

Abstract
Background and Purpose
Chronic alcohol consumption alters the gut–brain axis, but little is known about alcohol binge episodes on the functioning of the intestinal barrier. We investigated the influence of ethanol binges on bacterial translocation, gut inflammation and immunity, and tight junction (TJ) structure and the ability of the biolipid oleoylethanolamide (OEA) to prevent ethanol binge‐induced intestinal barrier dysfunction.
Experimental Approach
OEA was injected i.p. before repeated ethanol administration by oral gavage. Plasma, spleen, liver and mesenteric lymph nodes (MLN) were collected in sterile conditions for determination of bacterial load. Immune/inflammatory parameters, TJ proteins and apoptotic markers were determined in colonic tissue by RT‐PCR and Western blotting. TJ ultrastructure was examined by transmission electron microscopy.
Key Results
Ethanol binges induced bacterial translocation to the MLN (mainly) and spleen. Colonic tissues showed signs of inflammation, and activation of innate (Toll‐like receptor‐4) and adaptive (IgA) immune systems and TJ proteins (occludin and claudin‐3) were decreased after ethanol binges. Pretreatment with OEA reduced intestinal inflammation and immune activation and partially preserved the TJ structure affected by alcohol binges but had no effect on alcohol‐induced apoptosis. Ultrastructural analyses of colonic TJs revealed dilated TJs in all ethanol groups, with less electron‐dense material in non‐pretreated rats. The protective effects of i.p. OEA did not reduce bacterial translocation to the MLN. However, intragastric OEA administration significantly reduced plasma LPS levels and bacterial translocation to the MLN.
Conclusion and Implications
OEA‐based pharmacotherapies could potentially be useful to treat disorders characterized by intestinal barrier dysfunction, including alcohol abuse.
Abbreviations
- BEL
blood ethanol levels
- CFU
colony‐forming units
- iNOS
inducible nitric oxide synthase
- MLN
mesenteric lymph nodes
- OEA
oleoylethanolamide
- TJ
tight junction
- TLR4
Toll‐like receptor‐4
- ZO
zonula occludens
Introduction
Chronic alcohol consumption damages the gastrointestinal barrier, allowing the entry of microorganisms and bacterial products, such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5019, to inner organs and/or the systemic circulation. Alcohol can act in two ways: (i) via transepithelial mechanisms, whereby molecules pass directly through the epithelial cells, possibly because the cell membrane is weakened and damaged due to oxidative stress caused by ROS and (ii) via paracellular mechanisms, where molecules pass through the junctions between the epithelial cells by disrupting tight junction (TJ) proteins. Increased gut permeability allows bacteria and their toxins to leave the gut and infiltrate other organs through the bloodstream (reviewed in Bishehsari et al., 2017). This phenomenon, called leaky gut, has been demonstrated in experimental models of chronic alcohol intake and in alcohol‐dependent subjects (Keshavarzian et al., 2009; Leclercq et al., 2012). Alcohol affects mucosal immunity by suppressing one of the intestine's main lines of defence against bacteria, namely, the Paneth cells that secrete antibacterial compounds. Fewer antibacterial compounds result in additional intestinal bacterial overgrowth and allow endotoxin‐derived bacteria in the gut to trigger an inflammatory response (reviewed in Bishehsari et al., 2017). LPS recognition by the innate immune http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1754 (TLR4) initiates the release of a cascade of pro‐inflammatory mediators, which can reach the brain by several signalling pathways, ultimately inducing neuroinflammation, mood alterations and cognitive decline (Dantzer et al., 2008; Pascual et al., 2014).
A pattern of alcohol consumption in binge episodes has become popular with alcohol drinkers, with increased rates of morbidity (DHS, 2008). A single alcohol binge has been shown to induce a transient increase in serum endotoxin and bacterial DNA levels in healthy subjects (Bala et al., 2014). We have recently reported that repeated alcohol binges increase plasma LPS in rats (Antón et al., 2017a) and humans (Orio et al., 2017) and promote an inflammatory response that may have consequences for neurocognitive performance. However, the effects of binge drinking on intestinal barrier integrity, bacterial translocation and the mechanisms involved in gut barrier dysfunction have yet to be comprehensively investigated.
The gastrointestinal barrier is a continuous multilayer barrier, formed by a mucus layer, epithelial cells and innate or acquired immune factors that allows nutrient absorption from the lumen and restricts the passage of toxic compounds to the systemic circulation. The permeability of the barrier is partly regulated by TJs located in the apical part of the epithelial cells, which are formed by integral transmembrane proteins (occludin and claudins) spreading through the lipid bilayer so that one end contacts the interior of the cell and the other the exterior, and cytoplasmic proteins [zonula occludens (ZO)], which link the TJ to the actin cytoskeleton and form a binding site (Suzuki, 2013). The TJs then create a barrier, which regulates the paracellular movement of the molecules through the epithelial sheets, maintaining tissue homeostasis. A disruption of the intestinal barrier allows enhanced mucosal penetration of intestinal luminal toxic substances, pathogens and antigens that can lead to intestinal mucosal injury and inflammation. Intestinal barrier dysfunction may be either causative or consequential, since immune activation and pro‐inflammatory factors, such as the cytokines http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=958‐α, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4974 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4998, contribute to the barrier disruption increasing the gut permeability in a detrimental self‐propagating cycle (Keita and Soderholm, 2010; Du et al., 2016).
In previous studies, we demonstrated that pharmacological pretreatment with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2661 (OEA), a lipid mediator part of the acylethanolamide family, during the alcohol binge episodes has potent anti‐inflammatory, antioxidant and neuroprotective properties against the effects of alcohol and partially reduced alcohol binge‐induced increases in plasma LPS levels in rats (Antón et al., 2017a). In humans, we observed that regular alcohol binge drinkers show elevated plasma levels of LPS and inflammatory markers (Orio et al., 2017) and increased plasma levels of OEA and similar biolipids (Antón et al., 2017b), suggesting a homeostatic role for this biolipid in regulating alcohol responses (Bilbao et al., 2016). The main hypothesis that we tested in this study was whether the anti‐inflammatory actions of OEA may have a local action in the gut, regulating inflammation and barrier permeability.
Methods
Animals
Sixty‐eight adult (7–8 weeks old) male Wistar rats (Rattus Novergicus from Envigo, Barcelona, Spain) were used, weighing 250–300 g. We selected the same animal species and binge model as in our previous studies (Antón et al., 2017a) since this study is a continuation of this research. Animals were housed in groups of n = 3–4 (medium density) rats per cage on a reverse 12 h light/dark cycle under standard temperature and humidity conditions at the conventional SPF Animal Care Facility at the Complutense University of Madrid. Polysulfone cages were Eurostandard type IV (Tecniplast, Exton, PA, USA) with dimensions of 55.6 × 33.4 cm (lower space), 60.0 × 38.0 cm (upper space) and 19.5 cm high, with a surface area of 1875 cm2. Cages were filled with chopo ecopure chips6 premium (ENVIGO, Barcelona, Spain). Standard food (A04 SAFE, Scientific Animal Food and Engineering, Augy, France) and tap water were available ad libitum in the home cage. Animals were maintained under constant conditions for 10 days before the start of the experiments and under daily surveillance by veterinary staff and/or experimenters.
The animals were monitored during the experiment. When physical signs unrelated to alcohol intoxication were observed, the criteria of humane endpoint were applied. As humane endpoint criteria, we followed the main recommendations of Morton et al. (1999). In detail, changes in body weight, physical appearance and behaviour were scored as previously reported (Morton et al., 1999). All experimental protocols were approved and adhered to the guidelines of the Animal Welfare Committee of the Complutense University of Madrid (ethical approval reference: PROEX 420/15) according to European legislation (2010/63/UE). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010).
Randomization and blinding
The animals were randomly assigned to the experimental groups, and pharmacological treatments were counterbalanced in a random way. The animals were killed following the alternation of experimental groups, and the subjects were selected in a random manner inside a group. Blinding was used when possible. The experimenters were aware of the pharmacological treatments in each group. Tissue collection and preparation, quantification of blots and so on was done in a blind manner. TJ quantification was subjected to double blinding and done by three different investigators.
Drug administration
Animals were treated with intragastric (i.g.) ethanol three times per day using specific cannulae (16G needle, Fisher Scientific, Waltham, MA, USA), following a standard paradigm of a 4‐day binge alcohol intoxication protocol previously used by our group (Antón et al., 2017a) and by others (Obernier et al., 2002a, 2002b). Ethanol‐treated rats received an initial loading dose of 5 g·kg−1 of ethanol in a 30% solution (w.v‐1) and then a maximum of 3 g·kg−1 for subsequent doses, which were determined based on the animals' blood ethanol levels (BELs) and behaviour. Thus, the maximum theoretical dose received by an animal was 9 g·kg−1·day−1. In practice, the doses were titrated by observing behavioural signs related to high blood intoxication levels (i.e. loss of righting reflexes and limb ataxia) (Majchrowicz, 1975) and by measuring BELs at certain points during the binge protocol (2 h after the 15:00 h ethanol administration). In this study, the average dose of ethanol per rat was 8.07 g·kg−1·day−1. This repeated binge‐pattern ethanol paradigm maintained relatively constant intoxicating BEL in a range of sedation/ataxia (BEL average per group·day−1: ethanol 1995 ± 124 mg·L−1 and OEA + ethanol 2089 ± 88 mg·L−1) according to the 6‐point behavioural ethanol intoxication scale (Majchrowicz, 1975). The mortality in the ethanol groups was 9%, similar to that in other studies (Obernier et al., 2002a, 2002b; Antón et al., 2017a).
OEA (was synthesized as described by Giuffrida et al., 2000 and dissolved in 5% Tween‐80 in saline) was administered at 5 mg·kg−1 i.p. ten minutes before each administration of ethanol (first loading OEA dose was 10 mg·kg−1 i.p.). This protocol of OEA administration was previously used to demonstrate its anti‐inflammatory effect in binge drinking (Antón et al., 2017a). Additionally, OEA was also administered p.o. to examine its effect on bacterial translocation. For this OEA (dissolved in 1% carboxymethylcellulose in saline) was administered at 20 mg·kg−1 i.g. (3 mL·kg−1) 20 min before each ethanol administration using specific cannulae (16G needle, Fisher Scientific). This dose was chosen as previous studies had shown this to be an effective pharmacological dose of OEA p.o. (Nielsen et al., 2004; Zhou et al., 2012).
