

Abstract
The human distal gut is home to a rich and dense microbial community with representatives of all three domains of life which are intricately connected with our physiology and health. The combined genomes of these microbes, collectively called the human microbiome, vastly expand the metabolic capacities of our own genome, allowing us to break down and extract energy from dietary compounds that human enzymes cannot digest. In addition, the variable composition of these communities and their biotransformations might explain inter‐individual differences in toxicities, tolerances and efficacies for certain drugs. Recent advances in sequencing technologies and bioinformatics have provided exciting new insights into the genomes of our microbial symbionts, their functional capacities and the interactions between these microbes and their human host. This review summarizes the metabolic conversions of dietary components and pharmaceuticals that take place in the human distal gut, as well as their implications for human health.
Linked Articles
This article is part of a themed section on When Pharmacology Meets the Microbiome: New Targets for Therapeutics? To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v175.24/issuetoc
Abbreviations
- HMP
Human Microbiome Project
- SCFAs
short‐chain fatty acids
- spp.
species
- SRB
sulfate‐reducing bacteria
- TMA
trimethylamine
- TMAO
trimethylamine‐N‐oxide
The human gut microbiome
The human gut is home to 100 trillion microorganisms, most of which live in the colon, the distal part of the gastrointestinal tract. Collectively, these microbes are called the human gut microbiota or, if also referring to their genomes and the surrounding habitat, the human gut microbiome. The vast majority of these microorganisms are bacterial, although archaeal and fungal species are part of the gut microbiota as well (Human Microbiome Project Consortium, 2012; Hoffmann et al., 2013; Li et al., 2014). In addition, the intestinal microbiome includes eukaryotic viruses and bacteriophages, and although their numbers and diversity are much less well studied and not part of the usual estimate of 100 trillion microbes, they are likely to be present in high numbers as well (Robinson and Pfeiffer, 2014). Other human body sites such as mouth and skin are also colonized with microbes, but their numbers and densities are considerably outnumbered by those living in the distal gastrointestinal tract (Sender et al., 2016).
Symbiotic interactions, that is, close biological interactions between two or more different organisms, between multicellular eukaryotic life‐forms and microbes, date back to ancient times and probably played important roles in plant and animal evolution. It has been proposed that the phenotypes and functional capacities of eukaryotic hosts cannot be viewed separately from that of their indigenous microbiotas, but that instead both host and their symbionts have to be viewed as a network of inter‐genomic associations, or even a single biological unit (Bordenstein and Theis, 2015).
The main function of the mammalian gut microbiota is to assist with food digestion and energy extraction, by degrading dietary components such as fibre and cellulose that cannot be broken down and utilized by the host's functional capabilities alone. In addition, microbial colonization of the gut educates the host's immune system and helps to shape the correct development of anatomical structures in the intestinal tract (Round and Mazmanian, 2009). It has been estimated that the combined genomes of our gut microbiome encode for 500 times as many genes than the human genome (Li et al., 2014), and it is thus not surprising that this microbial community is capable of many more biochemical conversions and reactions than its human host. Our gut microbes are basically small biochemical factories, expanding our body's metabolic capacities many times (Grice and Segre, 2012).
The gut microbiome is intricately connected to other organs and our general health. There is accumulating evidence for bidirectional communication between the gut microbes and our brain, in a concept called the gut‐brain‐axis, in which microbes might have an influence on the host's brain development and behaviour, potentially either directly through microbial metabolites in the blood stream, or indirectly by, for example, changing the expression of certain genes (Dinan and Cryan, 2017).
Recent advances in high‐throughput DNA sequencing technologies have enabled us to assess not only the members of a microbial community by amplification of marker genes such as ribosomal RNA genes but also to analyse their full genomes and implied functional capabilities in much greater detail than before. The insights delivered by these sequencing surveys can now be complemented and expanded by other technologies such as transcriptomics, proteomics and metabolomics. Together, these novel ‘omics’ strategies have led to a much better understanding of the composition of the human‐associated microbial communities, the metabolic reactions they perform and their interactions with the human host (Franzosa et al., 2015).
Animal models, albeit artificial and not always an exact representative for human physiology, have made a considerable contribution to microbiome research as well (Kostic et al., 2013; Sonnenburg and Bäckhed, 2016). The blood of mice colonized with a normal microbiota contains dozens of metabolites not present in the blood of germ‐free mice, that is, animals delivered by Caesarean section and raised in sterile isolators, suggesting that the presence of a microbiome might have a large effect on the biochemistry of the host (Wikoff et al., 2009).
Here, we will provide a broad overview of the main metabolic processes performed by the human gut microbiota, focusing on dietary and xenobiotic ‘input’ molecules and microbial output molecules that are relevant for the physiology of the host. Many excellent reviews have been written about specific subtopics (Flint et al., 2012; Ursell and Knight, 2013; Koh et al., 2016; Louis and Flint, 2016; Spanogiannopoulos et al., 2016; Stilling et al., 2016; Zhang and Davies, 2016), but we will provide a basic overview of prevalent commensal species and the main metabolic processes they perform.
Composition of the healthy intestinal microbiome
The human gut microbiome consists of hundreds of microbial species, most of which belong to two bacterial phyla: Bacteroidetes and Firmicutes. Together, these two bacterial taxa constitute the vast majority of the gut community in stool samples, irrespective of diet or geographical location (Human Microbiome Project Consortium, 2012; Obregon‐Tito et al., 2015; Falony et al., 2016). Among the Bacteroidetes phylum, Bacteroides and Prevotella are the most abundant and prevalent genera (Table 1). Within Firmicutes, prevalence and abundance is highest for the Blautia, Eubacterium, Faecalibacterium, Roseburia and Ruminococcus genera (Falony et al., 2016; Zhernakova et al., 2016). Other less prominent but prevalent phyla are Actinobacteria, to which Bifidobacterium species belong, Fusobacteria (genus Fusobacterium) and Verrucomicrobia (genus Akkermansia). A novel phylum called Melainabacteria, related to Cyanobacteria, have recently been recognized as inhabitants of the human gut (Di Rienzi et al., 2013).
Table 1.
