

Abstract
Protein phosphatase 5 (PP5), a serine/threonine phosphatase, has a wide range of biological functions and exhibits elevated expression in tumor cells. We previously reported that pp5-deficient mice have altered ataxia-telangiectasia mutated (ATM)-mediated signaling and function. However, this regulation was likely indirect, as ATM is not a known PP5 substrate. In the current study, we found that pp5-deficient mice are hypersensitive to genotoxic stress. This hypersensitivity was associated with the marked up-regulation of the tumor suppressor tumor protein p53 and its downstream targets cyclin-dependent kinase inhibitor 1A (p21), MDM2 proto-oncogene (MDM2), and phosphatase and tensin homolog (PTEN) in pp5-deficient tissues and cells. These observations suggested that PP5 plays a role in regulating p53 stability and function. Experiments conducted with p53+/−pp5+/− or p53+/−pp5−/− mice revealed that complete loss of PP5 reduces tumorigenesis in the p53+/− mice. Biochemical analyses further revealed that PP5 directly interacts with and dephosphorylates p53 at multiple serine/threonine residues, resulting in inhibition of p53-mediated transcriptional activity. Interestingly, PP5 expression was significantly up-regulated in p53-deficient cells, and further analysis of pp5 promoter activity revealed that p53 strongly represses PP5 transcription. Our results suggest a reciprocal regulatory interplay between PP5 and p53, providing an important feedback mechanism for the cellular response to genotoxic stress.
Keywords: p53, cancer, gene knockout, protein serine/threonine phosphatase (PSP), protein phosphorylation, protein phosphatase 5, transcriptional regulation, tumorigenesis, posttranslational regulation, cell stress
Introduction
Maintenance of genomic stability is critical for cell growth and survival. Many genetic disorders, including most human cancers, are associated with some form of genomic instability. The tumor suppressor gene p53 is known to play a critical role in the maintenance of genomic stability in response to various cellular and genotoxic stress factors, including DNA cross-linking agents (1), oxidative stress (2), UV/ionizing irradiation (3), and persistent DNA damage (4). p53 is a transcription factor that can repress or induce many genes in response to genotoxic stress. Mdm2 is involved in an autoregulatory feedback loop that down-regulates p53 upon the conclusion of DNA repair for reentry into the cell cycle. Mdm2 functions in a ubiquitin ligase complex that is important for nuclear export of p53 and eventual destabilization of p53.
Upon encountering genotoxic stress, the ataxia-telangiectasia mutated (ATM)3/ATM and Rad3-related (ATR) kinases and CHK1/2 kinases are activated and subsequently phosphorylate p53. These phosphorylation events have been shown to prevent the interaction between p53 and MDM2 and subsequently lead to an increase in p53 protein level and activity (5, 6). In contrast to the well studied mechanism of p53 phosphorylation, dephosphorylation of p53 remains poorly understood. Several protein phosphatases, including protein phosphatase 2A (PP2A) (7–9), protein phosphatase 1 (PP1) (10, 11), protein phosphatase 1D (PPM1D; or Wip1) (12–14), and cell division cycle 14 (Cdc14) (15), have previously been implicated in the regulation of p53 phosphorylation via their phosphatase activities. There are 18 serine and threonine residues in p53 (16) that are phosphorylated in response to genotoxic and nongenotoxic stress (16–18). PP2A can dephosphorylate Ser-46 (9), and Wip1 may dephosphorylate Ser-15 (12, 13) of p53. Ser-46 is responsible for the induction of apoptotic genes and PTEN, whereas Ser-15 is associated with cell cycle arrest and Mdm2 (19). However, p53 mutants lacking some of these phosphorylation sites do not seem to overtly alter p53 function (20, 21), suggesting that the regulation of p53 phosphorylation is a highly complex event and may require other posttranslational modifications to achieve significant changes in biological activity (22).
Protein phosphatase 5 (PP5) is a serine/threonine phosphatase that contains a 34-amino-acid tetratricopeptide repeat domain that is known to mediate protein–protein interactions and serves as an autoinhibitory domain for the phosphatase activity of PP5 (23). The catalytic domain of PP5 is similar to those of other protein phosphatases, such as PP1, PP2A, and PP2B (24). PP5 is ubiquitously expressed and is believed to have multiple functions in several intracellular signaling networks, including cell cycle regulation (25) and cellular stress responses (26). Interestingly, several recent clinical studies have shown that PP5 is up-regulated in breast cancers, and the human PP5 gene resides at a chromosomal region that is frequently amplified in osteosarcoma patients (27). In a xenograft model, a 2-fold increase in PP5 protein levels significantly enhanced the growth rate of estrogen-dependent tumors (28). These studies suggest an important role for PP5 in tumorigenesis. Additionally, PP5 has been shown to be associated with several genotoxic stress–induced protein complex members, such as apoptosis signal-regulating kinase 1 (29, 30), the DNA-dependent Ser/Thr protein kinase DNA-PKcs (31), ATM/ATR (32–34), Raf1 (35), and the glucocorticoid receptor (36–38). Using a gene knockdown approach in cultured cells, Honkanen and co-workers (25, 39, 40) demonstrated that PP5 can act as a negative regulator of p53 function; however, the molecular mechanism by which PP5 regulates p53 function has not been closely analyzed.
Previously, we generated pp5-deficient mice and showed that pp5-deficient cells possess altered ATM-mediated signaling and function (32). However, this regulation was likely indirect as ATM is not known to be a PP5 substrate (41). In the current study, we identified p53 as a strong candidate substrate of PP5. Biochemical analyses demonstrated that PP5 was able to directly interact with p53 and dephosphorylate this protein at multiple Ser/Thr sites both in vitro and in vivo. p53 protein levels were significantly elevated in pp5-deficient cells and tissues and down-regulated in pp5-overexpressing cells compared with the levels in the normal controls. Subsequent analysis further demonstrated that hyperphosphorylation of p53 in pp5-deficient cells stabilized p53. In addition, we also identified two conserved putative p53-binding sites in the pp5 promoter region. Chromatin immunoprecipitation (ChIP) and luciferase assays confirmed that p53 is a potent transcriptional repressor of PP5. Compound p53+/−pp5−/− mutant mice exhibited significantly longer lifespans and later onset of tumorigenesis than mice that were double heterozygous for both genes (p53+/−pp5+/−). Collectively, our findings reveal a unique regulatory interplay between PP5 and p53, which likely constitutes a novel positive feedback mechanism involved in the cellular response to stress.
Results
Mice deficient in PP5 exhibit increased sensitivity to genotoxic stress and up-regulation of p53 protein levels
Our previous studies suggested that pp5-deficient cells exhibit defects in the G2/M cell cycle checkpoint in response to DNA damage (32). To further explore the mechanism of this phenomenon, we tested the biological response of the genotoxic reagent doxorubicin (DOX) using bone marrow low-density mononuclear (LDM) cells isolated from pp5-deficient (knockout (KO)) and sex-matched wildtype (WT) littermate mice. DOX was shown to be able to induce generation of superoxide and hydroxyl radicals, which can cause DNA oxidative damage, which subsequently leads to eventual cell cycle arrest and cellular apoptosis (42, 43). Using flow cytometry analysis, we found that fewer than 5% of the KO or WT LDM cells were apoptotic in the absence of DOX treatment (Fig. 1A). However, the percentage of apoptotic KO cells increased dramatically after DOX treatment (0.05 or 0.1 μg/ml; 12 h) compared with similarly treated WT cells (21.26 ± 0.49 and 41.94 ± 8.64% in PP5 KO cells versus 10.76 ± 1.22 and 25.21 ± 4.68% in WT cells, p < 0.001) (Fig. 1A). The result indicated that pp5-deficient bone marrow LDM cells are hypersensitive to DOX treatment. p53 is regarded as a critical sensor for cell stress and an important modulator of apoptosis in response to a range of stimuli (44, 45). The stress-induced apoptosis in hematopoietic progenitors is p53-dependent (46). To test whether PP5 is a regulator of p53 function, we examined whether p53 expression was altered in pp5-deficient LDM cells by Western blot analysis, which showed that PP5 mutant bone marrow cells had significantly higher p53 levels than the littermate controls (Fig. 1D). Next, we tested whether the increased cellular apoptosis observed in pp5-deficient bone marrow cells in response to DOX treatment was p53-dependent. RNA interference (RNAi) was used to knock down p53 by transducing WT and KO primary bone marrow cells with a retrovirus (pMSCV-eGFP/sh-p53) or a control virus carrying only eGFP (pMSCV-eGFP) (47). Following transduction, cells were sorted for eGFP to enrich the transduced cells and treated with DOX. Real-time quantitative RT-PCR was used to confirm the p53 levels in the control and p53 RNAi–treated cells along with the levels of the p53 downstream regulators MDM2 and p21. In both pp5-deficient and WT cells, p53 levels were efficiently reduced by RNAi, and this was accompanied by a reduction in MDM2 and p21 expression (Fig. 1C). We found that the increase in cell death in pp5-deficient cells in response to DOX treatment was diminished following p53 knockdown (Fig. 1B), further supporting the hypothesis that p53 mediates the biological function of PP5 in stress-induced apoptosis.
