

Abstract
Current molecular hypotheses have not yet delivered marketable treatments for Alzheimer’s disease (AD), arguably due to a lack of understanding of AD biology and an overreliance on conventional drug modalities. Protein–protein interactions (PPIs) are emerging drug targets, which show promise for the treatment of, e.g., cancer, but are still underexploited for treating neurodegenerative diseases. 14-3-3 binding to phosphorylated Tau is a promising PPI drug target based on its reported destabilizing effect on microtubules, leading to enhanced neurofibrillary tangle formation as a potential cause of AD-related neurodegeneration. Inhibition of 14-3-3/Tau may therefore be neuroprotective. Previously, we reported the structure-guided development of modified peptide inhibitors of 14-3-3/Tau. Here, we report further efforts to optimize the binding mode and activity of our modified Tau peptides through a combination of chemical synthesis, biochemical assays, and X-ray crystallography. Most notably, we were able to characterize two different high-affinity binding modes, both of which inhibited 14-3-3-binding to full-length PKA-phosphorylated Tau protein in vitro as measured by NMR spectroscopy. Our findings, besides producing useful tool inhibitor compounds for studying 14-3-3/Tau, have enhanced our understanding of the molecular parameters for inhibiting 14-3-3/Tau, which are important milestones toward the establishment of our 14-3-3 PPI hypothesis.
Keywords: 14-3-3, tau, protein−protein interactions, inhibitors, drug discovery, peptide chemistry
Introduction
Despite the best efforts of academia and pharma, there are currently no marketed drugs for AD. Nonetheless the drug development pipeline of agents to treat the underlying pathology of AD, so-called disease modify therapies (DMTs), is very active, comprising 70% of the 105 compounds in clinical trials.1 While the drug mechanisms of current agents in Phases I–III are heterogeneous, they can nonetheless be broadly classified as targeting either the amyloid cascade or the downstream pathophysiology, including the Tau pathway. Noticeably, more than half of the agents in Phase III are anti-amyloids compared to only 4% directly targeting Tau.1 The vast preponderance of all agents in the pipeline is either monoclonal antibodies or β-site amyloid precursor protein cleaving enzyme (BACE) inhibitors. Given that no treatment has yet been found, there is considerable incentive to address the relatively unexplored Tau pathway. The major one being that, in AD, the severity of cognitive decline is more correlated with the evolution of Tau neurofibrillary tangles (NFTs) than with that of amyloid deposits.2 However, the effective targeting of NFTs might require a shift to new molecular modalities,3 the solution to which could be found in previously intractable molecular targets. Protein–protein interactions (PPIs) are, by conventional standards, emerging drug targets,4−8 which show promise for the treatment of other disease types, such as cancer,9 but are as yet underexploited for treating neurodegenerative diseases, including AD, despite the clear potential.10 We therefore bring forward the PPI between 14-3-3 proteins and Tau, as a potential therapeutic target to treat AD.11−13
14-3-3 proteins are adapter proteins, existing in seven isoforms, β, ε, γ, η, σ, τ, and ζ,14−16 which bind to and modulate the function and folding of phosphorylated proteins.17 While abundantly expressed in the body, 14-3-3 proteins are most abundant in CNS compartments.18 14-3-3 proteins make important PPIs in diverse pathophysiological settings such as cancer19 and metabolic diseases,20 and in neurodegeneration. In the latter case, 14-3-3 proteins bind to a number of client proteins implicated in CNS diseases, among them Tau, α-synuclein, parkin, and LRRK2.21 Therefore, 14-3-3 proteins are fundamentally interesting targets for neurodegenerative drug therapy,13 in which either inhibition or stabilization of 14-3-3 PPIs may prove to be viable therapeutic strategies.22−25
Tau protein similarly exerts its multiple neuronal functions by binding a range of partners, the most well-documented being the binding and stabilizing of microtubules.26,27 This interaction, as is the case for many Tau interactions, is physiologically regulated by phosphorylation.28 However, hyperphosphorylation of Tau, associated with its aggregation inside neurons as paired helical filaments (PHF), are well-known hallmarks of AD.29 The Tau/14-3-3 association has been found to impact several aspects of the Tau pathway in neurodegeneration. First, 14-3-3ζ has been described to stimulate Tau phosphorylation by GSK3β kinase in cell model and in brain,30−32 and cAMP-dependent protein kinase in vitro.30 Additionally, 14-3-3ζ is reported to be associated with the neurofibrillary tangles composed of Tau PHF in AD brain extracts and to stimulate Tau aggregation in an in vitro assay.33,34
Our group has provided X-ray crystallographic evidence for the preferential, bivalent binding of 14-3-3 proteins to phosphoepitope sites, pS214 and pS324, of Tau with affinity in the μM range according to additional biochemical and biophysical data.11 We additionally showed that the 14-3-3/Tau interaction could be decreased through 14-3-3σ overexpression in SH-SY5Y cells.11 Taken together, these data led us to hypothesize that 14-3-3 binding to phosphorylated Tau (pTau) occurs at the expense of a stabilizing PPI between Tau and microtubules,11 which is concomitantly known to be decreased by the phosphorylation of Tau on S214.35 Small molecule stabilization of microtubules (MTs) is a promising therapeutic strategy to compensate for the loss of Tau-mediated stabilization.36,37 Small molecule inhibition of Tau aggregation, e.g., using cell permeable D-peptides,38 may also prove to be a complementary therapeutic strategy. Alternatively, inhibition of 14-3-3/Tau, e.g., through the targeted development of modified peptide inhibitors, could potentially exert a neuroprotective effect by decreasing its phosphorylation level, while increasing the pool of soluble Tau protein for the stabilization of microtubules and by preventing its aggregation, the 14-3-3 PPI hypothesis.
Toward testing our 14-3-3 PPI hypothesis, we previously demonstrated the potential to inhibit 14-3-3/Tau in vitro, with small molecules developed using a structure-guided approach.12 With the Tau-derived pS214 phosphopeptide epitope as the chemical starting point, we targeted chemical modifications specifically at the fusicoccin (FC) binding site of Mode III 14-3-3 PPIs39 to produce a potent inhibitor of 14-3-3ζ binding to full-length (fl)-pTau. In this paper, we report the results of studies in which we attempt structure-guided optimization of our Tau-derived 14-3-3 inhibitors. Most notably we were able to improve the activity of the lead compound from our previous study, and characterize two distinct high affinity modes of interaction by fluorescence polarization (FP) and isothermal calorimetry (ITC) measurements, which we correlate to an “open” and “closed” state based on seven new X-ray cocrystal structures. Both binding modes were shown to inhibit the binding of 14-3-3ζ to fl-pTau in a concentration-dependent manner, lending further weight to our 14-3-3 PPI hypothesis.
Results and Discussion
Rational Design of New 14-3-3 Inhibitors
In a previous study,8 we discovered that a synthetic derivative of a pS214-Tau peptide epitope modified at the C-terminus with a benzhydryl pyrrolidine moiety bound more strongly to the 14-3-3 protein than the unmodified Tau epitope. In the resulting crystal structure, the benzhydryl moiety, specifically, the pro-R phenyl ring, can be seen to occupy the fusicoccin (FC) pocket (Figure 1a), thus explaining the improved activity observed in the biochemical and biophysical assays. Closer inspection of the FC pocket identified a deep-lying pocket proximal to the pro-R phenyl group lined by seven amino acid residues derived from the same 14-3-3 protomer: Ser45, Tyr48, Lys49, Phe119, Lys122, Met123, and Tyr 127 (Figure 1a). Interestingly, a similar pocket is also present in other 14-3-3 protein–ligand crystal structure complexes (e.g., the 14-3-3-ERα complexed stabilized by FC, Figure 1b).22 The ortho and meta positions of the pro-R phenyl ring are located closest to the deep-lying pocket (Figure 1a). Based on these observations, we hypothesized that the affinity of our modified peptides might be further improved through structural variation of the benzhydryl pyrrolidine group, potentially addressing the deep-lying pocket in the process (Figure 1c). We therefore targeted the synthesis of a structurally diverse collection of monosubstituted benzhydryl pyrrolidine analogues (Tables 1–3), in which the phenyl substituent group differed in size and the polarity (i.e., H → Cl → Me → OMe → OCH2CH2OCH3, see Table 1).
Figure 1.

(a) Zoomed in perspective of deep-lying pocket, adjacent to the fusicoccin A (FC) pocket, present in previously published cocrystal structure of a synthetic Tau peptide, modified with a benzhydryl pyrrolidine moiety, bound to 14-3-3σΔC (PDB: 5HF3).12 The modified Tau peptide is depicted in white sticks, the protein surface and residues in orange, and the protein interior surface in dark gray. Pocket-forming amino acid residues are labeled. (b) The same deep-lying pocket present in the ternary complex of 14-3-3σΔC bound to the C-terminal ERα phospopeptide and FC. The ERα peptide is depicted in green sticks, FC is depicted in blue sticks, the protein surface and residues in orange, and the protein interior surface in dark gray. (c) Chemical structure of symmetric S-configured benzhydryl pyrrolidine and general structure of asymmetrically substituted benzhydryl pyrrolidines. Pro-R = Pro-R phenyl substituent, Pro-S = Pro-S phenyl substituent.
Table 1. Summary of Structures, Yields, and Diastereomeric Ratios (dr) for Pyrrolooxazolone (2.5a–g) and Benzhydryl Derivatives (2.6a–g) Described in Scheme 1.

Isolated yields of diastereomers either combined or separated by RP-HPLC (denoted I or II).
dr determined by comparing integral values in 1H NMR of crude.
Inseparable diastereomers.
Diastereomers separable by RP-HPLC (Supporting Information).
Table 3. Summary of Structures, and Associated IC50 (FP), and Kd Values for the Modified Tau Peptides 4.2a–g Described in Scheme 3b.

A mixture of diastereomers.
The confidence interval (CI) and ± standard error (SE) are reported.
Synthesis of Substituted Benzhydryl Pyrrolidine Derivatives
The synthesis of asymmetrically substituted benzhydryl pyrrolidine derivatives belonging to the generic structure depicted in Figure 1c was complicated by the absence of any literature precedent. Therefore, we elected for a synthesis based on the stereoretentive synthesis of symmetrically substituted (S)-2-diphenylmethylpyrrolidine (Scheme 1),40 starting from enantiopure l-proline ester, though cognizant of the lack of obvious stereocontrol in the formation of the asymmetric center at the benzhydryl carbon, and therefore the likely formation of diastereomers.
Scheme 1. Synthesis of Asymmetrically Substituted Benzhydryl Pyrrolidine Derivatives 2.6a–g (Table 1).