BEL determination and tissue/plasma collection
To verify intoxication, blood samples (20 μL) from rat tails were taken 120 min before the 15:00 h ethanol i.g. administration, and BELs were ascertained by using electrochemical detection of an enzymatic reaction with an AM1 Alcohol Analyzer (Analox Instruments, London, UK).
Samples were taken ~3 h after the last ethanol gavage following a lethal dose of sodium pentobarbital (300 mg·kg−1, i.p., Dolethal®, Spain). Blood was collected by cardiac puncture using trisodium citrate (3.15% w.v−1) as an anticoagulant. Plasma was obtained by blood centrifugation (2000 g) 15 min at 4°C and stored at −20°C until used. Samples of colon were used for biochemical analysis of intestinal barrier elements and gut inflammation. Specifically, the distal 8 cm portion of the colon was removed, cut longitudinally and frozen at −80°C until assayed or processed fresh for transmission electron microscopy. Mesenteric lymph nodes (MLN), liver and spleen were removed under sterile conditions and processed for bacterial translocation studies, as described below.
Bacterial translocation studies
Samples of the MLN, liver and spleen were weighed and homogenized in sterile saline. Aliquots (2 mL) of serial 10‐fold dilutions of the suspension were plated onto 5% blood agar and MacConkey's agar plates for recovery of aerobic bacteria and Brucella blood agar plates supplemented with vitamin K1 and hemin for anaerobic bacteria. After 24 and 48 h of incubation at 37°C, for aerobic and anaerobic cultures, respectively, colonies were counted. Quantitative culture results are expressed as the number of colony‐forming units (CFU)·g−1 of tissue. Any positive MLN, liver or spleen cultures were considered indicative of bacterial translocation from the intestinal lumen. Positive growth of bacteria only in the MLN may therefore provide the required information as these anatomical localizations are normally sterile in healthy individuals. Bacterial strains were identified by Gram stain, biochemical tests (oxidase, catalase and coagulase) and culture media (mannitol and bile esculin agar). Enterotube II (BD DIFCO™, Fisher Scientific, S.L., Madrid, Spain) was used for Gram‐negative bacteria identification.
RT‐PCR and Western blot analyses
RNA was prepared from samples of distal colon using TRIZOL® reagent (Invitrogen, Grand Island, NY, USA). Aliquots were converted to cDNA using random hexamer primers. Semi‐quantitative changes in mRNA levels were estimated by real‐time PCR in the presence of SYBR green probe using a 20‐l reaction in a Rotor‐Gene (Corbett Research®, Mortlake, NSW, Australia). mRNA expression was measured using a comparative quantification method based on the second differential maximum method to calculate single reaction efficiencies. This method calculates a ‘take off’ value for each individual sample based on the rate of change in fluorescence. A control sample is used as a calibrator, and relative amounts are calculated against this calibrator. The results were normalized with the housekeeping gene GAPDH present in each sample. Additional information and the primer sequences, accession numbers and other details can be found in the Supporting Information.
For Western blots, colonic samples were homogenized in 300 μL of PBS (pH = 7.4) mixed with a protease inhibitor cocktail (Complete, Roche®, Madrid, Spain) and RIPA buffer in a TissueLyser (Quiagen, Hilden, Germany). The frequency used was 50 oscillations for 4 min, followed by centrifugation at 12 000× g for 10 min at 4°C. After adjusting protein levels, homogenates were mixed with Laemmli sample buffer (Bio‐Rad®, Alcobendas, Madrid, Spain) containing β‐mercaptoethanol (50 μL·mL−1 of Laemmli), to get a final concentration of 2 mg·mL−1, and 15 uL was loaded into an electrophoresis gel. Proteins were blotted onto a nitrocellulose membrane (Amersham Ibérica®, Madrid, Spain) with a semidry transfer system (Bio‐Rad), incubated with specific primary and secondary antibodies (see Supporting Information) and revealed by use of the ECL™‐kit (Amersham Ibérica). Blots were imaged using an Odyssey® Fc System (Li‐COR Biosciences, Lincoln, Nebraska USA). The band intensities were quantified by densitometric analysis using ImageJ (NIH ImageJ® software, National Biosciences, Lincoln, NE, USA). Raw densitometric data in different blots were transformed as fold change of the control mean, expressed in arbitrary units of OD, and the housekeeping GAPDH or β‐actin proteins were used as loading controls.
Plasma LPS levels
Plasma LPS levels were determined using a colour‐based enzymatic reaction kit (Hycult Biotech, Uden, Netherlands), following the manufacturer's instructions. See Supporting Information.
Plasma and colonic IgA and IgM
Plasma and colonic IgA and IgM levels were measured by commercial ELISA‐based kits (Elabscience, Biotechnology Co., Ltd, Wuhan, China), following the manufacturer's instructions. Tissue data were normalized to the total amount of protein. Additional details are in the Supporting Information.
Pro‐inflammatory cytokine levels in the colon
Protein levels of TNF‐α in distal colon samples were determined by commercially available ELISA kit (RayBiotech®, Norcross, Georgia), with similar tissue preparation as that used for colonic Ig determinations and following the manufacturer's instructions. See Supporting Information for details.
Colon nitrites (NO2 −) levels
Samples of distal colon were homogenized in four volumes of PBS (pH = 7.4) mixed with a protease inhibitor cocktail (Complete, Roche) followed by centrifugation at 12 000× g for 10 min, and supernatants were used. Nitrites (NO2−) were measured using the Griess method: briefly, in an acidic solution with 1% sulphanylamide and 0.1% N‐(1‐Naphthyl) ethylenediamine, nitrites are converted into a pink compound that is measured spectrophotometrically at 540 nm in a microplate reader (Synergy 2; BioTek, Swindon, United Kingdom).
Protein assay
Protein levels were measured using Bradford's method based on the principle of protein–dye binding.
Transmission electron microscopy
Fresh colonic segments were fixed for 4 h in a solution of 4% paraformaldehyde/2% glutaraldehyde, cryoprotected overnight in PBS at 4°C, then postfixed in 1% osmium tetroxide and finally embedded in SPURR resin. Ultrathin sections (70 nm) were collected on 200 mesh cupper grids and stained with 2% uranyl acetate/3% lead citrate. Sections were examined under a JEOL JEM 1010 transmission electron microscope at the Spanish National Centre for Electron Microscopy (Madrid, Spain). To evaluate changes in TJs, the junctional regions of several sections of longitudinally sectioned villi per animal were examined by a member of the team blinded to the conditions of the experiment. We evaluated 5–15 TJs per animal (n = 4–6) in each experimental group. When possible, we measured the separation between the cell membranes of the adjacent epithelial cells along the length of the TJ complex [TJ length was 0.33 μm approximately (see results)] from the apical membrane and reported the maximum distance found between cell membranes for each TJ measured. Quantification was performed using the Image J software (NIH ImageJ, National Biosciences).
Statistical analyses
The data and statistical analyses comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Group sizes were designed taking into account the fact that some animals could be lost due to alcohol intoxication, since this binge ethanol protocol has been reported to induce a mortality rate of around 13–15% (Obernier et al., 2002a,b; Antón et al., 2017a). In order to correct for possible losses of animals, group sizes were unequal at the beginning of the experiment, with more subjects assigned to the ethanol‐treated groups. Group sizes were previously calculated using a specialized software (STATGRAPHICS Centurion XVI, UCM licence), considering the mortality induced by alcohol intoxication. Initial subjects were randomly assigned to the groups: control n = 9, OEA n = 9, ethanol n = 11 and ethanol + OEA n = 11. Two animals in the ethanol groups died during binge intoxication, as expected in view of our previous experience and the reported mortality with this alcohol binge protocol. The final sample was, therefore, control n = 9, OEA n = 9, ethanol n = 10 and ethanol + OEA n = 10 (Figure 7). To determine the large number of proteins and markers in the study, we randomly selected n = 6–8 per group for each marker. The number of animals was set to obtain a difference of 1.8 × sigma with a test potency of 80% with a level of significance of 0.05. Any variation in this size per group in the Western blot, PCR or ELISA was due to methodological pitfalls or outliers (trimming). ‘n’ represents biological replicates (number of animals) throughout the manuscript (and not technical replicates).
Figure 7.
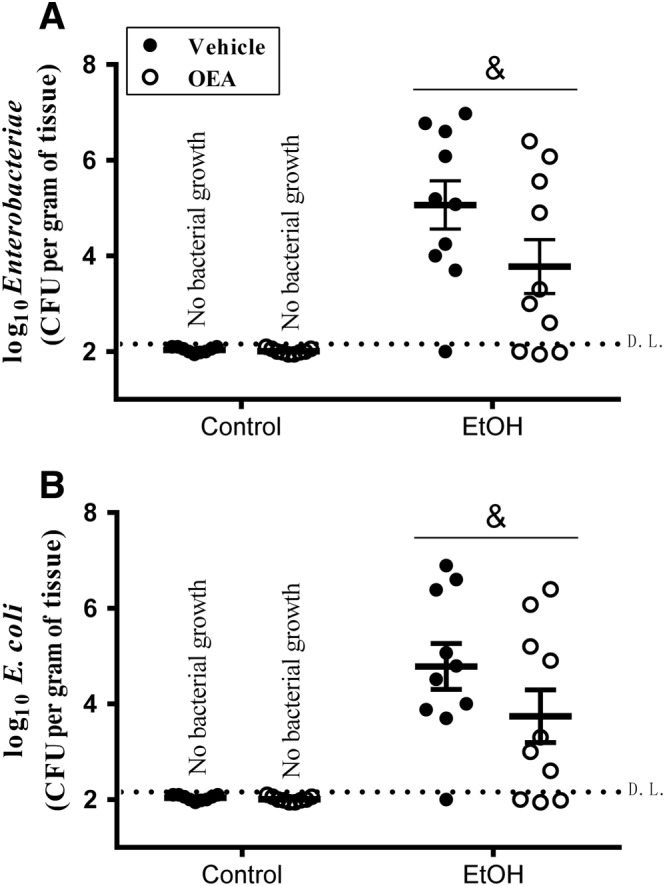
(A) Enterobacteriaceae and (B) Escherichia coli translocation to MLNs in rats exposed to the alcohol binge protocol and pharmacological OEA i.p. pretreatment. Dots indicate CFUs, and horizontal bars represent the mean. Animals with no bacterial growth are represented under the detection limit (D.L.; discontinued horizontal line). Data were corrected for tissue weight and expressed as log10 of mean (horizontal bars) ± SEM (vertical bars). (Control groups n = 6; EtOH groups n = 10). Bacteria were detected in ethanol binge rats but not in control animals, and there is a dependence between these variables (χ2 15.39, & P < 0.05). There were no significant differences between OEA‐ or vehicle‐pretreated animals within the ethanol groups (Student's t‐test, P > 0.05).