Abundant intestinal microbiota members and their metabolic conversions

The data shown here are compiled of data provided in a number of reviews (Louis and Flint, 2016; Magnúsdóttir et al., 2017; Desai et al., 2016; Koh et al., 2016; Ze et al., 2015; Blekhman et al., 2015; Reichardt et al., 2014; LeBlanc et al., 2013; Flint et al., 2012; Nakamura et al., 2010; Belenguer et al., 2006). Most Bacteroidetes and Firmicutes species are capable of producing vitamin B8, as reported by Magnúsdóttir et al. (2017). Genus and species names shown in bold were reported to have an abundance of over 1% in a cohort of 1135 Dutch individuals (Zhernakova et al., 2016)
The most commonly found representative of intestinal archaea is Methanobrevibacter smithii, which can be found in 25–95% of human stool samples (Hoffmann et al., 2013). In addition to bacteria and archaea, eukaryotes such as fungi are commonly detected in stool samples as well. In a study of 96 healthy volunteers, fungi were found in all stool samples, with Saccharomyces, Candida and Cladosporium as the most prevalent (Hoffmann et al., 2013).
Despite a myriad of studies on the composition of the human microbiome of healthy controls and that of diseased patients, and the relative uniform prevalence of the same bacterial phyla among healthy populations, it has been surprisingly hard to define exactly what constitutes a healthy microbiome. Large inter‐individual differences in microbial species composition and relative abundance among healthy subjects or populations from different geographical regions have made it challenging to identify the ‘core’ members of a healthy gut community (Lloyd‐Price et al., 2016).
Most large‐scale gut microbiota studies have been performed on North American and European subjects (Qin et al., 2010; Human Microbiome Project Consortium, 2012; Falony et al., 2016; Zhernakova et al., 2016). Even within these Western populations, it is hard to define a core microbiome. In a combined dataset of stool samples from nearly 4000 Belgian, Dutch, UK and US individuals, the core microbiome (taxa present in 95% of the samples) consisted of only 17 out of the 664 genera found (Falony et al., 2016). Gut microbiomes from other parts of the world often have a very different composition from those from Western individuals (Yatsunenko et al., 2012; Li et al., 2014; Obregon‐Tito et al., 2015; Nishijima et al., 2016), further narrowing this core (Falony et al., 2016). Many of these microbiota differences can be explained by variations in dietary intake. For example, Western diets high in animal protein and fat and low in fibre are associated with a higher relative abundance of Bacteroides, while Prevotella is more abundant in people consuming plant‐based, fibre‐rich diets (Yatsunenko et al., 2012; Obregon‐Tito et al., 2015).
Although it is hard to define the microbial taxa that constitute the healthy human microbiome, such worldwide studies have brought many new insights into the factors that determine its membership. The gut microbiome composition is believed to be the result of a combination of stochastic, lifestyle and host genetic variation. Diet, medicine use, age, health status and stool consistency were found to be the strongest environmental factors shaping the human gut microbiome (Falony et al., 2016; Zhernakova et al., 2016), while variations in the host genome play an important role as well (Blekhman et al., 2015; Goodrich et al., 2016).
Microbiome composition not only varies between individuals but also over time. A person's individual gut microbiome composition appears to be relatively stable in the absence of disease and dietary or lifestyle changes but can quickly and dramatically respond to an altered diet, international travel, food poisoning (David et al., 2014a,b) or antibiotic use (Dethlefsen and Relman, 2011). The fast response of the gut microbiome to such changes is thought to be the result of the rapid growth rate of many bacterial species and the regular large expulsion of gut contents (Sonnenburg and Bäckhed, 2016). In addition to these rapid temporal responses, age has been shown to affect the composition of the gut microbiome as well, albeit at a much slower rate (O'Toole and Jeffery, 2015).
Despite these temporal and population differences, patterns of gut microbial composition in health are starting to emerge. A dataset obtained from a healthy subset of nearly 1000 subjects from the uBiome citizen science cohort was used to define normal ranges of 28 microbial taxa (Almonacid et al., 2017).
Main functions of the gut microbiome
While human enzymes in the small intestine break down most dietary ingredients such as proteins, starch and fatty acids into absorbable smaller molecules, such as amino acids, and monosaccharides, not all molecules present in the diet can be digested in this part of the gastrointestinal tract. The human genome does not encode for enzymes that break down complex proteins and complex carbohydrates, that is, fibres and other plant‐derived polysaccharides. These molecules will therefore reach the colon largely intact, where they can be digested by the gut microbiome (Flint et al., 2012). In addition, the ability to degrade complex carbohydrates can be driven by the diet of the human population.
The most important function of the human microbiome in the distal gut is to extract energy from these otherwise indigestible dietary components (Flint et al., 2012). Not surprisingly, many metabolic processes in the colon lumen are dedicated to this task. Reconstructing the metabolic pathways of the different body sites sampled in the Human Microbiome Project (HMP) consortium showed site‐specific metabolic profiles (Human Microbiome Project Consortium, 2012). Human gut metabolic profiles as determined by metagenomic sequencing were characterized by glycosaminoglycan degradation, which was rare or absent in profiles from other body sites. This functionality was remarkably similar within gut samples from all HMP individuals despite large inter‐individual variations in microbial species composition (Lozupone et al., 2012) emphasizing that the composition of the gut microbiome is not as much about ‘Who is there?’, but about ‘What are they doing?’ This metabolic functional redundancy is likely to confer stability and resilience to the gut microbiota in the setting of dietary and environmental disturbances (Moya and Ferrer, 2016). Although inter‐individual metabolic capabilities are very similar overall, there are also studies showing population‐specific variations. Of note, Bacteroides plebeius strains from Japanese subjects harbour genes encoding for porphyranases and agarose (Hehemann et al., 2010). These genes are absent in other populations and are thought to have been acquired by B. plebeius by horizontal gene transfer from marine bacteria through the consumption of seaweed, thus highlighting the role of the environment as a selective force on the functional potential of the human microbiota.
In addition to the degradation of polysaccharides, other important functions of the gut microbiome include the synthesis of short‐chain fatty acids (SCFAs), specific lipopolysaccharides and certain vitamins and amino acids (Lloyd‐Price et al., 2016). A recent metabolic genome reconstruction of 773 members of the human gut microbiome genomes found genes encoding for 3200 unique chemical reactions, suggesting that this community encodes for hundreds or even thousands of metabolic pathways (Magnúsdóttir et al., 2017).