Figure 1.

Characterization of WT and PP5 KO mice after saline or DOX treatment. A, ablation of PP5 gives rise to bone marrow cells that are sensitive to p53-dependent apoptosis. WT and PP5 KO bone marrow (BM) cells were treated with DOX (0.05 and 0.1 μg/ml) for 12 h and then stained with annexin V and propidium iodide. Apoptotic cells (annexin V–positive cells) are indicated as a percentage of gated cells. Apoptotic cells are included in the graphical representation, which represents three independent experiments. B, WT and PP5 KO bone marrow low-density mononuclear cells were isolated and transduced with pMSCV or pMSCV-sh-p53. Following transduction, cells were sorted for eGFP using FACS to enrich for transduced cells, treated with DOX for 12 h, and analyzed by flow cytometry. C, real-time quantitative RT-PCR revealed that the expression of p53 and its downstream targets MDM2 and p21 decreased after sh-p53 transduction. sh-co, control. D, Western blot analysis showed the p53 expression levels in WT and PP5 KO bone marrow. E, sections from saline- or doxorubicin-treated WT and PP5 KO hearts stained with Sirius Red/Fast Green (scale bars, 50 μm).cardiomyocyte F, cardiomyocyte minimal fiber diameter (μm) measurements in WT and PP5 KO mice treated with saline or DOX. Values are presented as the mean ± S.D. (error bars) using Student's t test. * represents p < 0.05, ** represents p < 0.01, and *** represents p < 0.001.
To understand the physiological relevance of the interplay between PP5 and p53, an in vivo disease model of DOX-mediated cardiotoxicity was used as DOX treatment increases heart p53 protein levels and leads to cardiomyocyte atrophy (i.e. reduced size of cardiomyocytes) (48). A loss-of-function p53 mutant effectively blocked this DOX-mediated cardiomyocyte atrophy (48). To test whether PP5 mutant hearts were hypersensitive to DOX, 20 mg/kg DOX was administered to 2-month-old pp5-deficient and WT littermate control mice for 7 days, and saline was used as a control treatment (48). Based on the minimal diameters of the cardiomyocytes, we determined that the baseline size of the cardiomyocytes in pp5-deficient hearts was significantly smaller when compared with that in the WT controls (11.68 ± 0.54 μm in pp5-deficient cardiomyocytes versus 12.57 ± 0.28 μm in WT cardiomyocytes, n = 400 randomly selected cardiomyocytes/three animal hearts, p < 0.01), and this was associated with a higher level of p53 in pp5-deficient hearts than in WT hearts (Fig. 2, A and B). As predicted, DOX treatment was able to cause a significant reduction in the minimal diameter of the cardiomyocytes in pp5-deficient hearts compared with that in WT hearts (9.7 ± 1.51% reduction in pp5-deficient cardiomyocytes versus 7.7 ± 1.23% reduction in WT cardiomyocytes, n = 400 randomly selected cardiomyocytes/three animal hearts, p < 0.05; Fig. 1, E and F). This finding confirms the importance of PP5 in p53-mediated physiology.
Figure 2.

p53 levels and activity were elevated in pp5-deficient mice. A, Western blot analyses show that p53 expression was significantly elevated in the selected pp5-deficient tissues, thymus, spleen, heart, and liver. B, expression of the p53 target genes MDM2, PTEN, and p21 was increased in pp5-deficient mouse thymus (Thy) and heart. C, total and phospho-p53 (Ser-15) were detected in WT pp5−/− MEF cells using anti-p53 and anti-Ser-15 p53 antibodies. D, real-time quantitative RT-PCR indicated that the level of p53 mRNA remained unchanged in mutant MEF cells compared with the levels in the WT controls. p21 (E), Mdm2 (F), and Pten (G) mRNA levels were also increased in pp5-deficient MEF cells, as shown by real-time quantitative RT-PCR analysis of WT and pp5−/− cells. Values are expressed as the means ± S.D. (error bars) from three independent experiments. ** represents p < 0.01, and *** represents p < 0.001.
In addition to the elevated p53 expression levels in bone marrow (Fig. 1D), we also evaluated p53 protein levels in adult and embryonic tissues of WT and pp5-deficient mice, including the thymus, spleen, heart, and liver. As observed in bone marrow cells, p53 levels were significantly elevated in all the pp5-deficient tissues examined (Fig. 2A). Similarly, mouse embryonic fibroblasts (MEFs) isolated from pp5-deficient embryos (E12.5) had significantly elevated levels of p53 compared with control MEFs isolated from WT littermates (Fig. 2C). The phospho-p53 Ser-15 levels were also elevated (Fig. 2C).
To determine whether the increased levels of p53 in pp5-deficient mice enhanced the transcriptional activity, the expression levels of the downstream targets MDM2 (49, 50), PTEN (51), and p21 (52) were analyzed in thymus and heart samples via Western blotting. pp5-deficient tissues consistently exhibited higher levels of these p53 target genes than the WT controls (Fig. 2B). Moreover, the p21, Mdm2, and Pten mRNA levels in pp5-deficient MEFs were up-regulated compared with the levels in the WT control cells (Fig. 2, E, F, and G). However, the p53 mRNA levels were not altered in pp5-deficient cells (Fig. 2D), indicating that the elevation in p53 protein levels was likely due to the enhanced p53 stability in pp5-deficient cells.
PP5 deficiency reduces tumorigenesis in heterozygous p53+/− mice
Given that pp5-deficient mice exhibit increased p53 protein levels, we hypothesized that the PP5–p53 interplay may play a role in tumorigenesis. To test this hypothesis, we generated the compound mutant mice p53+/−pp5−/− and p53+/−pp5+/−. Both strains had one copy of p53, resulting in a moderate level of p53 expression, and one or no copies of pp5, resulting in moderate to no expression of PP5. Western blotting was used to confirm the significantly higher expression levels of p53 in p53+/−pp5−/− mice than in the pp5+/−p53+/− littermates (Fig. 3A) in the thymus, brain, and liver. The lifespan of p53+/−pp5−/− mice was significantly extended (Fig. 3B) with an increase in median lifespan observed from days 257 to 379. Both the p53+/−pp5+/− and p53+/−pp5−/− double mutant mice presented malignant tumors; however, delayed tumor onset was observed in p53+/−pp5−/− mice compared with p53+/−pp5+/− mice (Table 1). Tumor types were analyzed and are listed in Table S1. Therefore, the data further suggest that PP5 is a functional regulator of p53.
Figure 3.

PP5 deficiency enhances survival in p53+/− mice. A, Western blot analyses showed that p53 expression was significantly elevated in pp5−/−p53+/− tissues relative to the expression in pp5+/−p53+/− tissues. B, survival curves of pp5+/−p53+/− (n = 15) and pp5−/−p53+/− (n = 21) mouse cohorts.
Table 1.