The data and figures in the text are expressed as scatterplots, with mean ± SEM. The study of the composition of the microbiota in bacterial translocation has methodological limitations. We used two different approaches to visualize and analyse data from microbiological culture media; there were groups with no bacterial growth. We first included a minimum bacterial presence (1 CFU·mL−1) corrected to g of tissue for each animal, which is under the technique's detection limit. Statistical analyses were performed on groups with bacterial translocation (We excluded experimental groups with a total absence of bacterial growth.). We then included all groups and created contingency tables to study the dependence between the variables ethanol or treatment and bacterial presence, analysing the data with Fisher's exact test and χ2.
All data were subjected to the Kolmogorov–Smirnov normality test and then parametric or non‐parametric analyses were chosen accordingly. If the data were not normally distributed, a non‐parametric test (Mann–Whitney test to compare two groups or Kruskal–Wallis test with Dunn's post hoc test to compare three groups) was performed. If the data were found to follow a Gaussian distribution, parametric tests were done (Student's two‐tailed t‐test for comparisons between two groups or one/two‐way ANOVA). The data in ANOVA were first checked for inhomogeneity of variances by Barlett's test and transformed to log10 when appropriate. The data from PCR were normalized to the mean value of the control group (mean value shown as 1) and presented as fold change of the control mean. The data in Western blots were presented as fold change of the control mean, expressed in arbitrary units. Normalizations in Western blot and PCR were done in order to compare samples run in different blots/sets and to control for unwanted sources of variation. Data subjected to ANOVA were followed by post hoc tests only when the F value attained P < 0.05 and there was no significant inhomogeneity of variances. Two‐way ANOVA was used to compare the factors [alcohol/water] versus [OEA/vehicle]. The Bonferroni post hoc test in the two‐way ANOVA was run only when a significant interaction (F value P < 0.05) was found between factors. Correlation analyses were performed using Spearman's rank coefficients (r). A P value < 0.05 was set as the threshold for statistical significance. The data were analysed using GraphPad Prism 5 for Windows, version 5.01 (GraphPad Software, Inc., La Jolla, CA, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b).
Results
Ethanol binge induces Escherichia coli translocation to different organs
Bacteria CFUs were found mainly in the MLN but also in the spleen, whereas there was no presence of bacteria in liver or blood after ethanol binge treatment. Specifically, 83% and 50% of ethanol‐treated animals presented bacterial CFUs in MLN and spleen respectively. No bacteria were found in any tissue from control animals. Species isolated included Gram‐negative (Escherichia coli, Proteus vulgaris and Klebsiella spp.), Gram‐positive (Enterococcus spp. and coagulase‐negative Staphylococcus) and anaerobic bacteria (Clostridium spp). Among them, E. coli represented the majority of bacteria isolated (Figure 1). In this way, ethanol binge induced E. coli translocation to the MLN and spleen in 83% and 33% of animals respectively.
Figure 1.
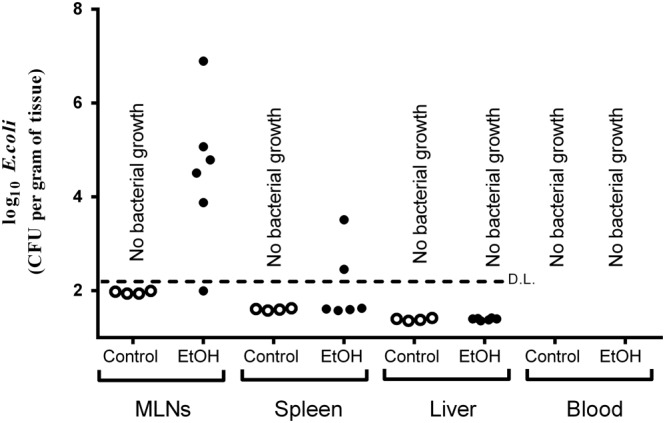
Bacteria CFUs of Escherichia coli in mesenteric lymph nodes (MLNs), spleen, liver and blood after ethanol binge exposure. Ethanol binge administration induced E. coli translocation to MLN in 83% of animals in the ethanol group and to the spleen in 33% of animals. Animals with no bacterial growth are represented under the detection limit (D.L.; discontinued horizontal line). Data are corrected by tissue weight (except blood samples) and expressed as log10. exploratory (pilot) study: control group n = 4; EtOH group n = 6.
This exploratory experiment indicated that ethanol binges induce bacterial translocation, so in the following experiments we studied the integrity of the intestinal barrier to test the pharmacological effects of OEA.
Colonic inflammation after ethanol binge administration and repeated OEA pretreatment
Ethanol binge administration induced the up‐regulation of TLR4 in colonic tissue (Figure 2). There was an overall effect of the alcohol on mRNA expression of TLR4 (Figure 2A; F (1, 22) = 19.16; P < 0.05) that was not prevented by OEA pretreatment. However, we observed an interaction (ethanol administration × OEA pretreatment) on TLR4 protein levels (Figure 2B; F (1, 23) = 8.21; P < 0.05), and post hoc analysis determined that ethanol‐treated animals showed increased TLR4 expression compared with controls, and OEA pretreatment counteracted this effect (Figure 2B).
Figure 2.
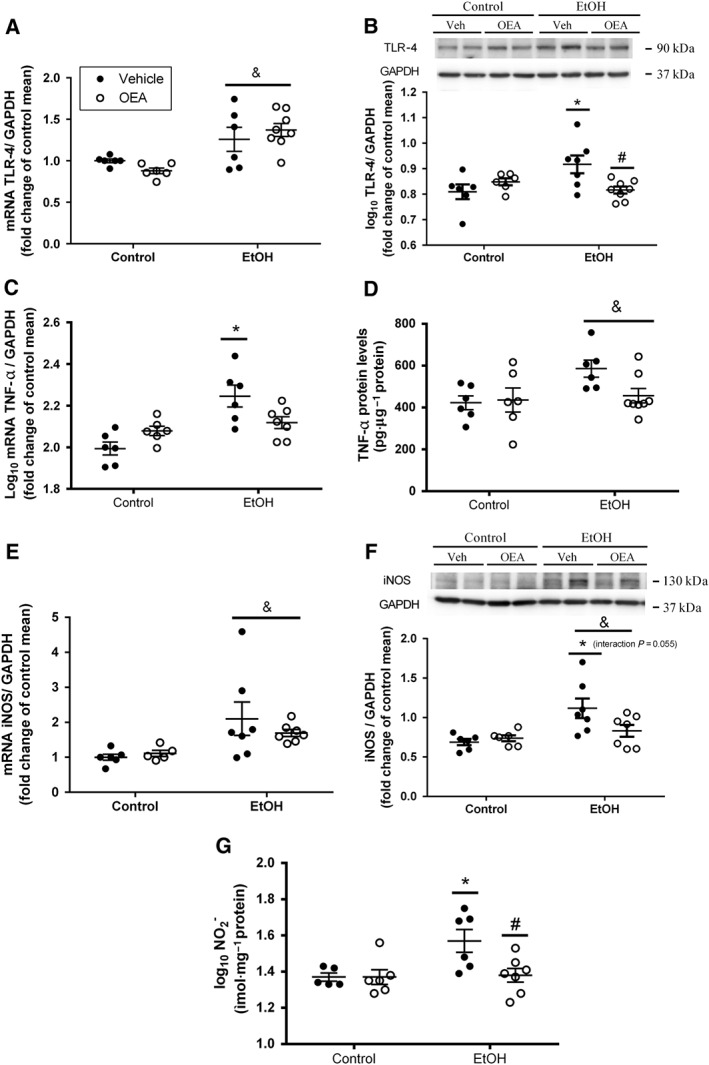
Activation of inflammatory components in distal colon after binge ethanol administration and effects of OEA i.p. pretreatment. (A) Relative mRNA levels of innate immunity receptor TLR4; (B) protein levels of TLR4; (C) relative mRNA levels of TNF‐α; (D) protein levels of TNF‐α; (E) relative mRNA levels of iNOS; (F) protein levels of iNOS; (G) quantification of nitrite levels in homogenized colonic samples. Results are presented as biological samples [Control + Vehicile (Veh) n = 6; Control + OEA n = 6; EtOH + Veh n = 7; EtOh + OEA n = 8] with the mean ± SEM. Overall ethanol effect: & P < 0.05 (two‐way ANOVA). Different from control group in the same condition: *P < 0.05; differences between ethanol and ethanol + OEA groups: # P < 0.05 (two‐way ANOVA, Bonferroni post hoc test after interaction).
TNF‐α mRNA (Figure 2C) and protein levels (Figure 4D) were up‐regulated after ethanol binge (F (1, 21) = 17.40, P < 0.05 and F (1, 22) = 4.76, P < 0.05 respectively). There was an interaction between alcohol and OEA at the mRNA level (F (1, 21) = 9.22; P < 0.05). Post hoc analyses identified a difference between the control and the alcohol group that was not found in the alcohol + OEA group (Figure 2C).
Figure 4.
