

Abstract
TORC1 (target of rapamycin complex) integrates complex nutrient signals to generate and fine-tune a growth and metabolic response. Npr1 (nitrogen permease reactivator) is a downstream effector kinase of TORC1 that regulates the stability, activity, and trafficking of various nutrient permeases including the ammonium permeases Mep1, Mep2, and Mep3 and the general amino acid permease Gap1. Npr1 exerts its regulatory effects on Mep1 and Mep3 via Par32 (phosphorylated after rapamycin). Activation of Npr1 leads to phosphorylation of Par32, resulting in changes in its subcellular localization and function. Here we demonstrate that Par32 is a positive regulator of TORC1 activity. Loss of Par32 renders cells unable to recover from exposure to rapamycin and reverses the resistance to rapamycin of Δnpr1 cells. The sensitivity to rapamycin of cells lacking Par32 is dependent on Mep1 and Mep3 and the presence of ammonium, linking ammonium metabolism to TORC1 activity. Par32 function requires its conserved repeated glycine-rich motifs to be intact but, surprisingly, does not require its localization to the plasma membrane. In all, this work elucidates a novel mechanism by which Npr1 and Par32 exert regulatory feedback on TORC1.
INTRODUCTION
Optimal growth of the cell depends on rapid adaptation to changes in nutrient quality and availability. Nitrogen is one of the essential nutrients because it is required for synthesis of DNA and amino acids in all living organisms. Saccharomyces cerevisiae is able to utilize a wide range of nitrogen-containing compounds such as ammonium ions, urea, and various amino acids. They have a comprehensive toolkit to discern the quantity and quality of the available nitrogen sources. Some nitrogen sources, such as ammonium ions, glutamine, and glutamate, are considered “preferred” due to the relative ease of assimilation and use from an energetic point of view (Godard et al., 2007; Ljungdahl and Daignan-Fornier, 2012; Conrad et al., 2014). By contrast, urea, proline, and arginine are not easily assimilated and are hence considered “nonpreferred.” In the presence of preferred nitrogen sources, the expression of genes required for uptake and metabolism of nonpreferred nitrogen sources is repressed, a transcriptional program known as nitrogen catabolite repression (NCR), though the extent is strain specific (Ljungdahl and Daignan-Fornier, 2012; Fayyad-Kazan et al., 2016). There are approximately 90 target genes for the NCR pathway, including the general amino acid permease Gap1; the ammonium permeases Mep1, Mep2, and Mep3; and the proline permease Put4 (Broach, 2012). In general, these genes are not expressed in the presence of preferred nitrogen sources but are strongly up-regulated in the presence of nonpreferred nitrogen sources or low levels of ammonium ions (Marini et al., 1997).
TORC1 (target of rapamycin complex) is the master regulator of cellular nitrogen metabolism (Cardenas et al., 1999; Loewith et al., 2002; De Virgilio and Loewith, 2006; Zhang et al., 2018). Nutrient availability is relayed to TORC1 by a complex and intricate network of upstream regulators and nutrient sensors (Loewith and Hall, 2011). Amino acid availability is sensed, for example, by the EGO Complex and Pib2, which are required for TORC1 reactivation after nitrogen starvation or after exposure to rapamycin, a direct physical inhibitor of TORC1 (Kim and Cunningham, 2015; Michel et al., 2017; Tanigawa and Maeda, 2017; Varlakhanova et al., 2017).
Under conditions of nutrient starvation, TORC1 is inactive, which leads to expression of genes required for scavenging, transporting, and utilization of poor sources of nitrogen. TORC1 inactivation also results in activation of Npr1 (nitrogen permease reactivator) kinase, which stabilizes nutrient permeases at the plasma membrane (Schmidt et al., 1998; MacGurn et al., 2011). Npr1 regulates the transport and stability of Gap1, which is a broad-spectrum amino acid scavenger (De Craene et al., 2001; Merhi and Andre, 2012), and positively regulates the activity of ammonium permeases Mep1, Mep2, and Mep3. It posttranslationally controls Mep2 activity by phosphorylation of an autoinhibitory C-terminal domain (Boeckstaens et al., 2014). Npr1 also activates Mep1 and Mep3 permeases by phosphorylation of the poorly characterized protein Par32 (Boeckstaens et al., 2015). Par32 has previously been reported to inhibit the activity of Mep1 and Mep3. It was shown to be localized to the plasma membrane and to interact with Mep1 and Mep3 when Npr1 is inactive. It was proposed that Par32 may be a physical plug that inhibits flux of ammonium ions through Mep1 and Mep3 (Boeckstaens et al., 2015).
Here we report that Par32 is required for reactivation of TORC1 after exposure to rapamycin. Par32 contains four repetitions of a conserved glycine-rich motif (GRGGAGNI) distributed throughout its primary sequence, and these motifs are essential for Par32 function in modulation of TORC1. Par32-dependent regulation of TORC1 is ammonium dependent. Npr1 mediates inhibition of Par32 and thus prevents TORC1 reactivation. These findings suggest that dysregulated ammonium transport can be inhibitory for TORC1 reactivation.
We demonstrate that Par32 subcellular localization is responsive to both nitrogen and glucose. Absence of Npr1 results in Par32 enrichment within the nucleus, in addition to its previously observed plasma membrane association. The latter is independent of Mep1 and Mep3 and depends on regions within the Par32 C-terminus. Surprisingly, deletion of the C-terminus did not abrogate Par32 function in TORC1 reactivation or its role in Npr1-dependent regulation of the NCR gene Gap1 in response to ammonium. Hence Par32 does not require plasma membrane association for its function and may regulate ammonium metabolism via a novel mechanism not involving direct physical blocking of plasma membrane-associated permeases.
Together, we show that Npr1 and Par32 exert a novel regulatory feedback on TORC1 via regulation of ammonium transport.
RESULTS
TORC1 activity is controlled by a complex network of upstream regulatory factors that link environmental cues, such as nutrient availability, to cell growth and metabolism. A pioneering study (Dubouloz et al., 2005) identified upstream activators of TORC1 by screening the yeast gene knockout collection for mutants that exhibit a defect in recovery from rapamycin-induced inhibition of TORC1. Eight mutants were obtained, which included activators of TORC1, such as components of the EGO Complex and Pib2, all of which were subsequently validated by several studies (Binda et al., 2009; Gong et al., 2011; Bonfils et al., 2012; Jeong et al., 2012; Zhang et al., 2012; Kim and Cunningham, 2015; Powis et al., 2015; Kira et al., 2016; Varlakhanova et al., 2017). One of the additional hits that was not characterized further was Δydl172c, an open reading frame (ORF) of unknown function.
The YDL172C ORF fully overlaps with the C-terminal 155 codons of the ORF of YDL173W, which encodes Par32 (phosphorylated after rapamycin). We hypothesized that, since Par32 is regulated by rapamycin, it is part of the TORC1 signaling network. Deletion of Par32, indeed, resulted in a severe defect in recovery from rapamycin (Figure 1A). Introduction of a plasmid containing PAR32 and its regulatory sequences into Δpar32 cells fully rescued this defect (Figure 1A). To confirm that the deficit in Δpar32 cells is TORC1 dependent, we used a previously characterized constitutively active Tor1 mutant, Tor1 L2134M (Takahara and Maeda, 2012; Kingsbury et al., 2014). Introduction of TOR1 L2134M into Δpar32 cells resulted in partial rescue of the growth defect after exposure to rapamycin (Supplemental Figure S1A), confirming that Par32 is a component of the TORC1 signaling pathway. Cells lacking Pib2, a known upstream activator of TORC1 (Varlakhanova et al., 2017), were rescued by TOR1 L2134M, as expected.
FIGURE 1:

Par32 is a positive regulator of TORC1 signaling, and its subcellular localization is regulated by nutrient availability. (A) Δpar32 cells are unable to recover from exposure to rapamycin. Exponentially growing cells (OD600 0.6–0.8) were untreated or treated with rapamycin (200 ng/ml in YPD) for 5 h at 30°C. Cells were then washed and plated on YPD. Cells were imaged after incubation for 2 d at 30°C. The leftmost spot in each case corresponds to 2 μl of a culture with OD600 0.5. Spots to the right of this correspond to 2 μl of sequential fivefold dilutions. Where indicated, PAR32 was expressed from a cen/ars plasmid. (B) Resistance to rapamycin of Δnpr1 cells is mediated by Par32. Cells were treated, washed, and plated as in A. (C) Rapamycin treatment or nutrient availability effects subcellular localization of Par32-EGFP. W303A or Δnpr1 cells expressing Par32-EGFP growing under the indicated conditions for 3 h were stained with 10 μM FM 4-64 for 30 min on ice prior to visualization. The main images show the green channel (Par32-EGFP), and the insets show the same field but with the green and red (FM 4-64) channels merged. (D) Quantification of the changes in plasma membrane association or nuclear enrichment of Par32-EGFP in W303A cells. The ratios of plasma membrane to cytosolic Par32-EGFP (left chart) or of nuclear to cytosolic Par32-EGFP (right chart) were determined by quantification of the background-corrected fluorescence intensities. The boxes within the plotted data show the second and third quartiles separated by the median of the data. Whiskers show the maximum and minimum, and the mean is shown with an “x.” Differences in membrane/cytosol ratios among the different growth conditions in W303A cells were significantly heterogeneous (one-way analysis of variance [ANOVA]: F5,35 = 6.47, hence p = 0.0002). Differences in nucleus/cytosol ratios were also significantly heterogeneous (one-way ANOVA: F5,42 = 18.28, hence p = 1.33 × 10–9). Selected significantly different pairs of means, as assessed by the post-hoc Tukey HSD test, are indicated (*p < 0.05; **p < 0.01). (E) As for D but in Δnpr1 cells. Differences in membrane/cytosol ratios among the different growth conditions in Δnpr1 cells were significantly heterogeneous (one-way ANOVA: F5,39 = 10.09, hence p = 2.96 × 10–6). Differences in nucleus/cytosol ratios were also significantly heterogeneous (one-way ANOVA: F5,39 = 7.33, hence p = 6.35 × 10–5). Selected significantly different means are indicated as in D. Scale bars, 5 μm.