In brief, the addition of phenylmagnesium bromide to commercial Weinreb amide 2.1, followed by acid workup produced benzoylpyrrolidine 2.2 in a 47% yield over the two steps (Scheme 1). Deprotection of the Boc group yielded the substituted pyrrolidine 2.3, which was followed by protection of the amine group as the ethyl carbamate 2.4. At this juncture, treatment of 2.4 with a range of structurally diverse substituted Grignard reagents (Table 1) produced a small library of pyrrolooxazolones 2.5a–g, in isolated yields ranging from 25 to 86%. The Grignard reagents used to make pyrrolooxazolones 2.5c and 2.5d are derived from 1-bromo-2-(2-methoxyethoxy)benzene and 1-bromo-3-(2-methoxyethoxy)benzene, respectively, which could be themselves prepared in one step from commercial compounds (Supporting Information). Pyrrolooxazolones 2.5a−g were converted to the corresponding asymmetrically substituted benzhydryl pyrrolidines 2.6a–g using palladium catalyzed hydrogenation conditions, a step which had been shown to occur without loss of steropurity for the synthesis of (S)-2-diphenylmethylpyrrolidine, as evidenced by chiral HPLC and X-ray analysis.40 For the series 2.5a–g, we were unable to detect more than one diastereomer by either NMR or LC-MS. For the series 2.6a–g by contrast, analogues 2.6c–f yielded diastereomers, which were separable by reverse-phase (RP) HPLC, with combined yields in the range of 33–71% and diastereomeric ratios (drs) in the range 63:37–86:14 (Table 1). We were unable to separate the diastereomers formed in the case of analogues 2.6a, 2.6b, and 2.6 g.
Synthesis of the First Library of Modified Tau Peptides
With the library of asymmetrically substituted pyrrolidine derivatives in hand, we proceeded with the synthesis of the corresponding modified Tau peptides. The synthesis of 3.2a has been described in a previous communication.12 The appearance of the R-epimer in the cocrystal structure of 3.2a with 14-3-3σ, likely caused by racemization of the threonine α-stereocenter during the synthesis, was unexpected because the S-epimer was observed to bind 14-3-3σ in cocrystal structures for analogous structures reported in the same study, prepared using the same synthetic strategy.12 An inspection of all structural data, superposed, suggested that the bulky benzhydryl moiety disfavors binding of the S-epimer of 3.2a through a steric clash between the benzhydryl group and the threonine side chain residue (an effect presumably absent in the case of the R-epimer). To test this hypothesis, and specifically probe the steric and stereochemical preferences of the modified Tau peptides for 14-3-3 binding, we synthesized a library of analogous modified Tau peptides in which the C-terminal l-Thr (3.2a) had been systematically replaced by either Gly (3.2b) or a short series of other S- and R-configured natural amino acid residues with different side chains, l-Ala (3.2c), d-Ala (3.2d), l-Val (3.2e), and d-Val (3.2f); Scheme 2.
Scheme 2. Synthesis of Modified Tau Peptides 3.2a–f (Table 2) and 4.2a–g (Table 3).

For the synthesis of 3.2a–f, X = l-Thr (3.2a), Gly (3.2b), l-Ala (3.2c), d-Ala (3.2d), l-Val (3.2e), d-Val (3.2f); see Table 2. For the synthesis of 4.2a-g, R = see Table 3.
Partially protected Tau peptides 3.1a–f were first synthesized as described for a previous synthesis of 3.1a (referred to as 3b in ref (12)) and characterized by LC-MS analysis (Supporting Information). Each partially protected peptide was then coupled to (S)-benzhydryl pyrrolidine using PyClock as coupling reagent followed by resin cleavage and deprotection of the side-chain protecting groups (TFA, TIS, H2O) to afford 3.2a–f in yields of 8–36% after purification by RP-HPLC (Scheme 2). While we could not conclusively exclude the possibility of diastereomer impurities in the final compounds, all modified Tau peptides were purified by RP-HPLC on an optimized gradient (see Methods). Please refer to the Supporting Information for LC-MS spectra of all compounds after purification.
Biochemical Evaluation of First Library of Modified Tau Peptides
We next investigated the activity of our first library of modified Tau peptides in a competitive FP assay (Table 2 and Figure S34) and compared their activities to that of our reference compound, 3.2a. The FP data shows that under the specific assay conditions used, all new modified peptides (3.2b–f) inhibit binding of the competitor FAM-labeled diphosphorylated Tau competitor peptide to 14-3-3ζ, with IC50 values in the same low micromolar range as the reference, 3.2a (Table 2).
Table 2. Summary of Structures, and Associated IC50 (FP), and Kd values for the Modified Tau Peptides 3.2a–f, the Synthesis of Which Is Described in Scheme 2a.
| FP |
ITC |
||||
|---|---|---|---|---|---|
| derivative | amino acid residue, X (Scheme 2) | IC50/μM | 95% CI | Kd/μM | ±SE |
| 3.2a | l-Thr | 8.1 | 7.3–8.9 | 5.4 | 0.4 |
| 3.2b | Gly | 5.9 | 5.2–6.8 | 5.6 | 0.5 |
| 3.2c | l-Ala | 5.3 | 4.7–5.8 | 3.3 | 0.7 |
| 3.2d | d-Ala | 6.2 | 5.8–6.6 | 3.0 | 0.6 |
| 3.2e | l-Val | 6.2 | 5.8–6.5 | 2.2 | 0.7 |
| 3.2f | d-val | 7.6 | 7.2–8.1 | 3.4 | 0.6 |
The confidence interval (CI) and ± standard error (SE) are reported.
ITC measurements were next performed in duplicate on modified Tau peptides 3.2a–f to determine their association constant (Ka), stoichiometry (N), and enthalpy (ΔH) and entropy change (ΔS) on binding to 14-3-3 (Supporting Information). Although the two sets of duplicate measurements are consistent with one another, one set of data are used here for a quantitative comparison of the different analogues. Collectively, the calculated Kd values for analogues 3.2a–f (Table 2) are of the same magnitude as the IC50 values determined by FP. The stoichiometries of binding (N) are also all approximately 1.0, which indicates a 1:1 binding between modified peptide and protein. Individually, the Kd (2.2–5.6 μM) and ΔG (−7.5 to −8.1 kcal mol–1) values for each modified peptide are also very similar across the series, which indicates that they all bind with similar affinity to 14-3-3. In the two cases where the activities of l- and d-isomers could be directly compared, for Ala and Val analogues, similar binding data was measured, e.g., compare the data for the l-Ala analogue, 3.2c (Kd = 3.3 μM, ΔG = −7.8 kcal mol–1), and the d-Ala analogue, 3.2d (Kd = 3.0 μM, ΔG = −7.8 kcal mol–1). There is additional evidence that increasing sterics and hydrophobicity at the C-terminal amino acid produce marginal gains in activity in the series Gly/3.2b (Kd = 5.6 μM, ΔG = −7.5 kcal mol–1) → l-Ala/3.2c (Kd = 3.3 μM, ΔG = −7.8 kcal mol–1) → l-Val/3.2e (Kd = 2.2 μM, ΔG = −8.1 kcal mol–1). The thermodynamic binding parameters, ΔH and −TΔS, are also similar for all Tau peptide inhibitors in this library (Supporting Information), with the ΔH value in the range −1.7 to −2.5 kcal mol–1, and the TΔS value in the range 5.0–6.6 kcal mol–1. Notably, the Kd, ΔH, TΔS, and ΔG values are near identical bearing either threonine (3.2a) or glycine (3.2b) at the C-terminus of the modified Tau peptide. Collectively, the similar FP and ITC data imply that the analogues 3.2a–f are binding to 14-3-3 with a similar binding mode, the hypothesis being that the benzhydryl group in each case binds the FC pocket in a mode similar to the one observed in the crystal structure for 3.2a (Figure 1a).12 Furthermore, increasing the steric bulk and hydrophobicity produces a marginal increase in activity, while inverting the stereochemistry of the C-terminal amino acid residue importantly has no significant effect on the modified Tau peptide’s binding and inhibitory properties.
Synthesis of the Second Library of Modified Tau Peptides
In view of the measured equipotency and similar thermodynamic profiles of the l-Thr (3.2a) and Gly analogues (3.2b), we elected to prepare a second library of modified Tau peptides based on 3.2b. We reasoned that replacing l-Thr with Gly would simplify the synthesis while retaining the ability to explore the FC pocket, potentially addressing the adjacent deep-lying pocket (Figure 1a) through the introduction of asymmetrically substituted benzhydryl pyrrolidine derivatives. A second library of modified Tau peptides, 4.2a–g, was therefore synthesized, of which the details are outlined in Scheme 2 and the structures summarized in Table 3. For some of the derivatives, two diastereomers were separated by RP-HPLC, denoted I and II, in yields of between 3 and 32% for the individual diastereomers, and overall combined isolated yields of between 34 and 57% (see Methods).
Biochemical Evaluation of Second Library of Modified Tau Peptides
As for the first library, we used FP and ITC to characterize the 14-3-3 binding properties of our second library of modified Tau peptides. The findings of this short study are summarized in Table 3. The FP data (Figure S35) shows that all analogues from this library function as competitive inhibitors of 14-3-3σ with IC50 values in the range 2.8–12.1 μM. Our ITC data show that all analogues bind to 14-3-3σ with Kd values in the range 2.7–20.6 μM and ΔG values between −6.7 and −7.9 kcal mol–1. The most active analogue in the series was the 2-(methoxyethoxy)- derived 4.2c-I (Kd = 2.7 μM/ ΔG = −7.9 kcal mol–1) and the least active, its regioisomer, analogue 4.2d-II (Kd = 20.6 μM/ ΔG = −6.7 kcal mol–1). Besides 4.2c-I, analogue 4.2e-I (Kd = 3.6 μM/ΔG = −7.7 kcal mol–1) and methoxy-derivative 4.2f-I (Kd = 4.7 μM/ΔG = −7.5 kcal mol–1) were also strong binders.
X-ray Crystallography Studies
To provide a molecular explanation for the high affinity binding of our new modified tau peptide inhibitors, the cocrystal structures of symmetrically substituted benzhydryl derivatives, 3.2d (1.70 Å), and 3.2e (2.00 Å), and asymmetrically substituted variants 4.2b (1.50 Å), 4.2c-I (1.40 Å), 4.2e-I (1.25 Å), 4.2f-I (1.40 Å) and 4.2f-II (1.45 Å) bound to 14-3-3σΔC were solved to their respective resolutions in parentheses (Figure 2 and Supporting Information). Figure 2a depicts a superimposition of the three modified Tau peptides, 3.2a (white),123.2d (purple), and 4.f-II (green), in their 14-3-3-bound state, in which all three peptides clearly bind with a similar extended mode within the amphipathic groove of the protein. However, the structural differences between the three peptides at the C-terminus is observed to perturb the positioning of the peptide backbone and the leucine side-chain C-terminal to the phosphoserine residue. This general trend is representative for all cocrystallized modified Tau peptides. In Figures 2B–I, a panel comparing zoomed-in perspectives of the FC pocket of the seven different cocrystallized modified Tau peptides can be seen: the two benzhydryl-derived (3.2d and 3.2e) and five asymmetrically substituted analogues (4.2b, 4.2c-I, 4.2e-I, 4.2f-I, and 4.2f-II), compared to 3.2a. In contrast to analogues 3.2d and 3.2e, in which the pyrrolidine group were logically determined to be S-configured (derived from commercial (S)-2-(diphenylmethyl)pyrrolidine), all five asymmetrically substituted analogues were unexpectedly observed to be R-configured at the α-carbon of the pyrrolidine ring, counter to our expectations (Figure 2e–i).40 Interestingly as well, all of the five peptides modified with an asymmetrically substituted benzhydryl group bound in a similar “open” state, which contrasted with the “closed” state observed in the case of the three symmetric benzhydryl analogues. A closer examination of this data reveals that the oxygen of the methoxy substituent on 4.2f-I (Figure 2h) is sufficiently close (3.1 Å) to engage in a hydrogen bond with the side chain of residue Asn-42–an interaction witnessed previously between the sugar ring-oxygen of the natural product fusicoccin A (FC) and Asn-42 of 14-3-3σ for stabilization of TASK-3 (PDB: 5D3F; 2.7 Å)23 and 14-3-3ζ for stabilization of CFTR (PDB: 3P1Q; 3.2 Å).41 By comparison, an analogous hydrogen bond interaction is absent from the crystal structure of the less potent diastereomer 4.2f-II (Figure 2i). The ITC data for diastereomers 4.2f-I and 4.2f-II indicate that their 2-fold difference in affinity by FP and 3.5-fold difference by ITC is entropically driven (Figure 3 and Table S2 in the Supporting Information). An explanation for the potency of analogue 4.2c-I, the most potent of the series, can be observed in the crystal structure in which the methoxyethoxy side chain clearly addresses the FC pocket (Figure 2f). A more detailed discussion of all the cocrystal structures can be found in the Supporting Information.