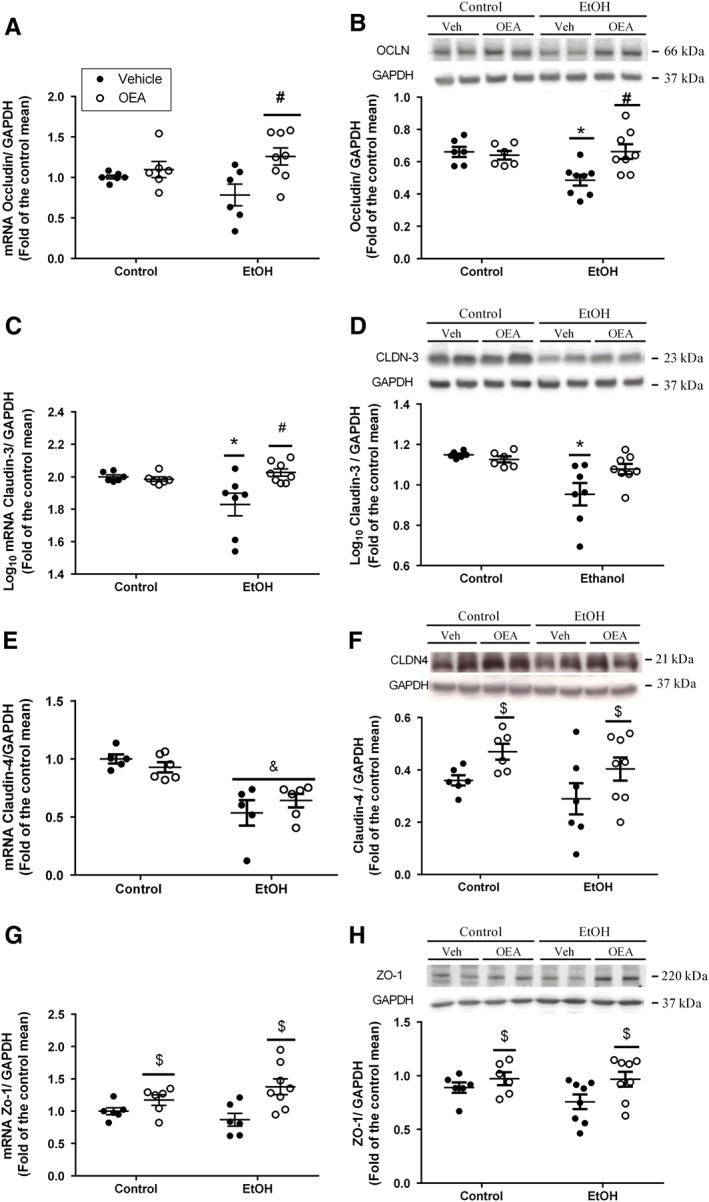
Alterations in colonic tight junction elements forming the intestinal barrier by ethanol binge administration and OEA i.p. pretreatment. Relative mRNA levels (left panels) and Western blot analyses (right panels) of (A, B) occludin, (C, D) claudin‐3, (E, F) claudin‐4 and (G,H) ZO‐1. Results are presented as biological samples [Control + Vehicle (Veh) n = 6; Control + OEA n = 6; EtOH + Veh n = 8; EtOh + OEA n = 8] with the mean ± SEM. Overall OEA effect: $ P < 0.05 (two‐way ANOVA). Different from control group in the same condition: *P < 0.05; differences between ethanol and ethanol + OEA groups: # P < 0.05 (two‐way ANOVA, Bonferroni post hoc test after interaction).
Inducible http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1250 (iNOS) and stable NO metabolites (nitrites) were measured as indexes of nitrosative stress. Ethanol binge administration induced a robust elevation in iNOS mRNA (Figure 2E; alcohol effect: F (1,22) = 8.78; P < 0.05) and protein expression (Figure 2F; F (1, 22) = 9.92; P < 0.05). OEA pretreatment did not counteract this increase in iNOS mRNA (no interaction and no OEA effect: (F (1, 22) = 0.046; n.s.), but there was an interaction (alcohol × OEA pretreatment) very close to significance (F (1, 22) = 4.09, P = 0.055) at the protein level (Figure 2F). We also measured nitrites, the final and stable product of NO and potential inducers of biomolecular nitrosation and lipid peroxidation. Nitrite levels were clearly elevated after ethanol binge (alcohol × OEA interaction: F (1, 20) = 4.44; P < 0.05). This increase in nitrites observed in the ethanol group versus controls was completely prevented by OEA pretreatment (Figure 2G).
IgM and IgA levels in colon and plasma after ethanol binge administration and OEA pretreatment
Igs are secreted to attack the antigens present in the structure of some microorganisms such as bacteria. IgM is one of the first lines of defence endogenously generated, mainly in blood and lymphatic tissue, to combat infections, whereas IgA is more abundant in mucosal membranes, including the gastrointestinal tract.
Ethanol binge treatment decreased the colonic and blood levels of IgM (Figure 3A, B; overall ethanol effect: F (1, 21) = 12.27, P < 0.05; F (1,22) = 4.88, P < 0.05, respectively) regardless of OEA pretreatment (no interaction and no OEA effect: F (1,21) = 0.34, n.s.; F (1,22) = 1.89, n.s. respectively). However, regarding colonic IgA levels (Figure 3C), we observed an interaction between ethanol × OEA (F (1,22) = 5.99, P < 0.05), and post hoc analyses revealed that ethanol binge induced a robust increase in IgA expression in the colon that was totally abolished by OEA pretreatment. Plasma levels of IgA (Figure 3D) were also increased after ethanol (overall effect: F (1, 23) = 8.65, P < 0.05), regardless of the OEA pretreatment (no interaction and no OEA effect: F (1,23) = 0.876, P > 0.05, n.s.).
Figure 3.
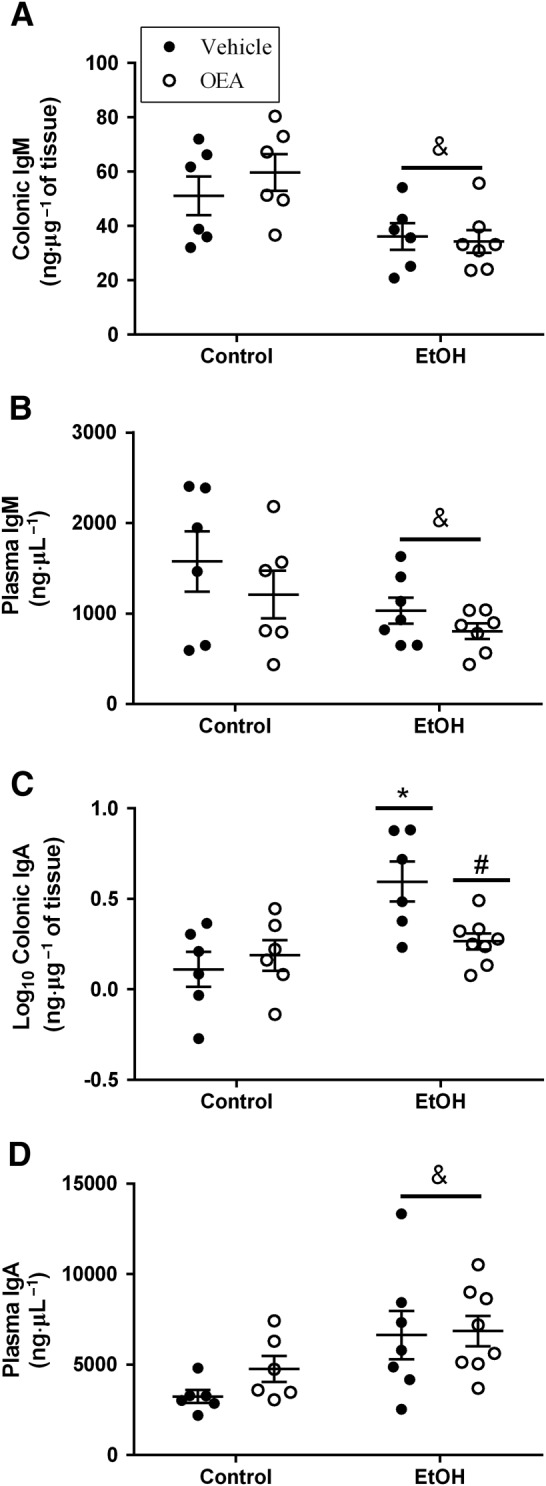
Expression of colonic and plasma IgM and IgA induced by ethanol binge exposure and effects of OEA i.p. pretreatment. (A) Colonic IgM levels; (B) plasma IgM levels; (C) colonic IgA levels; and (D) plasma IgA levels. Results are expressed as biological samples [Control + Vehicle (Veh) n = 6; Control + OEA n = 6; EtOH + Veh n = 7; EtOH + OEA n = 8] with mean (horizontal line) ± SEM (vertical line). Overall ethanol effect: & P < 0.05 (two‐way ANOVA). Different from control group in the same condition: *P < 0.05 and differences between ethanol and ethanol + OEA groups: # P < 0.05 (two‐way ANOVA, Bonferroni post hoc test after interaction).
Intestinal barrier integrity after alcohol binge administration and effects of OEA pretreatment
We checked the integrity of colonic membrane‐bound (occludin and claudins) and scaffolding (ZO) proteins after the bacterial translocation process. Occludin and claudin‐3 are highly expressed in colonic tissue. A reduction in their expression would be indicative of loss of integrity of the intestinal barrier.
We found interactions (ethanol × OEA pretreatment) in occludin mRNA and protein levels (Figure 4A,B; F (1, 22) = 4.09, P = 0.05 and F (1, 24) = 6.97, P < 0.05, respectively). Alcohol binge administration reduced the expression of occludin at the protein level (Figure 4B) but the decrease did not reach significance at the mRNA level (Figure 4A). We observed a significant up‐regulation of occludin mRNA in animals pretreated with OEA within the ethanol‐treated groups (Figure 4A), and, similarly, OEA counteracted the ethanol‐induced reduction in occludin protein (Figure 4B).
There were also interactions between alcohol × OEA pretreatment in claudin‐3 mRNA (Figure 4C; F (1, 23) = 7.18, P < 0.05) and protein levels (Figure 4D; F (1, 23) = 4.74, P < 0.05). Whereas alcohol reduced claudin‐3 mRNA and protein, OEA pretreatment was able to clearly counteract this effect on mRNA (Figure 4C) but not protein (Figure 4D) expression. Claudin‐4 mRNA decreased in both ethanol groups (Figure 4E; F (1, 18) = 32.11; P < 0.05), and OEA pretreatment increased claudin‐4 (Figure 4F; F (1, 23) = 6.425, P < 0.05) and claudin‐1 (data not shown; F (1, 21) = 11.45; P < 0.05) protein levels in both control and ethanol‐treated animals.
Ethanol binge administration did not modify ZO‐1 mRNA or protein levels (Figure 4G; F (1,22) = 0.13; n.s.; Figure 4H; F (1,24) = 1.07; n.s., respectively), although an overall effect of OEA pretreatment on mRNA (F (1,22) = 11.55; P < 0.05) and protein levels (F (1,24) = 4.93; P < 0.05) was found.