Previous studies demonstrated that phosphorylation of Par32 on exposure to rapamycin is mediated by the kinase Npr1 (Boeckstaens et al., 2015). Deletion of Npr1 resulted in increased resistance to rapamycin (Figure 1B) (Schmidt et al., 1998). Simultaneous deletion of Par32 and Npr1, however, abolished this resistance. This suggests that Par32 acts downstream of Npr1 in conferring resistance to rapamycin.
Par32 subcellular localization depends on nutrient availability
We next evaluated the subcellular distribution of Par32 in cells exposed to rapamycin. In untreated cells, Par32 tagged with EGFP at its C-terminus was distributed throughout the cell (Figure 1C). Treatment with rapamycin resulted in a shift of Par32 away from the plasma membrane (Figure 1, C and D). In Δnpr1 cells, the shift was inhibited, suggesting that Npr1 activation is required for the deenrichment of Par32 from the plasma membrane in response to rapamycin treatment. Surprisingly, Par32 exhibited a strong enrichment within the nucleus in Δnpr1 cells, even when untreated (Figure 1, C–E) (nucleus:cytosol ratio of Par32-EGFP in W303A cells: 1.17 ± 0.20; in Δnpr1 cells: 1.44 ± 0.08; t = 3.09; p = 0.009). The nuclear enrichment persisted in cells treated with rapamycin (nucleus:cytosol ratio of Par32-EGFP in rapamycin-treated W303A cells: 0.95 ± 0.45; in Δnpr1 cells: 1.66 ± 0.25; t = 3.92; p = 0.002).
Because rapamycin partially mimics nitrogen starvation, we next evaluated the changes in Par32 subcellular localization in response to a 3 h starvation by depletion of either nitrogen or carbon sources or combinations of these. Par32 was primarily cytosolic on nitrogen starvation, similarly to treatment with rapamycin (Figure 1, C and D). This localization depended on the presence of active Npr1 (Figure 1, C and E). In Δnpr1 cells under conditions of nitrogen starvation, Par32 significantly enriched at the plasma membrane (W303A membrane to cytosol ratio in synthetic defined (SD)–N: 0.86; Δnpr1 membrane to cytosol in SD–N: 1.56; t = 5.36; p = 0.0005) (Figure 1, D and E). Strikingly, glucose starvation resulted in Par32 enrichment within the nucleus but not at the plasma membrane, regardless of the presence or absence of Npr1 (Figure 1, C–E). Nuclear enrichment of Par32-EGFP in response to glucose starvation cannot be prevented or reversed by additional starvation for nitrogen, whereas simultaneous glucose starvation and rapamycin treatment fully prevented nuclear enrichment (Figure 1, C–E).
We next examined the timelines for the observed nutrient-dependent changes in Par32-EGFP subcellular localization by comparing an early (15 min) timepoint to 3 h. Assessing early and later responses distinguishes between signaling events versus those requiring alterations in gene expression. Deenrichment of Par32-EGFP from the plasma membrane in response to nitrogen starvation was observed after 15 min in W303A but not in Δnpr1 cells (Supplemental Figure S1B), highlighting the rapid nature of the Npr1-dependent response. Of note, in Δnpr1 cells, enrichment of Par32-EGFP at the plasma membrane was only observed at the 3-h timepoint, suggesting a transcriptional response may be required for the enhanced membrane localization of Par32-EGFP. This may reflect synthesis of the plasma membrane-associated nutrient transporters that are synthesized during nitrogen starvation. The changes in Par32-EGFP nuclear localization in response to glucose starvation were also apparent only after 3 h, suggesting a transcriptional response rather than signaling events (Supplemental Figure S1B).
The observed changes in Par32-EGFP subcellular localization observed on 3-h glucose starvation could be a response to lack of a carbon source in general or a specific response to the absence of glucose. To distinguish these possibilities, we evaluated the subcellular localization of Par32-EGFP in cells grown in media containing various carbon sources (glucose, ethanol/glycerol, and galactose). Par32 enriched within the nucleus in cells grown on ethanol/glycerol and galactose (Supplemental Figure S2). Hence, the nuclear localization is a consequence of the absence of glucose rather than a generalized response to carbon starvation. Glucose, therefore, represses nuclear enrichment or accumulation of Par32 or, alternatively, promotes export of Par32 from the nucleus. Taken together, these observations suggest that the presence of nitrogen or glucose has different effects on the subcellular localization of Par32.
The Par32 conserved GRGGAGNI motifs are not required for plasma membrane localization but are indispensable for nuclear accumulation
To determine which regions of Par32 are responsible for its nutrient-dependent changes in subcellular localization, we assessed the subcellular localization of a series of Par32 mutants. Saccharomyces cerevisiae Par32 has four copies of the glycine-rich motif Gly-Arg-Gly-Gly-Ala-Gly-Asn-Ile (GRGGAGNI), distributed throughout its primary sequence (Figure 2A). Previous work in the Σ1278b strain of S. cerevisiae demonstrated that a 4R-A mutation of Par32, where the Arg residue in each repetition of the motif is mutated to Ala, results in cytosolic protein, both in the presence and absence of Npr1 (Boeckstaens et al., 2015). In W303A cells, mutation of all four repetitions of GRGGAG to AAAAAA (Par32 4x mut) did not alter its plasma membrane enrichment in nutrient-rich conditions (Figures 2, B and C, and 1, C and D) (Par32 membrane to cytosol, 1.22; Par32 4x mut membrane to cytosol, 1.44; p = 0.34). Par32 4x mut, however, redistributed to the cytosol under conditions of nitrogen starvation (Figure 2, B and C). In Δnpr1 cells, under conditions of nitrogen starvation, Par32 4x mut remained enriched at the plasma membrane, like Par32 with intact motifs (Par32 membrane to cytosol 1.56 vs. Par32 4x mut membrane to cytosol 1.77; p = 0.28). These results suggest that the GRGGAG motifs are not necessary for membrane association. Surprisingly, in marked contrast to wild-type Par32, Par32 4x mut did not accumulate within the nucleus under any of the conditions tested (Figures 2, B and C, and 1, C and D, and Supplemental Figure S3). Under glucose starvation, Par32 4x mut, instead of nuclear enrichment like wild-type Par32, displayed significantly increased plasma membrane enrichment in both wild-type and Δnpr1 cells (p < 0.01 in wild-type cells; p < 0.01 in Δnpr1 cells), suggesting a potential link between nuclear and plasma membrane fractions. Taken together, the 4x GRGGAGNI repeats are responsible for the nuclear enrichment of Par32 but are not required for its plasma membrane localization.
FIGURE 2:

Subcellular localizations of Par32 4x mut and Par32 Δ288-295 in W303A and Δnpr1 cells under various nutrient conditions. (A) Schematic of the primary structure of Par32 indicating, to scale, the four GRGGAGNI motifs, the lysine stretch and the position of K291. (B) W303A or Δnpr1 cells expressing Par32 4x mut-EGFP were grown, stained with FM 4-64, and imaged as in Figure 1C. (C) The ratios of plasma membrane to cytosolic Par32 4x mut-EGFP (left chart) or of nuclear to cytosolic Par32 4x mut-EGFP (right chart) were determined by quantification of the background-corrected fluorescence intensities as in Figure 1D. Differences in the Par32 4x mut-EGFP membrane/cytosol ratio among the different growth conditions in W303A were significantly heterogeneous (one-way ANOVA: W303A, F2,17 = 7.25, hence p = 0.005). Significantly different pairs of means, as assessed by the post-hoc Tukey HSD test, are indicated (*p < 0.05; **p < 0.01). Differences in means of the nucleus/cytosol ratio of W303A cells expressing Par32 4x mut-EGFP were not significant (one-way ANOVA: W303A, F2,20 = 2.00, hence p = 0.16). Differences in means of the membrane/cytosol or nucleus/cytosol ratios of Δnpr1 cells expressing Par32 4x mut-EGFP were not significant (membrane/cytosol, F2,15 = 1.28, hence p = 0.31; nucleus/cytosol, F2,21 = 0.51, hence p = 0.61). (D) W303A or Δnpr1 cells expressing Par32 Δ288-295-EGFP were treated and imaged as in B. (E) The ratios of plasma membrane to cytosolic (left charts) or nuclear to cytosolic (right charts) Par32 Δ288-295-EGFP were determined as in C. Differences in the means of the Par32 Δ288-295-EGFP membrane/cytosol ratio among the different growth conditions in W303A and Δnpr1 cells were not significant (W303A, F2,12 = 0.83, hence p = 0.46; Δnpr1, F2,16 = 0.19, hence p = 0.83). However, differences in the means of the Par32 Δ288-295-EGFP nucleus/cytosol ratios among the growth conditions in W303A and Δnpr1 cells were significant (W303A, F2,25 = 5.97, hence p = 0.008; Δnpr1, F2,15 = 4.70, hence p = 0.026). Significantly different pairs of means, as assessed by the post-hoc Tukey HSD test, are indicated (*p < 0.05; **p < 0.01). Scale bars, 5 μm.