Distribution of mice studied for survival and tumor spectrum
| Genotype |
||
|---|---|---|
| p53+/−pp5+/− | p53+/−pp5−/− | |
| Number of mice | 15 | 21 |
| Number of animals analyzed by necropsy | 14 | 18 |
| Animals with metastasis | 11 (78%) | 10 (55%) |
PP5 interacts with p53 and dephosphorylates phospho-p53 at multiple Ser/Thr sites
Phosphorylation of p53 was shown to increase the stability of the p53 protein (16). To test the hypothesis that PP5 acts as a serine/threonine phosphatase that dephosphorylates p53, HEK293T cell lysate was immunoprecipitated with an anti-p53 antibody, and the immunoprecipitates were incubated in the absence or presence of purified PP5 for 30 min at 30 °C. Incubation was followed by Western blot analysis using antibodies that recognized specific phosphorylated residues in p53 (i.e. Ser-9, Ser-15, Ser-20, Ser-37, and Ser-46). As shown in Fig. 4A, we found that PP5 was able to dephosphorylate multiple sites in p53 in vitro, namely Ser-15, Ser-20, and Ser-37. Interestingly, we observed dephosphorylation at Ser-46, which is a site that is important for the induction of apoptotic genes (53). In contrast, the phosphatase Wip1 can dephosphorylate p53 at only Ser-15 (54). It was previously demonstrated that DNA-dependent protein kinase (DNA-PK) phosphorylates p53 at Ser-15 (55). We directly tested whether PP5 could dephosphorylate DNA-PK–induced phospho-p53 Ser-15. DNA-PK was first incubated with purified p53 in kinase reaction buffer for 30 min. After heating at 65 °C to inactivate the kinase, purified recombinant PP5 was added to the reaction. The level of p53 phosphorylation was monitored by Western blot analysis using anti-phospho-p53 (Ser-15) and anti-p53 antibodies. As shown in Fig. 4B, phospho-p53 Ser-15 levels were dramatically reduced following the addition of PP5. To further test the observed phenomenon in vivo, we transfected a constitutively active form of PP5 (PP5ca), which harbored a 13-amino-acid truncation at the C terminus, into WT and pp5-deficient MEF cells. PP5ca expression reduced the relative levels of total p53 and phospho-p53 in WT and pp5-deficient cells (Fig. 4C). Furthermore, Western blot analysis of total p53 and phospho-p53 was performed in transgenic mice overexpressing PP5ca. Our data demonstrated that both total p53 and phospho-p53 levels were dramatically reduced in the PP5ca transgenic mice compared with the levels in the littermate nontransgenic control (Fig. 4D).
Figure 4.

PP5 directly dephosphorylates phospho-p53 at multiple sites and interacts with p53. A, p53 immunoprecipitates were incubated in the absence (−) or presence (+) of purified PP5 for 30 min at 30 °C followed by Western blot analysis with antibodies against phosphorylated p53 in different tissues. B, purified PP5 dephosphorylated p53 at Ser-15 in vitro. Lane 1 represents the untreated p53 control. Following treatment with DNA-PK and ATP, p53 (lane 2) was incubated with purified PP5 (lane 3). Anti-Ser-15 p53 and anti-p53 monoclonal antibodies were used for Western blotting analyses. C, in constitutively active PP5-overexpressing MEF cells, the phospho-p53 levels were significantly lower than the levels in the WT and PP5 mutant cells. D, the total p53 and phospho-p53 levels were significantly decreased in the PP5ca transgenic mice. The interaction of p53 and PP5 was examined by co-IP (E) and GST pulldown assays (F). G, PP5 KO increased the half-life of p53. Four hours after 4-gray ionizing radiation treatment, the WT and PP5 KO MEF cells were treated with 200 g/ml CHX for the indicated times. Lysates were prepared and analyzed by Western blotting for p53 using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a loading control. H, levels of p53 were quantified by densitometry, and optical density was plotted as the percentage of p53 protein remaining. The p53 band intensity was normalized to the glyceraldehyde-3-phosphate dehydrogenase band intensity and then to the t = 0 controls. IB, immunoblotting; Tg, transgenic.
In addition, we performed both coimmunoprecipitation (co-IP) and GSH S-transferase (GST) pulldown assays to determine whether PP5 binds to p53. WT p53 was cotransfected with PP5 in H1299 cells. As shown in Fig. 4E, in the co-IP Western blot assay, PP5 was pulled down by the p53 antibody. The GST pulldown assay also demonstrated a direct interaction between PP5 and p53 (Fig. 4F). Taken together, our data suggest a novel function for PP5 as a phosphatase that directly regulates p53 dephosphorylation at multiple residues.
It is known that p53 protein levels are primarily regulated via various posttranslational modifications. Previously, the phosphorylation of p53 was thought to be associated with p53 stability and transcriptional activity (29, 30). N-terminal phosphorylation might interfere with the interaction of p53 with MDM2, leading to stabilization of p53 (6, 56). For example, the rate of degradation increased following a serine-to-alanine mutation on residue 15 of human p53, preventing phosphorylation at this site (57). In response to various stress signals, p53 is phosphorylated by a series of kinases, such as ATM/ATR, CHK1, and CHK2, which prolongs the p53 protein half-life from minutes to hours (58). To determine whether the p53 half-life was similarly increased in pp5-deficient cells, we treated WT and pp5-deficient MEF cells with cycloheximide (CHX) to inhibit protein synthesis and examined the total p53 protein levels at various time points posttreatment (Fig. 4G). As expected, the absence of PP5 significantly prolonged the half-life of p53. The half-life of p53 in WT cells was ∼32 min but was ∼56 min in PP5 KO cells (Fig. 4H). These data confirmed that the stability of p53 increased in pp5-deficient cells.
PP5 expression is negatively regulated by p53
PP5 has been shown to be up-regulated in tumor cells (26, 59). In an effort to determine the underlying mechanism, a survey of potential transcriptional binding sites for the human and mouse pp5 promoters was performed. Two conserved p53-binding sites were identified within the pp5 promoter region (Fig. 5A). p53 binds specifically to a consensus DNA sequence consisting of two copies of the 10-bp motif 5′-(A/G)(A/G)(A/G)C(A/T)(T/A)G(T/C)(T/C)(T/C)-3′ separated by 0–13 bp (60). This sequence has been observed in many p53 regulatory genes, including p21/Waf1, Mdm2, Bax, Gadd45, and Pcna. To determine whether p53 can regulate PP5 expression, we compared PP5 expression levels between WT and p53-deficient MEFs by Western blotting. Our data showed that PP5 expression was up-regulated in p53-deficient MEFs compared with the expression in WT cells (Fig. 5B). We also compared PP5 expression between WT and p53-deficient thymus and spleen. As shown in Fig. 5C, the PP5 protein levels were also significantly higher in the p53-deficient organs than in the WT organs. To determine whether the up-regulation was due to the increase in pp5 mRNA levels in p53 mutant cells, we analyzed the mRNA expression level of pp5 in WT and KO livers using quantitative RT-PCR. As shown in Fig. 5F, pp5 mRNA expression was significantly higher in the p53 mutant liver than in the WT liver (Fig. 5F), which was consistent with the protein level measurements (Fig. 5E). Furthermore, when p53 was transfected into p53-deficient MEFs, the PP5 expression level was greatly reduced (Fig. 5D). Taken together, these data suggest that p53 acts as a negative regulator of PP5.
Figure 5.

p53 negatively regulates PP5 expression. A, two putative consensus p53-binding sites were identified in the PP5 gene promoter region, and alignment analyses demonstrated that these p53-binding sites are highly conserved among mouse, rat, human, and chimpanzee. EX1–EX3, exons 1–3. B and C, Western blot analyses of the up-regulation of PP5 in p53 KO cells (B) and tissues (C). D, in p53 KO MEF cells, PP5 expression was repressed following p53 transfection. E and F, Western blot and real-time quantitative RT-PCR analyses of WT and pp5−/− mice liver samples. G, the binding of p53 to the pp5 promoter was determined by a ChIP assay. IgG and agarose beads served as negative controls. H, luciferase (Luc) assays demonstrated that the pp5 promoter (pro) was negatively regulated by p53 but not by mutant p53. Values represent the means ± S.D. (error bars) of three independent experiments. ** represents p < 0.01, and *** represents p < 0.001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
To determine whether p53 directly binds to the consensus sites identified above in the pp5 promoter region, we performed a ChIP assay using p53-overexpressing H1299 cells (a well known human cell line that is deficient in endogenous p53). As shown in Fig. 5G, p53-specific ChIP bands were readily amplified in anti-p53 immune complexes, and both consensus p53-binding sites were detected by PCR analysis. To further confirm the importance of the pp5 promoter region, 1.5 kb of the mouse pp5 promoter was subcloned into the promoter-less luciferase expression vector pGL3 (Promega). In parallel, H1299 cells were cotransfected with pPP5-promoter-Luc plus GL4-Renilla and either pcDNA-WT-p53, the pcDNA vector control, or pcDNA-mutant-p53 (codon 173, GTG→GTA), which was mutated in the DNA-binding domain. As shown in Fig. 5H, WT p53 strongly repressed luciferase activity, but mutant p53 (codon 173, GTG→GTA) and the pcDNA vector control did not exhibit similar repression. To further understand whether this activity depended on p53-binding sites (Fig. 5A), a mutant pp5 promoter–luciferase construct was generated that lacked the p53-binding sites. The luciferase assay showed that the repressive regulatory ability of p53 was lost after removal of the p53-binding sites from the pp5 promoter (Fig. 5H). These data suggested that p53 binds directly to the pp5 promoter and functions as a negative regulator of pp5 transcription.