Ultrastructural analysis of colonic TJs in ethanol binge‐treated animals and effects of OEA pretreatment
In order to test the state and recovery of colonic TJs after the bacterial translocation process, we applied transmission electron microscopy for ultrastructural analysis of TJ complexes in the colon. Figure 5 shows selected micrographs of the total TJs quantified for each experimental group (n = 25–42). TJs are the membrane bilayers localized in the apical zone, near the brush border that are indicated by arrows in the figure. Qualitative analyses revealed that control animals (Figure 5A) showed an intact apical intercellular ultrastructure, whereas ethanol‐treated animals (Figure 5B) showed dilated (larger) intercellular spaces and disrupted TJs (TJs contained less electron‐dense material) in areas indicated by the arrows. OEA‐pretreated ethanol binge animals (Figure 5C) showed similar dilated TJs as those in the vehicle pretreated ethanol rats, although the TJ electron‐dense material in those appears to be more preserved than in these, which may suggest that some TJ proteins are maintained to some extent. Control animals with OEA pretreatment (Figure 5D) were morphologically indistinguishable from controls. The microvilli or the surface of the enterocytes may appear different in the micrographs due to the fixation process. This study did not analyse the differences between the different experimental groups in terms of the presence of microvilli, enterocyte surface and desmosome structure. Figure 5E shows a representation of the apical structure, with the TJ, desmosome and microvilli and their correlates in a real micrograph.
Figure 5.
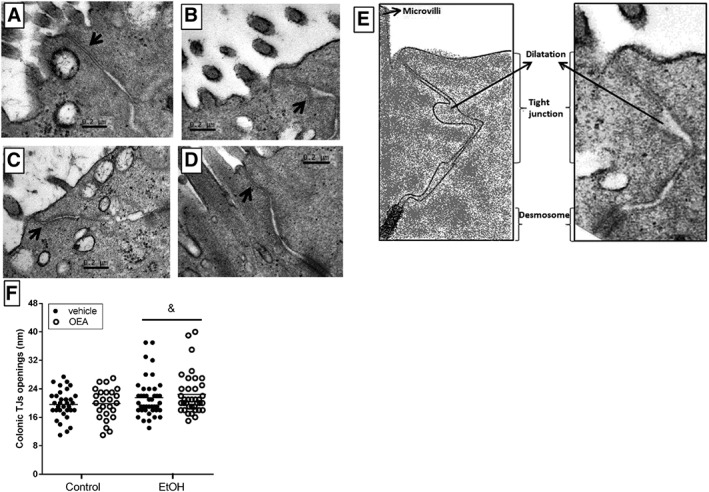
Transmission electron micrographs of colonic TJs in epithelial cells from: (A) control animals (n = 6), (B) ethanol binge‐treated animals (n = 4); (C) OEA i.p. pretreated ethanol animals (n = 4); (D) OEA control animals (n = 4); (E) schematic representation of the apical structure of the gut; (F) quantification of TJs per experimental group (control: n = 33; EtOH n = 42; EtOH + OEA n = 37; OEA n = 25). Ethanol binge‐treated animals showed dilated and disrupted TJs (arrow) compared with controls, which maintain the TJs ultrastructure preserved. Similar openings in TJs were observed in OEA‐pretreated ethanol animals versus ethanol treated rats (ethanol effect: & P < 0.05, two‐way ANOVA), although the presence of electron‐dense material was greater in that group. No difference between control and OEA‐pretreated control animals was observed. For quantitative analysis, two outliers in the ethanol group (51 and 89 nm) and one in the EtOH + OEA group (63 nm) were excluded. Arrows indicate TJ ultrastructure.
Quantitative analyses (Figure 5F) showed that control animals had an average distance of 20 nm between adjacent membranes along the length of the TJ, with a maximum opening of 28 nm detected in a control animal. Ethanol‐treated animals presented some dilated TJs (alcohol effect: F (1, 133) = 6220, P < 0.05) regardless of the OEA i.p. treatment (no interaction found: F (1,133) = 0, 1862, P > 0.05). The length of the TJs in each experimental group was also quantified: control: 0.30 ± 0.01 μm; EtOH: 0.36 ± 0.02 μm; EtOH + OEA: 0.35 ± 0.02 μm; and OEA: 0.32 ± 0.02 μm, with no significant effects among groups (Kruskal–Wallis test P > 0.05).
Apoptotic markers in colonic samples after ethanol binge administration and OEA pretreatment
In order to test whether ethanol induced the translocation of bacteria through an alternative mechanism other than the paracellular pathway (i.e. a transcellular pathway), we studied the markers of enterocyte apoptosis in colonic samples. We observed that repeated ethanol binge administration induced an up‐regulation of pro‐apoptotic molecules http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5078 (FasL) (Figure 6A; F (1, 19) = 4690, P < 0.05) and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1625 (Figure 6C; F (1, 20) = 4879, P < 0.05) compared to controls, implying apoptotic damage in enterocytes. We also examined procaspase‐9, as shown in Figure 6B: whereas ethanol groups revealed a reduction in procaspase‐9 levels (F (1, 19) = 5262, P < 0.05), OEA pretreatment had a clear incremental effect on these levels in both the ethanol and control groups (F (1, 19) = 13,76, P < 0.05), suggesting that OEA pretreatment may interfere with the conversion of procaspase‐9 to the active caspase‐9, although the effect in our study was insufficient to produce any significant changes in caspase‐9.
Figure 6.
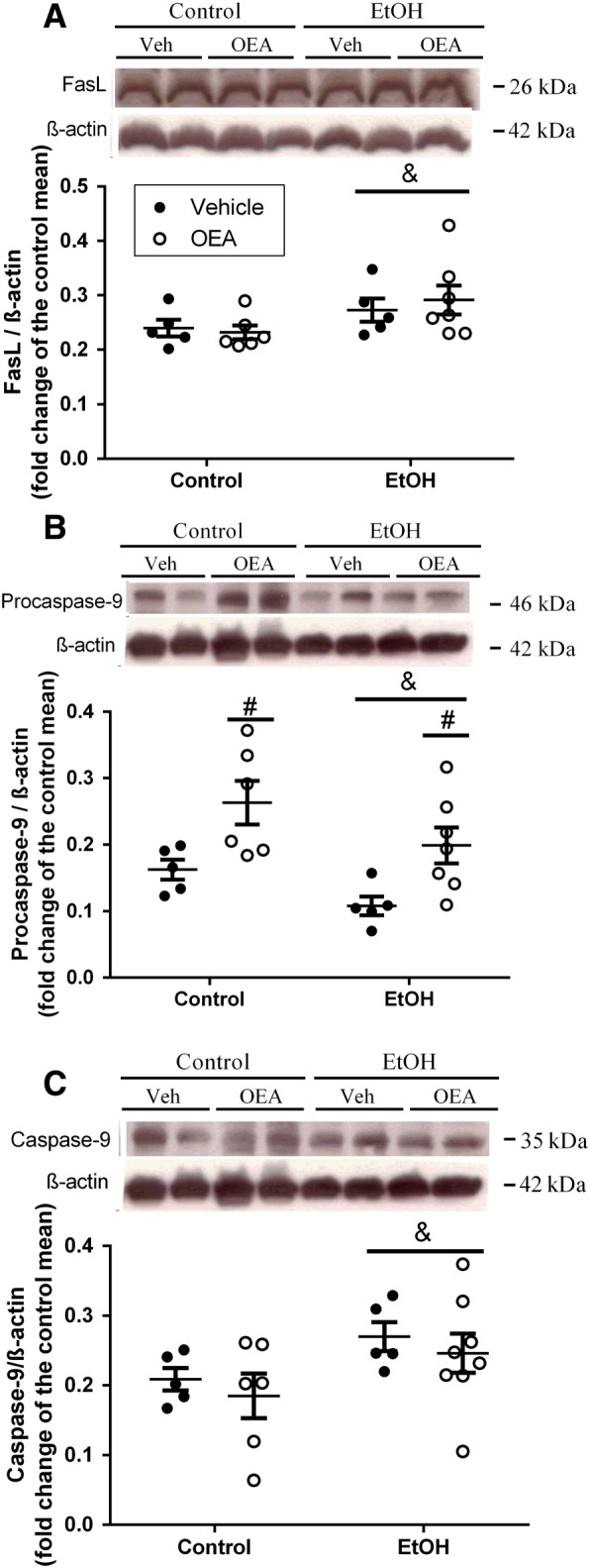
Apoptotic markers in colonic samples after ethanol binge administration and OEA pretreatment. (A) FasLigand (FasL); (B) Procaspase‐9; (C) Caspase‐9. Results are presented as biological samples [Control + Vehicle (Veh) n = 5; Control + OEA n = 6; EtOH + Veh n = 5; EtOh + OEA n = 7] with the mean ± SEM. Overall ethanol effect: & P < 0.05 and overall OEA effect # P < 0.05 (two‐way ANOVA).
Effects of repeated OEA administration on ethanol binge‐induced bacterial translocation to MLNs
Once it was determined that alcohol binge induced the translocation of bacteria to the MLN (Figure 1) and that OEA exerted partial protective effects on the intestinal barrier (Figures 2, 3, 4, 5), we studied the bacterial growth in MLN in animals pretreated with OEA i.p. As expected, there was no evidence of bacterial translocation to the MLN in control animals, whereas the ethanol groups showed translocation of Enterobacteriaceae (Figure 7A), mainly E. coli (Figure 7B), to the MLNs. The comparison between the groups with bacterial growth revealed no differences due to OEA pretreatment (Student's t‐test). Ethanol binge exposure induced Enterobacteriaceae (mainly E. coli) translocation to MLNs in 90% of ethanol‐treated animals, whereas bacterial growth in MLN was present in 70% of OEA‐pretreated animals (Figure 7). Statistical analyses indicated a dependence between the presence of bacteria in MLN and the ethanol treatment (χ2 = 15.39, P < 0.05), but no significant dependence between OEA i.p. pretreatment and bacterial presence in ethanol‐treated animals (χ2 = 1.250; n.s.) (Table 1).
Table 1.
Contingency tables to study the dependence between the variables ethanol/pharmacological pretreatment and the bacterial presence in MLNs
| (a) | Bacterial presence (n) | No bacterial presence (n) | Dependence between EtOH and presence of bacteria? | Dependence between OEA i.p. pretreatment and bacterial presence? |
|---|---|---|---|---|
| Control | 0 | 9 | Yes *P < 0.05 χ2 = 15.39, d.f. = 1 | – |
| Ethanol | 9 | 1 | No P > 0.05, χ2 = 1.250, d.f. = 1 | |
| EtOH + OEA i.p. | 7 | 3 | ||
| OEA i.p. | 0 | 9 | – |
| (b) | Bacterial presence (n) | No bacterial presence (n) | Dependence between EtOH and presence of bacteria? | Dependence between p.o. OEA pretreatment and bacterial presence? |
|---|---|---|---|---|
| Control | 0 | 6 |
Yes *P < 0.05 χ2 = 7.875, d.f. = 1 |
– |
| Ethanol | 6 | 2 |
Yes *P < 0.05, χ2 = 1.250, d.f. = 1 |
|
| EtOH + p.o. OEA | 1 | 5 | ||
| OEA p.o. | 0 | 6 | – |
Results indicate that ethanol binge drinking is associated with bacterial translocation in MLNs. The presence of MNL bacteria in ethanol‐treated animals is not affected by the (a) OEA i.p. pretreatment but it is affected by the (b) p.o. OEA pretreatment. n = number of animals.