Figure 2.

(a) Overlay of the cocrystal structures of the modified peptides 3.2a (white) (PDB: 5HF3)123.2d (purple) (PDB: 6FI5), and 4.2f-II (green) (PDB: 6FBW) bound to 14-3-3σΔC. (b–i) Zoom-in of FC pocket for the cocrystal structures of the modified peptides 3.2a (b) (PDB: 5HF3),123.2d (c) 3.2e (d) (PDB: 6FI4), 4.2b (e) (PDB: 6FBY), 4.2c-I (f) (PDB: 6FAW), 4.2e-I (g) (PDB: 6FAW), 4.2f-I (h) (PDB: 6FAV), and 4.2f-II (i) bound to 14-3-3σΔC. The stereochemical assignment (S and R notation) of the C-terminal amino acid and benzhydryl pyrrolidine solved in complex with the 14-3-3 protein have been added to each panel.
Figure 3.
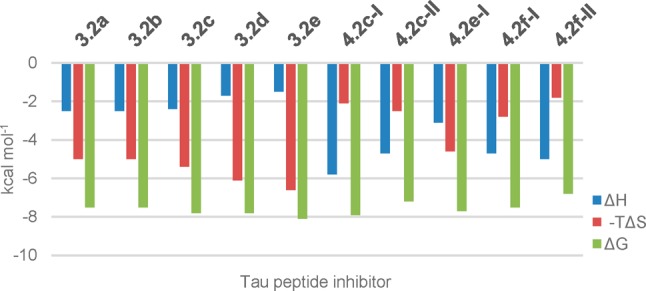
Comparison of enthalpic (ΔH, blue) and entropic contributions (-TΔS, red) to the Gibbs free energy of binding (ΔG, green) at 310 K for modified Tau peptides 3.2a, 3.2b, 3.2e, and 4.2f-I. For a comprehensive comparison of thermodynamic parameters for all modified Tau peptides see the Supporting Information (Table S2).
Stereochemical Outcome of Modified Tau Peptide Synthesis
Logically, the R-configuration of the d-Ala-derived modified Tau peptide was found in the 14-3-3/3.2d cocrystal structure (Figure 2c). Interestingly, the R-epimer was also observed in the cocrystal structure of the Val-derived modified Tau peptide 3.2e (Figure 2d), which parallels the result obtained previously12 for the l-Thr-derived modified Tau peptide 3.2a (Figure 2a). While the origin of the epimer formation in both cases remains unclear (both syntheses began with enantiomerically pure l-amino acids), it is hypothesized that the S-epimer of β-branched amino acids is sterically less favored for binding at the peptide C-terminus than the corresponding R-epimer. Significantly, the introduction of a glycine residue at the same C-terminal position of our peptides, which will not be dependent on epimerization, did not significantly affect the affinity of our modified Tau peptides. The electron densities clearly show that the R-epimer is bound to 14-3-3 for each of the five asymmetrically substituted analogues (4.2b, 4.2c-I, 4.2e-I, 4.2f-I, and 4.2f-II), which was unexpected because the synthesis of the corresponding monosubstituted benzhydryl pyrrolidine in each case commenced with S-configured l-proline, and was based on a enantioretentive synthesis of (S)-2-(diphenylmethyl)pyrrolidine.40 However, we did not monitor the stereochemical outcome at each step during the synthesis of our monosubstituted benzhydryl pyrrolidine derivatives.
Analysis of Thermodynamic Parameters of Binding
A clear difference was observed between the thermodynamic parameters for the asymmetrically substituted and those measured for the symmetrically substituted benzhydryl pyrrolidine-modified Tau peptides. In the case of the asymmetrically substituted analogues, the enthalpy change (ΔH) was in general more negative and the entropy change (-TΔS) more positive than for the symmetrically substituted benzhydryl pyrrolidine-modified Tau peptides. For example, while 3.2e (Kd = 2.2 μM, ΔG = −8.1 kcal) and 4.2f-I gave similar binding parameters (Kd = 4.7 μM, ΔG = −7.5 kcal mol–1), their corresponding thermodynamic components significantly diverge (Figure 4). In the case of 3.2e, the entropic factor (−TΔS= −6.6 kcal mol–1) contributed significantly more to the free energy of binding than the enthalpic factor (ΔH = −1.5 kcal mol–1), while for analogue 4.2f-I, the opposite was true (i.e., ΔH = −4.7 kcal mol–1 and −TΔS= −2.8 kcal mol–1). The same divergent behavior was observed across all Tau peptide inhibitors reported in Tables 2 and 3 such that compound binding could be classified as being either predominantly entropically driven (i.e., the symmetric benzhydryl analogues 3.2a–f) or enthalpically driven (i.e., the asymmetric analogues 4.2a–g).
Figure 4.
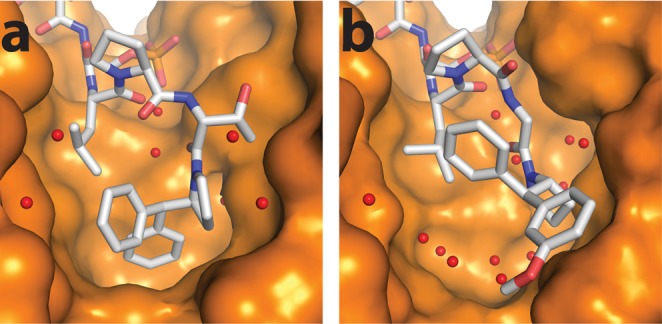
Crystal structure of (a) 3.2a at 1.8 Å resolution (PDB: 5HF3) and (b) 4.2f-I (PDB: 6FAV) at 1.4 Å resolution, in complex with 14-3-3σ. A difference in the amount of water molecules (red spheres) in the pocket can be observed, which may explain the difference in entropy observed in the ITC data.
A structural explanation for the two classifications of ITC data could be found in the crystallography data (Figure 4). The predominantly entropically driven binding profile characterized by ITC could be explained by the “closed” binding mode characterized by X-ray crystallography (illustrated by 3.2a, Figure 4a), in which the S-configured symmetric benzhydryl moiety is observed to fill the hydrophobic FC-binding pocket better. In this mode, the reduced solvent-exposed hydrophobic surface area results in fewer ordered water molecules (as evidenced in Figure 4a), which drives binding through a gain in entropy. By contrast, the predominantly enthalpically driven binding profile (illustrated by 4.2f-I, Figure 4b) could be explained by a more “open” binding mode in the X-ray crystal structure, in which the now R-configured asymmetrically substituted benzhydryl moiety is found in a more solvent-exposed state, which permits the binding of more ordered water molecules (as evidenced in Figure 4b), resulting in a comparatively lower entropic contribution on binding.
A similar correlation between the ITC and the crystallography data for all other solved structures was observed, which suggests that all derivatives bearing the same, either enthalpic or entropic, binding profile by ITC (whether cocrystallized or not with the 14-3-3 protein) bind via the aforedescribed “closed” or “open” binding modes by X-ray crystallography. The reason for the two different binding modes is likely two fold: first, the difference in R and S configuration between the asymmetrically substituted benzhydryl pyrrolidine moieties (R-configured), which all bind in the “open” state, and the S-configured symmetric benzhydryl pyrroldines, which bind in the “closed” state. Second, a steric clash between the asymmetrically substituted benzhydryl group and the protein surface may disfavor a “closed” binding mode, which the ligand would relieve by adopting the more “open” binding state. In view of the ITC data for the Gly-derivative 3.2b, the suggestion is the benzhydryl group of this derivative binds in a similar “closed” state as observed for e.g. 3.2a, 3.2d, and 3.2e. This result would suggest the added conformational freedom introduced by the C-terminal glycine residue would apparently not significantly influence the binding mode of 3.2b, and by extension analogues from within the 4.2 series. While the modifications at the C-terminus of our modified Tau peptides do not address the targeted deep-lying pocket of the 14-3-3 protein (Figure 1C), as hypothesized, they have enabled the potent targeting of chemically distinct sites within the adjacent FC pocket.
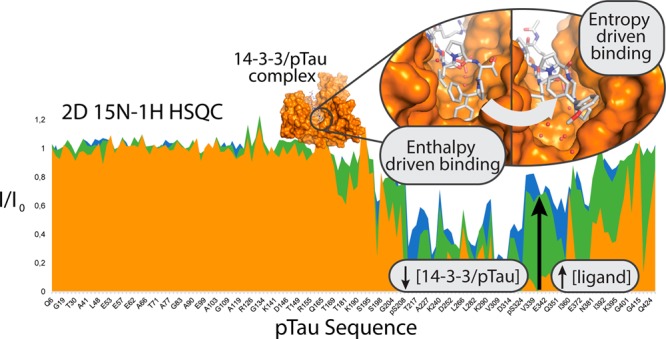
Inhibition of the Interaction of 14-3-3 with Full-Length Phosphorylated Tau Protein
We next investigated the potential of the three most potent analogues toward the inhibition of the interaction between 14-3-3ζ and full-length PKA-phosphorylated Tau (pTau). From the analogues tested, one (3.2e), binds to 14-3-3 via an “open” binding mode, while the other two, (4.2c-I and 4.2e-I), via the “closed” binding mode. NMR spectroscopy was used to assess the modulation of this PPI in solution, using chemical shift perturbation mapping, based on the assigned resonances of the phosphorylated Tau 1H–15N 2D spectrum.42−45 The 2D 15N–1H HSQC spectra of 15N-labeled pTau in the presence of unlabeled 14-3-3ζ were acquired with or without each of the inhibitors. The intensities of the correlation peaks corresponding to specific amino acid residues along pTau sequence (I) were monitored in each experiment and compared to the intensities of the corresponding correlation peaks in the spectrum of pTau alone (I0). The binding of 14-3-3ζ to pTau led to peak broadening and consequently, to the decrease of the (I/I0) ratio for resonances corresponding specifically to residues located in the binding region of 14-3-3 (Figure 5a and 5b). The addition of 4.2e-I to the solution containing 15N pTau/14-3-3ζ led to a dose-dependent recovery of the intensity of these same resonances (Figure 5a and 5b). This effect is well illustrated by the resonances corresponding to the amide groups of the PKA-phosphorylated serines of Tau that showed intensity recovery with addition of each analogue (Figures 5b and S36). The same set of 2D experiments was performed for inhibitors 3.2e and 4.2c-I resulting in a similar I/I0 profile (Figures 5, S37, and S38), which confirmed the capacity of these analogues to decrease the formation of the complex 14-3-3ζ/pTau. To get further insights on the inhibitory effect of these analogues, a series of 1H spectra of 4.2e-I was recorded in the presence of increasing concentrations of 14-3-3ζ (Figure 5c). By monitoring one well-isolated resonance of the spectrum of the small-molecule, a concentration dependent intensity decrease upon the addition of 14-3-3ζ was observed, which can be attributed to the interaction, as the sharp signal of the free ligand get broadened when complexed with the protein. Additionally, the spectrum of 4.2e-I was recorded in the presence and absence of pTau, which did not reveal any sign of interaction (Figure S39) as both spectra were identical. Based on these results, it can be concluded that 3.2e, 4.2c-I, and 4.2e-I inhibited the interaction pTau/14-3-3ζ in a concentration dependent manner, by binding to 14-3-3ζ and competing with pTau.