Metabolism of human milk in infants
The microbiome and metabolic reactions of the human infant gut are distinct from those of the adult gut. Colonization of the infant gut starts immediately after birth, in a process that is thought to involve initial seeding with vaginal and skin microbes derived from the mother, which during the first months of life are gradually replaced with strains derived from other sources, with larger shifts in microbial composition around the time of weaning or around antibiotic treatment (reviewed in Mueller et al., 2015).
Human milk is exceptionally rich in lactose, fatty acids and hundreds of different types of oligosaccharides consisting of different combinations of sugar moieties connected through a variety of glycosidic bonds, some of which are sialylated (Smilowitz et al., 2014). Like dietary fibres in the adult gastrointestinal tract, the milk oligosaccharide glycoside and other bonds cannot be lysed by human genome‐encoded enzymes, and the infant relies on bacteria to digest these compounds. The microbes needed to digest them are thought to be vertically transmitted from mother to infant through the milk (Mueller et al., 2015). These bacteria, in particular Bifidobacterium infantis, Bacteroides thetaiotaomicron and Bacteroides fragilis, are abundant in the gut microbiota of most exclusively breastfed infants in the first months of life (Yatsunenko et al., 2012; Bäckhed et al., 2015). The genomes of these species are well equipped for the digestion of the oligosaccharides present in human milk, encoding for several receptors, intracellular and extracellular glycoside hydrolases and sialidases that can digest the many sugar components of human milk oligosaccharides (Sela et al., 2011).
When breastfeeding stops and solid foods are introduced, the infant gut microbiota starts a trajectory towards a more adult‐like composition characterized by an increase in the abundance of Bacteroides, Clostridium, Faecalibacterium and Ruminococcus (Bäckhed et al., 2015; Mueller et al., 2015). The composition of the infant gut microbiota continues to increasingly resemble that of an adult until it reaches maturation at 3–4 years of age (Yatsunenko et al., 2012).
Microbial fermentation in the adult distal gut
In the adult gut, undigested dietary fibre, carbohydrates and proteins are fermented and further metabolized by the microbial communities in the caecum and colon (Figure 1). The main end products of the fermentation of complex carbohydrates are SCFAs, such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1058, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1062 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1059, and gases, such as CO2, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9532 and NH3. SCFAs are volatile fatty acids with one to six carbon atoms in straight or branched‐chain conformations (den Besten et al., 2013; Koh et al., 2016; Ríos‐Covián et al., 2016). In the human colon, acetate (C2), propionate (C3) and butyrate (C4) are the most abundant, collectively accounting for over 90% of SCFAs (Ríos‐Covián et al., 2016). These three compounds are present in the ratio 60:20:20 respectively (den Besten et al., 2013).
Figure 1.
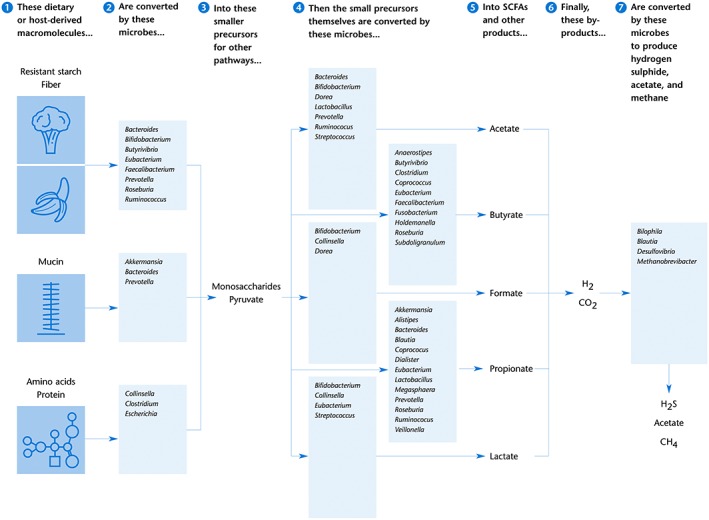
Main microbial fermentation pathways in the human gut. Boxes show the bacterial and archaeal genera involved in the digestion of macromolecules and the generation of SCFAs and other small molecules. The main species within those genera performing these reactions within the human gut are shown in Table 1. This graph is a simplified scheme; not all conversions and cross‐feedings could be shown here. For example, acetate and lactate can be used by some gut bacteria as a precursor to produce butyrate. Compiled from data provided by Louis and Flint (2016); Magnúsdóttir et al. (2017); Desai et al. (2016); Koh et al. (2016); Ríos‐Covián et al. (2016); Ze et al. (2015); Blekhman et al. (2015); Reichardt et al. (2014); LeBlanc et al. (2013); Flint et al. (2012); Nakamura et al. (2010); Belenguer et al. (2006).
Fermentation starts with the digestion of plant‐derived complex polysaccharides that were not digested by human enzymes in the small intestine. These dietary fibres include glycans such as cellulose, pectin and amylose, which consist of polymers of monosaccharide units. Glycoside hydrolases, the enzymes that can break down the bonds connecting these polymers, are found in specific distal gut microorganisms such as Bacteroides, Bifidobacterium, Clostridium, Prevotella, Roseburia and Ruminococcus spp. (Table 1) (Flint et al., 2012). The bacterial species that have this functionality are well equipped for this task. The genome of B. thetaiotaomicron in particular contains 172 glycosyl hydrolases and 20 sugar‐specific transporters (Xu et al., 2003). Ruminococcus bromii, another abundant member of the human gut microbiome, has a specialized genome with 21 glycoside hydrolases (Ze et al., 2015). The genomes of Bifidobacterium species contain a large number of carbohydrate‐modifying enzymes as well (Pokusaeva et al., 2011). Regardless of bacterial species or enzymes, the breakdown of dietary polysaccharides leads to the generation of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4809, an important metabolic intermediate that forms the starting point for numerous anabolic pathways, many of which lead to the synthesis of SCFAs (Koh et al., 2016; Stilling et al., 2016).