Correlations between Escherichia coli presence in MLN, plasma LPS and IgA plasma levels
Levels of LPS in plasma were evaluated as an indirect measure of gut permeability to endotoxins. As we previously demonstrated (Antón et al., 2017a), this protocol of ethanol binge administration significantly increased the levels of LPS in plasma. In the present study, the levels of LPS increased by about 57% versus controls, and OEA‐pretreated ethanol animals showed an increase of 27% in LPS levels. Statistical analyses reflected an overall alcohol effect (F (1,29) = 5.308, P < 0.05), regardless of OEA pretreatment (no interaction found: F (1,29) = 0.2001, P > 0.05), similar to our previously published results.
We observed positive correlations between E. coli presence in MLN with LPS levels in plasma (r = 0.556) and plasma IgA (r = 0.551), as well as correlations between plasma LPS and IgA levels themselves (r = 0.549).
Comparison between i.p. and p.o. administration of OEA in ethanol‐induced translocation of LPS and bacteria
The main objective of our current study was to discern the possible protective mechanisms of OEA in the intestinal barrier that could be related to the anti‐inflammatory and protective effects observed in our previous work using i.p. administration of OEA (Antón et al., 2017a). However, we further hypothesized here that p.o. administration of OEA could be even more effective at preserving the integrity of the colonic barrier. Thus, we measured plasma LPS levels and bacterial translocation under OEA i.g. treatment.
Figure 8A contains a schematic representation of the sites of action of the different routes of OEA administration. Figure 8B shows that ethanol binge administration induced E. coli translocation to MLNs in 75% of ethanol‐treated animals, whereas only 16% of p.o. OEA‐pretreated animals presented bacterial growth in MLN. The Fisher's exact test (χ2) indicated a dependence between pretreatment with p.o. OEA and the presence/absence of bacteria in MLNs (χ2 = 4.667; d.f. = 1; P < 0.05). The comparison between groups with bacterial growth revealed that pretreatment with p.o. OEA was able to reduce bacterial translocation in ethanol binge‐treated animals (Mann–Whitney test).
Figure 8.
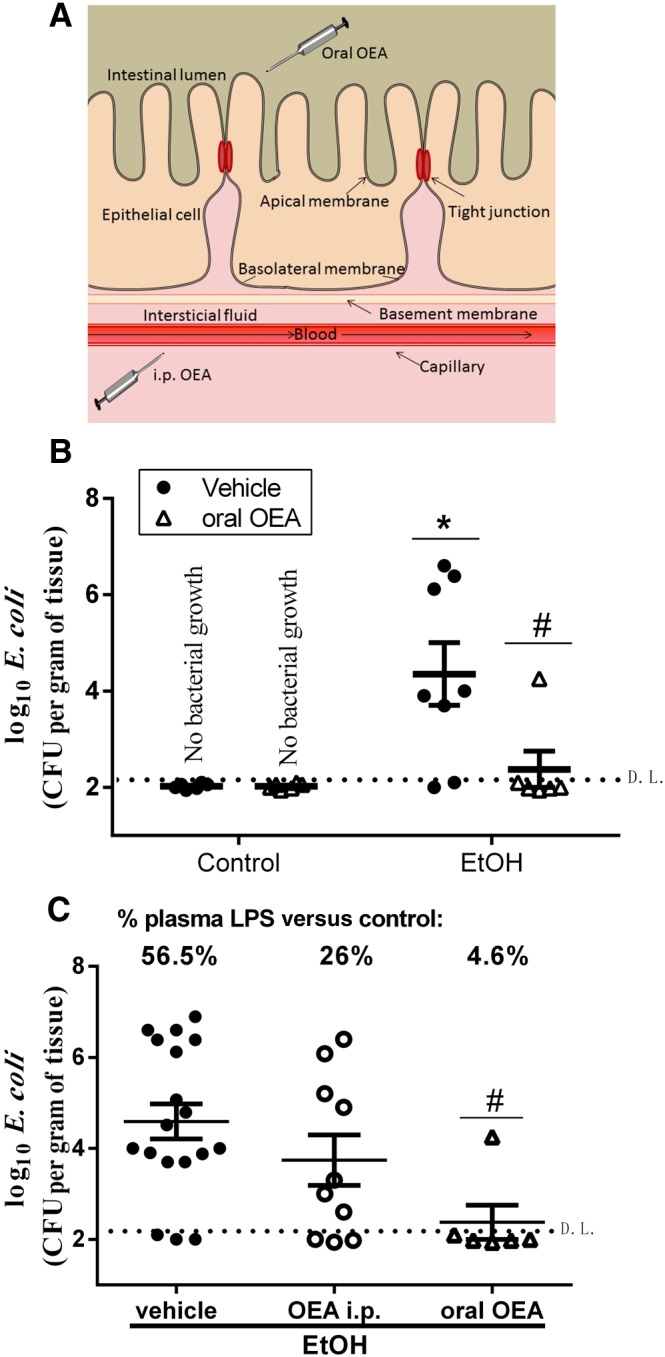
Effects of p.o. OEA administration on ethanol‐induced bacterial and LPS translocation in MLN. (A) Schematic representation of the different routes of administration of OEA (i.p. and p.o.). (B) Effect of p.o. OEA pretreatment on ethanol‐induced bacterial translocation in MLN. Data are expressed as biological samples [controls n = 6, OEA n = 6, EtOH +Vehicle (Veh) n = 8, EtOH + oral OEA n = 6] with mean ± SEM. (C) Comparison between effects of i.p. and p.o. OEA pretreatments on bacterial translocation (scatter plots) and on percentage increase in LPS in plasma (upper panel). Data are expressed as biological replicates (EtOH + Veh n = 18, EtOH + i.p. OEA n = 10, EtOH + oral OEA n = 6) and the mean ± SEM. Animals with no bacterial growth are represented under the detection limit (D.L.; discontinued horizontal line). Different from control group: *P < 0.05. Different from EtOH + Veh: # P < 0.05 [(B) Mann–Whitney; (C) Kruskal–Wallis with Dunn's post hoc test].
Figure 8C shows the comparison between the effects of different routes of OEA administration on bacterial translocation (scatter plots) and the % increase in plasma LPS levels over the controls in each experimental group (upper panel, Figure 8B). OEA p.o. is effective at reducing E. coli translocation in MLN in ethanol‐treated animals (Kruskal–Wallis test = 8.3, P < 0.05; Dunn's post hoc EtOH vs. oral OEA, P < 0.05), and there is a dependence between the route of administration and the bacteraemia in MLN (χ2 = 4.267; d.f. = 1; P < 0.05). We also observed a trend in the reduction of alcohol‐induced increases in plasma LPS levels after the pretreatment with OEA that was clearly more pronounced with the p.o. OEA; indeed, the LPS levels in p.o. OEA‐treated animals were near the control values. These results are in agreement with the bacterial translocation data presented in the scatter plots of Figure 8B.
Discussion
The main findings of this study were as follows: (i) ethanol binge episodes induced local gut inflammation and alterations in colonic TJ complexes, disrupting the intestinal barrier and allowing the passage of bacterial products (LPS) to plasma and bacterial translocation to the MLN (mainly) and spleen but not to the liver or bloodstream. (ii) Pretreatment with OEA i.p. reduced local gut inflammation and immune activation and to some extent preserved the structure of the colonic TJ proteins altered by ethanol binges but did not block ethanol‐induced apoptosis. However, these effects did not alter bacterial translocation to the MLN after ethanol exposure. (iii) OEA p.o. had a stronger effect, drastically reducing the entry of LPS into the bloodstream and inhibiting the translocation of bacteria to the MLNs. These results suggest that pharmacological treatment with OEA i.p. may partially preserve the integrity of the intestinal barrier after the bacterial translocation induced by alcohol and that p.o. OEA may be more powerfully protective against ethanol binge‐induced bacterial translocation. The different mechanisms of the effects of i.p. and p.o. OEA are discussed, but should be studied in more detail in future research.
There is growing evidence indicating that alcohol abuse alters intestinal permeability to toxic substances, although the direct mechanisms are not completely understood. A recent study showed that mice fed an alcohol‐containing diet exhibited decreased apical junctional proteins leading to colonic hyper‐permeability (Forsyth et al., 2017). Inflammation may exacerbate barrier dysfunction in a synergistic manner. For example, TLR4 and the pro‐inflammatory cytokine TNF‐α have been shown to modulate colonic TJ permeability (Li et al., 2013; Al‐Sadi et al., 2016), and anti‐IL‐6 antibody treatment improved ileum barrier function and reduced inflammation after a combined binge treatment and burn injury (Zahs et al., 2013). Here, we provide in vivo evidence that repeated ethanol binges induce an up‐regulation of the colonic TLR4 and pro‐inflammatory cytokines as well as modifications in the intestinal TJs ultrastructure and connection proteins. The source of this inflammation could be a combined effect of the i.g. ethanol administration and the presence of bacterial products such as LPS, since both are TLR4 agonists. Recent studies suggest that physiologically relevant concentrations of LPS (0–1 ng·mL−1) cause an increase in colonic epithelial TJ permeability that was mediated by the up‐regulation of myosin light‐chain kinase in a mechanism dependent on the TLR‐4/MyD88 signal transduction pathway (Nighot et al., 2017). Both long‐term (Tamai et al., 2002) and acute ethanol administration (Tamai et al., 2000) enhance small intestine absorption of LPS, and we demonstrated that this also occurs after repeated binge episodes (Antón et al., 2017a and current study).
The anti‐inflammatory actions of OEA against alcohol binge have been described both peripherally and within the brain (Antón et al., 2017a). The i.p. administration of OEA reduced TLR4 expression and the production of pro‐inflammatory cytokines and oxidative/nitrosative stress in the frontal cortex after repeated ethanol binges (Antón et al., 2017a) or LPS administration (Sayd et al., 2014). In the current study, we showed that this anti‐inflammatory profile extended to local actions in the colon, since we observed reduced levels of colonic TLR4, TNF‐α, iNOS and nitrites after OEA i.p. pretreatment in ethanol binge animals. It has been shown that alcohol‐induced colonic barrier dysfunction is mediated by an up‐regulation of iNOS, NO overproduction and oxidation/nitration of cytoskeletal proteins in vitro (Banan et al., 2000 ) and in an animal model of alcoholic liver injury (Tang et al., 2009). We also showed a partial effect of OEA inhibiting ethanol binge‐induced iNOS and NO expression in vivo, so local anti‐inflammatory actions of OEA on TLR4, cytokines and iNOS may contribute to a partial protective effect on intestinal barrier dysfunction, since inflammation has been shown to compromise epithelial barrier integrity in the colon and small intestine (Du et al., 2016).