The C-terminus of Par32 has a stretch of eight lysine residues (at positions 276–283), followed by 12 final resides that contain an additional lysine (K291) (Figure 2A). We made two deletion constructs: Par32 Δ276-295, which deletes all lysines as well as the final 12 residues, and Par32 Δ288-295, which leaves the stretch of lysines from 276 to 283 intact but removes K291. Par32 Δ288-295 did not localize to the plasma membrane under any conditions tested (Figure 2, D and E). For comparison, in Δnpr1 cells under nitrogen starvation, when Par32 maximally associated with the plasma membrane, Par32 Δ288-295 minimally associated (Par32 membrane to cytosol, 1.56, vs. Par32 Δ288-295 membrane to cytosol, 0.49; p < 1.7 × 10–6). However, Par32 Δ288-295 still enriched in the nucleus under all conditions (Figure 2, D and E, and Supplemental Figure S3) (e.g., in synthetic complete (SC) in W303A cells: Par32 nucleus to cytosol, 1.17, vs. Par32 Δ288-295 nucleus to cytosol, 1.70; p = 0.008). Par32 Δ276-295 behaved similarly (unpublished data).
To identify the specific amino acids responsible for membrane recruitment, we separately mutated either K291 to alanine (K291A), asparagine 295 to alanine (N295A), as well as several other residues (unpublished data). Whereas N295A behaved similarly to nonmutated protein under all conditions tested, K291A was required for plasma membrane localization: this construct lacked any detectable plasma membrane enrichment under all conditions, even in the absence of Npr1 (Figure 3, A–D). Hence, K291 is a critical determinant for the plasma membrane association of Par32. Interestingly, nuclear enrichment of Par32 K291A (unpublished data) was not different from Par32 in SC (t = 1.63, hence p = 0.12), whereas Par32 Δ288-295 was enriched in the nucleus compared with Par32 (t = 2.97, hence p = 0.008). As both of these constructs display no plasma membrane enrichment, these data suggest that lack of plasma membrane association does not directly translate into enhanced nuclear enrichment.
FIGURE 3:

Subcellular localizations of Par32 K291A and Par32 N295A in W303A and Δnpr1 cells under nutrient-rich and nitrogen-starvation conditions. (A, C) W303A or Δnpr1 cells expressing Par32 K291A-EGFP (A) or Par32 N295A-EGFP (C) were grown in SC or SD–N, stained with FM 4-64, and imaged as in Figure 1C. (B) The ratio of plasma membrane to cytosolic Par32 K291A-EGFP was determined as in Figure 1D. Differences in the means of the Par32 K291A-EGFP membrane to cytosol ratios when grown in SC or SD–N were not significant for W303A and Δnpr1 cells (two-tailed t test: W303A, 11 degrees of freedom, t = 0.15, hence p = 0.89; Δnpr1, 10 degrees of freedom, t = 2.19, hence p > 0.05). (D) The ratio of plasma membrane to cytosolic Par32 N295A-EGFP. Differences in the means of the Par32 N295A-EGFP membrane/cytosol ratios when grown in SC or SD–N were not significant for W303A and Δnpr1 cells (two-tailed t-test: W303A, 23 degrees of freedom, t = 1.08 hence p = 0.29; Δnpr1, 9 degrees of freedom, t = 1.91, hence p = 0.09). Scale bars, 5 μm.
The Par32 GRGGAGNI motifs are required for Npr1-independent posttranslational modification of Par32
Phosphorylation of Par32 by active Npr1 can be detected by a significant shift in electrophoretic mobility (Hughes Hallett et al., 2014; Boeckstaens et al., 2015; Varlakhanova et al., 2017). We compared the mobility shifts of Par32, C-terminally tagged with 3xHA, after nitrogen or glucose starvation, on rapamycin treatment, or on combinations of these. Under nutrient-rich conditions, Par32 runs as a doublet (Figure 4A). Nitrogen starvation or rapamycin treatment resulted in a significant Npr1-dependent shift: no shifts were observed in extracts prepared from Δnpr1 cells (Figure 4A). Glucose starvation did not alter the mobility of Par32. Notably, while simultaneous nitrogen and glucose starvation did not result in a mobility shift, glucose starvation with rapamycin treatment resulted in a shift comparable to treatment with rapamycin alone. This shift was Npr1-dependent (Figure 4A). The differences between nitrogen starvation and rapamycin treatment suggest that nitrogen availability may affect Npr1 activation independently of TORC1.
FIGURE 4:

Par32 electrophoretic mobility shifts are defined by nutrient availability. (A) Mobility shifts of Par32-3xHA under various nutrient conditions in W303A and Δnpr1 cells. W303A or Δnpr1 cells expressing the indicated 3xHA-tagged construct were grown under the indicated conditions for 3 h prior to cell lysis and blotting with an anti-HA antibody. Pgk1 was used as a loading control. (B) As in A but using nitrogen starvation for the indicated times. (C) As in B but using glucose starvation for the indicated times. (D) W303A or Δnpr1 cells expressing various Par32-3xHA constructs grown in the indicated conditions for 3 h. Pgk1 was used as a loading control.
We assessed the Par32 mobility shifts under conditions of nitrogen or glucose starvation at two different timepoints: 15 min and 3 h (Figure 4, B and C). After 15 min of nitrogen starvation, Par32 was modified in an Npr1-dependent manner to an extent similar to that observed after 3 h (Figure 4B), corresponding to the rapid deenrichment of Par32 we observed from the plasma membrane. Glucose starvation did not result in a change in Par32 mobility after 3 h. Interestingly, after 15 min of glucose starvation, however, Par32 ran as a single band in both W303A and Δnpr1 cells, suggesting transient Npr1-independent loss of a modification changes on glucose starvation (Figure 4C). Consistent with the timecourse observed in our subcellular localization studies, we propose that this modification is likely to be restored when Par32 enters the nucleus.
We next compared the mobility shifts of the various Par32 constructs used earlier to identify the regions required for the mobility shifts. Par32 4x mut was phosphorylated under conditions of nitrogen starvation but, notably, ran as a single band in Δnpr1 extracts, under all conditions (Figure 4D, compare Par32 4x mut-3xHA to Par32-3xHA). Based on our data, the upper band in the doublet depends on nuclear localization of Par32. Consistently, Par32 4x mut, which lacks nuclear localization, runs as a single band.
The point mutants Par32 K291A and Par32 N295A behaved similarly to Par32. As Par32 and Par32 K291A have different subcellular distributions, the plasma membrane localization of Par32 is not a requirement for Npr1-dependent mobility shifts. Both C-terminal truncations of Par32, Δ288-295 and Δ276-295, ran as a single band. Therefore, the extreme C-terminus of Par32 harbors either the direct site of modification responsible for the appearance of the doublet or this region is required for this modification. Interestingly, of all the mutants tested, Par32 Δ276-295 did not become phosphorylated on nitrogen starvation (Figure 4D). Taken together, these observations suggest that the region from 276 to 288 is required for the action for Npr1 on Par32 and therefore is likely required for Npr1 recruitment.
The Par32 4x GRGGAGNI motifs, but not plasma membrane association, are necessary for recovery from rapamycin
We identified sequence at the C-terminus of Par32 to be required for its association with the plasma membrane, based on subcellular localization studies (Figures 1–3). We next asked whether plasma membrane enrichment or localization affects Par32 function in TORC1 signaling. To this end, we assessed recovery from exposure to rapamycin using W303A and Δpar32 cells carrying various PAR32 constructs on a cen/ars plasmid. All of the mutants that showed a deficiency in plasma membrane localization by microscopy (Par32 K291A, Par32 Δ288-295, and Par32 Δ276-295) were still able to support recovery from exposure to rapamycin (Figure 5A). Only Par32 4x mut failed to rescue (Figure 5A). These results indicate that plasma membrane localization is not required for Par32 function within the TORC1 signaling pathway. However, one or more of the repeated GRGGAGNI motifs are indispensable for Par32-dependent regulation of TORC1 signaling.
