reduced ST2+, IL-5+ and IL-13+ ILC2 percentages were observed in Ahr−/−Il33−/− mice (Figures 5A, and 5C to 5F). The production of IL-5 and IL-13 by ILC2s in Ahr−/−Il33−/− mice were still higher than Il33−/− mice (Figures 5D to 5F). Together, these data suggest that enhancement of ILC2 function in Ahr−/− mice is at least in part dependent on the IL-33-ST2 pathway.
Figure 5. Ahr negatively regulates ILC2 function through IL-33-ST2 pathway and suppresses Gfi1 recruitment to the Il1rl1 locus.
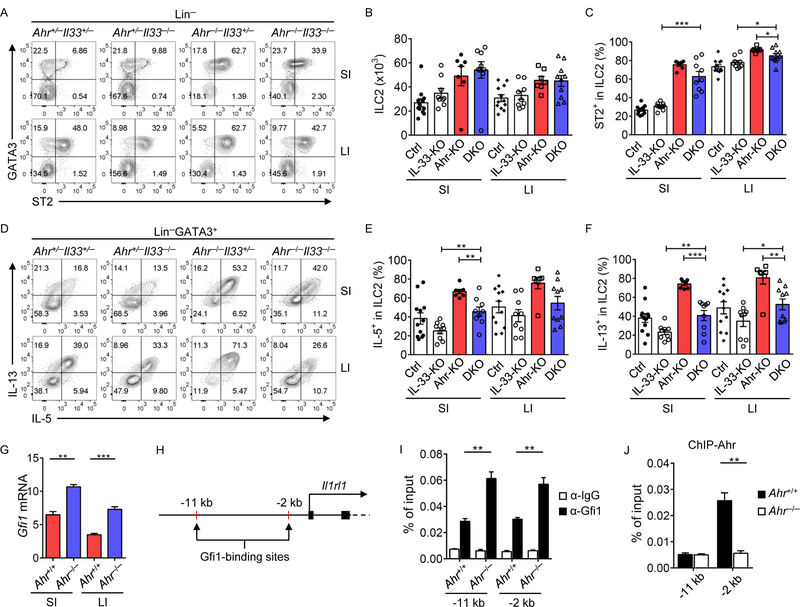
(A to F) FACS analyses of ST2 and GATA3 expression after gating on Lin− cells (A), and IL-5 and IL-13 expression after gating on Lin−GATA3+ cells (D) in the small intestine (SI) and large intestine (LI) of littermate mice of indicated genotypes. Data are representative of nine independent experiments. Absolute numbers of ILC2s (Lin−GATA3+) (B), percentages of ST2+ (C), IL-5+ (E), and IL-13+ (F) cells in ILC2s (Lin−GATA3+) in SI and LI of littermate mice of indicated genotypes (Ctrl: Ahr+/+ or Ahr+/−; IL-33 KO: Il33−/−; Ahr KO: Ahr−/−; DKO: Ahr−/−Il33−/−). Data are shown as mean ± SEM (n=7–12 per group). (G) mRNA of Gfi1 in ILC2s of littermate Ahr+/+ or Ahr−/− mice was determined by realtime RT-PCR. Data are representative of two independent experiments, and are shown as mean ± SEM (n=3). (H) Gfi1-binding sites at the Il1rl1 locus identified by ChIP-seq. (I and J) ILC2s from LI of littermate Ahr+/+ or Ahr−/− mice were expanded in vitro, and subjected to Gfi1 or Ahr ChIP assay. Enrichment of Gfi1 (I) or Ahr (J) at the Il1rl1 locus was determined by real-time PCR. Data are representative of two independent experiments, and are shown as mean ± SEM (n=3). Rabbit IgG isotype antibody was used as a negative control.
Gfi1 has been shown to regulate ST2 expression in ILC2s through direct binding to Il1rl1 (ST2) locus (Spooner et al., 2013). Consistent with RNA-seq data, the mRNA of Gfi1 was increased in Ahr−/− ILC2s (Figure 5G). Accordingly, more recruitment of Gfi1 was detected by ChIP-PCR assay to two binding sites of the Il1rl1 locus in Ahr-deficient ILC2s (Figures 5H and 5I). Enrichment of Ahr was observed at −2 kb site but not −11 kb upstream of the transcription start site at Il1rl1 locus (Figure 5J), suggesting that Ahr may directly suppress ST2 transcription.
Ahr ablation enhances anti-helminth immunity
Type 2 immunity is important for the host to control worm infection (Pelly et al., 2016; Schneider et al., 2018). To determine the role of Ahr in anti-helminth immunity, we infected Ahr−/− and littermate control (Ahr+/+ or Ahr+/−) mice with Heligmosomoides polygyrus bakeri (Figures 6A and 6B). A statistically significant decrease of worm burden in the lumen, as well as worm eggs in the feces were observed in Ahr−/− mice 12 days after inoculation (Figures 6A and 6B), while variation among individual mice was observed. Similarly, Ahr−/−Rag1−/− mice also had reduced worm recovery compared to littermate control (Ahr+/+Rag1−/− or Ahr+/−Rag1−/−) mice (Figures 6C and 6D). Ahr−/−Rag1−/− mice mounted stronger ILC2 responses with marked increase of ILC2s and higher IL-5 and IL-13 expression, consistent with elevated eosinophils (Figures 6E to 6H). In addition, Resistin-like molecule β (RELM β) expression that has been shown to be involved in protection against infection by parasitic helminth (Artis et al., 2004) was upregulated in the gut of Ahr−/−Rag1−/− mice during H. polygyrus bakeri infection (Figure 6I).
Figure 6. Ahr deficiency leads to better control of helminth infection with enhanced ILC2 immunity.