Discussion
In this study, we report the discovery of a novel PP5–p53 interplay with implications for p53-mediated apoptosis and tumorigenesis. Both bone marrow LDM cells and cardiomyocytes from pp5-deficient mice were studied and found to be hypersensitive to DOX treatment. Interestingly, p53 expression was significantly elevated in various tissues of pp5-deficient mice, indicating the importance of PP5 in disease development. Using an siRNA interference approach to knock down p53, we showed, for the first time, that p53 mediates the biological function of PP5 in stress-induced apoptosis. Consistent with elevated p53 expression in pp5-deficient mice, the p53 downstream genes p21 and Pten were significantly up-regulated in terms of mRNA and protein expression levels in pp5-deficient mice compared with the levels in the WT controls (Fig. 2, B–G). Notably, a comparison of survival led to a promising result: the p53+/−pp5−/− mice survived 122 days longer, on average, than the p53+/−pp5+/− mice (Fig. 3B). Using in vitro biochemical analyses, we demonstrated that PP5 directly interacts with p53 and dephosphorylates phospho-p53 at multiple Ser/Thr sites. Furthermore, consensus p53-binding sites were identified within the pp5 promoter region by bioinformatics analysis, and the role of these sites in PP5 repression was confirmed via a ChIP assay (Fig. 5). Taken together, our data indicate that these interactions represent a positive feedback loop between p53 and PP5 in response to cellular and genotoxic stress.
p53 has been found to have multiple functions in the maintenance of genome integrity, cellular apoptosis, senescence, cell cycle control, metabolism, stem cell reprogramming, and autophagy. As p53 is a stress-induced transcription factor, we explored the interactions between PP5 and p53 during genotoxic stress by examining p53-induced apoptosis in pp5-deficient mice. The results indicated that high p53 levels are accompanied by hypersensitivity to DOX treatment in pp5-deficient mice. Different p53 functions are regulated by posttranslational modifications, including acetylation, phosphorylation, methylation, ubiquitination, SUMOylation, and O-GlcNAcylation (18). Phosphorylation of Ser or Thr sites is critical for the regulation of the degradation, stabilization, and transcriptional activity of p53 (5). Our in vitro phosphorylation experiments indicated that PP5 dephosphorylates phospho-p53 at not only Ser-15 but also some additional sites in vitro (Fig. 4A) in contrast to Wip1, which exclusively targets Ser-15 (54). Previous studies demonstrated via suppression of pp5 expression using an antisense pp5 oligonucleotide that PP5 appears to function as a negative regulator of p53 at Ser-15, which is consistent with our findings. However, they did not observe a decrease in the phosphorylation level of p53 following overexpression of PP5 (25). Several in vitro studies have demonstrated that PP5 usually exhibits low phosphatase activity under normal conditions (23), which is likely due to the interaction between the small autoinhibitory domain at the C terminus (residues 490–499) and the tetratricopeptide repeat domain. This interaction prevents potential substrate access to the PP5 catalytic domain (62). It has been shown that PP5 can be activated ∼10-fold in vitro via a 13-amino-acid C-terminal truncation (62, 63). Therefore, the discrepancies between previous findings and the current study likely result from our usage of PP5ca rather than WT PP5. In our experimental system, PP5ca maintained the total p53 levels at a significantly low value relative to the levels in the control. These results were obtained both in vitro and in vivo (Fig. 4, C and D).
p53-deficient mice are developmentally viable, but these mice exhibit reduced survival due to the development of various tumors within 10 months of age, including lymphomas, sarcomas, carcinoma, and osteosarcoma (64). p53 heterozygous mice also develop tumors but at a later age. Approximately 50% of heterozygous p53 mice develop tumors by 18 months of age. By 2 years, >95% of heterozygous mice die of tumors in contrast to a death rate of only ∼20% in their WT littermates (65). Our studies on pp5-deficient mice have revealed the role of PP5 in regulating the functions of p53, especially stability and activity. We hypothesized that PP5 could functionally regulate p53-mediated tumorigenesis. To test this hypothesis, we produced p53+/−pp5+/− and p53+/−pp5−/− double mutant mice and measured survival. Reduced p53 levels in p53+/− mice can lead to increased genomic instability, which increases the likelihood of the development of somatic p53-null cells (66). We found that the p53 levels in the p53+/−pp5−/− mice were enhanced enough to either prevent or reduce tumorigenesis, consequently increasing longevity by an average of 122 days (Fig. 3B).
Beyond its function as a transcriptional activator, p53 also functions as a transcriptional repressor (67). We observed that PP5 expression was up-regulated in MEFs and in tissues of p53-deficient mice (Fig. 5, B and C). It has been reported that there are two distinct types of repression mediated by p53: those that require consensus p53-binding elements and those that do not require such elements. Polo-like kinase 1 (68) and Cdc25c (69) are critical mitotic checkpoint genes that are subject to p53-mediated repression. Similar to Polo-like kinase 1 and Cdc25c, the pp5 promoter contains a consensus p53-binding element. Our experiment using promoter-driven luciferase and ChIP assays demonstrated that p53 directly binds to the pp5 promoter, resulting in PP5 repression (Fig. 5, G and H).
In summary, our work shows that PP5 is a protein phosphatase that is capable of directly regulating p53 phosphorylation, stability, and function and that the expression of PP5 is negatively regulated by p53 (Fig. 6). This novel regulatory interplay may provide the feedback necessary for altering the response of p53 in response to cellular stress.
Figure 6.
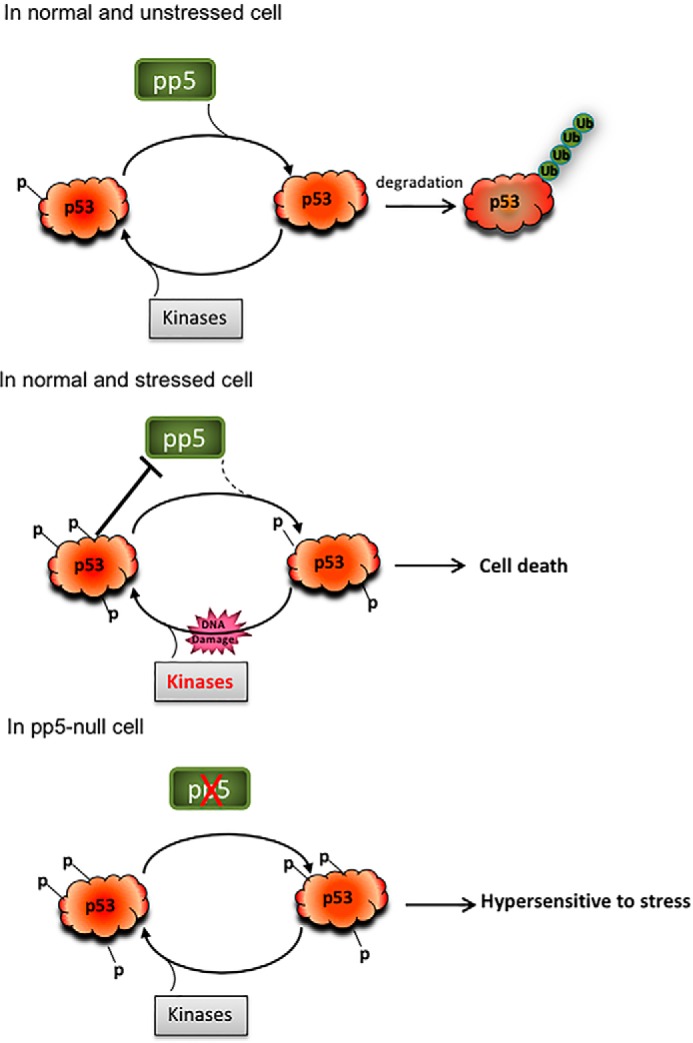
Schematic model showing the interplay between p53 and PP5 in response to stress and the role of PP5 in p53-mediated cellular functions. Ub, ubiquitin.