FIGURE 5:

Characterization of Par32 and its various mutants in response to rapamycin exposure. (A) Δpar32 cells expressing Par32 4x mut do not recover from exposure to rapamycin. Exponentially growing W303A or Δpar32 cells (OD600 0.6–0.8) expressing the indicated PAR32 construct on a cen/ars plasmid were treated with rapamycin (200 ng/ml in YPD) for 5 h at 30°C. Cells were then washed and plated on YPD. Cells were imaged after incubation for 2 d at 30°C. The leftmost spot in each case corresponds to 2 μl of a culture with OD600 0.5. Spots to the right of this correspond to 2 μl of sequential fivefold dilutions. (B) Nuclear targeting of Par32 4x mut does not rescue growth after rapamycin exposure. W303A or Δpar32 cells expressing the indicated constructs (NLS, SV40 Large T Antigen nuclear localization sequence) were, where indicated, untreated or treated with rapamycin (200 ng/ml in YPD) for 5 h at 30°C prior to washing and plating on YPD. Plates were imaged after 2 d at 30°C. (C) Deletion of Mep1 and Mep3 rescues the Δpar32 rapamycin-sensitive growth phenotype. The indicated strains were treated with rapamycin and plated as in A. (D) The sensitivity of cells lacking Par32 is ammonium dependent. The experiment was performed as in C but cells were grown overnight in SD–ammonium and plated on either SC or SD–ammonium.
Since the Par32 4x mut exhibited a defect in nuclear accumulation when compared with wild-type Par32, we reasoned that the inability of the Par32 4x mut to recover from exposure to rapamycin may be due to its inability to accumulate within the nucleus. We therefore tagged wild-type Par32 and Par32 4x mut with the nuclear localization sequence (NLS) from the SV40 Large T antigen. These fusion constructs were indeed robustly localized to the nucleus, with or without rapamycin treatment (Supplemental Figures S4A and S4B). Δpar32 cells expressing NLS-Par32 recovered from exposure to rapamycin as efficiently as those expressing Par32 (Figure 5B). Δpar32 cells expressing NLS-Par32 4x mut still failed to recover from exposure to rapamycin (Figure 5C). This suggests that the defect of Par32 4x mut is not simply due to defective transport to the nucleus. The four GRGGAGNI motifs are therefore likely to be required for the recruitment of an as yet unidentified partner that translocates to the nucleus. This may be how Par32 exerts its function.
Par32 has previously been reported to be an inhibitor of the ammonium permeases Mep1 and Mep3 (Boeckstaens et al., 2015). We therefore asked whether rapamycin sensitivity associated with loss of Par32 could be attributed to Mep1 and Mep3. Cells lacking both Mep1 and Mep3 were not altered in their ability to recover from exposure to rapamycin compared with W303A cells (Figure 5C). Whereas Δpar32 cells did not recover from exposure to rapamycin, Δmep1Δmep3Δpar32 triple-knockout cells recovered like W303A (Figure 5C). Surprisingly, simultaneous deletion of Mep1 and Mep3 in a Δnpr1 background did not prevent plasma membrane association of Par32 (Supplemental Figure S4C). This suggests that Par32 interaction with the plasma membrane is mediated via a different mechanism.
Expression of the Mep transporters is regulated by two transcription factors, Gln3 and Gat1 (Marini et al., 1997). Since the rapamycin sensitivity of Δpar32 cells depended on the presence of Mep1 and Mep3, we evaluated how deletion of their transcriptional regulators Gat1 or Gln3 would affect recovery of Δpar32 from exposure to rapamycin. Additional deletion of either Gat1 or Gln3 did not rescue the recovery of Δpar32 cells from exposure to rapamycin (Supplemental Figure S5, A and B). This suggests that Mep1 and Mep3 expression in the W303 background may depend on both Gat 1 and Gln3 or that there are additional factors controlling expression of these Meps in this strain.
Mep1 and Mep3 are both high-capacity, low-affinity ammonium transporters (Marini et al., 1997). To evaluate whether the sensitivity of Δpar32 cells to rapamycin depends on ammonium transport into the cell, we determined whether cells could recover from exposure to rapamycin in SC and SD–ammonium. In SD–ammonium, Δpar32 or Δnpr1Δpar32 cells recovered to an extent comparable to that observed with W303A cells, confirming the role of ammonium in the rapamycin sensitivity of cells lacking Par32 (Figure 5D). This establishes a likely relationship among Npr1, Par32, and ammonium metabolism in feedback regulation of TORC1. In addition, Δnpr1 cells maintained their increased resistance to rapamycin exposure, even the absence of ammonium, and this resistance was significantly diminished by additional deletion of Par32, suggesting an ammonium-independent component of TORC1 regulation by Npr1 and Par32 in addition to an ammonium-dependent one.
Par32 regulates expression of the general amino acid permease Gap1
Previously, hierarchical clustering of genetic interaction profiles assembled from whole genome synthetic genetic array data predicted that Par32 functions within the Gap1 sorting pathway, based on its inclusion in a cluster with genes known to be involved in this pathway, such as MTC5, LST4, and SLM4, among others (Costanzo et al., 2010). The current version of the Cell Map (https://thecellmap.org) identifies two temperature-sensitive alleles of the essential gene LST8 as having the highest Pearson correlation coefficients with PAR32 (PCC for lst8-6, 0.213; PCC for lst8-15, 0.203) (Usaj et al., 2017). Lst8 is a component of the Tor signaling pathway and associates with both Tor1 and Tor2 and is found in both TORC1 and TORC2 complexes (Aylett et al., 2016; Karuppasamy et al., 2017). Lst8 functions in sorting of the general amino acid permease Gap1 and is also a negative regulator of the retrograde pathway, which allows expression of certain enzymes of the TCA cycle even in the presence of glucose (Chen and Kaiser, 2003). Additional evidence that PAR32 might indeed be involved in regulation of Gap1 is that Npr1 is a known regulator of the localization and stability of permeases, including Gap1 (De Craene et al., 2001; MacGurn et al., 2011; Merhi and Andre, 2012).
We therefore evaluated the sorting and expression levels of Gap1 in cells lacking Par32 in the presence and absence of nitrogen. In W303A cells grown in nutrient-rich and nitrogen-replete medium (ammonium-replete, SC), Gap1 was expressed at low levels. Any Gap1 that was expressed was sorted to the vacuole and none was detected on the plasma membrane (Figure 6). Δpar32 cells behaved similarly to W303A when grown in SC. Gap1 was derepressed in Δnpr1 cells in ammonium-replete conditions, as expected from previous reports that demonstrate NCR derepression in Δnpr1 cells (Crespo et al., 2004; Feller et al., 2006). However, in Δnpr1 cells, Gap1 was still sorted to the vacuole, as in W303A cells. Simultaneous deletion of Npr1 and Par32 resulted in abrogation of the effects on Gap1 expression observed in cells lacking only Npr1: expression of Gap1 was once again similar to that observed in W303A cells (Figure 6).
FIGURE 6:

Subcellular localization of the general amino acid permease Gap1 in Δpar32 cells. Indicated strains expressing Gap1-GFP were grown in SC or under nitrogen starvation conditions (SD–N) for 5 h and stained with 10 μM FM 4-64 for 30 min on ice prior to visualization. Scale bar, 5 μm.
In W303A cells, under conditions of nitrogen starvation (SD–N), Gap1 expression was strongly up-regulated and was trafficked mostly to the plasma membrane (Figure 6). Nitrogen-starved Δpar32 cells had lower levels of Gap1, when compared with W303A cells, and also exhibited a slight sorting defect: although a significant component of the Gap1 pool reached the plasma membrane, some was detected in punctate structures. Interestingly, any Gap1 that reached the plasma membrane was unequally distributed with a marked preference for the membranes of developing buds.
Nitrogen starvation of Δnpr1 cells resulted in high expression levels and sorting defects of Gap1 (Figure 6), as previously reported (De Craene et al., 2001). Simultaneous loss of Npr1 and Par32, on nitrogen starvation, resulted in limited expression and severe sorting defects (Figure 6).