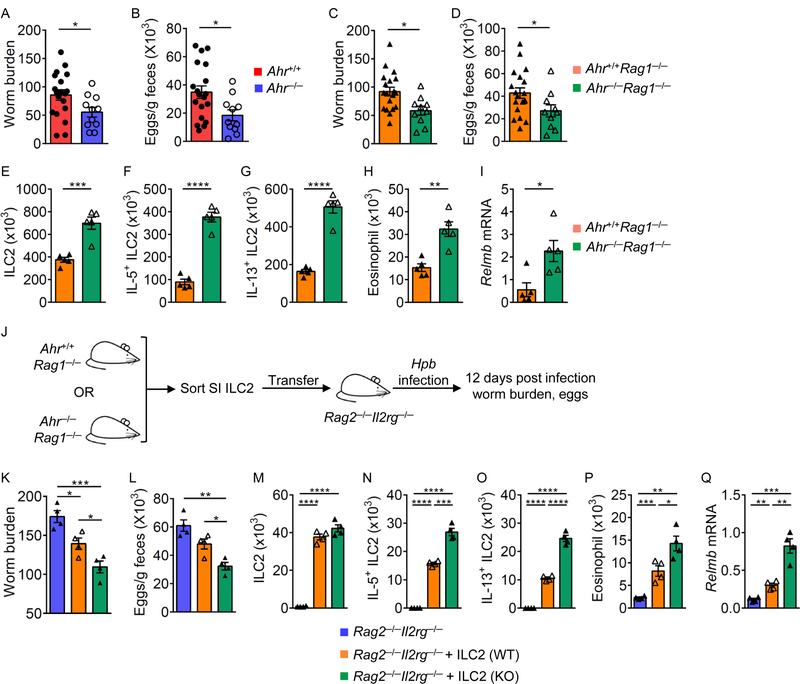
Littermate Ahr+/+ and Ahr−/− (A and B), or Ahr+/+Rag1−/− and Ahr−/−Rag1−/− (C to I) mice were inoculated with infective H. polygyrus bakeri larvae. Adult worms in the small intestines (SI) of Ahr+/+ and Ahr−/− mice (A), or Ahr+/+Rag1−/− and Ahr−/−Rag1−/− mice (C), and eggs in the feces of Ahr+/+ and Ahr−/− mice (B), or Ahr+/+Rag1−/− and Ahr−/− Rag1−/− mice (D) were counted at 12 days after inoculation. Data are shown as mean ± SEM (n=19 for Ahr+/+ or Ahr+/+Rag1−/−; n=11 for Ahr−/−; n=10 for Ahr−/−Rag1−/−). (E to I) Absolute numbers of ILC2s (Lin−GATA3+) (E), IL-5+ ILC2s (F), IL-13+ ILC2s (G), and eosinophils (CD11c−CD11b+SiglecF+SSChi) (H) in the small intestine (SI) of Ahr+/+Rag1−/− and Ahr−/−Rag1−/− mice. mRNA of Relmb in the proximal part of SI from Ahr+/+Rag1−/− and Ahr−/−Rag1−/− mice (I). Data are shown as mean ± SEM (n=5 per group). (J to Q) SI ILC2s (1 × 105) sorted from Ahr+/+Rag1−/− or Ahr−/−Rag1−/− littermate mice were transferred into Rag2−/−Il2rg−/− littermate mice respectively. Two days after transfer, these mice were inoculated with infective H. polygyrus bakeri larvae, and the mice were analyzed 12 days post infection. Experimental design (J), and the adult worm counts in SI (K), eggs in the feces (L) of Rag2−/−Il2rg−/− mice without ILC2s transfer or receiving ILC2s from Ahr+/+Rag1−/− (WT) or Ahr−/−Rag1−/− (KO) mice. Absolute numbers of ILC2s (Lin−GATA3+) (M), IL-5+ ILC2s (N), IL-13+ ILC2s (O), and eosinophils (P) in SI of indicated mice. mRNA of Relmb in the proximal part of SI from indicated mice determined by realtime RT-PCR (Q). Data are shown as mean ± SEM (n=4 per group).
It has been shown that lack of IL-22 impairs the worm expulsion upon Nippostrongylus brasiliensis and Trichuris muris infections (Turner et al., 2013). Thus, the defect of IL-22-producing ILC3s in Ahr-deficient mice might compromise the ILC2-mediated worm clearance. To validate the cell-intrinsic function of ILC2s in anti-parasite immunity, the Ahr-sufficient or deficient ILC2s were transferred into Rag2−/−Il2rg−/− mice and H. polygyrus bakeri infection was conducted with the recipients (Figure 6J). Compared to those receiving Ahr-sufficient ILC2s, Rag2−/−Il2rg−/− mice receiving Ahr-deficient ILC2s had enhanced anti-helminth immunity with less worm and egg counts (Figures 6K to 6Q). Together, these data demonstrate that similar to the steady state, Ahr deficiency results in enhanced ILC2 function during helminth infection that enhances protective immunity in the gut.
Gain-of-function of Ahr suppresses ILC2s but enhances ILC3s to better control C. rodentium infection
Expression of AhrΔPAS-B (CA-Ahr) in mice promoted gut RORγt+ ILC3s frequencies and numbers (Figures 7A to 7C). Further analysis of ILC2s in Ahr+/+ and AhrdCAIR/+ mice showed that CA-Ahr suppressed ILC2 numbers and ST2 expression (Figures 7D to 7G). Similarly, Ahr activation by treatment with the ligand 5,11-Dihydroindolo[3,2-b]carbazole-6-carboxaldehyde (FICZ) led to downregulation of ILC2s and their ST2 expression (Figures S7G and S7H). The modest effect of Ahr activation to suppress ILC2s by the gain-of-function approaches is presumably due to activation of Ahr by endogenous ligands that are already abundantly present in the host. Consistent with this notion, mice fed with Ahr ligand-depleted diet (AIN-76A) showed minimal impact on Ahr expression (Figure S7I) or ST2+ ILC2 in the gut (Figures S7J and S7K). Together, these data suggest that Ahr activation regulates the ILC2-ILC3 balance in the gut by suppressing ILC2s while promoting ILC3s.