Figure 5.

4.2e-I inhibits the binding of pTau to 14-3-3ζ in a concentration-dependent manner. (A) Selected enlarged regions of the overlaid 1H–15N HSQC spectra showing the intensity recovery of the correlation peaks corresponding to the amide groups of the PKA-phosphorylated Serines after the addition of 4.2e-I. The spectra are shown superimposed to the pTau spectrum, which is colored in black. With the addition of a 3-fold excess of 4.2e-I (considering 14-3-3ζ concentration) it is possible to remark the intensity recovery of the weakest epitopes (pS208, pS356 and pS416) and finally, with the addition of a 10-fold, it is possible to detect pS214 and pS324. (B) Plot of the ratios of the bound (I)/free (I0) 1H–15N correlation peak intensities of full length pTau 60 μM (y axis) versus the amino acid sequence (x axis) in the presence of 14-3-3ζ 120 μM (red plot); 14-3-3ζ 120 μM + 4.2e-I 360 μM (green plot) and 14-3-3ζ 120 μM + 4.2e-I 1200 μM (blue plot). A total of 155 correlation peak intensities are shown. The x axis is not proportional. The domains of full-length pTau (N-ter for N-terminal; PRD for Proline-Rich Domain; MTBR for Microtubule Binding Region; C-ter for C-terminal) and the phosphorylation sites (S208, S214, S324, S356, and S416) are identified below the x axis. (C) Section of overlaid 1H spectra of 4.2e-I 150 μM alone (blue) and in the presence of 14-3-3ζ 10 μM (red), 30 μM (green), 75 μM (purple), and 150 μM (gold). The enlarged region shows a well-isolated resonance corresponding to a 4.2e-I proton.
Conclusions
Here, we describe the synthesis of a novel 17-membered collection of modified Tau peptide inhibitors, targeting increased binding affinity at the fusiccocin A (FC) pocket and proximal deep-lying pocket. In the case of the peptides bearing a symmetric, S-configured benzhydryl pyrroldine group, exchanging the C-terminal Thr residue during the synthesis with a number of other l- and d-amino acid residues at the C-terminus of the Tau peptide, including nonchiral Gly, did not produce a significant change in the IC50 and Kd and associated thermodynamic parameters. This result suggested that the C-terminal amino acid in the modified Tau peptide can be flexibly replaced by amino acids bearing different side chains and stereochemistry. All resulting Tau peptide analogues (Tables 2 and 3) were active inhibitors of 14-3-3/Tau. Interestingly, using a combination of FP, ITC, and X-ray crystallography data, we characterized two binding modes for our modified Tau peptides: one “closed” entropically driven state, the other an “open” enthalpic state at the peptide C-terminus. The difference between the two binding modes is most likely caused by the inverted stereochemistry at the α-carbon between the commercial S-configured symmetric benzhydryl pyrrolidine (“closed”) and the synthetic R-configured asymmetric benzhydryl pyrrolidine (“open”) (an unexpected outcome of the synthesis) and steric factors induced by monosubstitution of the benzhydryl group. While neither of these two modes were capable of addressing the deep-lying pocket (Figure 1c), both inhibited 14-3-3-binding to full-length PKA-phosphorylated Tau protein in vitro. Considering the manner in which the Tau-derived phosphoepitope has been studied presently, it could also be envisaged as a noncovalent tether to investigate new chemotypes to address the FC pocket, thus potentially opening the door to new nonphosphorylated inhibitors or novel stabilizers of Mode III 14-3-3 PPIs.
Methods
Synthesis of Substituted Benzhydryl Pyrrolidine Derivatives
General Methods
Unless otherwise stated, all solvents employed were commercially available and used without purification. Water was purified using a Millipore purification train. Dry solvents were obtained from a MBRAUN Solvent Purification System (MB-SPS-800). Deuterated solvents were obtained from Cambridge Isotope laboratories. All reagents were commercially available, supplied by Sigma-Aldrich, and used without purification. NMR data were recorded on a Bruker Cryomagnet for NMR spectroscopy (400 MHz for 1H NMR and 100 MHz for 13C NMR). Proton experiments were reported in parts per million (ppm) downfield of TMS. All 13C spectra were relative to the residual chloroform signal (77.16 ppm). 1H NMR spectra are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublets, td = triplet of doublets), integration, and coupling constant (J) in Hertz (Hz). Analytical liquid chromatography coupled with mass spectrometry (LC-MS) was performed on a C4 Jupiter SuC4300A 150 × 2.0 mm column using H2O with 0.1% formic acid (FA) and acetonitrile with 0.1% FA, in general with a gradient of 5% to 100% acetonitrile in H2O in 15 min (connected to a Thermo Fischer LCQ Fleet ion trap mass spectrometer). Preparative high pressure liquid chromatography (HP-LC) was performed on a Gemini S4 110A 150 × 21.20 mm column using H2O with 0.1% F.A. and Acetonitrile with 0.1% FA. Silica column chromatography was performed manually using silica gel with particle size 60–200 μm (60 Å). Reaction progress was monitored by thin-layer chromatography using Merck TLC silica gel 60 F254 plates.
tert-Butyl (S)-2-Benzoylpyrrolidine-1-carboxylate (2.2)
To an oven-dried 100 mL two-necked flask was added magnesium turnings (1.18 g, 48.4 mmol) and anhydrous tetrahydrofuran (THF, 10 mL) under argon pressure. A small amount of iodine was added followed by the slow addition of bromobenzene (3.80 g, 24.2 mmol). The reaction was slowly heated using a heat gun to start the reaction and it was then stirred at room temperature for 30 min. Subsequently, the reaction was cooled down to 0 °C, at which time the Weinreb amide 2.1 (2.50 g, 9.7 mmol) in THF (6 mL) was added slowly. The resultant mixture was stirred at 0 °C for 3 h, and then quenched with saturated NH4Cl (15 mL) and extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The product was purified by column chromatography, eluting with heptane/EtOAc 72:28 v/v to yield tert-butyl (S)-2-benzoylpyrrolidine-1-carboxylate (2.2) as a white powder (1.24 g, 4.50 mmol, 47%). Silica gel TLC Rf = 0.23 (heptane/EtOAc 72:28 v/v); LC-MS (ESI): calcd for C16H21NO3 [M+H-BOC]+: 176.10, observed 176.25, LC, Rt = 6.76 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 8.02–7.91 (m, 2H), 7.62–7.52 (m, 1H), 7.51–7.41 (m, 2H), 5.37–5.16 (m, 1H), 3.73–3.42 (m, 2H), 2.40–2.24 (m, 1H), 2.02–1.85 (m, 3H), 1.47 (s, 9H); 13C NMR (100 MHz, CDCl3): δ 198.92, 154.45, 135.29, 133.19, 128.70, 128.69, 128.52, 128.51, 79.78, 61.36, 46.81, 29.82, 28.21, 28.20, 28.19, 24.18.
(S)-Phenyl(pyrrolidin-2-yl)methanone (2.3)
The t-Boc-protected amine 2.2 (1.14 g, 4.1 mmol) was dissolved in dichloromethane (25 mL) and a solution of trifluoroacetic acid (10 mL) (70:30 v/v) was added. The reaction was stirred at room temperature for 2 h. The solvent was evaporated, yielding (S)-phenyl(pyrrolidin-2-yl)methanone (2.3) (718 mg, 4.1 mmol, 99%), which was directly used without purification. LC-MS (ESI): calcd for C11H13NO [M + H]+: 176.10, observed 176.25, LC, Rt = 2.38 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.98 (d, J = 7.6 Hz, 2H), 7.72 (t, J = 7.6 Hz, 1H), 7.60–7.52 (m, 2H), 5.56–5.47 (m, 1H), 3.69–3.52 (m, 2H), 2.80–2.68 (m, 1H), 2.30–2.18 (m, 1H), 2.16–1.97 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 193.67, 135.79, 131.26, 129.33, 129.32, 129.21, 129.19, 63.18, 47.47, 30.22, 24.32.
Ethyl (S)-2-Benzoylpyrrolidine-1-carboxylate (2.4)
(S)-Phenyl(pyrrolidin-2-yl)methanone (2.3) (500 mg, 2.58 mmol) was dissolved in methanol (25 mL). K2CO3 (1.58 g, 11.4 mmol) was added followed by ethyl chloroformate (340 mg, 3.1 mmol) and the reaction mixture was stirred for 24 h at room temperature. The reaction mixture was quenched with NH4Cl (15 mL) and extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with brine, dried over Na2SO4, and filtered. Evaporation of solvent afforded ethyl (S)-2-benzoylpyrrolidine-1-carboxylate (2.4) (501 mg, 2.0 mmol, 71%) as a yellow oil. LC-MS (ESI): calcd for C14H17NO3 [M + H]+: 248.12, observed 248.08, LC, Rt = 5.89 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 8.04–7.91 (m, 2H), 7.63–7.53 (m, 1H), 7.52–7.43 (m, 2H), 5.44–5.19 (m, 1H), 4.27–3.90 (m, 2H), 3.77–3.43 (m, 2H), 2.44–2.22 (m, 1H), 2.02–1.86 (m, 3H), 1.28 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 198.31, 155.15, 135.06, 133.30, 128.73, 128.73, 128.53, 128.52, 61.39, 61.15, 47.00, 30.90, 24.25, 14.77.