Materials and methods
Mice
The present study used pp5 KO and littermate WT mice maintained in a C57BL/6 background. Adult mice received two intraperitoneal injections of DOX (10 mg/kg) or vehicle (saline) at 3-day intervals and were euthanized 7 days after the initial injection. p53+/− male mice in a C57BL/6 background were crossed with pp5+/− females to generate p53+/−pp5+/−, and then crossing of p53+/−pp5+/− males and females was performed to generate p53+/−pp5+/− and p53+/−pp5−/− mice. Male p53+/−pp5+/− and p53+/−pp5−/− mice were kept under a standard light/dark regimen (12-h light/12-h dark) for further studies. All animal experiments were conducted in accordance with the “Guide for the Care and Use of Laboratory Animals” and were approved by the Animal Care and Research Advisory Committee of the Institute of Laboratory Animal Sciences, Chinese Academy of Medical Sciences, and the Indiana University School of Medicine.
Cell lines and culture
MEF cells were obtained from WT and pp5-deficient (KO) embryos at day 13.5 of gestation (70). H1299 and p53−/− MEFs were gifts from Dr. Hu Lu. Cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum.
In vitro phosphorylation and dephosphorylation of p53
Purified recombinant p53 was obtained from Dr. Lindsey Mayo. DNA-PK was purchased from Promega (catalog number 9PIV581). PP5 cDNA was cloned into pET21 with BamHI/EcoRI, and the GST fusion protein was expressed, purified, and cleaved as described using B-PER GST Fusion Protein Purification kits (Thermo Fisher Scientific). p53 was phosphorylated in vitro by DNA-PK according to the manufacturer's instructions. After heating at 65 °C to inactivate the kinase, purified recombinant PP5ca was added to the reaction with phosphatase assay buffer (50 mm Tris, 4 mm MnCl2, 4 mm MgCl2, 1 mm EGTA, 0.1% 2-mercaptoethanol, pH 7.6). The level of phosphorylated p53 was assessed by Western blot analysis using anti-phospho-p53 (Ser-15) and anti-p53 antibodies. The p53 immunoprecipitates were incubated in the absence (−) or presence (+) of purified PP5 for 30 min at 30 °C followed by Western blot analysis with antibodies against different p53 phosphorylation sites.
Coimmunoprecipitation and in vivo GST pulldown
Approximately 1 mg of total protein obtained from cotransfected H1299 cells was coimmunoprecipitated using a Pierce co-IP kit (catalog number 26149) following the manufacturer's instructions. Anti-p53 (DO-1, sc-126, Santa Cruz Biotechnology, Inc.) and anti-IgG (A7007, Beyotime, China) were used for antibody immobilization and to pull down p53. Anti-PP5 antibody (H-170, sc-67039, Santa Cruz Biotechnology, Inc.) was used to detect the interaction between p53 and PP5. For the in vivo GST pulldown assay, equal amounts of GST and GST-pp5 expression vectors were transfected into HEK293T cells as indicated. Cell lysates were incubated with GSH-Sepharose beads, and the amount of endogenous p53 pulled down was assayed by Western blotting using a p53 antibody (DO-1).
p53 half-life
p53 protein half-life studies were performed as described by McVean et al. (71). WT and pp5−/− MEF cells were treated with ionizing radiation for 4 h. CHX (2 μg/ml) was then added to inhibit further protein synthesis. Cells were harvested in radioimmune precipitation assay buffer 15, 30, 45, and 60 min after CHX treatment. Aliquots containing 100 μg of total protein were analyzed by Western blotting.
Bone marrow cell culture and retroviral transduction
Bone marrow LDM cells from pp5 KO and WT mice were purified using a Ficoll gradient as described previously (72). After 24 h of prestimulation, the cells were treated with 0, 0.01, and 0.1 μg/ml DOX (Sigma) for 12 h. The treated cells were stained with annexin V–allophycocyanin (BD Pharmingen) followed by flow cytometry according to the manufacturer's instructions. Ecotropic retroviral supernatants (pMSCV and pMSCV-p53) were prepared using Eco-Phoenix packaging cells. Bone marrow cells were then transferred and sorted as described previously (61) and treated with DOX as described above.
Histology
Hearts were harvested, cryoprotected in 30% sucrose, embedded, and sectioned at 10 μm using standard techniques. To quantitate minimal cardiomyocyte fiber diameters, images from Sirius Red/Fast Green–stained sections were captured, digitized, and analyzed with NIH ImageJ software as described previously (48). At least 400 randomly selected cardiomyocytes from each animal were analyzed.
Gel electrophoresis and Western blotting
Samples were resolved on denaturing SDS gels. Transfer of the samples to Immobilon-P® membranes and immunoblotting were performed as described previously (26). Primary antibodies were used to detect the following targets: p53, p53 Ser-15, p53 Ser-20, PP5, p21, and PTEN. The blots were then incubated with the appropriate peroxidase-conjugated secondary antibodies followed by detection using enhanced chemiluminescence.
Quantitative RT-PCR
Total RNA was isolated from mouse tissues or cells using TRIzol (Invitrogen). First-strand cDNA was synthesized by using the Transcriptor First-Strand cDNA Synthesis kit (Roche Applied Science) using 1 μg of RNA as a template according to the manufacturer's instructions. Real-time PCR was performed using a LightCycler 480 with LightCycler 480 SYBR Green I Master Mix (Roche Applied Science). The relative expression levels of the PCR products were normalized to Rpl7. The primers are listed in Table 2.
Table 2.
Primers used for real-time PCR
| Primer name | Sequence |
|---|---|
| Mouse Rpl7_F | CAGAGTTGAAGGTGAAGCGCCTG |
| Mouse Rpl7_R | TCCTTGCCATCCTGGCCATC |
| Mouse Mdm2_F | TCTGGACTCGGAAGATTACAGCCTG |
| Mouse Mdm2_R | GGAGGATTCATTTCATTGCATGAGG |
| Mouse Pten_F | TCACCTGGATTACAGACCCGTGG |
| Mouse Pten_R | CACAGGCAATGGCTGAGGGAAC |
| Mouse p21_F | CACGTGGCCTTGTCGCTGTCTT |
| Mouse p21_R | CGTGGGCACTTCAGGGTTTTCT |
| Mouse p53_F | GAAGTCCTTTGCCCTGAACTGCC |
| Mouse p53_R | GTTTACGCCCGCGGATCTTG |
| Human RPL7_F | AGCTGAAGATCAAGCGCCTGAGAA |
| Human RPL7_R | TTGCCATCCTCGCCATTCGA |
| Human PP5_F | TCTACGGTTTCGAGGGTGAGGTG |
| Human PP5_R | TGAACAGGCCTCCGTGCATG |
| Human p21_F | TGTCTTGTACCCTTGTGCCTCGC |
| Human p21_R | ATCAGCCGGCGTTTGGAGTG |
| Human MDM2_F | GAAAGCCTGGCTCTGTGTGTAATAAGG |
| Human MDM2_R | AGATCCGGATTCGATGGCGTC |
| Human p53_F | GCCCATCCTCACCATCATCACAC |
| Human p53_R | TGTTGTTGGGCAGTGCTCGC |
ChIP and luciferase assay
ChIP assays were performed using the ChIP Assay kit (Millipore). The lysates were immunoprecipitated with either rabbit IgG or an anti-p53 antibody (DO-1). The primers used in the PCRs are shown in Fig. 5A. The mouse pp5 promoter containing the p53-binding site was amplified from genomic DNA with the following primers: pr F, 5′-TAATGGTACCGCCTTGAATGCCACATGGAAGAA-3′; pr R, 5′-ATCAGATCTAAACATTATCCACCCCAGCCCC-3′. The amplified fragment was then cloned into the pGL3-luciferase vector (Promega) between the KpnI and BglII sites. The mutant mouse pp5 promoter, which lacked two p53-binding sites (Fig. 5), was synthesized by Thermo Fisher Scientific (Shanghai, China) and subcloned into the pGL3-luciferase vector between the KpnI and BglII sites. Luciferase activity was analyzed 48 h after transfection, and transfection efficiency was normalized with a Renilla expression vector.