Figure 7. Activation of Ahr suppresses ILC2s but enhances ILC3s to protect the host from C. rodentium infection.
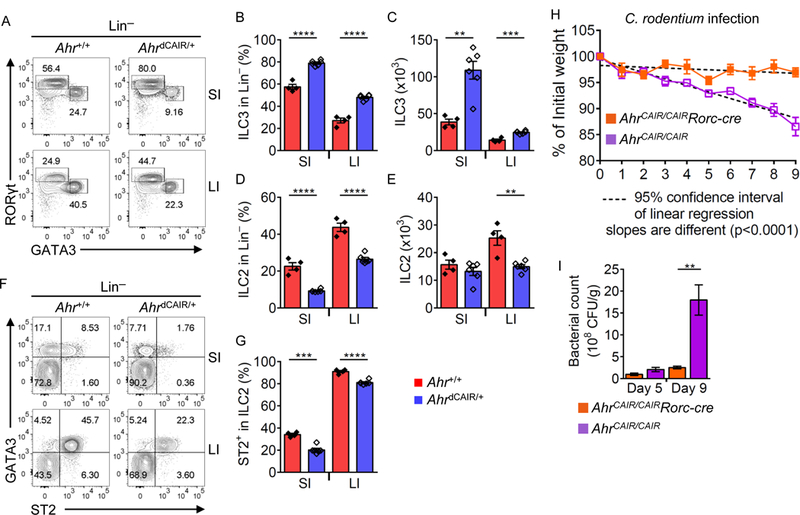
(A) FACS analysis of GATA3 and RORγt expression by Lin− LPLs in the small intestine (SI) and large intestine (LI) of Ahr+/+ or AhrdCAIR/+ littermate mice. Data are representative of two independent experiments. Percentages of ILC3s (Lin−RORγt+) (B) and ILC2s (Lin−GATA3+) (D) in Lin−SI and LI LPLs of Ahr+/+ or AhrdCAIR/+ littermate mice. Absolute numbers of ILC3s (Lin−RORγt+) (C) and ILC2s (Lin−GATA3+) (E). Data are shown as mean ± SEM (n=4–6 per group). (F) FACS analysis of ST2 and GATA3 expression after gating on Lin− cells in SI and LI of Ahr+/+ or AhrdCAIR/+ littermate mice. Data are representative of two independent experiments. (G) Percentage of ST2+ cells in ILC2s (Lin−GATA3+) of SI and LI of Ahr+/+ or AhrdCAIR/+ littermate mice. Data are shown as mean ± SEM (n=4–6 per group). (H and I) AhrCAIR/CAIR or AhrCAIR/CAIRRorc-cre mice were inoculated with C. rodentium. (H) Body weight changes were monitored at the indicated time points. (I) Bacterial counts (CFU) were measured at day 5 or day 9 after inoculation and normalized to per gram of feces. Data are shown as mean ± SEM (n=6 for AhrCAIR/CAIR, n=5 for AhrCAIR/CAIRRorc-cre).
We and others have shown that Ahr expression by RORγt+ ILC3s is necessary for their function to control mucosal infection of C. rodentium (Kiss et al., 2011; Lee et al., 2011; Qiu et al., 2012). However, it is unknown if Ahr expression in RORγt+ cells is sufficient for the host to control this bacterial infection. To this end, we developed AhrCAIR/CAIRRorc-cre mice in which CA-Ahr was expressed only in RORγt+ cells but not in other cell types (Figure S7L). AhrCAIR/CAIR phenocopied Ahr−/− mice and had a marked reduction of IL-22+ ILC3s (Ye et al., 2017). However, AhrCAIR/CAIRRorc-cre mice had similar IL-22+ ILC3s compared to Ahr-sufficient mice (Figure S7M). When inoculated with C. rodentium, AhrCAIR/CAIR mice could not control the bacteria in the gut resulting in a progressive loss in body weight and death at day 9 of the infection (Figure 7H and data not shown), consistent with a compromised ILC3 compartment. Strikingly, compared to AhrCAIR/CAIR mice, AhrCAIR/CAIRRorc-cre mice were completely protected from C. rodentium infection revealed by less weight loss, and reduced bacterial load (Figures 7H and 7I). These data show that CA-Ahr expression in RORγt+ cells is sufficient to mount protective ILC3 immunity against mucosal bacteria, demonstrating the key role of Ahr in regulating the ILC2-ILC3 balance and gut immunity.
Discussion
The molecular mechanisms underlying the balance of ILC subsets in the tissues are poorly understood. Here, we showed that the expression of Ahr in gut ILCs represent an adaptation to external environment and is essential for intestinal immunity through regulation of the ILC2-ILC3 balance.
Previous studies showed that enhancer landscapes characterizing T cell lineages are pre-established and strongly influenced by environmental stimuli (Samstein et al., 2012; Shih et al., 2016; Xu and Smale, 2012). We hypothesized that the unique chromatin events (enhancers) are specified in gut ILC2s by environmental cues and are responsible for promoting tissue-specific Ahr expression in gut ILC2s. We speculated that one candidate of such stimuli would be microbiota that are abundantly present in the gut. The fact that fewer changes of the chromatin at the Ahr locus and unaltered expression of Ahr in gut ILC2s from germfree mice supports a model that the microbiota may play a dispensable role for Ahr transcription in ILC2s, consistent with the microbiota-independent mechanism of Ahr expression in gut Treg cells (Ye et al., 2017). Our data also suggested that deprivation of Ahr dietary ligand had minimal or modest impact on Ahr expression or activity in ILC2s. Together, these data suggest that a complex interaction among microbiota, diet, and most likely other unidentified environmental factors in the gut may specify Ahr expression in intestinal immune cells. Among the gut ILC2-specific chromatin events at the Ahr locus, the remodeling of Peak+54 was dependent on Ahr, indicating that Ahr facilitates its own gene chromatin accessibility. Despite high expression of Ahr in gut Treg cells (Ye et al., 2017), Peak+54 was absent at the Ahr locus, and accordingly the transcription of Ahr was unaffected in Ahr-deficient Treg cells. These data support a cell type-specific positive feedback role of Ahr in its own transcription in ILC2s. Our attempt to determine the role of Peak+54 as an enhancer in the standard reporter assay using transient transfection did not yield conclusive data, consistent with its function in chromatin remodeling presumably not involved in transient transfection assays (Zhou et al., 2007). Thus, the function of Peak+54 needs to be studied at an organismal level by generation of mice with a mutation in this element and subsequent evaluation of immune responses (e.g., ILC2 compartment) under steady-state physiological conditions and in disease.