General Procedure for the Synthesis of Pyrrolooxazolones 2.5a–g
To an oven-dried, either 50 or 100 mL two-necked flask were added the magnesium turnings and anhydrous THF under an argon atmosphere. A catalytic (spatula end) amount of iodine was added, followed by slow addition of the substituted bromobenzene neat via Hamilton gastight syringe. The reaction was gently heated to initiate the reaction, and stirred at room temperature for 30 min. Subsequently, the reaction was cooled down to 0 °C and a 1 mL solution of 2.4 in THF introduced through slow dropwise addition. The resulting mixture was stirred at 0 °C for 2 h, and then warmed to 70 °C. With the exception of analogue 2.5a, which was heated for 3 h and worked up directly, in the general case, the reaction was monitored by measuring LC-MS on small aliquots of the reaction, and heated for 24 h. For the workup, the reaction mixture was cooled to room temperature, quenched through addition of sat. aq. NH4Cl (5 mL), and extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with brine, dried over Na2SO4, filtered and then concentrated in vacuo. Except with the synthesis of analogues 2.5a, 2.5b, and 2.5 g, which proceeded directly to the purification step, the crude material was dissolved in methanol (3 mL) and treated with either 100 mg NaOH (2.5c and 2.5d) or 500 mg KOH (2.5e and 2.5f) to promote further ring-closure to the pyrrolooxazolone. In these latter cases, the reaction was stirred at room temperature for 24 h and worked up as described previously. The isolated crude was purified by either silica gel column chromatography (2.5a, 2.5b, 2.5c, 2.5d, and 2.5 g) or reversed-phase flash chromatography on a Biotage Isolera system (2.5e and 2.5f) to yield the pure pyrrolooxazolone after evaporation of elution solvents under reduced pressure.
Pyrrolooxazolone 2.5a
The synthesis was performed in accordance with the general method using magnesium turnings (39 mg, 1.6 mmol) suspended in 5 mL of anhydrous THF, 2-bromotoluene (138 mg, 0.81 mmol), and 2.4 (95 mg, 384 μmol). The product was purified by silica gel column chromatography, eluting with heptane/EtOAc 70:10 v/v to yield pyrrolooxazolone 2.5a as a yellow oil (129 mg, 330 mmol, 86%). Silica gel TLC Rf = 0.30 (heptane/EtOAc 70:30 v/v); LC-MS (ESI): calcd for C19H19NO2 [M + H]+: 294.14, observed 294.17, LC, Rt = 7.08 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.55–7.50 (m, 1H), 7.34–7.26 (m, 5H), 7.22–7.13 (m, 3H), 4.81 (dd, J = 9.0, 6.2 Hz, 1H), 3.69–3.59 (m, 1H), 3.28–3.19 (m, 1H), 2.11 (s, 3H), 1.95–1.76 (m, 2H), 1.63–1.49 (m, 1H), 1.28–1.16 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 159.64, 140.15, 139.03, 138.69, 133.00, 128.65, 128.21, 128.20, 127.65, 125.87, 125.87, 125.52, 125.24, 87.98, 67.66, 45.35, 27.96, 26.00, 21.56.
Pyrrolooxazolone 2.5b
The synthesis was performed in accordance with the general method using magnesium turnings (39 mg, 1.6 mmol) suspended in 5 mL of anhydrous THF, 3-bromotoluene (138 mg, 0.81 mmol), and 2.4 (95 mg, 384 μmol). The product was purified by silica gel column chromatography, eluting with heptane/EtOAc 70:30 v/v to yield pyrrolooxazolone 2.5b as a yellow oil (46 mg, 157 μmol, 41%). Silica gel TLC Rf = 0.29 (heptane/EtOAc 70:30 v/v); LC-MS (ESI): calcd for C19H19NO2 [M + H]+: 294.14, observed 294.08, LC, Rt = 7.11 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.42–7.28 (m, 7H), 7.25–7.21 (m, 1H), 7.12 (d, J = 7.5 Hz, 1H), 4.54 (dd, J = 10.5, 5.5 Hz, 1H), 3.78–3.68 (m, 1H), 3.29–3.20 (m, 1H), 2.34 (s, 3H), 2.04–1.62 (m, 3H), 1.18–1.06 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 160.48, 143.24, 140.43, 138.34, 129.07, 128.39, 128.28, 128.28, 127.63, 126.58, 125.45, 125.44, 122.90, 85.91, 69.24, 46.02, 28.98, 24.93, 21.59.
Pyrrolooxazolone 2.5c
The synthesis was performed in accordance with the general method using magnesium turnings (49 mg, 2.0 mmol) suspended in 4 mL of anhydrous THF, 1-bromo-2-(2-methoxyethoxy)-benzene (280 mg, 0.81 mmol), and 2.4 (80 mg, 324 μmol). The product was purified by silica gel column chromatography, eluting with heptane/EtOAc 60:40 v/v to yield pyrrolooxazolone 2.5c as a yellow oil (65 mg, 184 μmol, 57%). Silica gel TLC Rf = 0.34 (heptane/EtOAc 60:40 v/v); LC-MS (ESI): calcd for C21H23NO4 [M + H]+: 354.16, observed 354.00, LC, Rt = 6.83 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.71 (dd, J = 7.9, 1.7 Hz, 1H), 7.52–7.46 (m, 2H), 7.34–7.27 (m, 3H), 7.25–7.18 (m, 1H), 7.03–6.90 (m, 2H), 4.85 (dd, J = 9.0, 6.2 Hz, 1H), 4.23–4.16 (m, 2H), 3.81–3.69 (m, 2H), 3.69–3.61 (m, 1H), 3.41 (s, 3H), 3.19–3.09 (m, 1H), 2.03–1.93 (m, 1H), 1.90–1.80 (m, 2H), 1.39–1.25 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 159.53, 154.38, 140.42, 131.80, 129.39, 127.83, 127.83, 127.58, 127.18, 126.39, 126.39, 121.38, 112.03, 86.80, 70.74, 68.46, 66.96, 58.78, 45.24, 28.76, 25.81.
Pyrrolooxazolone 2.5d
The synthesis was performed in accordance with the general method using magnesium turnings (49 mg, 2.0 mmol) suspended in 4 mL of anhydrous THF, 1-bromo-3-(2-methoxyethoxy)-benzene (280 mg, 0.81 mmol), and 2.4 (100 mg, 404 μmol). The product was purified by silica gel column chromatography, eluting with heptane/EtOAc 60:40 v/v to yield pyrrolooxazolone 2.5d as a yellow oil (114 mg, 323 μmol, 80%). Silica gel TLC Rf = 0.24 (heptane/EtOAc 60:40 v/v); LC-MS (ESI): calcd for C21H23NO4 [M + H]+: 354.16, observed 353.92, LC, Rt = 6.68 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.40–7.27 (m, 6H), 7.15–7.08 (m, 2H), 6.88–6.83 (m, 1H), 4.52 (dd, J = 10.4, 5.5 Hz, 1H), 4.11–4.07 (m, 2H), 3.77–3.66 (m, 3H), 3.43 (s, 3H), 3.28–3.19 (m, 1H), 2. 02–1.78 (m, 2H), 1.75–1.65 (m, 1H), 1.18–1.04 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 160.33, 158.89, 144.89, 140.18, 129.54, 128.29, 128.28, 127.71, 125.43, 125.43, 118.40, 113.83, 113.14, 85.80, 70.97, 69.22, 67.26, 59.21, 45.99, 28.96, 24.95.
Pyrrolooxazolone 2.5e
The synthesis was performed in accordance with the general method using magnesium turnings (49 mg, 2.0 mmol) suspended in 4 mL anhydrous THF, 2-bromoanisole (151 mg, 0.81 mmol), and 2.4 (100 mg, 404 μmol). The product was purified by reversed-phase flash chromatography on a Biotage Isolera system, eluting with a gradient of 5–100% ACN in H2O to yield pyrrolooxazolone 2.5e as a yellow oil (40 mg, 129 mmol, 32%). LC-MS (ESI): calcd for C19H19NO3 [M + H]+: 310.14, observed 310.08, LC, Rt = 6.49 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.67 (dd, J = 7.8, 1.7 Hz, 1H), 7.41–7.27 (m, 5H), 7.25–7.20 (m, 1H), 7.06–6.91 (m, 2H), 4.72 (dd, J = 8.7, 6.2 Hz, 1H), 3.86 (s, 3H), 3.72–3.60 (m, 1H), 3.21–3.09 (m, 1H), 1.98–1.90 (m, 1H), 1.89–1.80 (m, 2H), 1.45–1.32 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 159.32, 155.28, 140.27, 131.63, 129.49, 127.95, 127.94, 127.68, 127.10, 126.29, 126.28, 121.34, 111.63, 87.13, 68.72, 55.21, 45.19, 28.68, 25.92.
Pyrrolooxazolone 2.5f
The synthesis was performed in accordance with the general method using magnesium turnings (49 mg, 2.0 mmol) suspended in 4 mL of anhydrous THF, 3-bromoanisole (151 mg, 0.81 mmol), and 2.4 (100 mg, 404 μmol). The product was purified by reversed-phase flash chromatography on a Biotage Isolera system, eluting with a gradient of 5–100% ACN in H2O to yield pyrrolooxazolone 2.5f as a yellow oil (31 mg, 100 μmol, 25%). LC-MS (ESI): calcd for C19H19NO3 [M + H]+: 310.14, observed 310.17, LC, Rt = 6.43 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.42–7.26 (m, 6H), 7.12 (d, J = 8.0 Hz, 1H), 7.06 (t, J = 2.2 Hz, 1H), 6.84 (dd, J = 8.0, 2.4 Hz, 1H), 4.53 (dd, J = 10.5, 5.5 Hz, 1H), 3.78 (s, 3H), 3.75–3.68 (m, 1H), 3.32–3.18 (m, 1H), 2.04–1.80 (m, 2H), 1.77–1.67 (m, 1H), 1.20–1.05 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 160.36, 159.72, 144.93, 140.21, 129.56, 128.30, 128.30, 127.72, 125.42, 125.42, 118.07, 113.44, 112.23, 85.83, 69.29, 55.32, 45.99, 28.98, 24.95.
Pyrrolooxazolone 2.5g
The synthesis was performed in accordance with the general method using magnesium turnings (49 mg, 2.0 mmol) suspended in 4 mL of anhydrous THF, 3-bromophenyl isopropyl ether (217 mg, 1.0 mmol), and 2.4 (100 mg, 404 μmol). The product was purified by silica gel column chromatography, eluting with CH2Cl2/heptane 80:20 v/v to yield pyrrolooxazolone 2.5 g as a yellow oil (117 mg, 347 μmol, 86%). Silica gel TLC Rf = 0.29 (DCM/heptane 80:20 v/v); LC-MS (ESI): calcd for C21H23NO3 [M + H]+: 338.17, observed 338.08, LC, Rt = 7.12 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.42–7.26 (m, 5H), 7.26–7.22 (m, 1H), 7.10–7.03 (m, 2H), 6.82 (dd, J = 8.0, 2.0 Hz, 1H), 4.57–4.47 (m, 2H), 3.78–3.67 (m, 1H), 3.29–3.19 (m, 1H), 2.00–1.80 (m, 2H), 1.76–1.66 (m, 1H), 1.30 (dd, J = 7.8, 6.1 Hz, 6H), 1.18–1.04 (m, 1H); 13C NMR (101 MHz, CDCl3): δ 160.39, 158.00, 144.94, 140.27, 129.52, 128.29, 128.27, 127.67, 125.45, 125.44, 117.91, 114.95, 114.19, 85.83, 69.89, 69.27, 45.99, 28.99, 24.95, 22.00, 21.98.