Decreased steady-state levels of Gap1 in Δpar32 cells could be due to either decreased expression of Gap1 or increased degradation. Gap1 is known to be degraded by ubiquitin-dependent trafficking to the vacuole via multivesicular bodies. EGFP is stable in the vacuolar acidic environment, and hence increased degradation would be associated with increased vacuolar signal compared with W303A cells, which is not the case. This suggests that Par32 may instead regulate Gap1 expression. To evaluate activity of the GAP1 promoter in Δpar32 cells, we constructed a reporter based on one used previously (Chen and Kaiser, 2003). Here, the GAP1 promoter region and codons 1–53 of Gap1 were fused to codons 10–1024 of Escherichia coli lacZ on a cen/ars plasmid. We evaluated β-galactosidase activity as a readout for expression from the GAP1 promoter in various strains in nitrogen-replete or depleted conditions. The results corresponded to what we observed by microscopy. In SC, W303A and Δpar32 cells had low levels of Gap1 (Figure 7A). By contrast, Δnpr1 cells exhibited significantly higher levels of Gap1 (W303A, 7.69 A.U.; Δpar32, 2.11 A.U.; Δnpr1, 84.20 A.U.; p < 0.01). Δnpr1Δpar32 cells had Gap1 levels similar to W303A and Δpar32 cells. In SD–N, Gap1 levels were increased to 107.74 A.U. in W303A, as expected. Δpar32 also showed a robust increase in expression (59.23 A.U., not significant). In Δnpr1 cells, Gap1 levels were further increased (to 237.46 A.U.; p < 0.01). In Δnpr1Δpar32 cells, Gap1 levels were significantly lower than in Δnpr1 cells (81.90 A.U.; p = <0.05). These results together indicate that Par32 mediates the Npr1 effect on Gap1 expression.
FIGURE 7:

Par32 and Npr1 regulate expression of Gap1 in SC-grown cells. (A) Strains, as indicated, expressing the Gap1-lacZ reporter construct were grown in SC or under nitrogen starvation (SD–N, 5 h) conditions. Cells were then lysed, and β-galactosidase activity was determined. For cells grown in SC (filled boxes, mean ± s.d., n = 9–13 independent experiments), the mean β-galactosidase activity levels for the indicated strains were significantly heterogeneous (one-way ANOVA; F3,36 = 47.96, hence p = 1.17 × 10–12). Significant differences between pairs, as determined using the post-hoc Tukey HSD test, are indicated above the relevant pairs using full lines. Mean β-galactosidase activity levels for cells grown in SD–N (open boxes, n = 5–8 independent experiments) were also significantly heterogeneous (one-way ANOVA; F3,21 = 6.74, hence p = 0.0023). Significant differences between relevant pairs are indicated with dotted lines (*p < 0.05; **p < 0.01). (B) The four GRGGAG motifs in Par32 are required for Gap1 expression. Δnpr1Δpar32 cells expressing the Gap1-lacZ reporter construct and the indicated Par32 constructs were grown in SC and compared with W303A and Δnpr1 cells expressing the reporter. Cells were lysed, and β-galactosidase activity was determined. The mean β-galactosidase activity levels were significantly different (one-way ANOVA; F7,28 = 12.27, hence p = 4.18 × 10–7; n = 3–9 independent experiments). Significant differences between relevant pairs, as determined using the post-hoc Tukey HSD test, are indicated (*p < 0.05; **p < 0.01). (C) Regulation of Gap1 expression in ammonium-replete conditions (SC) by Npr1, and Par32 is independent of Mep1 and Mep3. The indicated strains expressing the Gap1-lacZ reporter construct were grown in SC and lysed prior to determination of β-galactosidase activity. The means of β-galactosidase activity levels for the various strains were significantly different (one-way ANOVA; F6,46 = 37.44, hence p = 4.44 × 10–16). Significant differences between selected pairs, as determined using the post-hoc Tukey HSD test, are indicated (**p < 0.01). (D) Par32-mediated repression of Gap1 expression is ammonium dependent. The experiment was performed as in C, but the cells were grown in SD–ammonium. The means of β-galactosidase activity levels for the various strains were significantly different (one-way ANOVA F3,8 = 19.36, hence p = 0.0005). Significant differences between selected pairs, as determined using the post-hoc Tukey HSD test, are indicated (**p < 0.01).
To determine which regions of Par32 are required for the increased Gap1 expression observed in Δnpr1 cells in SC medium, we assessed Gap1 expression, using our Gap1-lacZ fusion, in Δnpr1Δpar32 cells expressing various PAR32 constructs on cen/ars plasmids. Expression of PAR32, PAR32 Δ288-295 and the point mutants PAR32 N295A and PAR32 K291A in Δnpr1 Δpar32 cells rescued expression of Gap1 (PAR32, p < 0.01; PAR32 Δ288-295, p < 0.05; PAR32 N295A, p < 0.01; PAR32 K29, p < 0.01) to levels comparable to that seen in Δnpr1 cells alone. Expression of PAR32 4x mut failed to rescue altogether (Figure 7B). This result suggests that Par32 function in Δnpr1 cells does not require its localization to the plasma membrane, whereas the GRGGAGNI motifs are essential for Gap1 expression in the Δnpr1 background.
Deletion of Mep1 and Mep3 did not increase Gap1 expression when compared with W303A (W303A, 7.70 A.U.; Δmep1Δmep3, 15.09 A.U.; not significant) (Figure 7C). This suggests that the upregulation of Gap1 expression in ammonium-replete conditions (SC) in Δnpr1 cells cannot be solely attributed to inhibition of Mep1 and Mep3.
To determine whether Par32 mediates the effects of ammonium on inhibition of Gap1 expression in Δnpr1 cells, we assayed Gap1 expression in cells grown in SC medium lacking ammonium sulfate (SD–ammonium). Gap1 expression in Δnpr1 cells was increased further in SD–ammonium as compared with SC (SC: 84.2 ± 33.1 A.U.; SD–ammonium: 200.4 ± 45.9; mean ± standard deviation [s.d.]; t = 4.8 hence p = 0.007) (Figure 7D). Strikingly, the repression of Gap1 expression observed in Δnpr1Δpar32 cells grown in SC (Figure 7C) was not observed in SD–ammonium: here, Gap1 expression was the same in Δnpr1 and Δnpr1Δpar32 cells (Δnpr1 cells in SD–ammonium: 200.4 ± 45.9; Δnpr1Δpar32 cells in SC–ammonia: 192.8 ± 40.4 A.U.; mean ± s.d.; not significant) (Figure 7D). Taken together, therefore, Par32 inhibits the ammonium-dependent repression of Gap1 expression.
DISCUSSION
TORC1 is the master regulator of cell growth, integrating complex signals of nutrient availability into appropriate anabolic and growth responses. TORC1 activity is modulated by the combined effects of an extensive array of positive and negative regulators. Par32 is a poorly characterized protein that is heavily phosphorylated in response to inhibition of TORC1. Here we demonstrate a novel ammonium-dependent function for Par32 in reactivation of TORC1 after exposure to rapamycin (Figures 1 and 5). We further demonstrate that the positive regulation of TORC1 by Par32 does not require its plasma membrane association. Instead, it requires that the GRGGAGNI motifs distributed through its primary sequence are intact. Our subcellular localization studies reveal that these same motifs are necessary for the nuclear localization of Par32.
We show that the effects of Par32 on TORC1 activity are dependent on ammonium as well as the presence of the ammonium permeases Mep1 and Mep3 (Figures 5 and 7). Deletion of Mep1 and Mep3 does not affect recovery from rapamycin but does rescue the Δpar32 rapamycin sensitivity. We also demonstrate that Par32 is an effector of Npr1 in ammonium-rich conditions. Deletion of Npr1 results in up-regulation of expression of Gap1 in nitrogen-rich conditions, which would otherwise be repressive for NCR target genes. Additional deletion of Par32 completely reverses this effect (Figure 7) and this reversal is ammonium dependent. Therefore, Par32 functions to regulate the cell’s ammonium metabolism, which is in accordance with previous reports (Dubois and Grenson, 1979; Boeckstaens et al., 2015).
A previous report demonstrated Par32-dependent regulation of ammonium transport via Mep1 and Mep3 ammonium permeases. Par32 was shown to be accumulated at the plasma membrane in the presence of a poor nitrogen source and in the absence of Npr1 kinase. It was therefore proposed that Par32 functions as a physical inhibitor of Mep1 and Mep3 at the plasma membrane. In our studies, Par32 also associated with plasma membrane during nitrogen starvation and in the absence of Npr1. This association was, however, Mep1 and Mep3 independent, but was dependent on the C-terminal regions of Par32. Deletion of the C-terminal regions of Par32 abolished its plasma membrane association but surprisingly did not abrogate Par32 function in TORC1 regulation or ammonium-mediated repression of the NCR target gene Gap1.
In addition to plasma membrane localization, we observed a striking previously unreported nuclear enrichment of Par32 in Δnpr1 cells in response to nitrogen starvation (Figure 1, C–E). Glucose starvation also resulted in nuclear enrichment of Par32 in both W303A and Δnpr1 cells (Figure 1, C–E). Whereas nuclear localization was intact for the functional C-terminal truncations of Par32, Par32 4xmut failed to localize to the nucleus under all conditions tested (Figure 2, B–E). This raises the possibility that the Par32 GRGGAGNI motifs are required for the recruitment of a yet-to-be identified interactor and mediator of Par32 function in the regulation of ammonium transport or metabolism. The lack of nuclear localization of Par32 4x mut further suggests that such an interactor translocates to or from the nucleus in a nutrient-dependent manner. The nature of this interactor is to be determined. In bacteria and archaea, the control of nitrogen transport and metabolism is well understood and is mediated by the highly conserved family of trimeric PII proteins, which control the activities of a range of transporters, enzymes, and transcription factors involved in nitrogen and carbon metabolism and directly interact with their targets (Forchhammer, 2004; Merrick, 2014). PII proteins, in turn, are regulated by posttranslational modifications that modify their interactions with their targets. PII proteins and their modifiers serve as direct sensors of cellular nutrient and energy status by binding small molecules that report on these, such as 2-oxoglutarate and glutamine (Huergo et al., 2013). For example, the E. coli PII protein GlnK directly inhibits the ammonium permease AmtB by physical interaction, and this binding is regulated by nutrient-dependent posttranslational modification (Conroy et al., 2007). Fungi and higher eukaryotes do not have PII proteins. However, Par32 has been suggested to perform a PII-like function for Mep1 and Mep3, but our studies demonstrate that plasma membrane association of Par32 is not a requirement for its regulation of ammonium metabolism. Furthermore, fungal Par32 proteins consists of a variable number (three to six) of stringently conserved GRGGAG motifs embedded in an otherwise low complexity sequence. Par32 is therefore unlikely to form a direct physical plug and may interact or regulate those permeases via a more indirect route.