Gfi1 is a positive regulator of ILC2s (e.g., promoting ST2 expression) (Spooner et al., 2013). However, the data that Gfi1 binds to Peak+54 and promotes the expression of Ahr, a negative regulator of ILC2s, suggest a self-limiting mechanism to prevent exuberant ILC2 responses. Co-occupancy of Ahr and Gfi1 at the Il1rl1 promoter, and increased Gfi1 expression and recruitment to the Il1rl1 promoter in Ahr-deficient ILC2s favor a model where Ahr keeps the ILC2 effector functions in check by suppressing Gfi1-ST2 pathway; for example, through the counteraction of Gfi1 at the Il1rl1 promoter.
Ablation of the IL-33-ST2 pathway has been shown to relieve experimental colitis, including the 2,4,6-trinitrobenzenesulfonic acid (TNBS)- and dextran sulfate sodium (DSS)-induced colitis models in mice (Sedhom et al., 2013; Zhu et al., 2015). Consistently, increased IL-33 and soluble ST2 have been shown in the colons of inflammatory bowel disease patients (Pastorelli et al., 2010), in line with the proinflammatory nature of type 2 immunity (e.g., IL-13) (Neurath, 2014). However, a recent report has shown that the ILC2-associated effector molecule Areg participates in tissue repair in the gut (Monticelli et al., 2015). ILC2s can also express IL-9 to promote epithelial cell maintenance in the lung (Mohapatra et al., 2016). These data support a model where ILC2s at different stages of disease and/or some subset of ILC2s (i.e., Areg+ ILC2s or IL-9+ ILC2s) may have protective function in tissue damage. Areg+IL-5− or Areg+IL-13− cells were unaffected in Ahr-deficient mice. In addition, IL-9 expression was unchanged in Ahr-deficient ILC2s (data not shown). These data indicate that Ahr may play a differential role in regulation of ILC2 subsets that express distinct cytokines.
The plasticity exists in certain ILCs as they can convert into other subsets under different conditions (Colonna, 2018). Lack of Bcl11b in ILC2s leads to loss of ILC2s while gain of ILC3 features (Califano et al., 2015). Given the role of Ahr in regulation of ILC3 maintenance, it remains to be determined whether Ahr deficiency in ILC3s could convey its plasticity into ILC2s; however, this hypothesis needs to be carefully tested, such as using a fate-mapping genetic approach.
Genetic ablation of Ahr enhanced gut ILC2 function, consistent with enhanced immunity to adult H. polygyrus bakeri infection. Whether Ahr plays a similar role in other helminth infections remains to be determined. Ahr deficiency has been shown to cause compromised immunity against C. rodentium due to a loss of IL-22+ ILC3s (Kiss et al., 2011; Lee et al., 2011; Qiu et al., 2012). Strikingly, Ahr expression in RORγt+ cells alone is sufficient to maintain a functional ILC3 compartment and control C. rodentium infection. Because Ahr plays a differential role in various immune cells, pharmacological modulation of Ahr expression and/or activity may affect host immunity in a complex manner. Inhibition of Ahr promotes ILC2s that are beneficial for the host to mount an efficient immunity against worm infection but may also contribute to type 2-associated gut inflammation, such as ulcerative colitis and food allergy (Neurath, 2014; Stefka et al., 2014; Tordesillas et al., 2017). On the other hand, Ahr activation may promote anti-bacterial immunity through enhancement of ILC3s and control autoimmunity via promoting Treg cell functions (Gutierrez-Vazquez and Quintana, 2018; Zhou, 2016).
Although Ahr is expressed highly in mouse gut ILC2s, it remains to be determined whether this expression pattern is also evident in human. Specifically, careful comparison of Ahr expression in ILCs among different human tissues is needed. Published highthroughput single cell sequencing data show the existence of Ahr in human tonsil ILC2s; however, the level of its expression is lower than that in tonsil ILC3s (Bjorklund et al., 2016). In addition, Ahr mRNA is detectable in ILC2s from human peripheral blood mononuclear cells (PBMC), and TNF-like ligand 1A (TL1A) together with IL-25 and IL-33 can upregulate Ahr transcription in these cells (Lim et al., 2016). Thus, future efforts are needed to elucidate the molecular mechanisms by which Ahr is expressed in different tissue milieu under the steady state and/or during inflammation. Understanding how Ahr regulates the gut ILC balance may provide new therapeutic opportunities in disease treatment and/or prevention.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Liang Zhou (liangzhou497@ufl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All the mice in this study were maintained in Specific Pathogen Free (SPF) facilities at the University of Florida. Mice of both sexes were littermates and were 6–8 weeks old unless otherwise indicated. Ahr−/− (Fernandez-Salguero et al., 1995), AhrCAIR (Ye et al., 2017), Rorc-cre and Rorcgfp/gfp (Eberl and Littman, 2004; Sun et al., 2000), Il33−/− mice (Pichery et al., 2012), and Il5RFP-Cre mice (Nussbaum et al., 2013) were previously described. Rag2−/−Il2rg−/− mice were purchased from Taconic Farms. Ahrf/f mice, Vav1-cre and EIIa-cre mice were purchased from Jackson Laboratory. All studies with mice were approved by the Animal Care and Use Committee of the University of Florida.