Surveillance against pathogens is carried out by the innate and the adaptive immune systems. LPS or other pathogen‐associated molecular patterns are first recognized by innate immunity receptors, such as TLR4, inducing local inflammatory mediators that may activate T and B lymphocytes to produce IgA and IgM as part of adaptive immune system activation. Overproduction of local inflammatory mediators may compromise the integrity of the intestinal barrier, allowing the migration of immune components to the bloodstream or vice versa (Keita and Soderholm, 2010). Thus, increased plasma IgA levels may be considered as a peripheral marker of inflammation. Here, we showed that repeated ethanol binges induced an up‐regulation of plasma IgA expression that correlated with bacterial translocation and plasma LPS levels.
Still more pronounced was the elevation in intestinal IgA after ethanol treatment, which was completely blocked by OEA pretreatment, suggesting that OEA blocks both the immune (TLR4) and adaptive (IgA) components of the intestinal immune system. IgA is a major immunoglobulin isotype in the intestine and plays a role in gut homeostasis, protecting the epithelium from pathogenic microorganisms and regulating the gut microbiome (Inamine and Schnabl, 2018). Acute alcohol has been demonstrated to increase the number of IgA‐immunoreactive cells by a mechanism involving endogenous NO (Budeč et al., 2007), which is in accordance with our data. The effects of ethanol binge on IgM production were unexpected, since the drug decreased plasma and gut IgM levels. The reason for this result is at the moment unknown, and further research is needed to understand this adaptation to alcohol binge exposure.
Our results show that repeated ethanol binges alter the integrity of some colonic TJ proteins, such as claudin‐3 and occludin (and claudin‐4 mRNA). OEA pretreatment modulated the colonic expression of occludin and claudin‐3 in alcohol‐administered animals, showing protective actions against alcohol binge‐induced disruption of the intestinal barrier. In this sense, an occludin deficiency in mice promoted ethanol‐induced epithelial TJ alterations and colonic barrier dysfunction (Mir et al., 2016). OEA pretreatment increased the expression of ZO‐1, claudin‐1 and claudin‐4 under control and alcohol conditions. This possible defensive effect of OEA in physiological conditions is very interesting and deserves further investigation. A modulatory role of OEA on the intestinal barrier under control conditions has been suggested in vitro (Karwad et al., 2017).
It is unclear whether the disruption of TJs observed in this study was sufficient to induce bacterial translocation. E. coli has an average size of 0.4 × 1 μm, but it should be noted that we observed the structure of the TJ complexes at one specific moment after the translocation. Bacterial size appears not to be the limiting factor for translocation, since enteric bacteria have been shown to differ in their fibrogenic capability to alter barrier integrity. These differences appear to be linked to the type of inflammatory response they produce. Whereas E. coli strains from healthy controls did not disrupt TJs, E. coli producing α‐haemolysin in inflammatory‐related diseases such as ulcerative colitis can cause a rapid loss of TJ integrity, which contributes to the pathophysiology of the disease (Mirsepasi‐Lauridsen et al., 2016). The bacterial passage to the inner organs may therefore depend synergistically on different factors such as the inflammatory conditions and the TJ ultrastructure. The influence of pathogenic and commensal bacteria on epithelial barrier function also has severe implications in promoting bacterial translocation, and further work is thus necessary to elucidate how bacterial presence after repeated ethanol consumption modulates TJ permeability. In any case, in our study, we visualized TJ complexes when the bacterial translocation had already taken place, so we cannot exclude the possibility that the openings of the TJ complexes were larger at the specific moment of translocation. We can only demonstrate the consequences of the bacterial passage on TJ structure. Apical junctional complex structure can be dynamic, and the precise location of some of the component parts may influence the final outcome (Karwad et al., 2017). We observed that in animals pretreated with OEA i.p. the intestinal barrier was preserved at the level of inflammation, immune activation and certain TJ proteins
Recent evidence suggests that elevated inflammation in the gut combined with high levels of nitrosative stress could affect epithelial junctional complexes such as TJ proteins and contribute to gut leakiness and serum endotoxin levels (Cho et al., 2018). TNF‐α, through its receptor TNFR1, is known to contribute to the disruption of TJs and associated dysbiosis (Chen et al., 2015). Our results concur with these discoveries, since we observed elevated TNF‐α levels, iNOS expression and nitrite production in ethanol‐treated animals, together with disruption of TJ proteins such as claudin‐3 and occludin.
In spite of the promising protective effects of OEA in the gut barrier, we did not observe any significant reductions in plasma LPS or bacterial translocation to the MLN after its administration i.p. We cannot rule out the possibility that the passage of bacteria after ethanol binges could be related to other mechanisms that affect cell viability. Oxidative stress‐induced damage to the cell membrane could explain the leakage of bacterial products through transepithelial mechanisms. However, recent in vitro studies indicate that the influence of OEA on intestinal permeability is not related to pore formation, destruction of the epithelial monolayer or cell viability effects. OEA (apically applied) induces changes in the proteins that facilitate cell adhesion such as F‐actin, indicating morphological changes in the filament arrangements (Karwad et al., 2017). However, it is unclear how the change in apical morphology may connect to changes in cell–cell junctional complexes and render the interactions tighter. Our current study focused on the integrity of TJ proteins rather than on the structure of the microvilli and desmosome or the enterocyte surface. Nevertheless, we measured certain factors related to other mechanisms that could explain the bacterial leakage, such as the apoptosis of enterocyte cells. We found that ethanol binges increased FasL and caspase‐9 in colonic samples, signalling enterocyte apoptotic damage. Whereas OEA inhibited nitrosative stress, inflammation and some TJ protein degradation, it failed to inhibit apoptosis, which could explain the absence of a significant effect on bacterial translocation after i.p. treatment. Other markers of enterocyte viability such as Krt20 should be verified in future studies.
Unlike i.p. treatment, i.g. administration of OEA was highly effective in reducing LPS and bacterial leakage, suggesting that the action from the apical side of the epithelial cells is stronger than from the basal side. This concurs with a recent in vitro study showing opposing actions of OEA on intestinal hyperpermeability when applied to the apical or basolateral compartments (Karwad et al., 2017). In this study, the ionic conductance of the paracellular pathway was measured in human Caco‐2 cells, as a proxy for TJ integrity. Whereas OEA increased the transepithelial resistance of the Caco‐2 cell monolayer in a dose‐dependent manner when applied to the apical membrane – indicating a decrease in intestinal permeability – the application of OEA to the basolateral membrane decreased TEER, suggesting increased intestinal permeability. These opposing actions of OEA were apparently mediated by different receptors (TRPV1 and PPAR‐α respectively).
According to current knowledge, OEA is produced by the small intestine after feeding and is released into the blood as an incretin that regulates a number of body functions (Rodriguez de Fonseca et al., 2001), an action that in this experiment was simulated by i.p. OEA administration. Increased OEA release in the small intestine was observed in response to alcohol in vivo (Bilbao et al., 2016) and to inflammation in vitro (Karwad et al., 2017), suggesting a local role of OEA in regulating alcohol responses and intestinal permeability respectively. The idea that inflammation may trigger the release of OEA has been also proposed in our previous studies, in which we observed exacerbated inflammatory responses and increased plasma OEA levels in human alcohol binge drinkers (Orio et al., 2017; Antón et al., 2017b). Alcohol also induced neuroinflammation and OEA release in the frontal cortex of laboratory animals (Bilbao et al., 2016; Antón et al., 2017a). It is not clear whether OEA is synthesized from the enterocytes or the mucosal layer. The administration of OEA p.o. simulates a specific action on the mucosal side of the gut. The different results obtained with the two routes of administration indicate that the action of OEA is stronger from the apical side of the epithelial cells than from the basal side. The mechanisms involved in the action of oral OEA are unknown at present, and this study points to the need for further research in this area. OEA may permeate the macrophage‐containing submucosa and exert its anti‐inflammatory actions there. Alternatively, due to the lipid amine nature of OEA, changes in the structure of the lipid bilayer and the lipid raft composition could be future targets of investigation, since ethanol‐ or LPS‐induced translocation and clustering of TLR4 and signalling molecules into lipid rafts and lipid raft disrupting agents abolished the ethanol‐induced TLR4 signalling pathway (Fernandez‐Lizarbe et al., 2008).
In summary, alcohol binge episodes induced a dysfunction in the intestinal barrier characterized by an increased intestinal inflammatory response, activation of the innate and adaptive immune systems, loss of TJ protein complexes in the apical zone of the intestinal epithelium and apoptosis of enterocytes, which resulted in elevated plasma LPS levels and bacterial translocation to the MLN. Pretreatment with OEA i.p. ameliorated the local inflammatory and innate/adaptive immune responses, partially preventing the damage to TJ proteins induced by an alcohol binge but failed to block the apoptotic mechanisms associated with the transcellular pathway. As a consequence, OEA i.p. treatment did not inhibit alcohol binge‐induced bacterial translocation. However, p.o. administration of OEA significantly reduced LPS levels and bacterial leakage to the MLNs, indicating that the route of administration is an important variable in the protective effects of OEA in alcohol abuse. Our results have important implications for clinical practice, suggesting that OEA‐based pharmacotherapies, at least as a co‐adjuvant strategy, could be potentially useful to treat disorders characterized by intestinal barrier dysfunction, including alcohol abuse.
Author contributions
L.O. conceived the study and obtained the funding. M.A., A.R.G. and L.O. designed the study. B.G.B., M.A., A.R.G., A.B., A.P. and L.O. performed the drug administration, tissue collection and biochemical experiments. J.R.C., N.G. and M.L.G.L. were in charge of microbiological studies. F.R.F. provided synthesized OEA. J.C.L. provided methodological support and revised the manuscript. M.A., A.R.G. and L.O. analysed the data, and L.O. drafted the manuscript. All authors approved the final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Data S1 RT‐PCR, Westerm Blot and ELISA details.
Acknowledgements
This study has been supported by the Plan Nacional sobre Drogas ref: 2015/005 (Ministerio de Sanidad, Servicios Sociales e Igualdad) to L.O. A.R.G. is a recipient of a fellowship from Consejería de Educación, Juventud y Deporte (Comunidad de Madrid/Fondo Social Europeo).