Author contributions
J. W., W. Z., L. D. M., X. L., W. S., and W. Y. formal analysis; J. W., T. S., W. Z., L. D., H. G., L. Z., Z. Y., H. C., Q. Z., L. D. M., and X. L. methodology; J. W., L. F., Z. X., W. S., and W. Y. writing-original draft; T. S., W. Z., Z. Y., X. L., and W. Y. data curation; E. R. S., L. F., L. D. M., Z. X., D. X., X. L., W. S., and W. Y. resources; L. F., L. D. M., X. L., W. S., and W. Y. conceptualization; L. F., W. S., and W. Y. investigation; L. F., L. D. M., X. L., W. S., and W. Y. writing-review and editing; Z. X., D. X., and W. S. funding acquisition; W. S. and W. Y. supervision; W. S. and W. Y. project administration.
Supplementary Material
This work was supported by Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences Grant CIFMS2017-I2M-3-015, National Natural Science Foundation of China Grants NSFC81272273 (to W. Y.) and NSFC31571207 (to Z. X.), National Institutes of Health Grants R01HL181092 and P01HL134599 (to W. S.), and the Fountain Valley Life Science Fund (to D. X.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1.
- ATM
- ataxia-telangiectasia mutated
- ATR
- ATM and Rad3-related
- PP
- protein phosphatase
- Cdc
- cell division cycle
- PTEN
- phosphatase and tensin homolog
- DOX
- doxorubicin
- LDM
- low-density mononuclear
- KO
- knockout
- eGFP
- enhanced GFP
- MEF
- mouse embryonic fibroblast
- DNA-PK
- DNA-dependent protein kinase
- PP5ca
- constitutively active form of PP5
- co-IP
- coimmunoprecipitation
- GST
- GSH S-transferase
- CHX
- cycloheximide
- SUMO
- small ubiquitin-like modifier.
References
- 1. Pu Y., Lin P., Vaughan F. L., and Bernstein I. A. (1995) Appearance of interleukin 1α relates DNA interstrand cross-links and cytotoxicity in cultured human keratinocytes exposed to bis-(2-chloroethyl) sulfide. J. Appl. Toxicol. 15, 477–482 10.1002/jat.2550150609 [DOI] [PubMed] [Google Scholar]
- 2. Sakurai T., He G., Matsuzawa A., Yu G. Y., Maeda S., Hardiman G., and Karin M. (2008) Hepatocyte necrosis induced by oxidative stress and IL-1α release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 14, 156–165 10.1016/j.ccr.2008.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Feldmeyer L., Keller M., Niklaus G., Hohl D., Werner S., and Beer H. D. (2007) The inflammasome mediates UVB-induced activation and secretion of interleukin-1β by keratinocytes. Curr. Biol. 17, 1140–1145 10.1016/j.cub.2007.05.074 [DOI] [PubMed] [Google Scholar]
- 4. McKay B. C., Ljungman M., and Rainbow A. J. (1998) Persistent DNA damage induced by ultraviolet light inhibits p21waf1 and bax expression: implications for DNA repair, UV sensitivity and the induction of apoptosis. Oncogene 17, 545–555 10.1038/sj.onc.1201963 [DOI] [PubMed] [Google Scholar]
- 5. Gu W., and Roeder R. G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606 10.1016/S0092-8674(00)80521-8 [DOI] [PubMed] [Google Scholar]
- 6. Shieh S. Y., Ikeda M., Taya Y., and Prives C. (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325–334 10.1016/S0092-8674(00)80416-X [DOI] [PubMed] [Google Scholar]
- 7. Scheidtmann K. H., Mumby M. C., Rundell K., and Walter G. (1991) Dephosphorylation of simian virus 40 large-T antigen and p53 protein by protein phosphatase 2A: inhibition by small-t antigen. Mol. Cell. Biol. 11, 1996–2003 10.1128/MCB.11.4.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Messner D. J., Romeo C., Boynton A., and Rossie S. (2006) Inhibition of PP2A, but not PP5, mediates p53 activation by low levels of okadaic acid in rat liver epithelial cells. J. Cell. Biochem. 99, 241–255 10.1002/jcb.20919 [DOI] [PubMed] [Google Scholar]
- 9. Mi J., Bolesta E., Brautigan D. L., and Larner J. M. (2009) PP2A regulates ionizing radiation-induced apoptosis through Ser46 phosphorylation of p53. Mol. Cancer Ther. 8, 135–140 10.1158/1535-7163.MCT-08-0457 [DOI] [PubMed] [Google Scholar]
- 10. Takenaka I., Morin F., Seizinger B. R., and Kley N. (1995) Regulation of the sequence-specific DNA binding function of p53 by protein kinase C and protein phosphatases. J. Biol. Chem. 270, 5405–5411 10.1074/jbc.270.10.5405 [DOI] [PubMed] [Google Scholar]
- 11. Li D. W., Liu J. P., Schmid P. C., Schlosser R., Feng H., Liu W. B., Yan Q., Gong L., Sun S. M., Deng M., and Liu Y. (2006) Protein serine/threonine phosphatase-1 dephosphorylates p53 at Ser-15 and Ser-37 to modulate its transcriptional and apoptotic activities. Oncogene 25, 3006–3022 10.1038/sj.onc.1209334 [DOI] [PubMed] [Google Scholar]
- 12. Fiscella M., Zhang H., Fan S., Sakaguchi K., Shen S., Mercer W. E., Vande Woude G. F., O'Connor P. M., and Appella E. (1997) Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 94, 6048–6053 10.1073/pnas.94.12.6048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choi J., Nannenga B., Demidov O. N., Bulavin D. V., Cooney A., Brayton C., Zhang Y., Mbawuike I. N., Bradley A., Appella E., and Donehower L. A. (2002) Mice deficient for the wild-type p53-induced phosphatase gene (Wip1) exhibit defects in reproductive organs, immune function, and cell cycle control. Mol. Cell Biol. 22, 1094–1105 10.1128/MCB.22.4.1094-1105.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crescenzi E., Raia Z., Pacifico F., Mellone S., Moscato F., Palumbo G., and Leonardi A. (2013) Down-regulation of wild-type p53-induced phosphatase 1 (Wip1) plays a critical role in regulating several p53-dependent functions in premature senescent tumor cells. J. Biol. Chem. 288, 16212–16224 10.1074/jbc.M112.435149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li L., Ljungman M., and Dixon J. E. (2000) The human Cdc14 phosphatases interact with and dephosphorylate the tumor suppressor protein p53. J. Biol. Chem. 275, 2410–2414 10.1074/jbc.275.4.2410 [DOI] [PubMed] [Google Scholar]
- 16. Ashcroft M., Kubbutat M. H., and Vousden K. H. (1999) Regulation of p53 function and stability by phosphorylation. Mol. Cell. Biol. 19, 1751–1758 10.1128/MCB.19.3.1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Meek D. W., and Anderson C. W. (2009) Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 1, a000950 10.1101/cshperspect.a000950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dai C., and Gu W. (2010) p53 post-translational modification: deregulated in tumorigenesis. Trends Mol. Med. 16, 528–536 10.1016/j.molmed.2010.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mayo L. D., Seo Y. R., Jackson M. W., Smith M. L., Rivera Guzman J., Korgaonkar C. K., and Donner D. B. (2005) Phosphorylation of human p53 at serine 46 determines promoter selection and whether apoptosis is attenuated or amplified. J. Biol. Chem. 280, 25953–25959 10.1074/jbc.M503026200 [DOI] [PubMed] [Google Scholar]
- 20. Sluss H. K., Armata H., Gallant J., and Jones S. N. (2004) Phosphorylation of serine 18 regulates distinct p53 functions in mice. Mol. Cell. Biol. 24, 976–984 10.1128/MCB.24.3.976-984.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thompson T., Tovar C., Yang H., Carvajal D., Vu B. T., Xu Q., Wahl G. M., Heimbrook D. C., and Vassilev L. T. (2004) Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J. Biol. Chem. 279, 53015–53022 10.1074/jbc.M410233200 [DOI] [PubMed] [Google Scholar]