METHOD DETAILS
Isolation of Lymphocytes from Intestinal Lamina Propria, Lung, or Adipose Tissue, and Flow Cytometry
Isolation of intestinal lamina propria cells and flow cytometry were done as previously described (Qiu et al., 2012). CD16/32 antibody (eBioscience) was used to block the non-specific binding to Fc receptors before surface staining. To isolate lymphocytes from lung or adipose tissue, blood was perfused out of circulatory system. Lung or epididymal fat tissue was collected and digested in complete RPMI medium containing DNase I (150 μg/ml, Sigma) and collagenase IV (300 U/ml, Sigma) (for the lung) or collagenase II (2 μg/μl, Sigma) (for the fat) at 37°C in 5% CO2 incubator for 1–1.5 hours. The digested lung or fat tissues were smashed and filtered through 100 μm cell strainer. Mononuclear cells were then harvested from the interphase of an 80% and 40% Percoll gradient after a spin at 2500 rpm for 20 min at room temperature. Lymphocytes isolated from intestinal lamina propria were stained with antibodies against following makers: GATA3 (PE-Cy7), RORγt (PE), T-bet (PE-Cy7), Eomos (PerCP-eFluor 710), Ahr (eFluor 660), ST2 (PE), KLRG1 (PerCP-eFluor 710), CD127 (APC), IL-13 (Alexa Flour 488), IL-5 (Brilliant Violet 421), Areg (Biotin), IL-22 (APC), CD45.2 (PerCP-Cy5.5), CD45.2 (APC), CD45.1 (PerCP-Cy5.5), CD45.1 (APC-Cy7), CD90.2 (APC-eFluor 780), CD90.1 (APC-eFluor 780), CD127 (PE), CD25 (PE-Cy7), α4β7 (APC), Flt3 (PerCP-Cy5.5), CD117 (APC), Sca-1 (PE-Cy7), CD11c (PE-Cy7) and Siglec F (Alexa Fluor 647). Lymphocytes isolated from the lung or adipose tissue were stained with antibodies against following makers: GATA3 (PE-Cy7), Ahr (AHR eFluor 660), KLRG1 (PerCP-Cy5.5), CD127 (APC). Lineage marker mix (Lin) contained APC-Cy7 or APC-eFluor 780-CD3, CD5, CD19, B220, Ly6G, CD11b, CD11c, and Ter119. To sort CLP, CHILP, ILC2P, ILC2s and ILC3s, Lin contained FITC-CD3, CD5, CD19, B220, Ly6G, FcεR1, CD11c, CD11b, Ter119, NK1.1, and CD16/CD32. For intracellular staining of Ahr in ILC1s and NK cells, Lin contained APC-Cy7 or APC-eFluor 780-CD3, CD5, CD19 and B220. For nuclear transcription factor staining, cells were fixed and permeabilized with Foxp3 staining buffer Kit (eBioscience). For cytokine staining, cells were stimulated with 50 ng/ml PMA and 500 ng/ml ionomycin for 4 hours and Brefeldin A (2 µg/ml) was added 2 hours before cells were harvested. The live and dead cells were discriminated by Live and Dead violet viability kit (Invitrogen) or Zombie Aqua Fixable Viability Kit (Biolegend).
Quantitative Real-Time RT-PCR
RNA of sorted cells from the gut was isolated with Trizol reagent (Invitrogen). cDNA was synthesized using GoScript™ Reverse Transcription kit (Promega). Real-time RT-PCR was performed using SYBR Green (Biorad) and different primer sets (table S1).
Chromatin Immunoprecipitation (ChIP) Assay and ChIP-seq Tracks Generation
Sorted ILC2s (Lin−KLRG1+CD90+) were cultured with IL-2 (10 ng/ml), IL-7 (10 ng/ml) and IL-33 (10 ng/ml) in RPMI medium for 5 days. For ChIP of Ahr, cells were treated with FICZ (200 nM) for 4 hours before harvest. Cells were cross-linked with 1% formaldehyde for 15 minutes. Chromatin was sheared by sonication with Bioruptor Pico (30” on and 30” off for 25 cycles) and immunoprecipitated with anti-Ahr (Enzo Life Science), or anti-GATA3 (BD Biosciences), or anti-Gfi1 (Abcam) using iDeal ChIP-Seq Kit for transcription factors (Diagenode). Eluted DNA was used for real-time PCR analyses using specific primers (Table S1). For the GATA3 ChIP-seq track, the bedgraph file was generated to visualize the read density, with a bin size of 200 bps; the number of reads per bin is normalized against total read number (8408659 reads) and bin size (200 bp). For Gfi1 ChIP-seq, the read density was normalized to 10 million reads.
RNA-seq and Analysis
ILC2s (Lin−KLRG1+CD90+) were sorted by flow cytometry from the gut of littermate Ahr+/+Rag1−/− or Ahr−/−Rag1−/− mice. Two independent RNA-seq experiments were performed (i.e., two biological replicates for each group that contained RNA from pooled intestinal ILC2 from 6 to 8 mice). About 1 X 106 ILC2s were pooled and lysed in Trizol (Invitrogen). RNA was subsequently extracted with RNAeasy Mini Kit (Qiagen). Total RNA was treated with Ribo-zero kit and RNAseq libraries were generated using kit from Illumina. Barcoded samples were pooled and sequenced over 2 lanes on an Illumina HiSeq 2500 instrument (the University of Chicago Genomics Core) to produce 50 bp single-end reads. De-multiplexed raw data files from the sequencing core were analyzed for quality control using FastQC (Babraham Bioinformatics). Reads were mapped (with TopHat2 fast splice junction mapper for RNA-Seq) to the mm10 assembly of the Mus musculus genome (National Center for Biotechnology Information) and filtered for uniquely mapped reads (Kim et al., 2013). Genome visualization tracks (bedgraph files, scaled to 1 million reads) were uploaded to the University of California Santa Cruz (UCSC) Genome Browser for visual comparison of expression levels (Meyer et al., 2013; Patel and Jain, 2012). Quantitated relative mRNA expression levels (FPKM) were calculated based on exon regions using Cufflinks and the mm10 reference genome annotations (Trapnell et al., 2010). Significantly changed genes were identified by Cuffdiff (Trapnell et al., 2010). Protein-coding genes found to be significantly changed (q-value ≤ 0.05, and filtered on max FPKM ≥ 1) were used for pathway analysis with GSEA software and the Molecular Signature Database (MSigDB) (Subramanian et al., 2005).