Par32 nuclear accumulation was not observed in previous work (Boeckstaens et al., 2015). This difference may stem from the strain backgrounds used in our respective studies: while our studies were performed in W303A, the previous work used Σ1278b. There are several known differences between these two model strains, including differences in ammonium metabolism (Magasanik and Kaiser, 2002). Another difference between these strains is that Σ1278b is known to have a hyperactive ras/cyclic AMP/protein kinase A (PKA) pathway (Stanhill et al., 1999). The PKA pathway responds to glucose and regulates the signal transduction required for cell growth (Zurita-Martinez and Cardenas, 2005; Peeters, 2014). Under conditions of glucose starvation, the PKA pathway is less active (Rodkaer and Faergeman, 2014), due, in part, to activation of Snf1 (Nicastro et al., 2015). We hypothesize that hyperactive PKA in Σ1278b may be responsible for the absence of nuclear Par32. If correct, Par32 localization should be sensitive and responsive to glucose. Indeed, when we starved cells for glucose, we observed a robust nuclear accumulation of Par32 (Figure 1, C and D). In further support of this hypothesis, Δsnf1 cells, which are hyperactive in PKA signaling, did not exhibit any nuclear Par32, even in the absence of glucose (unpublished data). We therefore propose that Par32 functions at the interface between nitrogen and carbon metabolism and that these strains may have different set points in their regulatory networks. This may include the subcellular localization of putative mediators of Par32 function.
Given the functions of Npr1 and Par32 in ammonium metabolism, how do they regulate TORC1? If Par32 is indeed a general negative regulator of ammonium transporters, then one might expect that deletion of Par32 will result in unregulated high intracellular ammonium levels, especially after treatment with rapamycin, which is associated with an increased expression of ammonium permeases. In our work, deletion of Par32 resulted in ammonium-dependent inhibition of TORC1, demonstrating that dysregulated elevated ammonium levels contribute to rapamycin toxicity. Previous studies demonstrated that S. cerevisiae can display ammonium toxicity, and, in response, several amino acids, including glutamine and glutamate, are excreted from the cell via the SPS amino acid transporter system (Hess et al., 2006). These amino acids are required for reactivation of TORC1 by EGO Complex and Pib2. Hence, loss of Par32 may be associated with a drop in intracellular amino acids and a consequent inability to reactivate TORC1 (Figure 8).
FIGURE 8:
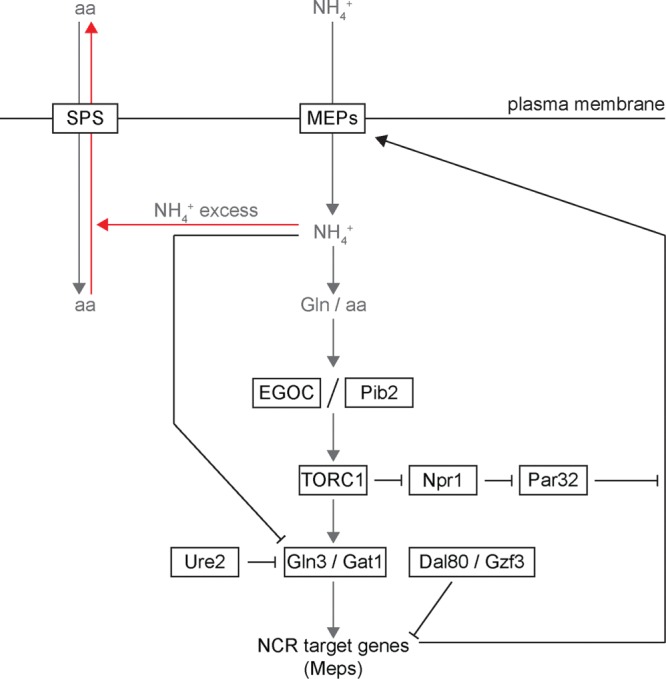
Schematic depicting the connection between ammonium metabolism and TORC1 signaling. Abbreviations: aa, amino acids; EGOC, EGO Complex; Gln, glutamine; NH4+, ammonium ion; Meps, Mep1, Mep2, and Mep3, ammonium permeases; NCR, nitrogen catabolite repression; SPS, SPS amino acid transporters. Arrows indicate positive relationships or regulation, and blunted lines represent inhibitory relationships.
Par32 is phosphorylated by the kinase Npr1, which itself is inhibited when TORC1 is active. Loss of Npr1 is associated with resistance to rapamycin, which depends on the presence of Par32. Active Npr1 thus inhibits TORC1 by negative regulation of Par32. To date, Npr1 has been considered to be a downstream effector of TORC1, which regulates the localization and stability of permeases. Our studies, together with other recent observations (Li et al., 2017), suggest an exciting mechanism by which Npr1 may exert its negative feedback on TORC1.
Taken together, our findings highlight an unexpected plasma membrane-independent role for Par32 in regulation of ammonium transport. This work illustrates how regulation of ammonium homeostasis by Npr1 and Par32 impinge on TORC1 activity thus providing a feedback loop to fine-tune the cell’s growth response to ammonium and glucose availability.
MATERIALS AND METHODS
Cloning and plasmids
Plasmids used in this work are listed in Supplemental Table S1. S cer. PAR32, containing the coding sequence and upstream and downstream fragments of 311 and 121 base pairs, respectively, was amplified from genomic DNA prepared from W303A/α diploids using the Yeast DNA Extraction Kit (Thermo Fisher Scientific, Pittsburgh, PA) and was introduced into the target vector by Gibson assembly (Gibson et al., 2009). To generate S cer. PAR32 4x mut (PAR32 4x GRGGAG-AAAAAA), a fragment of the PAR32 coding sequence, from E18 to R251, containing all four GRGGAG-AAAAAA mutations, was synthesized and was fused with overlapping separate fragments, encompassing the PAR32 upstream sequence and codons 1–22 of the open reading frame and codons 247–295 (end) of the open reading frame and the downstream sequence, respectively, by PCR-mediated overlap extension (Heckman and Pease, 2007). The full construct was then introduced into the appropriate vector by Gibson assembly. N-terminal NLS fusions (using the nuclear localization sequence from the SV40 Large T Antigen, PKKKRKV) were made by PCR-mediated overlap extension and Gibson assembly. To generate GAP1-lacZ, designed based on a construct used previously (Chen and Kaiser, 2003), a fragment encompassing a promoter region of 379 base pairs and codons 1–53 of the open reading frame of GAP1 was amplified from W303A/α genomic DNA. This was fused to codons 10–1024 of the lacZ open reading frame, amplified from E. coli strain BL21 and a terminator sequence of 300 base pairs taken from the S cer. VPS1 gene, by PCR-mediated overlap extension. All other mutants and fusion constructs were made using appropriate fragments by PCR-mediated overlap extension and Gibson assembly. All constructs were verified by sequencing. Primer and construct sequences are available on request.
Media
YPD (2% peptone, 1% yeast extract, 2% glucose, supplemented with l-tryptophan and adenine) was used for routine growth. Synthetic complete (SC; yeast nitrogen base, ammonium sulfate, 2% glucose, amino acids) or synthetic defined (SD; yeast nitrogen base, ammonium sulfate, 2% glucose, appropriate amino acid dropout) media were used prior to microscopy or to maintain plasmid selection as indicated. For sporulation, cells were successively cultured in YPA (2% potassium acetate, 2% peptone, 1% yeast extract) and SPO (1% potassium acetate, 0.1% yeast extract, 0.05% glucose). For nitrogen starvation, cells were grown in SD–N (0.17% yeast nitrogen base without amino acids and ammonium sulfate, 2% glucose). For glucose starvation, cells were grown in SD–glucose (yeast nitrogen base, ammonium sulfate, appropriate amino acid dropout). YPGAL consisted of 2% peptone, 1% yeast extract, and 2% galactose, supplemented with l-tryptophan and adenine). YPEG contained 2% peptone, 1% yeast extract, 3% ethanol, and 0.3% glycerol, supplemented with l-tryptophan and adenine. SD–ammonium omitted ammonium sulfate from the SC recipe.