General Procedure for the Synthesis of Asymmetrically Substituted Benzhydryl Pyrrolidine Analogues 2.6a–g
A 25 mL, glass round-bottomed flask was loaded with the pyrrolooxazolone precursor, dissolved in either pure ethyl acetate or a methanol/ethyl acetate solvent mixture, and treated with the catalyst, Pd/C (10%). The flask was flushed with hydrogen gas for 15 min, 50 μL of triethylamine was added to the reaction mixture, and the reaction stirred at room temperature under a hydrogen atmosphere for 24 h. The catalyst was filtered off by passing the reaction mixture through Celite, eluting with EtOAc. The solvent was removed under reduced pressure to obtain the benzhydryl pyrrolidine analogue. For analogues 2.6a, 2.6b, and 2.6 g, the material was used without further purification. Analogues 2.6c, 2.6d, 2.6e, and 2.6f were further purified by reversed-phase preparative HPLC to isolated separable diastereomers.
Benzhydryl Pyrrolidine Analogue 2.6a
The synthesis was performed in accordance with the general method using 2.5a (50 mg, 170 μmol) and Pd/C (10%) (25 mg) in methanol (1.5 mL) and ethyl acetate (0.5 mL) to form 2.6a (33 mg, 131 μmol, 77%), which was directly used without purification. LC-MS (ESI): calcd for C18H21N [M + H]+: 252.17, observed 252.17, LC, Rt = 4.94 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.42–7.28 (m, 7H), 7.25–7.21 (m, 1H), 7.12 (d, J = 7.5 Hz, 1H), 4.54 (dd, J = 10.5, 5.5 Hz, 1H), 3.78–3.68 (m, 1H), 3.29–3.20 (m, 1H), 2.34 (s, 3H), 2.04–1.62 (m, 3H), 1.18–1.06 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 160.48, 143.24, 140.43, 138.34, 129.07, 128.39, 128.28, 128.28, 127.63, 126.58, 125.45, 125.44, 122.90, 85.91, 69.24, 46.02, 28.98, 24.93, 21.59.
Benzhydryl Pyrrolidine Analogue 2.6b
The synthesis was performed in accordance with the general method using 2.5b (45 mg, 153 μmol) and Pd/C (10%) (25 mg) in methanol (1.5 mL) and ethyl acetate (0.5 mL) to form 2.6b (22 mg, 88 μmol, 59%), which was directly used without purification. LC-MS (ESI): calcd for C18H21N [M + H]+: 252.17, observed 252.25, LC, Rt = 4.52; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.36 (d, J = 7.2 Hz, 1H), 7.30–7.24 (m, 2H), 7.19–7.06 (m, 5H), 6.97 (d, J = 7.1 Hz, 1H), 3.95–3.83 (m, 1H), 3.77 (d, J = 10.2 Hz, 1H), 3.04–2.80 (m, 2H), 2.30 (s, 3H), 1.86–1.70 (m, 3H), 1.50–1.38 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 143.40, 143.24, 138.03, 128.86, 128.66, 128.66, 128.36, 128.07, 128.07, 127.19, 126.47, 124.95, 62.18, 57.88, 45.92, 30.57, 24.57, 21.51.
Benzhydryl Pyrrolidine Analogue 2.6c
The synthesis was performed in accordance with the general method using 2.5c (56 mg, 158 μmol) and Pd/C (10%) (35 mg) in methanol (1.9 mL) and ethyl acetate (0.6 mL) to form 2.6c, which was purified further by preparative HPLC (gradient of 15–40% ACN in 12 min.), to obtain the two different diastereomers 2.6c-I (2 mg, 7 μmol, 4%) and 2.6c-II (22 mg, 71 μmol, 44%) as pure compounds. 2.6c-I: LC-MS (ESI): calcd for C20H25NO2 [M + H]+: 312.19, observed 312.25, LC, Rt = 4.44 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.41 (d, J = 7.6 Hz, 2H), 7.30 (t, J = 7.6 Hz, 2H), 7.23–7.14 (m, 2H), 7.04 (d, J = 7.6 Hz, 1H), 6.93–6.79 (m, 2H), 4.84 (d, J = 8.3 Hz, 1H), 4.58–4.49 (m, 1H), 4.17–4.06 (m, 2H), 3.89–3.72 (m, 2H), 3.53 (s, 3H), 3.05–2.95 (m, 1H), 2.88–2.73 (m, 1H), 2.10–1.99 (m, 1H), 1.94–1.81 (m, 1H), 1.79–1.65 (m, 2H); 13C NMR (100 MHz, CDCl3): δ 155.47, 140.05, 130.00, 129.99, 129.29, 129.27, 128.74, 128.70, 128.41, 127.01, 121.42, 111.73, 70.57, 67.52, 61.60, 59.44, 47.41, 45.34, 29.50, 24.08. 2.6c-II: LC-MS (ESI): calcd for C20H25NO2 [M + H]+: 312.19, observed 312.33, LC, Rt = 4.55 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.37–7.27 (m, 5H), 7.23–7.16 (m, 2H), 6.95 (t, J = 7.6 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 4.56–4.44 (m, 2H), 4.14 (t, J = 4.7 Hz, 2H), 3.81–3.61 (m, 2H), 3.44 (s, 3H), 3.15 (m, 1H), 2.98 (m, 1H), 2.02 (m, 1H), 1.96–1.87 (m, 2H), 1.67 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 167.99, 155.41, 140.72, 129.94, 128.65, 128.60, 128.59, 128.28, 128.28, 126.92, 121.76, 111.95, 70.64, 66.81, 61.60, 59.06, 48.83, 45.35, 30.88, 24.06.
Benzhydryl Pyrrolidine Analogue 2.6d
The synthesis was performed in accordance with the general method using 2.5d (110 mg, 311 μmol) and Pd/C (10%) (50 mg) in methanol (2.6 mL) and ethyl acetate (0.9 mL) to form 2.6d, which was purified further by preparative HPLC (gradient of 15–40% ACN in 12 min.), to obtain two different diastereomers 2.6d-I (53 mg, 170 μmol, 55%) and 2.6d-II (16 mg, 51 μmol, 16%) as pure compounds. 2.6d-I: LC-MS (ESI): calcd for C20H25NO2 [M + H]+: 312.19, observed 312.25, LC, Rt = 4.14 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.40 (d, J = 7.7 Hz, 2H), 7.33–7.26 (m, 2H), 7.22–7.11 (m, 2H), 6.87–6.79 (m, 2H), 6.74 (d, J = 8.4 Hz, 1H), 4.17–4.10 (m, 2H), 4.05 (t, J = 4.4 Hz, 2H), 3.72 (t, J = 5.2, 2H), 3.44 (s, 3H), 2.75–2.66 (m, 2H), 1.92–1.83 (m, 2H), 1.81–1.71 (m, 1H), 1.68–1.57 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 159.01, 143.13, 140.43, 129.85, 128.90, 128.90, 128.11, 128.10, 127.15, 120.16, 114.34, 112.67, 70.98, 67.15, 62.11, 59.25, 54.75, 44.89, 30.76, 23.82. 2.6d-II: LC-MS (ESI): calcd for C20H25NO2 [M + H]+: 312.19, observed 312.25, LC, Rt = 4.40 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.26–7.16 (m, 6H), 7.11–7.00 (m, 2H), 6.76 (d, J = 8.1 Hz, 1H), 4.24–4.10 (m, 4H), 3.72 (t, J = 4.4 Hz, 2H), 3.42 (s, 3H), 2.91 (t, J = 7.1 Hz, 2H), 2.00–1.77 (m, 3H), 1.73–1.60 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 159.04, 130.18, 128.91, 128.91, 127.60, 127.60, 127.22, 120.56, 114.26, 113.91, 71.08, 67.20, 62.81, 59.19, 54.80, 45.37, 30.67, 23.71.
Benzhydryl Pyrrolidine Analogue 2.6e
The synthesis was performed in accordance with the general method using 2.5e (40 mg, 129 μmol) and Pd/C (10%) (30 mg) in methanol (1.0 mL) and ethyl acetate (0.6 mL) to form 2.6e, which was purified further by preparative HPLC (19% ACN in 15 min.), to obtain the two different diastereomers 2.6e-I (6 mg, 22 μmol, 17%) and 2.6e-II (10 mg, 37 μmol, 28%) as pure compounds. 2.6e-I: LC-MS (ESI): calcd for C18H21NO [M + H]+: 268.16, observed 268.25, LC, Rt = 3.67 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.38–7.32 (m, 2H), 7.24–7.07 (m, 5H), 6.92–6.77 (m, 2H), 4.61 (d, J = 11.3 Hz, 1H), 4.53–4.38 (m, 1H), 3.81 (s, 3H), 2.88–2.73 (m, 2H), 1.99–1.81 (m, 3H), 1.75–1.62 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 156.50, 139.97, 129.44, 128.66, 128.65, 128.41, 128.30, 128.24, 128.23, 127.00, 120.99, 111.15, 62.11, 55.45, 47.48, 45.49, 29.95, 23.70. 2.6e-II: LC-MS (ESI): calcd for C18H21NO [M + H]+: 268.16, observed 268.25, LC, Rt = 3.70 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.35 (dd, J = 7.6, 1.6 Hz, 1H), 7.26–7.20 (m, 4H), 7.20–7.11 (m, 2H), 6.88 (td, J = 7.5, 1.1 Hz, 1H), 6.77 (dd, J = 8.3, 1.1 Hz, 1H), 4.47 (d, J = 11.7 Hz, 1H), 4.45–4.35 (m, 1H), 3.74 (s, 3H), 2.88–2.68 (m, 2H), 2.01–1.83 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 157.06, 140.61, 128.61, 128.61, 128.44, 128.13, 128.12, 128.11, 127.95, 127.01, 120.83, 110.87, 61.72, 55.28, 48.28, 45.37, 30.83, 23.80.
Benzhydryl Pyrrolidine Analogue 2.6f
The synthesis was performed in accordance with the general method using 2.5f (30 mg, 97 μmol) and Pd/C (10%) (25 mg) in methanol (1.0 mL) and ethyl acetate (1.0 mL) to form 2.6f, which was purified further by preparative HPLC (20% ACN in 15 min.), to obtain the two different diastereomers 2.6f-I (7 mg, 26 μmol, 26%) and 2.6f-II (2 mg, 7 μmol, 7%) as pure compounds. 2.6f-I: LC-MS (ESI): calcd for C18H21NO [M + H]+: 268.16, observed 268.25, LC, Rt = 3.55 min. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.39 (d, J = 7.6 Hz, 2H), 7.31–7.27 (m, 2H), 7.21–7.13 (m, 2H), 6.84 (d, J = 8.0 Hz, 1H), 6.79 (t, J = 2.0 Hz, 1H), 6.72 (dd, J = 8.0, 2.0 Hz, 1H), 4.17–4.04 (m, 2H), 3.75 (s, 3H), 2.78–2.69 (m, 2H), 1.93–1.82 (m, 2H), 1.81–1.73 (m, 1H), 1.67–1.56 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 159.79, 143.41, 140.91, 129.83, 128.87, 128.86, 128.09, 128.08, 127.09, 119.95, 113.75, 112.07, 62.09, 55.18, 55.17, 44.96, 30.69, 23.92. 2.6f-II: LC-MS (ESI): calcd for C18H21NO [M + H]+: 268.16, observed 268.25, LC, Rt = 3.63 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.40–7.27 (m, 2H), 7.26–7.16 (m, 5H), 7.06 (d, J = 7.7 Hz, 1H), 7.02 (t, J = 2.1 Hz, 1H), 6.72 (dd, J = 8.3, 2.4 Hz, 1H), 4.24–4.10 (m, 2H), 3.82 (s, 3H), 2.86 (t, J = 7.2 Hz, 2H), 1.98–1.74 (m, 3H), 1.72–1.60 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 159.86, 141.86, 141.27, 130.08, 128.96, 128.94, 127.61, 127.60, 127.22, 120.37, 113.62, 113.36, 99.99, 62.66, 55.33, 54.88, 45.36, 30.82, 23.74.