Acetate is the most abundant SCFA in the distal gut. It is generated by gut microbes through two different metabolic routes. About two‐thirds of the acetate is produced from pyruvate as the result of complex carbohydrate fermentation, and this capacity is present in a wide range of enteric bacteria (Ríos‐Covián et al., 2016). The remaining acetate is made by acetogenic bacteria such as Blautia spp., which synthesize it from hydrogen (or http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4540) and carbon dioxide (see below) (Nakamura et al., 2010; den Besten et al., 2013; Koh et al., 2016; Ríos‐Covián et al., 2016).
Propionate formation from dietary carbohydrates and amino acids by gut bacteria mainly occurs through two pathways, the http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3637 pathway or the propanediol pathway (Louis and Flint, 2016). The succinate pathway is present in many Firmicutes/Negativicutes (including Veillonella, Dialister and Megasphaera spp.), in Bacteroidetes (Bacteroides, Prevotella and Alistipes) and in Verrucomicrobia (Akkermansia), and this is likely to be the most important route in the human gut (Reichardt et al., 2014). The propanediol pathway is found in Proteobacteria and Ruminococcus spp. (Reichardt et al., 2014).
Butyrate biosynthesis pathways have been found in several phylogenetic groups of gut bacteria and all involve the conversion of butyryl‐CoA to butyrate (Stilling et al., 2016). The majority of the gut butyrate producers do this via butyryl‐CoA:acetate CoA‐transferase; this pathway is present in, for example, Anaerostipes spp., Eubacterium spp., Faecalibacterium prausnitzii and Roseburia spp. Alternatively, butyrate can be synthesized via phophotransbutyrylase and butyrate kinase in the genera Butyrivibrio, Coprococcus and Subdoligranulum (den Besten et al., 2013; Louis and Flint, 2016; Ríos‐Covián et al., 2016).
Several gut bacteria produce http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2932, such as Bifidobacterium, Eubacterium, Lactobacillus and Streptococcus spp. Most of this lactate is quickly converted into butyrate by other bacteria including Eubacterium hallii and Anaerostipes caccae, so its concentration in human faeces is usually very low (Belenguer et al., 2006).
Like lactate, formate is present in low concentrations in the stool because it gets rapidly metabolized by other microbial species. Formate is produced by, for example, Bifidobacterium, Collinsella and Dorea spp, and can be used by Desulfovibrio and Methanobrevibacter spp. (Nakamura et al., 2010; Rey et al., 2013; Zhang and Davies, 2016).
When dietary fibre content is low, intestinal microbes will use less favourable nutrient sources such as dietary fats or proteins or even switch to digesting host‐secreted mucins (Desai et al., 2016; Koh et al., 2016). For example, Akkermansia muciniphila ferments host mucins to form propionate (Derrien et al., 2016). Digestion of host mucus glycoproteins can lead to erosion of the colonic mucus barrier, which in turn can lead to increased intestinal permeability and increased vulnerability for infections with pathogens (Desai et al., 2016).
SCFAs function in human physiology
SCFAs produced by the gut microbiota are important molecules that can exert a wide range of functions (Figure 2). The main functions of intestinal SCFAs are to serve as an energy source for intestinal cells, signalling molecules, modulators of water and electrolyte absorption and regulators of lipid metabolism and components of the intestinal immune system (reviewed in, e.g. den Besten et al., 2013; Koh et al., 2016; Louis and Flint, 2016; Ríos‐Covián et al., 2016; Stilling et al., 2016). In addition, SCFAs also modulate electrolyte and water absorption that can have various effects on organs beyond the gut. Therefore, SCFAs are key molecules in the communication between the host and microbiome, and they are believed to play important roles in health (Ríos‐Covián et al., 2016; Stilling et al., 2016).
Figure 2.
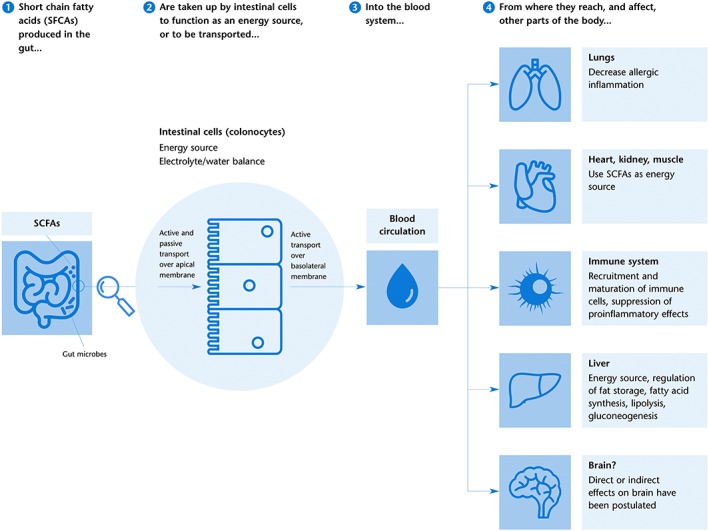
SCFAs effects on the gut and beyond. SCFAs are produced as the result of microbial fermentation in the distal gut (Figure 1) and absorbed by colonocytes through active and passive transport over the apical membrane. SCFAs are partly consumed by colonocytes as an energy source, while the remaining molecules are actively transported over the basolateral membrane and enter the blood circulation. From there, SCFA can affect processes in several peripheral organs by changing DNA transcription through the inhibition of histone deacetylation, binding to and activating GPCRs, or as metabolites in mitochondrial β‐oxidation. Effects of SCFAs, in particular butyrate, on the brain have been hypothesized, either directly by passing the blood–brain barrier or indirectly by effects on the peripheral nervous system. Graphic based on den Besten et al. (2013), Koh et al. (2016) and Stilling et al. (2016).
Clearly, SCFAs have many different functions, and the synthesis of these molecules is generally considered to beneficial for human physiology. Since SCFAs are the end products of microbial fibre degradation, their concentrations and ratios are closely associated with dietary fibre intake. Higher levels of SCFAs, in particular butyrate, and the bacteria that synthesize them are associated with reduced risk for various diseases, such as inflammatory bowel diseases, diabetes and intestinal cancer (Koh et al., 2016; Ríos‐Covián et al., 2016).