- 22. Kruse J. P., and Gu W. (2009) Modes of p53 regulation. Cell 137, 609–622 10.1016/j.cell.2009.04.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen M. X., and Cohen P. T. (1997) Activation of protein phosphatase 5 by limited proteolysis or the binding of polyunsaturated fatty acids to the TPR domain. FEBS Lett. 400, 136–140 10.1016/S0014-5793(96)01427-5 [DOI] [PubMed] [Google Scholar]
- 24. Barton G. J., Cohen P. T., and Barford D. (1994) Conservation analysis and structure prediction of the protein serine/threonine phosphatases. Sequence similarity with diadenosine tetraphosphatase from Escherichia coli suggests homology to the protein phosphatases. Eur. J. Biochem. 220, 225–237 10.1111/j.1432-1033.1994.tb18618.x [DOI] [PubMed] [Google Scholar]
- 25. Zuo Z., Dean N. M., and Honkanen R. E. (1998) Serine/threonine protein phosphatase type 5 acts upstream of p53 to regulate the induction of p21(WAF1/Cip1) and mediate growth arrest. J. Biol. Chem. 273, 12250–12258 10.1074/jbc.273.20.12250 [DOI] [PubMed] [Google Scholar]
- 26. Golden T., Swingle M., and Honkanen R. E. (2008) The role of serine/threonine protein phosphatase type 5 (PP5) in the regulation of stress-induced signaling networks and cancer. Cancer Metastasis Rev. 27, 169–178 10.1007/s10555-008-9125-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Atiye J., Wolf M., Kaur S., Monni O., Böhling T., Kivioja A., Tas E., Serra M., Tarkkanen M., and Knuutila S. (2005) Gene amplifications in osteosarcoma-CGH microarray analysis. Genes. Chromosomes Cancer 42, 158–163 10.1002/gcc.20120 [DOI] [PubMed] [Google Scholar]
- 28. Golden T., Aragon I. V., Rutland B., Tucker J. A., Shevde L. A., Samant R. S., Zhou G., Amable L., Skarra D., and Honkanen R. E. (2008) Elevated levels of Ser/Thr protein phosphatase 5 (PP5) in human breast cancer. Biochim. Biophys. Acta 1782, 259–270 10.1016/j.bbadis.2008.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morita K., Saitoh M., Tobiume K., Matsuura H., Enomoto S., Nishitoh H., and Ichijo H. (2001) Negative feedback regulation of ASK1 by protein phosphatase 5 (PP5) in response to oxidative stress. EMBO J. 20, 6028–6036 10.1093/emboj/20.21.6028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sekine Y., Hatanaka R., Watanabe T., Sono N., Iemura S., Natsume T., Kuranaga E., Miura M., Takeda K., and Ichijo H. (2012) The Kelch repeat protein KLHDC10 regulates oxidative stress-induced ASK1 activation by suppressing PP5. Mol. Cell 48, 692–704 10.1016/j.molcel.2012.09.018 [DOI] [PubMed] [Google Scholar]
- 31. Wechsler T., Chen B. P., Harper R., Morotomi-Yano K., Huang B. C., Meek K., Cleaver J. E., Chen D. J., and Wabl M. (2004) DNA-PKcs function regulated specifically by protein phosphatase 5. Proc. Natl. Acad. Sci. U.S.A. 101, 1247–1252 10.1073/pnas.0307765100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yong W., Bao S., Chen H., Li D., Sánchez E. R., and Shou W. (2007) Mice lacking protein phosphatase 5 are defective in ataxia telangiectasia mutated (ATM)-mediated cell cycle arrest. J. Biol. Chem. 282, 14690–14694 10.1074/jbc.C700019200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ali A., Zhang J., Bao S., Liu I., Otterness D., Dean N. M., Abraham R. T., and Wang X. F. (2004) Requirement of protein phosphatase 5 in DNA-damage-induced ATM activation. Genes Dev. 18, 249–254 10.1101/gad.1176004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang J., Bao S., Furumai R., Kucera K. S., Ali A., Dean N. M., and Wang X. F. (2005) Protein phosphatase 5 is required for ATR-mediated checkpoint activation. Mol. Cell. Biol. 25, 9910–9919 10.1128/MCB.25.22.9910-9919.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. von Kriegsheim A., Pitt A., Grindlay G. J., Kolch W., and Dhillon A. S. (2006) Regulation of the Raf-MEK-ERK pathway by protein phosphatase 5. Nat. Cell Biol. 8, 1011–1016 10.1038/ncb1465 [DOI] [PubMed] [Google Scholar]
- 36. Zhang Y., Leung D. Y., Nordeen S. K., and Goleva E. (2009) Estrogen inhibits glucocorticoid action via protein phosphatase 5 (PP5)-mediated glucocorticoid receptor dephosphorylation. J. Biol. Chem. 284, 24542–24552 10.1074/jbc.M109.021469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pazdrak K., Straub C., Maroto R., Stafford S., White W. I., Calhoun W. J., and Kurosky A. (2016) Cytokine-induced glucocorticoid resistance from eosinophil activation: protein phosphatase 5 modulation of glucocorticoid receptor phosphorylation and signaling. J. Immunol. 197, 3782–3791 10.4049/jimmunol.1601029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hinds T. D. Jr., Stechschulte L. A., Cash H. A., Whisler D., Banerjee A., Yong W., Khuder S. S., Kaw M. K., Shou W., Najjar S. M., and Sanchez E. R. (2011) Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid receptor and peroxisome proliferator-activated receptor-γ (PPARγ). J. Biol. Chem. 286, 42911–42922 10.1074/jbc.M111.311662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dean D. A., Urban G., Aragon I. V., Swingle M., Miller B., Rusconi S., Bueno M., Dean N. M., and Honkanen R. E. (2001) Serine/threonine protein phosphatase 5 (PP5) participates in the regulation of glucocorticoid receptor nucleocytoplasmic shuttling. BMC Cell Biol. 2, 6 10.1186/1471-2121-2-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Amable L., Grankvist N., Largen J. W., Ortsäter H., Sjöholm A., and Honkanen R. E. (2011) Disruption of serine/threonine protein phosphatase 5 (PP5:PPP5c) in mice reveals a novel role for PP5 in the regulation of ultraviolet light-induced phosphorylation of serine/threonine protein kinase Chk1 (CHEK1). J. Biol. Chem. 286, 40413–40422 10.1074/jbc.M111.244053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bakkenist C. J., and Kastan M. B. (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506 10.1038/nature01368 [DOI] [PubMed] [Google Scholar]
- 42. Liu L. F. (1989) DNA topoisomerase poisons as antitumor drugs. Annu. Rev. Biochem. 58, 351–375 10.1146/annurev.bi.58.070189.002031 [DOI] [PubMed] [Google Scholar]
- 43. Cummings J., Anderson L., Willmott N., and Smyth J. F. (1991) The molecular pharmacology of doxorubicin in vivo. Eur. J. Cancer 27, 532–535 10.1016/0277-5379(91)90209-V [DOI] [PubMed] [Google Scholar]
- 44. Schmitt C. A., Fridman J. S., Yang M., Baranov E., Hoffman R. M., and Lowe S. W. (2002) Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 1, 289–298 10.1016/S1535-6108(02)00047-8 [DOI] [PubMed] [Google Scholar]
- 45. Hemann M. T., Fridman J. S., Zilfou J. T., Hernando E., Paddison P. J., Cordon-Cardo C., Hannon G. J., and Lowe S. W. (2003) An epi-allelic series of p53 hypomorphs created by stable RNAi produces distinct tumor phenotypes in vivo. Nat. Genet. 33, 396–400 10.1038/ng1091 [DOI] [PubMed] [Google Scholar]
- 46. Freie B., Li X., Ciccone S. L., Nawa K., Cooper S., Vogelweid C., Schantz L., Haneline L. S., Orazi A., Broxmeyer H. E., Lee S. H., and Clapp D. W. (2003) Fanconi anemia type C and p53 cooperate in apoptosis and tumorigenesis. Blood 102, 4146–4152 10.1182/blood-2003-03-0971 [DOI] [PubMed] [Google Scholar]