Yeast genetic manipulation and molecular biology
Strains used in this work are listed in Supplemental Table S2. In general, gene deletions were generated in W303A/α diploids by homologous recombination and complete replacement of the target open reading frame using cassettes amplified from pFA6a-kanMX6, pFA6a-His3MX6 (Longtine et al., 1998), or pFA6-natMX4 (Goldstein and McCusker, 1999) flanked with sequence (40 base pairs) proximal to the open reading frame of the target gene. Diploids were subsequently sporulated by starvation in SPO. Following manual tetrad dissection, knockout haploids were validated by colony PCR, microscopy, and, in some cases, sequencing. Strains harboring more than one genomic modification were generated by mating and sporulation of appropriate parental strains, followed by extensive revalidation. Δgln3::HIS3 was made in W303A or Δpar32::NAT haploids. The standard PEG 3,350/lithium acetate/single-stranded carrier DNA protocol was used for yeast transformation (Gietz and Schiestl, 2007).
Analysis of growth by serial dilution
Cells were grown overnight in YPD, SD with the appropriate dropout for plasmid maintenance, or SD–ammonium, as relevant. Cells were then diluted and regrown to mid–logarithmic phase (OD600 0.6–0.8) in the appropriate medium at 30°C. Cells were then diluted to 0.5 OD600/ml and fivefold serial dilutions were made in water. Each dilution (2 μl) was spotted onto the indicated plates. Where relevant, cells were incubated for 5 h in YPD supplemented with 200 ng/ml rapamycin at 30°C. After extensive washing, cells were resuspended in fresh YPD and plated on YPD. All plates were incubated at 30°C for 2 d prior to imaging.
Western blotting
Protein extracts for Western blotting were obtained as described (Millen et al., 2009). Briefly, cells were lysed on ice by resuspension in 1 ml water supplemented with 150 μl 1.85 M NaOH and 7.5% β-mercaptoethanol. Protein was precipitated by addition of 150 μl 50% (wt/vol) trichloracetic acid. Pellets were washed twice with acetone, resuspended in 150 μl 1× SDS–PAGE buffer and were boiled for 5 min at 95°C. 3xHA was detected with an anti-HA mouse monoclonal antibody [HA.C5] (Abcam; ab18181). Pgk1 was detected with an anti-Pgk1 mouse monoclonal antibody [22C5D8] (Abcam; ab113687). These were detected using the IRDye680RD goat anti-mouse (Li-Cor; 926-68070) conjugated secondary antibody and the Odyssey system (Li-Cor).
Preparation of yeast for microscopy
Cells were grown overnight in YPD or SD with the appropriate dropout to maintain plasmid selection. Cells were then diluted in SC and grown to mid–logarithmic phase. For the various nutrient conditions, cells were then washed and transferred to the appropriate medium for the indicated times. For plasma membrane colocalization studies, cells were placed on ice and incubated with 10 μM FM 4-64 (Thermo Fisher Scientific) for 30 min, followed by immediate imaging. For rapamycin treatment, cells in YPD were treated for the indicated time with a final concentration of 200 ng/ml rapamycin (Thermo Fisher Scientific). Cells were plated onto No. 1.5 glass-bottomed coverdishes (MatTek Corporation, Ashland, MA) previously treated with 15 μl 2 mg/ml concanavalin-A (Sigma-Aldrich, St. Louis, MO).
Confocal microscopy and image analysis
Confocal images were acquired on a Nikon (Melville, NY) A1 confocal microscope, with a 100× Plan Apo 100× oil objective. NIS Elements Imaging software was used to control acquisition. Images were further processed using the Fiji distribution of ImageJ (Schindelin et al., 2012) or NIS Elements. The plasma membrane was defined as the region of maximum fluorescence intensity of FM 4-64. A custom ImageJ macro was used to fit an ellipse onto this region. Distribution of Par32-EGFP was quantified by dividing the background-corrected fluorescence intensity of Par32-EGFP at the plasma membrane with the averaged value (from a box of width 1 μm) of the background-corrected fluorescence intensity in a zone up to 0.2 μm from the plasma membrane. For determination of nucleus to cytosol ratios of Par32-EGFP, cells expressing an NLS-BFP marker were used to define the nucleus and the boundary between the nucleus and cytosol. Distribution of Par32-EGFP between the nucleus and cytosol was then quantified by dividing the background-corrected fluorescence intensity of Par32-EGFP signal in the nucleus (an averaged signal from a box of dimensions 0.5 × 0.2–0.25 μm positioned 0.5–0.55 μm from the nucleus-cytosol boundary) with the averaged value (using a box of same dimensions positioned in the cytosol 0.5–0.55 μm from the nucleus-cytosol boundary) of the background-corrected fluorescence in the cytosol.
β-Galactosidase assays
Cells expressing the Gap1-lacZ reporter construct and vector controls or the indicated PAR32 constructs were grown overnight in SD or SD–ammonium with the appropriate amino acid dropouts. The cells were then rediluted in the appropriate medium. For nitrogen starvation, cells were incubated in SD–N prior to measurement. OD600 values for each culture was measure prior to the assay. The assay was performed using the Yeast β-galactosidase Assay Kit (Thermo Fisher Scientific) in accordance with the manufacturer’s recommendations. Each experiment was done with three technical replicates and multiple (3–13) biological replicates.
Supplementary Material
ACKNOWLEDGMENTS
GAP1-GFP was a gift from Allyson O’Donnell (University of Pittsburgh). We thank Simon Watkins and the staff of the Center for Biologic Imaging (University of Pittsburgh School of Medicine). This work was supported by National Institutes of Health grant GM120102 (M.G.J.F.).
Abbreviations used:
- EGO
exit from rapamycin-induced growth arrest
- HA
hemagglutinin
- NCR
nitrogen catabolite repression
- Npr1
nitrogen permease reactivator
- Par32
phosphorylated after rapamycin
- SPS
Ssy1-Ptr3-Ssy5
- TCA
tricarboxylic acid cycle
- Tor1
target of rapamycin
- TORC1
target of rapamycin complex.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-03-0158) on August 29, 2018.
REFERENCES
- Aylett CH, Sauer E, Imseng S, Boehringer D, Hall MN, Ban N, Maier T. (2016). Architecture of human mTOR complex 1. Science , 48–52. [DOI] [PubMed] [Google Scholar]
- Binda M, Peli-Gulli MP, Bonfils G, Panchaud N, Urban J, Sturgill TW, Loewith R, De Virgilio C. (2009). The Vam6 GEF controls TORC1 by activating the EGO complex. Mol Cell , 563–573. [DOI] [PubMed] [Google Scholar]
- Boeckstaens M, Llinares E, Van Vooren P, Marini AM. (2014). The TORC1 effector kinase Npr1 fine tunes the inherent activity of the Mep2 ammonium transport protein. Nat Commun , 3101. [DOI] [PubMed] [Google Scholar]
- Boeckstaens M, Merhi A, Llinares E, Van Vooren P, Springael JY, Wintjens R, Marini AM. (2015). Identification of a novel regulatory mechanism of nutrient transport controlled by TORC1-Npr1-Amu1/Par32. PLoS Genet , e1005382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfils G, Jaquenoud M, Bontron S, Ostrowicz C, Ungermann C, De Virgilio C. (2012). Leucyl-tRNA synthetase controls TORC1 via the EGO complex. Mol Cell , 105–110. [DOI] [PubMed] [Google Scholar]
- Broach JR. (2012). Nutritional control of growth and development in yeast. Genetics , 73–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas ME, Cutler NS, Lorenz MC, Di Como CJ, Heitman J. (1999). The TOR signaling cascade regulates gene expression in response to nutrients. Genes Dev , 3271–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EJ, Kaiser CA. (2003). LST8 negatively regulates amino acid biosynthesis as a component of the TOR pathway. J Cell Biol , 333–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, Schothorst J, Kankipati HN, Van Zeebroeck G, Rubio-Texeira M, Thevelein JM. (2014). Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev , 254–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy MJ, Durand A, Lupo D, Li XD, Bullough PA, Winkler FK, Merrick M. (2007). The crystal structure of the Escherichia coli AmtB-GlnK complex reveals how GlnK regulates the ammonia channel. Proc Natl Acad Sci USA , 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al (2010). The genetic landscape of a cell. Science , 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo JL, Helliwell SB, Wiederkehr C, Demougin P, Fowler B, Primig M, Hall MN. (2004). NPR1 kinase and RSP5-BUL1/2 ubiquitin ligase control GLN3-dependent transcription in Saccharomyces cerevisiae. J Biol Chem , 37512–37517. [DOI] [PubMed] [Google Scholar]
- De Craene JO, Soetens O, Andre B. (2001). The Npr1 kinase controls biosynthetic and endocytic sorting of the yeast Gap1 permease. J Biol Chem , 43939–43948. [DOI] [PubMed] [Google Scholar]
- De Virgilio C, Loewith R. (2006). The TOR signalling network from yeast to man. Int J Biochem Cell Biol , 1476–1481. [DOI] [PubMed] [Google Scholar]
- Dubois E, Grenson M. (1979). Methylamine/ammonia uptake systems in saocharomyces cerevisiae: multiplicity and regulation. Mol Gen Genet , 67–76. [DOI] [PubMed] [Google Scholar]