In addition, butyrate and the other SCFAs might play a role in communication between the distal gut and more distant parts of the human body. SCFAs are detectable in peripheral blood, and it has been speculated that these small microbial molecules could even reach the brain. Here, they might exert effects on human mental state and behaviour, either directly or via other molecules, and as such could be the missing link in the gut/brain axis (Stilling et al., 2016).
The relative low amounts of fibre in Western diets as compared to diets of, for example, hunter‐gatherers, are likely to be associated with lower amounts and different types of SCFA. Although in vivo production and absorption of SCFA in humans are hard to measure, model systems have confirmed that caecal SCFA production is strongly dependent on dietary fibre content, and this might have important implications on human health (den Besten et al., 2013).
Hydrogen production and conversion
One of the byproducts formed during the anaerobic degradation of organic matter is hydrogen (H2) which is generated to dispose of reducing equivalents (Nakamura et al., 2010). If the concentration of excess hydrogen in the colon reaches high levels, the fermentation pathways will be inhibited. Thus, the presence of hydrogen‐consuming (hydrogenotrophic) microbes is needed to increase the efficiency of the fermentation process. There are several H2‐consuming members of the human gut microbiome, which can be categorized into three broad groups: acetogens, methanogens and sulfate/sulfite reducers (Nakamura et al., 2010; Rey et al., 2013).
Acetogens convert hydrogen into acetate by using CO2 as an electron acceptor. These include Blautia hydrogenotrophica, which synthesize acetate from hydrogen (or formate) and carbon dioxide via the Wood‐Ljungdahl pathway (Nakamura et al., 2010; den Besten et al., 2013; Koh et al., 2016; Ríos‐Covián et al., 2016). As mentioned above, this acetate‐generation pathway is different from the routes being used by a wide range of intestinal bacteria to directly ferment plant polysaccharides into acetate.
Methanogenesis, the conversion of hydrogen to methane, is a chemical process exclusively found in archaea, in which CO2 is reduced using hydrogen or formate as an electron donor. In the human gut, this reaction is predominantly performed by Methanibrevibacter smithii (Nakamura et al., 2010).
Hydrogen can be also converted to H2S by sulfate‐reducing bacteria (SRB), which use sulfate as the electron acceptor. SRB in the human gut are almost exclusively Desulfovibrio species, which are present in the colon of about one‐quarter of healthy US and European adults, with Desulfovibrio piger as their most common representative (Rey et al., 2013; Zhernakova et al., 2016). In the gut, the sulfate needed for this reaction is present in host mucins and in dietary components, since sulfate is used as an antioxidant in several food items (bread, dried fruit) or is used as a dietary supplement in the form of chondroitin sulfate. D. piger itself does not contain sulfatase genes to liberate the sulfate needed for the sulfate reduction and therefore relies on B. thetaiotaomicron, which produces sulfatases that release sulfate from host mucins or from chondroitin sulfate (Rey et al., 2013).
In the complex microbial environment of the human colon, the efficient removal of the hydrogen generated during the fermentation of plant polysaccharides is essential for continuous SCFA production. The presence of both hydrogen‐producers and hydrogen‐consumers is one of the many syntrophic (cross‐feeding) metabolic interactions found in the human gut, with sulfate reduction and methanogenesis the most commonly encountered pathways of hydrogen clearance in the colon (Ríos‐Covián et al., 2016; Stilling et al., 2016).
Vitamin synthesis
Another important function of the gut microbiota is the synthesis of essential nutrients such as amino acids and vitamins. These molecules are needed by the human body as a precursor for the synthesis of several enzymes or other compounds but cannot be made by humans themselves. Humans are therefore dependent on the presence of these essential molecules in our diet or supplements or on the capacity of our gut microbiome to synthesize them. Vitamins produced by the gut microbiome include http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2771 and most of the B‐vitamins (LeBlanc et al., 2013).
B‐vitamins are a group of molecules including http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4787 (vitamin H), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4563 (vitamin B9), http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4629 (vitamin B1) and cobalamin (vitamin B12) that are required in nucleotide and amino acid synthesis and metabolic processes. Most of these can be synthesized by members of the gut microbiota (LeBlanc et al., 2013). In a systematic search for the presence of biosynthesis pathways for eight B‐vitamins in the genomes of 256 common gut bacteria, about half of these genomes contained genes encoding the synthesis of at least one of these vitamins (Magnúsdóttir et al., 2015).
Vitamin K (menaquinone) can be produced by the gut microbiota as well. Genes encoding vitamin K biosynthesis pathways were recently found to be present in the genomes of 118 out of 254 gut bacteria, including Akkermansia, Bacteroides, Lactobacillus and Prevotella spp., but whether these bacteria actually produce vitamin K remains to be determined (Ravcheev and Thiele, 2016).
Additional conversions in the human colon
Other than the well‐studied metabolites discussed above, the human gut microbiome generates hundreds of other products, and many of these are thought to play roles in microbial–microbial or microbial–host interactions (Donia and Fischbach, 2015). Although the function of most of these molecules remains unknown as of now, ongoing research will probably uncover many new metabolites with interesting diagnostic and therapeutic applications. Here, we will list some recent discoveries in this field as a foretaste of the exciting possibilities to come.
First, Oxalobacter formigenes is an http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4538‐degrading anaerobic gut bacterium thought to protect against hyperoxaluria and kidney stones by decreasing the amount and absorption of oxalate in the gut (Siener et al., 2013). Oxalate is found in edible plants such as rhubarb, parsley and spinach. In the body, it can combine with calcium to form small crystals or larger stones that can clog the kidney tubules. Although the exact mechanisms behind the association of O. formigenes presence and reduced calcium oxalate stone formation are not yet known, this finding will lead to future kidney stone prevention and treatment strategies (Siener et al., 2013).
Another example of a recent discovery is that genetic lactose‐intolerance (i.e. a mutated gene encoding for the lactase enzyme) is associated with an increased abundance of Bifidobacterium spp. in stool. It was hypothesized that Bifidobacterium spp., which can metabolize lactose, allow a lactose‐intolerant person to consume milk products (Blekhman et al., 2015).