- 47. Hemann M. T., Zilfou J. T., Zhao Z., Burgess D. J., Hannon G. J., and Lowe S. W. (2004) Suppression of tumorigenesis by the p53 target PUMA. Proc. Natl. Acad. Sci. U.S.A. 101, 9333–9338 10.1073/pnas.0403286101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhu W., Soonpaa M. H., Chen H., Shen W., Payne R. M., Liechty E. A., Caldwell R. L., Shou W., and Field L. J. (2009) Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 119, 99–106 10.1161/CIRCULATIONAHA.108.799700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vogelstein B., Lane D., and Levine A. J. (2000) Surfing the p53 network. Nature 408, 307–310 10.1038/35042675 [DOI] [PubMed] [Google Scholar]
- 50. Ryan K. M., Phillips A. C., and Vousden K. H. (2001) Regulation and function of the p53 tumor suppressor protein. Curr. Opin. Cell Biol. 13, 332–337 10.1016/S0955-0674(00)00216-7 [DOI] [PubMed] [Google Scholar]
- 51. Stambolic V., MacPherson D., Sas D., Lin Y., Snow B., Jang Y., Benchimol S., and Mak T. W. (2001) Regulation of PTEN transcription by p53. Mol. Cell 8, 317–325 10.1016/S1097-2765(01)00323-9 [DOI] [PubMed] [Google Scholar]
- 52. el-Deiry W. S., Tokino T., Velculescu V. E., Levy D. B., Parsons R., Trent J. M., Lin D., Mercer W. E., Kinzler K. W., and Vogelstein B. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 10.1016/0092-8674(93)90500-P [DOI] [PubMed] [Google Scholar]
- 53. D'Orazi G., Cecchinelli B., Bruno T., Manni I., Higashimoto Y., Saito S., Gostissa M., Coen S., Marchetti A., Del Sal G., Piaggio G., Fanciulli M., Appella E., and Soddu S. (2002) Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat. Cell Biol. 4, 11–19 10.1038/ncb714 [DOI] [PubMed] [Google Scholar]
- 54. Takekawa M., Adachi M., Nakahata A., Nakayama I., Itoh F., Tsukuda H., Taya Y., and Imai K. (2000) p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 19, 6517–6526 10.1093/emboj/19.23.6517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Woo R. A., McLure K. G., Lees-Miller S. P., Rancourt D. E., and Lee P. W. (1998) DNA-dependent protein kinase acts upstream of p53 in response to DNA damage. Nature 394, 700–704 10.1038/29343 [DOI] [PubMed] [Google Scholar]
- 56. Riley T., Sontag E., Chen P., and Levine A. (2008) Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 9, 402–412 10.1038/nrm2395 [DOI] [PubMed] [Google Scholar]
- 57. Fiscella M., Ullrich S. J., Zambrano N., Shields M. T., Lin D., Lees-Miller S. P., Anderson C. W., Mercer W. E., and Appella E. (1993) Mutation of the serine 15 phosphorylation site of human p53 reduces the ability of p53 to inhibit cell cycle progression. Oncogene 8, 1519–1528 [PubMed] [Google Scholar]
- 58. Luo K., Li Y., Yin Y., Li L., Wu C., Chen Y., Nowsheen S., Hu Q., Zhang L., Lou Z., and Yuan J. (2017) USP49 negatively regulates tumorigenesis and chemoresistance through FKBP51-AKT signaling. EMBO J. 36, 1434–1446 10.15252/embj.201695669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Urban G., Golden T., Aragon I. V., Scammell J. G., Dean N. M., and Honkanen R. E. (2001) Identification of an estrogen-inducible phosphatase (PP5) that converts MCF-7 human breast carcinoma cells into an estrogen-independent phenotype when expressed constitutively. J. Biol. Chem. 276, 27638–27646 10.1074/jbc.M103512200 [DOI] [PubMed] [Google Scholar]
- 60. el-Deiry W. S., Kern S. E., Pietenpol J. A., Kinzler K. W., and Vogelstein B. (1992) Definition of a consensus binding site for p53. Nat. Genet. 1, 45–49 10.1038/ng0492-45 [DOI] [PubMed] [Google Scholar]
- 61. Yang Z., Kondo T., Voorhorst C. S., Nabinger S. C., Ndong L., Yin F., Chan E. M., Yu M., Würstlin O., Kratz C. P., Niemeyer C. M., Flotho C., Hashino E., and Chan R. J. (2009) Increased c-Jun expression and reduced GATA2 expression promote aberrant monocytic differentiation induced by activating PTPN11 mutants. Mol. Cell. Biol. 29, 4376–4393 10.1128/MCB.01330-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kang H., Sayner S. L., Gross K. L., Russell L. C., and Chinkers M. (2001) Identification of amino acids in the tetratricopeptide repeat and C-terminal domains of protein phosphatase 5 involved in autoinhibition and lipid activation. Biochemistry 40, 10485–10490 10.1021/bi010999i [DOI] [PubMed] [Google Scholar]
- 63. Oberoi J., Dunn D. M., Woodford M. R., Mariotti L., Schulman J., Bourboulia D., Mollapour M., and Vaughan C. K. (2016) Structural and functional basis of protein phosphatase 5 substrate specificity. Proc. Natl. Acad. Sci. U.S.A. 113, 9009–9014 10.1073/pnas.1603059113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dumble M. L., Donehower L. A., and Lu X. (2003) Generation and characterization of p53 mutant mice. Methods Mol. Biol. 234, 29–49 10.1385/1-59259-408-5:29 [DOI] [PubMed] [Google Scholar]
- 65. Venkatachalam S., Shi Y. P., Jones S. N., Vogel H., Bradley A., Pinkel D., and Donehower L. A. (1998) Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. EMBO J. 17, 4657–4667 10.1093/emboj/17.16.4657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Donehower L. A., Godley L. A., Aldaz C. M., Pyle R., Shi Y. P., Pinkel D., Gray J., Bradley A., Medina D., and Varmus H. E. (1995) Deficiency of p53 accelerates mammary tumorigenesis in Wnt-1 transgenic mice and promotes chromosomal instability. Genes Dev. 9, 882–895 10.1101/gad.9.7.882 [DOI] [PubMed] [Google Scholar]
- 67. Johnson R. A., Ince T. A., and Scotto K. W. (2001) Transcriptional repression by p53 through direct binding to a novel DNA element. J. Biol. Chem. 276, 27716–27720 10.1074/jbc.C100121200 [DOI] [PubMed] [Google Scholar]
- 68. McKenzie L., King S., Marcar L., Nicol S., Dias S. S., Schumm K., Robertson P., Bourdon J. C., Perkins N., Fuller-Pace F., and Meek D. W. (2010) p53-dependent repression of polo-like kinase-1 (PLK1). Cell Cycle 9, 4200–4212 10.4161/cc.9.20.13532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. St Clair S., Giono L., Varmeh-Ziaie S., Resnick-Silverman L., Liu W. J., Padi A., Dastidar J., DaCosta A., Mattia M., and Manfredi J. J. (2004) DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: one involves direct binding to the cdc25C promoter. Mol. Cell 16, 725–736 10.1016/j.molcel.2004.11.002 [DOI] [PubMed] [Google Scholar]
- 70. Yang Z., Wolf I. M., Chen H., Periyasamy S., Chen Z., Yong W., Shi S., Zhao W., Xu J., Srivastava A., Sánchez E. R., and Shou W. (2006) FK506-binding protein 52 is essential to uterine reproductive physiology controlled by the progesterone receptor A isoform. Mol. Endocrinol. 20, 2682–2694 10.1210/me.2006-0024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. McVean M., Xiao H., Isobe K., and Pelling J. C. (2000) Increase in wild-type p53 stability and transactivational activity by the chemopreventive agent apigenin in keratinocytes. Carcinogenesis 21, 633–639 10.1093/carcin/21.4.633 [DOI] [PubMed] [Google Scholar]
- 72. Munugalavadla V., Sims E. C., Borneo J., Chan R. J., and Kapur R. (2007) Genetic and pharmacologic evidence implicating the p85α, but not p85β, regulatory subunit of PI3K and Rac2 GTPase in regulating oncogenic KIT-induced transformation in acute myeloid leukemia and systemic mastocytosis. Blood 110, 1612–1620 10.1182/blood-2006-10-053058 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.