Transposase-Accessible Chromatin Sequencing (ATAC-seq) and Analysis
Sorted ILC2s (Lin−KLRG1+CD90+) from the gut were subjected to ATAC-seq according to the published protocol (Buenrostro et al., 2013) with a modification in the library purification step, which was described previously (Ye et al., 2017). ATAC-seq reads were mapped to the mouse genome (mm9) with bowtie2 (Langmead and Salzberg, 2012). The mapped reads were filtered using samtools (Li et al., 2009), keeping only the uniquely aligned reads, and bedgraph files (scaled to 10 million reads) were made with bedtools (Quinlan and Hall, 2010). ATAC-seq peak locations were identified using Homer (Heinz et al., 2010). Differentially-expressed ATAC-seq peaks were identified by first quantifying peak signal using HTSeq-count, and then using DESeq2 for differential analysis (Anders et al., 2015; Love et al., 2014). ATAC-seq signal heatmaps were generated by ngs.plot.r (Shen et al., 2014).
Retroviral Transduction of ILC2s
HEK293T cells were transfected with retroviral plasmid (MSCV-IRES-Thy1.1-Cre, MIT-Cre) and the packaging plasmid 10A1 using polyethylenimine (PEI). Viral supernatant was collected after transfection. Sorted ILC2s (Lin−KLRG1+CD90+) from the small intestine were cultured in the presence of IL-2, IL-7 and IL-33 (10 ng/ml each) for 1 day. The cells were centrifuged at 2500 rpm for 2 hr at 30 °C in 200 μl of virus supernatant containing 8 µg/ml polybrene (Sigma). The ILC2s were then cultured for 3 day before harvest.
Heligmosomoides polygyrus bakeri Infection
Mice were inoculated with 200 H. polygyrus bakeri infective third stage larvae (L3) in 200 µl sterile water by oral gavage. Worms were counted in the small intestinal lumen at days 12 after inoculation. Fecal pellets were collected, weighed, and homogenized in 2 ml of distilled H2O. Then, 2 ml of saturated NaCl solution was added to each tube, and 400 µl of the mixed solution was loaded onto McMaster 2-chamber egg counter. The worm eggs were counted within the grid and the eggs/gram feces was determined by the following formula:
The 26.67 in the formula comes from the total volume in which the feces is suspended (4 mL) divided by the volume within the grid (0.15 mL).
Citrobacter rodentium Infection and Colony-forming Units (CFUs)
Citrobacter rodentium (DBS100, ATCC51459) was cultured overnight in LB medium and the cell density was determined at OD600. 1010 CFUs of bacteria in 200 µl PBS was gavaged orally into each mouse. The body weight was monitored at indicated time points. Fecal contents were plated on MacConkey plates after serial dilution, and the CFUs of C. rodentium were counted and normalized to fecal weight after incubation of plate at 37°C for 24 hours.
In Vivo Ahr Ligand Treatment
Six day-old C57/BL6 wildtype littermate mice were treated with FICZ (0.5 μg/day) by intraperitoneal injection for 7 days. DMSO was used as a vehicle control.
Ahr Ligand Deficient Diet Treatment
Two females from the same trio-breeding cage (one male and two females) dropped pups at the same time and were separated into individual cages with the pups. Then the nursing dams were fed with the Ahr ligand-deficient diet (AIN-76A, Envigo) or normal control diet (7912, Envigo) for 4 weeks before the analysis.
Bone Marrow Transfer
Bone marrow from Ahr−/− (CD45.2/CD45.2) and wildtype (CD45.1/CD45.1) mice were mixed at 1:1 ratio, and 4×106 cells in total were intravenously injected into Rag2−/−Il2rg−/− mice irradiated at 550 rads twice with 5 hours interval. Recipient mice were treated with antibiotics (sulfamethoxazole and trimethoprim suspension, Hi-Tech Pharmacal) for 2 weeks after injection and were analyzed 8 weeks after transfer.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Methods
Unless otherwise noted, statistical analysis was performed with the unpaired two-tailed Student’s t test on individual biological samples with GraphPad Prism. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-sequencing and ATAC-sequencing data files reported in this paper is GEO: GSE119461. The accession number for the ChIP-seq of GATA3 in ILC2s is GEO: GSE71198 (Yagi et al., 2014). The accession number for the RNA-seq of GATA3-deficient ILC2s is GEO: GSE47851 (Zhong et al., 2016). The accession number for the ChIP-seq of Gfi1 in ILC2s is GEO: GSE50806, and the accession number for the microarray of Gfi1-deficient ILC2s is GEO: GSE45621 (Spooner et al., 2013). The accession number for the ATAC-seq of hematopoietic progenitors and ILCs is GEO: GSE77695 (Shih et al., 2016).
Supplementary Material
Highlights.