In contrast to the mostly beneficial gut microbial metabolites discussed above, other microbially synthesized molecules have been implicated in human disease. Dietary http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4780 (abundant in red meat) and phosphatidylcholine are metabolized by gut microbiota members into http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5521 (TMA). In the liver, TMA is converted into trimethylamine‐N‐oxide (TMAO), which is involved in atherosclerosis and is a risk factor for cardiovascular disease (Koeth et al., 2013; Spanogiannopoulos et al., 2016; Zhang and Davies, 2016). TMAO levels are higher in meat eaters than in vegetarians or vegans, and experiments in animals and human volunteers have suggested that the gut microbiota is likely to be responsible for the link between the consumption of red meat and cardiovascular disease (Koeth et al., 2013). Recently, TMA conversion has been assigned to particular Clostridia and Eubacterium spp. (Rath et al., 2017).
Drug metabolism
The vast combined array of chemical capabilities allows gut microbes to not only metabolize dietary and host components but also xenobiotics, that is, chemical substances that are not a natural part of an organism or its diet, such as drugs and pollutants. These biotransformations, most often reduction and hydrolysis, can result in three types of changes. They can activate drugs, inactivate drugs or make them more toxic. Currently, at least 50 different drug conversions performed by the human gut microbiota have been described and extensively reviewed (Ursell and Knight, 2013; Klaassen and Cui, 2015; Spanogiannopoulos et al., 2016; Wilson and Nicholson, 2017). Some well‐studied examples will be highlighted here briefly.
Certain medications, such as those with azo‐bonds, rely on gut microbial enzymes to activate them. These drugs are administered to patients as pro‐drugs, and microbial enzymes in the distal gut are needed to activate the compound into its effective form. Examples in this category include http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4840, a drug to treat rheumatoid arthritis and inflammatory bowel diseases and the laxative pro‐drug sodium picosulfate (Wilson and Nicholson, 2017).
In contrast, gut microbial enzymes can also inactivate certain drugs, making them less effective than anticipated. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4726, a drug used to treat congestive heart failure, can be inactivated by Eggerthella lenta strains that carry the cgr operon in their genome (Haiser et al., 2013). For people who carry such E. lenta strains, the drug will not be as effective as for patients whose microbiomes contain E. lenta strains without the cgr operon or no E. lenta at all.
A third category of gut microbial conversions can make drugs more toxic or interfere with the host's detoxification process. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5239 (paracetamol), a commonly used pain reliever and fever reducer worldwide, can induce severe hepatotoxicity when used in high amounts. However, acetaminophen toxicity has also been found in patients who consumed the drug at levels regarded as safe (Clayton et al., 2009). This variable tolerance of individuals for acetaminophen is thought to be dependent on the composition of the gut microbiome. In the liver and intestinal mucosa, acetaminophen is detoxified by sulfonation or glucuronidation by human enzymes. However, certain gut bacteria, such as Clostridium difficile, produce p‐cresol from dietary protein residues. This p‐cresol can be converted in the liver to p‐cresol‐sulfate, a process that competes with the detoxification of acetaminophen. Thus, individuals whose microbiota produces a lot of p‐cresol are not as good at detoxifying acetaminophen as others, and the drug will be more toxic for them (Clayton et al., 2009).
The chemotherapy prodrug http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6823, mainly used to treat colon cancer, is activated by hydrolysis by host enzymes to http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6925, inactivated by glucuronidation in the liver and excreted in the bile. Expression of the inactivation enzyme can be influenced by genetic variations in the human genome or the gut microbiome composition. Bacterial β‐glucuronidases in the colon can scavenge the glucuronic acid from the inactivated SN‐38 molecule and re‐activate the inactivated drug, which will lead to diarrhoea. Administration of irinotecan with selective bacterial glucuronidase‐inhibitors was found to be very effective in mice (Roberts et al., 2013).
Mycotoxins are poisonous small molecules produced by certain mould species that can be found as contaminants in food items such as peanuts, corn, spices and dried fruits. Deoxynivalenol is a toxin produced by Fusarium moulds and a frequent contaminant of cereals, often both in the toxic unconjugated form and a conjugated form. The gut microbiotas of some individuals hydrolyze the conjugated form to the toxic form, while those of other individuals can transform the toxic form into a less‐toxic compound, thus leading to differential responses to contamination with this mycotoxin (Gratz et al., 2013).
The examples listed above are just some of the many microbial conversions of xenobiotics described. Because of the large inter‐individual variations in gut microbial composition, the same compound in the same dose can have a very different effect, with toxicity and efficacy varying from person to person. This microbial contribution in drug metabolism is often overlooked in clinical studies. Knowledge of the composition and metabolic activity of an individual's gut microbiota can be extremely helpful in predicting that individual's response to certain drugs (Ursell and Knight, 2013; Klaassen and Cui, 2015).
Concluding remarks
This review has provided a broad overview of known biochemical reactions performed by microbes in the human gut and their importance for human physiology. Some important concepts are worth restating here.
Firstly, gut microbial species do not exist as single entities but rather interact with each other by either competing for the same resources or by collaborating through metabolic cross‐feeding, where one microbe's byproduct can be used as a substrate by another microbe (Flint et al., 2012; Donia and Fischbach, 2015; Zhang and Davies, 2016).
Secondly, important functions of the human gut microbiome such as polysaccharide breakdown and SCFA synthesis are not performed by one particular phylogenetic lineage but by polyphyletic guilds of microbial taxa. This functional redundancy might provide the host with a robust microbiota, where the removal of one member does not necessarily result in the loss of an essential functionality. Therefore, it is generally accepted that a diverse gut microbiota, that is, one containing a high count of microbial species, is associated with health (Human Microbiome Project Consortium, 2012; Lozupone et al., 2012).
Thirdly, the human gut microbiome might be responsible for some previously unexplained inter‐individual responses to medications or dietary components and different disease risks. The composition of the human microbiome not only varies between individuals as a function of host genetics, geographical, societal and environmental factors but also over time with changes in diet, travel, disease status and medication intake (Human Microbiome Project Consortium, 2012; David et al., 2014a,b; Blekhman et al., 2015; Obregon‐Tito et al., 2015; Falony et al., 2016; Zhernakova et al., 2016). Alongside the enormous functional capacity of the intestinal microbiome, this adds a tremendous amount of inter‐individual metabolic variation on top of that encoded by the human genome and an additional layer of intra‐individual variation (Lloyd‐Price et al., 2016).