Antón, M. , Rodríguez‐González, A. , Ballesta, A. , González, N. , del Pozo, A. , de Fonseca, F. R. , Gómez‐Lus, M. L. , Leza, J. C. , García‐Bueno, B. , Caso, J. R. , and Orio, L. (2018) Alcohol binge disrupts the rat intestinal barrier: the partial protective role of oleoylethanolamide. British Journal of Pharmacology, 175: 4464–4479. 10.1111/bph.14501.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Sadi R, Guo S, Ye D, Rawat M, Ma TY (2016). TNF‐α modulation of intestinal tight junction permeability is mediated by NIK/IKK‐α axis activation of the canonical NF‐κB pathway. Am J Pathol 186: 1151–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antón M, Alén F, Gómez de Heras R, Serrano A, Pavón FJ, Leza JC et al (2017a). Oleoylethanolamide prevents neuroimmune HMGB1/TLR4/NF‐kB danger signaling in rat frontal cortex and depressive‐like behavior induced by ethanol binge administration. Addict Biol 22: 724–741. [DOI] [PubMed] [Google Scholar]
- Antón M, Rodríguez‐González A, Rodríguez‐Rojo IC, Pastor A, Correas Á, Serrano A et al (2017b). Increased plasma oleoylethanolamide and palmitoleoylethanolamide levels correlate with inflammatory changes in alcohol binge drinkers: the case of HMGB1 in women. Addict Biol. https://doi.org/ 10.1111/adb.12580. [DOI] [PubMed] [Google Scholar]
- Bala S, Marcos M, Gattu A, Catalano D, Szabo G (2014). Acute binge drinking increases serum endotoxin and bacterial DNA levels in healthy individuals. PLoS One 9: e96864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banan A, Fields JZ, Decker H, Zhang Y, Keshavarzian A (2000). Nitric oxide and its metabolites mediate ethanol‐induced microtubule disruption and intestinal barrier dysfunction. J Pharmacol Exp Ther 294: 997–1008. [PubMed] [Google Scholar]
- Bilbao A, Serrano A, Cippitelli A, Pavón FJ, Giuffrida A, Suárez J et al (2016). Role of the satiety factor oleoylethanolamide in alcoholism. Addict Biol 21: 859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishehsari F, Magno E, Swanson G, Desai V, Voigt RM, Forsyth CB et al (2017). Alcohol and gut‐derived inflammation. Alcohol Res 38: 163–171. [PMC free article] [PubMed] [Google Scholar]
- Budeč M, Koko V, Todorović V, Marković D, Poštić M, Drndarević N et al (2007). Possible mechanism of acute effect of ethanol on intestinal IgA expression in rat. Int Immunopharmacol 7: 858–863. [DOI] [PubMed] [Google Scholar]
- Chen P, Stärkel P, Turner JR, Ho SB, Schnabl B (2015). Dysbiosis‐induced intestinal inflammation activates tumor necrosis factor receptor I and mediates alcoholic liver disease in mice. Hepatology 61: 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YE, Yu LR, Abdelmegeed MA, Yoo SH, Song BJ (2018). Apoptosis of enterocytes and nitration of junctional complex proteins promote alcohol‐induced gut leakiness and liver injury. J Hepatol 69: 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. 10.1111/bph.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9: 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEUTSCHE HAUPTSELLE FÜR SCHTFRAGUEN e.V. (DHS) (2008). Binge Drinking and Europe. DHS: Hamm. [Google Scholar]
- Du L, Kim JJ, Shen J, Dai N (2016). Crosstalk between inflammation and ROCK/MLCK signaling pathways in gastrointestinal disorders with intestinal hyperpermeability. Gastroenterol Res Pract 2016, 7374197: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Lizarbe S, Pascual M, Gascon MS, Blanco A, Guerri C (2008). Lipid rafts regulate ethanol‐induced activation of TLR4 signaling in murine macrophages. Mol Immunol 45: 2007–2016. [DOI] [PubMed] [Google Scholar]
- Forsyth CB, Shaikh M, Bishehsari F, Swanson G, Voigt RM, Dodiya H et al (2017). Alcohol feeding in mice promotes colonic hyperpermeability and changes in colonic organoid stem cell fate. Alcohol Clin Exp Res 41: 2100–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida A, Rodriguez de Fonseca F, Piomelli D (2000). Quantification of bioactive acylethanolamides in rat plasma by electrospray mass espectometry. Ann Biochem 280: 87–93. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamine T, Schnabl B (2018). Immunoglobulin A and liver diseases. J Gastroenterol 53: 691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwad MA, Macpherson T, Wang B, Theophilidou E, Sarmad S, Barrett DA et al (2017). Oleoylethanolamine and palmitoylethanolamine modulate intestinal permeability in vitro via TRPV1 and PPARα. FASEB J 31: 469–481. [DOI] [PubMed] [Google Scholar]
- Keita AV, Soderholm JD (2010). The intestinal barrier and its regulation by neuroimmune factors. Neurogastroenterol Motil 22: 718–733. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshavarzian A, Farhadi A, Forsyth CB, Rangan J, Jakate S, Shaikh M et al (2009). Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J Hepatol 50: 538–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclercq S, Cani PD, Neyrinck AM, Stärkel P, Jamar F, Mikolajczak M et al (2012). Role of intestinal permeability and inflammation in the biological and behavioral control of alcohol‐dependent subjects. Brain Behav Immun 26: 911–918. [DOI] [PubMed] [Google Scholar]
- Li X, Wang C, Nie J, Lv D, Wang T, Xu Y (2013). Toll‐like receptor 4 increases intestinal permeability through up‐regulation of membrane PKC activity in alcoholic steatohepatitis. Alcohol 47: 459–465. [DOI] [PubMed] [Google Scholar]
- Majchrowicz E (1975). Induction of physical dependence upon ethanol and the associated behavioral changes in rats. Psychopharmacologia 43: 245–254. [DOI] [PubMed] [Google Scholar]
- Mir H, Meena AS, Chaudhry KK, Shukla PK, Gangwar R, Manda B et al (2016). Occludin deficiency promotes ethanol‐induced disruption of colonic epithelial junctions, gut barrier dysfunction and liver damage in mice. Biochim Biophys Acta 1860: 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirsepasi‐Lauridsen HC, Du Z, Struve C, Charbon G, Karczewski J, Krogfelt KA et al (2016. Mar). Secretion of Alpha‐Hemolysin by Escherichia coli Disrupts Tight Junctions in Ulcerative Colitis Patients. Clin Transl Gastroenterol 7: e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton DB (1999). Humane Endpoints in Animal Experiments for Biomedical Research, Humane endpoints in animal experimentation for biomedical research: Ethical, legal and practical aspects The Royal Society Medical Press: London, pp. 5–12. [Google Scholar]
- Nielsen MJ, Petersen G, Astrup A, Hansen HS (2004). Food intake is inhibited by oral oleoylethanolamide. J Lipid Res 45: 1027–1029. [DOI] [PubMed] [Google Scholar]
- Nighot M, Al‐Sadi R, Guo S, Rawat M, Nighot P, Watterson MD et al (2017). Lipopolysaccharide‐induced increase in intestinal epithelial tight permeability is mediated by Toll‐like receptor 4/myeloid differentiation primary response 88 (MyD88) activation of myosin light chain kinase expression. Am J Pathol 187: 2698–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obernier JA, Bouldin TW, Crews FT (2002a). Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol Clin Exp Res 26: 547–557. [PubMed] [Google Scholar]
- Obernier JA, White AM, Swartzwelder HS, Crews FT (2002b). Cognitive deficits and CNS damage after a 4‐day binge ethanol exposure in rats. Pharmacol Biochem Behav 72: 521–532. [DOI] [PubMed] [Google Scholar]
- Orio L, Anton M, Rodriguez‐Rojo IC, Correas A, Garcia‐Bueno B, Corral M et al (2017). Young alcohol binge drinkers have elevated blood endotoxin, peripheral inflammation and low cortisol levels: neuropsychological correlations in women. Addict Biol. 10.1111/adb.12543. [DOI] [PubMed] [Google Scholar]
- Pascual M, Pla A, Minarro J, Guerri C (2014). Neuroimmune activation and myelin changes in adolescent rats exposed to high‐dose alcohol and associated cognitive dysfunction: a review with reference to human adolescent drinking. Alcohol Alcohol 49: 187–192. [DOI] [PubMed] [Google Scholar]
- Rodriguez de Fonseca F, Navarro M, Gomez R, Escuredo L, Nava F, Fu J et al (2001). An anorexic lipid mediator regulated by feeding. Nature 414: 209–212. [DOI] [PubMed] [Google Scholar]
- Sayd A, Anton M, Alen F, Caso JR, Pavon J, Leza JC et al (2014). Systemic administration of oleoylethanolamide protects from neuroinflammation and anhedonia induced by LPS in rats. Int J Neuropsychopharmacol 18 10.1093/ijnp/pyu111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T (2013). Regulation of intestinal epithelial permeability by tight junctions. Cell Mol Life Sci 70: 631–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai H, Kato S, Horie Y, Ohki E, Yokoyama H, Ishii H (2000). Effect of acute ethanol administration on the intestinal absorption of endotoxin in rats. Alcohol Clin Exp Res 24: 390–394. [PubMed] [Google Scholar]
- Tamai H, Horie Y, Kato S, Yokoyama H, Ishii H (2002). Long‐term ethanol feeding enhances susceptibility of the liver to orally administered lipopolysaccharides in rats. Alcohol Clin Exp Res 26: 75S–80S. [DOI] [PubMed] [Google Scholar]
- Tang Y, Forsyth CB, Farhadi A, Rangan J, Jakate S, Shaikh M et al (2009). Nitric oxide‐mediated intestinal injury is required for alcohol‐induced gut leakiness and liver damage. Alcohol Clin Exp Res 33: 1220–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahs A, Bird MD, Ramirez L, Choudhry MA, Kovacs EJ (2013). Anti‐IL‐6 antibody treatment but not IL‐6 knockout improves intestinal barrier function and reduces inflammation after binge ethanol exposure and burn injury. Shock 39: 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Yang L, Ma A, Zhang X, Li W, Yang W et al (2012). Orally administered oleoylethanolamide protects mice from focal cerebral ischemic injury by activating peroxisome proliferator‐activated receptor a. Neuropharmacology 63: 242–249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 RT‐PCR, Westerm Blot and ELISA details.