Benzhydryl Pyrrolidine Analogue 2.6g
The synthesis was performed in accordance with the general method using 2.5 g (95 mg, 282 μmol) and Pd/C (10%) (50 mg) in ethyl acetate (3.0 mL) to form 2.6 g (67 mg, 227 μmol, 81%), which was used directly without further purification. LC-MS (ESI): calcd for C20H25NO [M + H]+: 296.19, observed 296.25, LC, Rt = 4.16 min; 1H NMR (400 MHz, CDCl3): δ (ppm) 7.40–7.26 (m, 4H), 7.22–7.12 (m, 2H), 6.90–6.80 (m, 2H), 6.72–6.65 (m, 1H), 4.56–4.44 (m, 1H), 3.90–3.78 (m, 1H), 3.72 (dd, J = 10.3, 2.4 Hz, 1H), 3.07–2.82 (m, 2H), 1.87–1.70 (m, 3H), 1.49–1.39 (m, 1H), 1.31 (dd, J = 6.1, 1.6 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 157.85, 145.01, 143.43, 129.35, 128.65, 128.65, 128.04, 128.03, 126.47, 120.28, 116.19, 112.98, 69.66, 62.15, 58.22, 45.98, 30.48, 24.61, 22.08, 22.04.
General Procedure for the Synthesis of Partially Protected Tau Peptides 3.1a–f
The partially protected Tau peptides 3.1a–f were prepared in accordance with a previous published synthesis of 3.1a.12 In each case, the peptides were synthesized on a 50 μmol scale by automated peptide synthesis (Intavis MultiPep RSi) using a Fmoc SPPS strategy performed on a 2-chlorotrityl resin (Iris Biotech GmbH and AGTC Bioproducts) preloaded with the corresponding C-terminal amino acid. The Fmoc-protected amino acid building blocks (4.2 eq., 0.5 M, Novabiochem) were dissolved in N-methyl-2-pyrrolidone (NMP) and coupled sequentially to the resin using N,N-diisopropylethylamine (DIPEA, 8 equiv, prepared as 1.6 M stock solution in NMP, Biosolve) and (2-(1H-benzotriazol-1-yl)-1,1,3,3tetramethyluronium hexafluorophosphate (HBTU, 4 equiv, 0.4 M stock solution in NMP, Biosolve). Each amino acid coupling was repeated once to ensure complete conversion. Fmoc-deprotection was performed using 20% piperidine in NMP (twice per cycle). The peptide N-terminus was N-acetylated prior to resin cleavage using Ac2O/pyridine/NMP (1:1:3). Resin cleavage of the partially protected peptide was performed using 30% hexafluoroisopropanol (HFIP, Sigma-Aldrich) in CH2Cl2 (1 mL per 100 mg resin, 1 × 20 min, 1 × 10 min.) and the organic solvents removed to dryness by rotary evaporation. The isolated material was then redissolved in acetonitrile/water/0.1% trifluoroacetic acid (TFA) and lyophilized to obtain a white powder. The yields of the crude partially protected peptides were typically >90% yield. The partially protected peptides were characterized by The partially protected peptides were characterized by Analytical Liquid Chromatography coupled with Mass Spectrometry (LC-MS), via one of the following methods:
Method A: using a C4 Jupiter SuC4300A 150 × 2.0 mm column using H2O with 0.1% formic acid (FA) and acetonitrile with 0.1% FA with a gradient of 5% to 100% acetonitrile in H2O in 15 min (connected to a Thermo Fischer LCQ Fleet ion trap mass spectrometer.
Method B: using a C18 Atlantis T3 5 μm 150 × 1 mm column using H2O with 0.1% trifluoroacetic acid (TFA) and acetonitrile with 0.1% TFA with a gradient of 5% to 100% acetonitrile in H2O in 15 min (connected to a Thermo Finnigan LCQ Deca XP MAX mass spectrometer) (see the Supporting Information for LC-MS spectra).
General Procedure for the Synthesis of Modified Tau Peptides 3.2b–f and 4.2a–g
The modified Tau inhibitors 3.2b–f and 4.2a–g were prepared in accordance with a previously published synthesis of 3.2a.12 A 25 mL round-bottomed flask fitted with a magnetic stirring bar was charged with the partially protected Tau peptide (3.1b-f, 1.1 equiv). The peptide was dissolved in N,N-dimethylformamide (DMF, 0.06 M) and treated with 6-chloro-benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorosphate (PyClock) (Novabiochem, 1.5 equiv). The reaction mixture was stirred for 15 min, which was followed by the sequential addition of N,N-diisoproylethylamine (DIPEA, 5 equiv) and 1 equiv of either (S)-2-(diphenylmethyl)pyrrolidine, for the synthesis of modified tau peptides 3.2a–f, or benzhydryl pyrrolidine analogues 2.6a–g, for the synthesis of modified tau peptides 4.2a–g. The reaction was left to stir, conversion monitored by LC-MS, and then worked up after 24 h by evaporating the organic solvent under reduced pressure to obtain the crude material. Deprotection of the side-chain protecting groups was performed by stirring the crude material for 3 h in a 95/2.5/2.5 (v/v) mixture of TFA/H2O/triisopropylsilane (TIS) (0.016 M). The peptide was then precipitated into 40 mL ice-cold diethyl ether (Et2O), stored at −30 °C for 10 min, centrifuged at 2500 rpm for 10 min and the supernatant decanted. Fresh ice-cold Et2O (40 mL) was added to pellet, and the subsequent step repeated. The crude pellet was dissolved in H2O/ACN + 0.1% TFA and then purified by reversed-phase HPLC using a C18 column (Atlantis T3 prep OBD, 19 × 150 mm) using the optimized gradient conditions described below beside each modified peptide. After purification the aqueous organic solvent mixture was removed by lyophilization to obtain the modified Tau inhibitors 3.2 a-f and 4.2a-g, typically as white amorphous powders. Afterward, the modified tau peptides were characterized by analytical liquid chromatography coupled with mass spectrometry (LC-MS) using a C18 Atlantis T3 5 μm 150 × 1 mm column using H2O with 0.1% TFA and acetonitrile with 0.1% TFA with a gradient of 5% to 100% acetonitrile in H2O in 15 min (connected to a Thermo Finnigan LCQ Deca XP MAX mass spectrometer) (see the Supporting Information for LC-MS spectra). In the case of Tau inhibitors 4.2c–f, two diastereomers (I and II) were separated, which were studied separately in the subsequent biochemical and X-ray crystallography studies.
Modified Tau Peptide 3.2a
The synthesis was performed in accordance with the general method using (S)-2-(diphenylmethyl)pyrrolidine (9 mg, 37 μmol) and purification performed by preparative reversed-phase HPLC using a linear gradient of 40–45% MeCN to form 3.2a (3 mg, 3 μmol, 8%). LC-MS (ESI): calcd for C52H78N11O14P1 [M + H]+: 1112.55, observed 1112.6, LC, Rt = 7.62 min.
Modified Tau Peptide 3.2b
The synthesis was performed in accordance with the general method using (S)-2-(diphenylmethyl)pyrrolidine (10 mg, 42 μmol) and purification performed by preparative reversed-phase HPLC using a linear gradient of 40–45% MeCN to form 3.2b (11 mg, 10 μmol, 24%). LC-MS (ESI): calcd for C50H74N11O13P1 [M + H]+: 1068.52, observed 1068.5, LC, Rt = 7.87 min.
Modified Tau Peptide 3.2c
The synthesis was performed in accordance with the general method using (S)-2-(diphenylmethyl)pyrrolidine (10 mg, 42 μmol) and purification performed by preparative reversed-phase HPLC using a linear gradient of 40–45% MeCN to form 3.2c (16 mg, 15 μmol, 36%). LC-MS (ESI): calcd for C51H76N11O13P1 [M + H]+: 1082.54, observed 1082.6, LC, Rt = 8.00 min.
Modified Tau Peptide 3.2d
The synthesis was performed in accordance with the general method using (S)-2-(diphenylmethyl)pyrrolidine (10 mg, 42 μmol) and purification performed by preparative reversed-phase HPLC using a linear gradient of 40–45% MeCN to form 3.2d (13 mg, 12 μmol, 29%). LC-MS (ESI): calcd for C51H76N11O13P1 [M + H]+: 1082.54, observed 1082.6, LC, Rt = 8.02 min.
Modified Tau Peptide 3.2e
The synthesis was performed in accordance with the general method using (S)-2-(diphenylmethyl)pyrrolidine (8 mg, 34 μmol) and purification performed by preparative reversed-phase HPLC using a linear gradient of 40–45% MeCN to form 3.2e (10 mg, 9 μmol, 26%). LC-MS (ESI): calcd for C53H80N11O13P1 [M + H]+: 1110.57, observed 1110.7, LC, Rt = 8.82 min.
Modified Tau Peptide 3.2f
The synthesis was performed in accordance with the general method using (S)-2-(diphenylmethyl)pyrrolidine (8 mg, 34 μmol), and purification performed by preparative reversed-phase HPLC using a linear gradient of 40–45% MeCN to form 3.2f (4 mg, 4 μmol, 12%). LC-MS (ESI): calcd for C53H80N11O13P1 [M + H]+: 1110.57, observed 1110.7, LC, Rt = 8.83 min.
Modified Tau Peptide 4.2a
The synthesis was performed in accordance with the general method using benzhydryl pyrrolidine analogue 2.6a (7 mg, 28 μmol), and purification performed by preparative reversed-phase HPLC using a linear gradient of 38–43% MeCN to form 4.2a as an inseparable mixture of diastereomers (9 mg, 8 μmol, 29%). LC-MS (ESI): calcd for C51H76N11O13P1 [M + H]+: 1082.54, observed 1082.6, LC, Rt = 8.38 min.
Modified Tau Peptide 4.2b
The synthesis was performed in accordance with the general method using benzhydryl pyrrolidine analogue 2.6b (8 mg, 32 μmol), and purification performed by preparative reversed-phase HPLC using a linear gradient of 37–42% MeCN to form 4.2b as an inseparable mixture of diastereomers (5 mg, 5 μmol, 16%). LC-MS (ESI): calcd for C51H76N11O13P1 [M + H]+: 1082.54, observed 1082.6, LC, Rt = 8.42 min.
Isomeric Modified Tau Peptide 4.2c-I
The synthesis was performed in accordance with the general method using isomeric benzhydryl pyrrolidine analogue 2.6c-I (2 mg, 6 μmol), and purification performed by preparative reversed-phase HPLC using a linear gradient of 37–42% MeCN to form 4.2c-I (1 mg, 1 μmol, 17%). LC-MS (ESI): calcd for C53H80N11O15P1 [M + H]+: 1142.56, observed 1142.7, LC, Rt = 8.05 min.