Finally, there are still many microbial biotransformations in the human gut that are poorly understood or remain to be characterized (Donia and Fischbach, 2015). Molecular surveys of the human gut have recently discovered novel lineages such as the Melainabacteria, which are probably capable of performing yet‐uncharacterized metabolic routes (Di Rienzi et al., 2013). Even for well‐studied gut microbiome members such as Escherichia coli and B. thetaiotaomicron, many genes have not yet been assigned to a known function. In addition, genomic analysis is likely to reveal strain‐level variations in metabolic capacities within species as well, as shown above for E. lenta. With the rapidly increasing amount of metagenomic data obtained from human gut samples, there is still a lot of knowledge to be gathered.
Such expected new insights of yet‐to‐be discovered metabolic functions and inter‐individual differences are likely to greatly contribute to the personalized medicine field. In the near future, analysis of a patient's gut microbiome will be an integral part of personal clinical care and pharmaceutical development.
Nomenclature of ligands
Key ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016).
Conflict of interest
All authors of this paper are employees of uBiome in San Francisco, CA, USA, and have received stock options as well as other compensation.
Bik, E. M. , Ugalde, J. A. , Cousins, J. , Goddard, A. D. , Richman, J. , and Apte, Z. S. (2018) Microbial biotransformations in the human distal gut. British Journal of Pharmacology, 175: 4404–4414. 10.1111/bph.14085.
References
- Almonacid DE, Kraal L, Ossandon FJ, Budovskaya YV, Cardenas JP, Bik EM et al (2017). 16S rRNA gene sequencing and healthy reference ranges for 28 clinically relevant microbial taxa from the human gut microbiome. PLoS ONE 12: e0176555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva‐Datchary P et al (2015). dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17: 690–703. [DOI] [PubMed] [Google Scholar]
- Belenguer A, Duncan SH, Calder AG, Holtrop G, Louis P, Lobley GE et al (2006). Two routes of metabolic cross‐feeding between Bifidobacterium adolescentis and butyrate‐producing anaerobes from the human gut. Appl Environ Microbiol 72: 3593–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud D‐J, Bakker BM (2013). The role of short‐chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res 54: 2325–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blekhman R, Goodrich JK, Huang K, Sun Q, Bukowski R, Bell JT et al (2015). Host genetic variation impacts microbiome composition across human body sites. Genome Biol 16: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordenstein SR, Theis KR (2015). Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PLoS Biol 13: e1002226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK (2009). Pharmacometabonomic identification of a significant host‐microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci U S A 106: 14728–14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Materna AC, Friedman J, Campos‐Baptista MI, Blackburn MC, Perrotta A et al (2014a). Host lifestyle affects human microbiota on daily timescales. Genome Biol 15: R89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE et al (2014b). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505: 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien M, Belzer C, de Vos WM (2016). Akkermansia muciniphila and its role in regulating host functions. Microb Pathog 106: 171–181. [DOI] [PubMed] [Google Scholar]
- Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M et al (2016). A dietary fiber‐deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167: 1339–1353.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, Relman DA (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A 108: 4554–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzi SC, Sharon I, Wrighton KC, Koren O, Hug LA, Thomas BC et al (2013). The human gut and groundwater harbor non‐photosynthetic bacteria belonging to a new candidate phylum sibling to Cyanobacteria. Elife 2: e01102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinan TG, Cryan JF (2017). Gut‐brain axis in 2016: brain‐gut‐microbiota axis – mood, metabolism and behaviour. Nat Rev Gastroenterol Hepatol 14: 69–70. [DOI] [PubMed] [Google Scholar]
- Donia MS, Fischbach MA (2015). HUMAN MICROBIOTA. Small molecules from the human microbiota. Science 349:1254766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falony G, Joossens M, Vieira‐Silva S, Wang J, Darzi Y, Faust K et al (2016). Population‐level analysis of gut microbiome variation. Science 352: 560–564. [DOI] [PubMed] [Google Scholar]
- Flint HJ, Scott KP, Duncan SH, Louis P, Forano E (2012). Microbial degradation of complex carbohydrates in the gut. Gut Microbes 3: 289–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, Hsu T, Sirota‐Madi A, Shafquat A, Abu‐Ali G, Morgan XC et al (2015). Sequencing and beyond: integrating molecular “omics” for microbial community profiling. Nat Rev Microbiol 13: 360–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, Ober C et al (2016). Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe 19: 731–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz SW, Duncan G, Richardson AJ (2013). The human fecal microbiota metabolizes deoxynivalenol and deoxynivalenol‐3‐glucoside and may be responsible for urinary deepoxy‐deoxynivalenol. Appl Environ Microbiol 79: 1821–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Segre JA (2012). The human microbiome: our second genome. Annu Rev Genomics Hum Genet 13: 151–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ (2013). Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science 341: 295–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehemann J‐H, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G (2010). Transfer of carbohydrate‐active enzymes from marine bacteria to Japanese gut microbiota. Nature 464: 908–912. [DOI] [PubMed] [Google Scholar]
- Hoffmann C, Dollive S, Grunberg S, Chen J, Li H, Wu GD et al (2013). Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS ONE 8: e66019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen CD, Cui JY (2015). Review: mechanisms of how the intestinal microbiota alters the effects of drugs and bile acids. Drug Metab Dispos 43: 1505–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT et al (2013). Intestinal microbiota metabolism of L‐carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 19: 576–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh A, De Vadder F, Kovatcheva‐Datchary P, Bäckhed F (2016). From dietary fiber to host physiology: short‐chain fatty acids as key bacterial metabolites. Cell 165: 1332–1345. [DOI] [PubMed] [Google Scholar]
- Kostic AD, Howitt MR, Garrett WS (2013). Exploring host‐microbiota interactions in animal models and humans. Genes Dev 27: 701–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc JG, Milani C, de Giori GS, Sesma F, van Sinderen D, Ventura M (2013). Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol 24: 160–168. [DOI] [PubMed] [Google Scholar]
- Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S et al (2014). An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol 32: 834–841. [DOI] [PubMed] [Google Scholar]
- Lloyd‐Price J, Abu‐Ali G, Huttenhower C (2016). The healthy human microbiome. Genome Med 8: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis P, Flint HJ (2016). Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol 19: 29–41. [DOI] [PubMed] [Google Scholar]
- Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R (2012). Diversity, stability and resilience of the human gut microbiota. Nature 489: 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnúsdóttir S, Ravcheev D, de Crécy‐Lagard V, Thiele I (2015). Systematic genome assessment of B‐vitamin biosynthesis suggests co‐operation among gut microbes. Front Genet 6: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnúsdóttir S, Heinken A, Kutt L, Ravcheev DA, Bauer E, Noronha A et al (2017). Generation of genome‐scale metabolic reconstructions for 773 members of the human gut microbiota. Nat Biotechnol 35: 81–89. [DOI] [PubMed] [Google Scholar]
- Moya A, Ferrer M (2016). Functional redundancy‐induced stability of gut microbiota subjected to disturbance. Trends Microbiol 24: 402–413. [DOI] [PubMed] [Google Scholar]
- Mueller NT, Bakacs E, Combellick J, Grigoryan Z, Dominguez‐Bello MG (2015). The infant microbiome development: mom matters. Trends Mol Med 21: 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Lin HC, McSweeney CS, Mackie RI, Gaskins HR (2010). Mechanisms of microbial hydrogen disposal in the human colon and implications for health and disease. Annu Rev Food Sci Technol 1: 363–395. [DOI] [PubMed] [Google Scholar]
- Nishijima S, Suda W, Oshima K, Kim S‐W, Hirose Y, Morita H et al (2016). The gut microbiome of healthy Japanese and its microbial and functional uniqueness. DNA Res 23: 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obregon‐Tito AJ, Tito RY, Metcalf J, Sankaranarayanan K, Clemente JC, Ursell LK et al (2015). Subsistence strategies in traditional societies distinguish gut microbiomes. Nat Commun 6: 6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole PW, Jeffery IB (2015). Gut microbiota and aging. Science 350: 1214–1215. [DOI] [PubMed] [Google Scholar]
- Pokusaeva K, Fitzgerald GF, van Sinderen D (2011). Carbohydrate metabolism in Bifidobacteria. Genes Nutr 6: 285–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C et al (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath S, Heidrich B, Pieper DH, Vital M (2017). Uncovering the trimethylamine‐producing bacteria of the human gut microbiota. Microbiome 5: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravcheev DA, Thiele I (2016). Genomic analysis of the human gut microbiome suggests novel enzymes involved in quinone biosynthesis. Front Microbiol 7: 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt N, Duncan SH, Young P, Belenguer A, McWilliam Leitch C, Scott KP et al (2014). Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J 8: 1323–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey FE, Gonzalez MD, Cheng J, Wu M, Ahern PP, Gordon JI (2013). Metabolic niche of a prominent sulfate‐reducing human gut bacterium. Proc Natl Acad Sci U S A 110: 13582–13587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ríos‐Covián D, Ruas‐Madiedo P, Margolles A, Gueimonde M, de Los Reyes‐Gavilán CG, Salazar N (2016). Intestinal short chain fatty acids and their link with diet and human health. Front Microbiol 7: 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AB, Wallace BD, Venkatesh MK, Mani S, Redinbo MR (2013). Molecular insights into microbial β‐glucuronidase inhibition to abrogate CPT‐11 toxicity. Mol Pharmacol 84: 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson CM, Pfeiffer JK (2014). Viruses and the microbiota. Annu Rev Virol 1: 55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, Mazmanian SK (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9: 313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela DA, Li Y, Lerno L, Wu S, Marcobal AM, German JB et al (2011). An infant‐associated bacterial commensal utilizes breast milk sialyloligosaccharides. J Biol Chem 286: 11909–11918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender R, Fuchs S, Milo R (2016). Revised estimates for the number of human and bacteria cells in the body. PLoS Biol 14: e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siener R, Bangen U, Sidhu H, Hönow R, von Unruh G, Hesse A (2013). The role of Oxalobacter formigenes colonization in calcium oxalate stone disease. Kidney Int 83: 1144–1149. [DOI] [PubMed] [Google Scholar]
- Smilowitz JT, Lebrilla CB, Mills DA, German JB, Freeman SL (2014). Breast milk oligosaccharides: structure‐function relationships in the neonate. Annu Rev Nutr 34: 143–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg JL, Bäckhed F (2016). Diet‐microbiota interactions as moderators of human metabolism. Nature 535: 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanogiannopoulos P, Bess EN, Carmody RN, Turnbaugh PJ (2016). The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat Rev Microbiol 14: 273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stilling RM, van de Wouw M, Clarke G, Stanton C, Dinan TG, Cryan JF (2016). The neuropharmacology of butyrate: the bread and butter of the microbiota‐gut‐brain axis? Neurochem Int 99: 110–132. [DOI] [PubMed] [Google Scholar]
- Ursell LK, Knight R (2013). Xenobiotics and the human gut microbiome: metatranscriptomics reveal the active players. Cell Metab 17: 317–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC et al (2009). Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A 106: 3698–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson ID, Nicholson JK (2017). Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl Res 179: 204–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC et al (2003). A genomic view of the human‐Bacteroides thetaiotaomicron symbiosis. Science 299: 2074–2076. [DOI] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez‐Bello MG, Contreras M et al (2012). Human gut microbiome viewed across age and geography. Nature 486: 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ze X, Ben David Y, Laverde‐Gomez JA, Dassa B, Sheridan PO, Duncan SH et al (2015). unique organization of extracellular amylases into amylosomes in the resistant starch‐utilizing human colonic firmicutes bacterium Ruminococcus bromii. MBio 6: e01058–e01015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LS, Davies SS (2016). Microbial metabolism of dietary components to bioactive metabolites: opportunities for new therapeutic interventions. Genome Med 8: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhernakova A, Kurilshikov A, Bonder MJ, Tigchelaar EF, Schirmer M, Vatanen T et al (2016). Population‐based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352: 565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]