- Dubouloz F, Deloche O, Wanke V, Cameroni E, De Virgilio C. (2005). The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell , 15–26. [DOI] [PubMed] [Google Scholar]
- Fayyad-Kazan M, Feller A, Bodo E, Boeckstaens M, Marini AM, Dubois E, Georis I. (2016). Yeast nitrogen catabolite repression is sustained by signals distinct from glutamine and glutamate reservoirs. Mol Microbiol , 360–379. [DOI] [PubMed] [Google Scholar]
- Feller A, Boeckstaens M, Marini AM, Dubois E. (2006). Transduction of the nitrogen signal activating Gln3-mediated transcription is independent of Npr1 kinase and Rsp5-Bul1/2 ubiquitin ligase in Saccharomyces cerevisiae. J Biol Chem , 28546–28554. [DOI] [PubMed] [Google Scholar]
- Forchhammer K. (2004). Global carbon/nitrogen control by PII signal transduction in cyanobacteria: from signals to targets. FEMS Microbiol Rev , 319–333. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, 3rd, Smith HO. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods , 343–345. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH. (2007). Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc , 35–37. [DOI] [PubMed] [Google Scholar]
- Godard P, Urrestarazu A, Vissers S, Kontos K, Bontempi G, van Helden J, Andre B. (2007). Effect of 21 different nitrogen sources on global gene expression in the yeast Saccharomyces cerevisiae. Mol Cell Biol , 3065–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH. (1999). Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast , 1541–1553. [DOI] [PubMed] [Google Scholar]
- Gong R, Li L, Liu Y, Wang P, Yang H, Wang L, Cheng J, Guan KL, Xu Y. (2011). Crystal structure of the Gtr1p-Gtr2p complex reveals new insights into the amino acid-induced TORC1 activation. Genes Dev , 1668–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman KL, Pease LR. (2007). Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc , 924–932. [DOI] [PubMed] [Google Scholar]
- Hess DC, Lu W, Rabinowitz JD, Botstein D. (2006). Ammonium toxicity and potassium limitation in yeast. PLoS Biol , e351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huergo LF, Chandra G, Merrick M. (2013). P(II) signal transduction proteins: nitrogen regulation and beyond. FEMS Microbiol Rev , 251–283. [DOI] [PubMed] [Google Scholar]
- Hughes Hallett JE, Luo X, Capaldi AP. (2014). State transitions in the TORC1 signaling pathway and information processing in Saccharomyces cerevisiae. Genetics , 773–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong JH, Lee KH, Kim YM, Kim DH, Oh BH, Kim YG. (2012). Crystal structure of the Gtr1p(GTP)-Gtr2p(GDP) protein complex reveals large structural rearrangements triggered by GTP-to-GDP conversion. J Biol Chem , 29648–29653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuppasamy M, Kusmider B, Oliveira TM, Gaubitz C, Prouteau M, Loewith R, Schaffitzel C. (2017). Cryo-EM structure of Saccharomyces cerevisiae target of rapamycin complex 2. Nat Commun , 1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A, Cunningham KW. (2015). A LAPF/phafin1-like protein regulates TORC1 and lysosomal membrane permeabilization in response to endoplasmic reticulum membrane stress. Mol Biol Cell , 4631–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsbury JM, Sen ND, Maeda T, Heitman J, Cardenas ME. (2014). Endolysosomal membrane trafficking complexes drive nutrient-dependent TORC1 signaling to control cell growth in Saccharomyces cerevisiae. Genetics , 1077–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kira S, Kumano Y, Ukai H, Takeda E, Matsuura A, Noda T. (2016). Dynamic relocation of the TORC1-Gtr1/2-Ego1/2/3 complex is regulated by Gtr1 and Gtr2. Mol Biol Cell , 382–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yan G, Liu S, Jiang T, Zhong M, Yuan W, Chen S, Zheng Y, Jiang Y, Jiang Y. (2017). Target of rapamycin complex 1 and Tap42-associated phosphatases are required for sensing changes in nitrogen conditions in the yeast Saccharomyces cerevisiae. Mol Microbiol , 938–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungdahl PO, Daignan-Fornier B. (2012). Regulation of amino acid, nucleotide, and phosphate metabolism in Saccharomyces cerevisiae. Genetics , 885–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Hall MN. (2011). Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics , 1177–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. (2002). Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell , 457–468. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast , 953–961. [DOI] [PubMed] [Google Scholar]
- MacGurn JA, Hsu PC, Smolka MB, Emr SD. (2011). TORC1 regulates endocytosis via Npr1-mediated phosphoinhibition of a ubiquitin ligase adaptor. Cell , 1104–1117. [DOI] [PubMed] [Google Scholar]
- Magasanik B, Kaiser CA. (2002). Nitrogen regulation in Saccharomyces cerevisiae. Gene , 1–18. [DOI] [PubMed] [Google Scholar]
- Marini AM, Soussi-Boudekou S, Vissers S, Andre B. (1997). A family of ammonium transporters in Saccharomyces cerevisiae. Mol Cell Biol , 4282–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merhi A, Andre B. (2012). Internal amino acids promote Gap1 permease ubiquitylation via TORC1/Npr1/14-3-3-dependent control of the Bul arrestin-like adaptors. Mol Cell Biol , 4510–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick M. (2014). Post-translational modification of P II signal transduction proteins. Front Microbiol , 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AH, Hatakeyama R, Kimmig P, Arter M, Peter M, Matos J, De Virgilio C, Kornmann B. (2017). Functional mapping of yeast genomes by saturated transposition. Elife , e23570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millen JI, Krick R, Prick T, Thumm M, Goldfarb DS. (2009). Measuring piecemeal microautophagy of the nucleus in Saccharomyces cerevisiae. Autophagy , 75–81. [DOI] [PubMed] [Google Scholar]
- Nicastro R, Tripodi F, Gaggini M, Castoldi A, Reghellin V, Nonnis S, Tedeschi G, Coccetti P. (2015). Snf1 phosphorylates adenylate cyclase and negatively regulates protein kinase A-dependent transcription in Saccharomyces cerevisiae. J Biol Chem , 24715–24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters KTJ. (2014). Glucose sensing and signal transduction in Saccharomyces cerevisiae . In: Molecular Mechanisms in Yeast Carbon Metabolism, ed. J Piškur and C Compagno, Berlin: Springer-Verlag, 21–56. [Google Scholar]
- Powis K, Zhang T, Panchaud N, Wang R, De Virgilio C, Ding J. (2015). Crystal structure of the Ego1-Ego2-Ego3 complex and its role in promoting Rag GTPase-dependent TORC1 signaling. Cell Res , 1043–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodkaer SV, Faergeman NJ. (2014). Glucose- and nitrogen sensing and regulatory mechanisms in Saccharomyces cerevisiae. FEMS Yeast Res , 683–696. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods , 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Beck T, Koller A, Kunz J, Hall MN. (1998). The TOR nutrient signalling pathway phosphorylates NPR1 and inhibits turnover of the tryptophan permease. EMBO J , 6924–6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanhill A, Schick N, Engelberg D. (1999). The yeast ras/cyclic AMP pathway induces invasive growth by suppressing the cellular stress response. Mol Cell Biol , 7529–7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahara T, Maeda T. (2012). Transient sequestration of TORC1 into stress granules during heat stress. Mol Cell , 242–252. [DOI] [PubMed] [Google Scholar]
- Tanigawa M, Maeda T. (2017). An in vitro TORC1 kinase assay that recapitulates the Gtr-independent glutamine-responsive TORC1 activation mechanism on yeast vacuoles. Mol Cell Biol , e00075-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usaj M, Tan Y, Wang W, VanderSluis B, Zou A, Myers CL, Costanzo M, Andrews B, Boone C. (2017). TheCellMap.org: a web-accessible database for visualizing and mining the global yeast genetic interaction network. G3 (Bethesda) , 1539–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varlakhanova NV, Mihalevic MJ, Bernstein KA, Ford MGJ. (2017). Pib2 and the EGO complex are both required for activation of TORC1. J Cell Sci , 3878–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Peli-Gulli MP, Yang H, De Virgilio C, Ding J. (2012). Ego3 functions as a homodimer to mediate the interaction between Gtr1-Gtr2 and Ego1 in the ego complex to activate TORC1. Structure , 2151–2160. [DOI] [PubMed] [Google Scholar]
- Zhang W, Du G, Zhou J, Chen J. (2018). Regulation of sensing, transportation, and catabolism of nitrogen sources in Saccharomyces cerevisiae. Microbiol Mol Biol Rev , e00040-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurita-Martinez SA, Cardenas ME. (2005). Tor and cyclic AMP-protein kinase A: two parallel pathways regulating expression of genes required for cell growth. Eukaryot Cell , 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.