Unique Ahr gene chromatin features mark gut adaptation of Ahr expression in ILC2s
Ahr inhibits ILC2 function in a cell-intrinsic manner
Genetic ablation of Ahr enhances gut ILC2 function during anti-helminth immunity
Cell-specific activation of Ahr enhances gut ILC3 function in anti-bacterial immunity
Acknowledgements
We thank the entire Zhou lab for help and suggestions, specifically Drs. Yetao Wang and Lifeng Xiong in studying ILC3 regulation and ILC2P, respectively. We thank Drs. Jakob von Moltke and Richard Locksley to share Il5RFP-cre mice. We thank Flow Cytometry Facility at the University of Florida. This work was in part made possible from an NIH instrumentation grant (1S10 OD021676–01). We thank Genomics Facility at University of Chicago for service and assistance. The work was supported by the National Institutes of Health (AI089954 and DK105562 L.Z.). Liang Zhou is a Pew Scholar in Biomedical Sciences, supported by the Pew Charitable Trusts, and an Investigator in the Pathogenesis of Infectious Disease, supported by Burroughs Wellcome Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the
Declaration of interests
The authors declare no competing interests.
References
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artis D, and Spits H (2015). The biology of innate lymphoid cells. Nature 517, 293–301. [DOI] [PubMed] [Google Scholar]
- Artis D, Wang ML, Keilbaugh SA, He W, Brenes M, Swain GP, Knight PA, Donaldson DD, Lazar MA, Miller HR, et al. (2004). RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proceedings of the National Academy of Sciences of the United States of America 101, 13596–13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund AK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, Sandberg R, and Mjosberg J (2016). The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol 17, 451–460. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Califano D, Cho JJ, Uddin MN, Lorentsen KJ, Yang Q, Bhandoola A, Li H, and Avram D (2015). Transcription Factor Bcl11b Controls Identity and Function of Mature Type 2 Innate Lymphoid Cells. Immunity 43, 354–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, and Colonna M (2015). Aryl hydrocarbon receptor: Linking environment to immunity. Seminars in immunology 27, 310–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M (2018). Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity. Immunity 48, 1104–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez VS, Cervantes-Barragan L, Robinette ML, Bando JK, Wang Y, Geiger TL, Gilfillan S, Fuchs A, Vivier E, Sun JC, et al. (2016). Transforming Growth Factor-beta Signaling Guides the Differentiation of Innate Lymphoid Cells in Salivary Glands. Immunity 44, 1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denney L, Byrne AJ, Shea TJ, Buckley JS, Pease JE, Herledan GMF, Walker SA, Gregory LG, and Lloyd CM (2015). Pulmonary Epithelial Cell-Derived Cytokine TGF-beta 1 Is a Critical Cofactor for Enhanced Innate Lymphoid Cell Function. Immunity 43, 945–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbo M, Crinier A, Vely F, and Vivier E (2017). Innate lymphoid cells: major players in inflammatory diseases. Nature reviews. Immunology 17, 665–678. [DOI] [PubMed] [Google Scholar]
- Eberl G, and Littman DR (2004). Thymic origin of intestinal alphabeta T cells revealed by fate mapping of RORgammat+ cells. Science 305, 248–251. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, Rudikoff S, Ward JM, and Gonzalez FJ (1995). Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 268, 722–726. [DOI] [PubMed] [Google Scholar]
- Guo X, Qiu J, Tu T, Yang X, Deng L, Anders RA, Zhou L, and Fu YX (2014). Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity 40, 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Vazquez C, and Quintana FJ (2018). Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 48, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim TY, Hwang YY, Scanlon ST, Zaghouani H, Garbi N, Fallon PG, and McKenzie AN (2016). Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat Immunol 17, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepworth MR, Monticelli LA, Fung TC, Ziegler CG, Grunberg S, Sinha R, Mantegazza AR, Ma HL, Crawford A, Angelosanto JM, et al. (2013). Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 498, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, and Kishimoto T (2008). Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proceedings of the National Academy of Sciences of the United States of America 105, 9721–9726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, and Diefenbach A (2011). Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334, 1561–1565. [DOI] [PubMed] [Google Scholar]
- Klose CSN, and Artis D (2016). Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol 17, 765–774. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Cella M, McDonald KG, Garlanda C, Kennedy GD, Nukaya M, Mantovani A, Kopan R, Bradfield CA, Newberry RD, and Colonna M (2011). AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol 13, 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Bostick JW, and Zhou L (2017). Regulation of Innate Lymphoid Cells by Aryl Hydrocarbon Receptor. Frontiers in immunology 8, 1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Heller JJ, Bostick JW, Lee A, Schjerven H, Kastner P, Chan S, Chen ZE, and Zhou L (2016). Ikaros Inhibits Group 3 Innate Lymphoid Cell Development and Function by Suppressing the Aryl Hydrocarbon Receptor Pathway. Immunity 45, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim AI, Menegatti S, Bustamante J, Le Bourhis L, Allez M, Rogge L, Casanova JL, Yssel H, and Di Santo JP (2016). IL-12 drives functional plasticity of human group 2 innate lymphoid cells. J Exp Med 213, 569–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer LR, Zweig AS, Hinrichs AS, Karolchik D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, et al. (2013). The UCSC Genome Browser database: extensions and updates 2013. Nucleic acids research 41, D64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra A, Van Dyken SJ, Schneider C, Nussbaum JC, Liang HE, and Locksley RM (2016). Group 2 innate lymphoid cells utilize the IRF4-IL-9 module to coordinate epithelial cell maintenance of lung homeostasis. Mucosal Immunol 9, 275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DMW, and Artis D (2015). IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proceedings of the National Academy of Sciences of the United States of America 112, 10762–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurath MF (2014). Cytokines in inflammatory bowel disease. Nature reviews. Immunology 14, 329–342. [DOI] [PubMed] [Google Scholar]
- Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, Thornton EE, Krummel MF, Chawla A, Liang HE, and Locksley RM (2013). Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, Englezakis A, Barlow JL, Hams E, Scanlon ST, et al. (2014). MHCII-Mediated Dialog between Group 2 Innate Lymphoid Cells and CD4(+) T Cells Potentiates Type 2 Immunity and Promotes Parasitic Helminth Expulsion. Immunity 41, 283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastorelli L, Garg RR, Hoang SB, Spina L, Mattioli B, Scarpa M, Fiocchi C, Vecchi M, and Pizarro TT (2010). Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proceedings of the National Academy of Sciences of the United States of America 107, 8017–8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RK, and Jain M (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PloS one 7, e30619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelly VS, Kannan Y, Coomes SM, Entwistle LJ, Ruckerl D, Seddon B, MacDonald AS, McKenzie A, and Wilson MS (2016). IL-4-producing ILC2s are required for the differentiation of TH2 cells following Heligmosomoides polygyrus infection. Mucosal Immunol 9, 1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, and Girard JP (2012). Endogenous IL-33 Is Highly Expressed in Mouse Epithelial Barrier Tissues, Lymphoid Organs, Brain, Embryos, and Inflamed Tissues: In Situ Analysis Using a Novel Il-33-LacZ Gene Trap Reporter Strain. J Immunol 188, 3488–3495. [DOI] [PubMed] [Google Scholar]
- Qiu J, Guo X, Chen ZM, He L, Sonnenberg GF, Artis D, Fu YX, and Zhou L (2013). Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity 39, 386–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Heller JJ, Guo XH, Chen ZME, Fish K, Fu YX, and Zhou L (2012). The Aryl Hydrocarbon Receptor Regulates Gut Immunity through Modulation of Innate Lymphoid Cells. Immunity 36, 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, and Weiner HL (2008). Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453, 65–71. [DOI] [PubMed] [Google Scholar]
- Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, Gilfillan S, and Colonna M (2015). Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol 16, 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein RM, Arvey A, Josefowicz SZ, Peng X, Reynolds A, Sandstrom R, Neph S, Sabo P, Kim JM, Liao W, et al. (2012). Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 151, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, O’Leary CE, von Moltke J, Liang HE, Ang QY, Turnbaugh PJ, Radhakrishnan S, Pellizzon M, Ma A, and Locksley RM (2018). A Metabolite-Triggered Tuft Cell-ILC2 Circuit Drives Small Intestinal Remodeling. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedhom MAK, Pichery M, Murdoch JR, Foligne B, Ortega N, Normand S, Mertz K, Sanmugalingam D, Brault L, Grandjean T, et al. (2013). Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut 62, 1714–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, and Nestler E (2014). ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC genomics 15, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih HY, Sciume G, Mikami Y, Guo L, Sun HW, Brooks SR, Urban JF Jr., Davis FP, Kanno Y, and O’Shea JJ (2016). Developmental Acquisition of Regulomes Underlies Innate Lymphoid Cell Functionality. Cell 165, 1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoni Y, and Newell EW (2018). Dissecting human ILC heterogeneity: more than just three subsets. Immunology 153, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg GF, and Artis D (2015). Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med 21, 698–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spooner CJ, Lesch J, Yan D, Khan AA, Abbas A, Ramirez-Carrozzi V, Zhou M, Soriano R, Eastham-Anderson J, Diehl L, et al. (2013). Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat Immunol 14, 1229–1236. [DOI] [PubMed] [Google Scholar]
- Stefka AT, Feehley T, Tripathi P, Qiu J, McCoy K, Mazmanian SK, Tjota MY, Seo GY, Cao S, Theriault BR, et al. (2014). Commensal bacteria protect against food allergen sensitization. Proceedings of the National Academy of Sciences of the United States of America 111, 13145–13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger B, Di Meglio P, Gialitakis M, and Duarte JH (2014). The aryl hydrocarbon receptor: multitasking in the immune system. Annual review of immunology 32, 403–432. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZM, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, Mebius RE, and Littman DR (2000). Requirement for ROR gamma in thymocyte survival and lymphoid organ development. Science 288, 2369–2373. [DOI] [PubMed] [Google Scholar]
- Tordesillas L, Berin MC, and Sampson HA (2017). Immunology of Food Allergy. Immunity 47, 32–50. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JE, Stockinger B, and Helmby H (2013). IL-22 mediates goblet cell hyperplasia and worm expulsion in intestinal helminth infection. PLoS pathogens 9, e1003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, and Stockinger B (2008). The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109. [DOI] [PubMed] [Google Scholar]
- Xu J, and Smale ST (2012). Designing an enhancer landscape. Cell 151, 929–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi RJ, Zhong C, Northrup DL, Yu F, Bouladoux N, Spencer S, Hu GQ, Barron L, Sharma S, Nakayama T, et al. (2014). The Transcription Factor GATA3 Is Critical for the Development of All IL-7R alpha-Expressing Innate Lymphoid Cells. Immunity 40, 378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Qiu J, Bostick JW, Ueda A, Schjerven H, Li S, Jobin C, Chen ZE, and Zhou L (2017). The Aryl Hydrocarbon Receptor Preferentially Marks and Promotes Gut Regulatory T Cells. Cell reports 21, 2277–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LH, Shin JH, Haggadone MD, and Sunwoo JB (2016). The aryl hydrocarbon receptor is required for the maintenance of liver-resident natural killer cells. J Exp Med 213, 2249–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong C, Cui K, Wilhelm C, Hu G, Mao K, Belkaid Y, Zhao K, and Zhu J (2016). Group 3 innate lymphoid cells continuously require the transcription factor GATA-3 after commitment. Nat Immunol 17, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L (2016). AHR Function in Lymphocytes: Emerging Concepts. Trends in immunology 37, 17–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Nazarian AA, Xu J, Tantin D, Corcoran LM, and Smale ST (2007). An inducible enhancer required for Il12b promoter activity in an insulated chromatin environment. Molecular and cellular biology 27, 2698–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JF, Yang FL, Sang LX, Zhai JB, Zhang XQ, Yue D, Li SJ, Li Y, Lu CL, and Sun X (2015). IL-33 Aggravates DSS-Induced Acute Colitis in Mouse Colon Lamina Propria by Enhancing Th2 Cell Responses. Mediat Inflamm [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.