Isomeric Modified Tau Peptides 4.2c-II
The synthesis was performed in accordance with the general method using isomeric benzhydryl pyrrolidine analogue 2.6c-II (9 mg, 29 μmol), and purification performed by preparative reversed-phase HPLC using a linear gradient of 37–42% MeCN to form 4.2c-II (7 mg, 6 μmol, 21%). LC-MS (ESI): calcd for C53H80N11O15P1 [M + H]+: 1142.56, observed 1142.6, LC, Rt = 8.35 min.
Isomeric Modified Tau Peptide 4.2d-I
The synthesis was performed in accordance with the general method using isomeric benzhydryl pyrrolidine analogue 2.6d-I (12 mg, 39 μmol), and purification performed by preparative reversed-phase HPLC using a linear gradient of 37–42% MeCN to form 4.2d-I (1 mg, 1 μmol, 3%). LC-MS (ESI): calcd for C53H80N11O15P1 [M + H]+: 1142.56, observed 1142.8, LC, Rt = 8.07 min.
Isomeric Modified Tau Peptides 4.2d-II
The synthesis was performed in accordance with the general method using isomeric benzhydryl pyrrolidine analogue 2.6d-II (4 mg, 13 μmol), and purification performed by preparative reversed-phase HPLC using a linear gradient of 35–40% MeCN to form 4.2d-II (4 mg, 4 μmol, 31%). LC-MS (ESI): calcd for C53H80N11O15P1 [M + H]+: 1142.56, observed 1142.7, LC, Rt = 8.23 min.
Isomeric Modified Tau Peptide 4.2e-I
The synthesis was performed in accordance with the general method using isomeric benzhydryl pyrrolidine analogue 2.6e-I (4 mg, 15 μmol), and purification performed by preparative reversed-phase HPLC using a linear gradient of 37–42% MeCN to form 4.2e-I (4 mg, 4 μmol, 27%). LC-MS (ESI): calcd for C51H76N11O14P1 [M + H]+: 1098.53, observed 1098.9, LC, Rt = 7.60 min.
Isomeric Modified Tau Peptides 4.2e-II
The synthesis was performed in accordance with the general method using isomeric benzhydryl pyrrolidine analogue 2.6e-II (7 mg, 26 μmol), and purification performed by preparative reversed-phase HPLC using a linear gradient of 35–40% MeCN to form 4.2e-II (6 mg, 5 μmol, 19%). LC-MS (ESI): calcd for C51H76N11O14P1 [M + H]+: 1098.53, observed 1098.7, LC, Rt = 8.32 min.
Isomeric Modified Tau Peptide 4.2f-I
The synthesis was performed in accordance with the general method using isomeric benzhydryl pyrrolidine analogue 2.6f-I (6 mg, 22 μmol), and purification performed by preparative reversed-phase HPLC using a linear gradient of 37–42% MeCN to form 4.2f-I (8 mg, 7 mmol, 32%). LC-MS (ESI): calcd for C51H76N11O14P1 [M + H]+: 1098.53, observed 1098.7, LC, Rt = 8.38 min.
Isomeric Modified Tau Peptides 4.2f-II
The synthesis was performed in accordance with the general method using isomeric benzhydryl pyrrolidine analogue 2.6f-II (2 mg, 8 μmol), and purification performed by preparative reversed-phase HPLC using a linear gradient of 35–40% MeCN to form 4.2f-II (2 mg, 2 μmol, 25%). LC-MS (ESI): calcd for C51H76N11O14P1 [M + H]+: 1098.53, observed 1098.7, LC, Rt = 8.28 min.
Modified Tau Peptide 4.2g
The synthesis was performed in accordance with the general method using benzhydryl pyrrolidine analogue 2.6 g (11 mg, 37 μmol), and purification performed by preparative reversed-phase HPLC using a linear gradient of 40–45% MeCN to form 4.2 g as an inseparable mixture of diastereomers (1 mg, 1 μmol, 3%). LC-MS (ESI): calcd for C53H80N11O14P1 [M + H]+: 1126.56, observed 1126.7, LC, Rt = 8.68 min.
Biochemical Evaluation of Modified Tau Peptides 3.2a–f and 4.2a–g
Fluorescence Polarization Assays
FP assays were performed using 100 nM FAM-labeled tethered Tau peptide (5,6-FAM)-RTP(ps)LPTG(GGS)3GSKCG(pS)LGNIHHK in buffer containing 10 mM HEPES (pH 7.4), 10 mM NaCl, 0.1% (v/v) Tween20, and 0.1% (w/v) bis(trimethylsilyl)acetamide (BSA). First, a dilution series of His-14-3-3ζ to the labeled peptide was made in order to obtain a Kd value and select an appropriate concentration for the subsequent inhibition experiments. For the determination of IC50 values, dilution series of the inhibitor peptides were made to a solution containing 100 nM labeled peptide and 10 μM His-14-3-3ζ (EC80). The assays were performed in Corning black, round-bottom, low-volume, 384 microwell plates (ref 4514). The polarization was measured by use of a filter-based microplate reader (Tecan Infinite F500) using a fluorescein filter set (λex: 485 nm/20 nm, λem: 535 nm/25 nm) (10 reads per well). All experiments were performed in triplicate. To obtain Kd and IC50 values, the resulting curve was fitted by use of GraphPad Prism 6.0 for Windows (GraphPad Software Inc., La Jolla, CA).
Isothermal Titration Calorimetry
ITC experiments were performed on a Malvern ITC200 isothermal titration calorimeter (Microcal Inc.). The peptide and protein were separately dissolved in ITC-buffer containing 25 mM HEPES (pH 7.5), 100 mM NaCl, 10 mM MgCl2 and 0.5 mM TCEP (Tris(2-carboxyethyl)phosphine). In the sample cell, a solution of 0.1 mM 14-3-3ζ protein was placed and the syringe was loaded with a solution of 1 mM peptide inhibitor, which was titrated stepwise into the cell with 2 μL aliquots (with a delay of 180 s between each titration). For each measurement, a series of 19 injections was performed using the following settings: reference power, 5 μCal/s; initial delay, 60 s; stirring speed, 750 rpm; temperature, 37 °C. All measurements were performed in duplicate. The data was analyzed using Origin 7.0 software. A nonlinear regression analysis was performed, using a single-site binding model with varying stoichiometry (N), association constant (Ka = 1/Kd), and molar binding enthalpy (ΔH) to determine the thermodynamic parameters.
X-ray Crystallography Studies
Sitting Drop Crystallization of 14-3-3σ-Peptide Complexes
The 14-3-3σ protein used for crystallization was truncated C-terminally after the Thr-231 residue to enhance crystallization, called 14-3-3σΔC. Crystallization was attempted for all Tau inhibitors 3.2a–f and 4.2a–g with 14-3-3σΔC. The 14-3-3σΔC-peptide complex was dissolved in crystallization buffer (20 mM HEPES (pH 7.5), 2 mM MgCl2, 2 mM BME) at a 1:1.5 and 1:5 molar ratio with a resulting protein concentration of 12.5 mg/mL and this incubated overnight at 4 °C. A sitting drop crystal screen was set up on a 96-wells plate (Corning pZero 3550). First, 75 μL of the optimized 14-3-3σΔC screening conditions (5% glycerol, 0.19 M CaCl2, varying concentration of PEG400 (24%–29% (v/v)), 0.095 M HEPES at varying pH of 7.1, 7.3, 7.5 and 7.7) were pipetted into the reservoirs. Then, by use of a pipetting robot (Mosquito), 500 nL drops were made by pipetting 250 nL of the screening conditions into 250 nL of protein–peptide complex. The plate was covered with an airtight foil, and incubated at 4 °C. Crystals were harvested after 1–12 weeks and were flash-cooled in liquid nitrogen before measuring.
Data Collection and Processing
Diffraction data for 3.2a–d were collected at the Petra(III) synchrotron in Hamburg, and data for 4.2b, 4.2c-I, 4.2e-I, 4.2f-I, and 4.2f-II were collected on a Rigaku Compact HomeLab beamline (home source). The data was processed using iMosflm46 in the ccp4i package or XDS and the structures were phased by molecular replacement (using PDB: 4Y3B) in Phenix Phaser.47 Refinement and model building were done with Phenix.refine48 and Coot49 software packages. Figures were made by PyMol (DeLano Scientific LLC, version 0.99rc6).
NMR Spectroscopy Studies on Full-Length PKA-Phosphorylated Tau Protein
15N Labeled Tau Protein Expression and Purification
E. coli BL21 cells were transformed with the pET15b vector carrying the longest Tau isoform (2N4R, 441 amino acid residues). A 20 mL preculture in Luria–Bertani (LB) medium containing 100 μg/mL ampicillin was grown overnight at 37 °C and was used to inoculate a 1 L culture in M9 minimal medium supplemented with 4 g/L Glucose, 1 g/L 15NH4Cl and 0.4 g/L 15N rich-medium (Isogro 15N, Isotec). The culture was grown at 37 °C to an OD600 of 0.8 and induced with 0.5 mM IPTG. Incubation was continued for 4 h at 37 °C and the culture was then harvested by centrifugation. Purification of recombinant Tau protein was achieved by first heating the sample at 75 °C for 15 min followed by a cation exchange chromatography. The pure fractions were further buffer exchanged with 50 mM ammonium bicarbonate using a desalting column before lyophilization. Protein concentration was estimated by absorption at 280 nm. Detailed protocols can be found in refs (50) and (51).
PKA Catalytic Subunit Expression and Purification
E. coli BL21 cells were transformed with the pet15b Vector (Addgene plasmid # 14921) carrying the gene coding for the N-terminally His-tagged PKA catalytic subunit alpha from M. musculus. A 20 mL preculture in Luria–Bertani (LB) medium containing 100 μg/mL ampicillin was grown overnight at 37 °C and was used to inoculate a 1 L culture in LB medium. The culture was grown at 20 °C to an OD600 of 0.8 and induced with 0.5 mM IPTG. Incubation was continued for 20 h at 20 °C and the culture was then harvested by centrifugation. The His-tagged PKA catalytic subunit alpha was purified by affinity chromatography using a Ni-NTA column (GE Healthcare, Uppsala, Sweden) and buffer-exchanged with 250 mM potassium phosphate, 0.1 mM DTT pH 6.5 on a desalting column. The protein was aliquoted, flash frozen in liquid nitrogen and stored at −80 °C.
In Vitro Tau Phosphorylation
Recombinant 15N labeled Tau protein (100 μM) was incubated with 1.5 μM recombinant PKA catalytic subunit (PKAc) at 30 °C for 3 h, in a buffer consisting of 50 mM Hepes pH 8.0, 5 mM ATP, 12.5 mM MgCl2, 50 mM NaCl, 5 mM DTT, 1 mM EDTA. The reaction was heat-inactivated at 75 °C for 15 min and the solution was centrifuged in order to eliminate the precipitated PKAc. Buffer exchange with 50 mM ammonium carbonate on a desalting column was performed before lyophilization.
15N–1H HSQC Spectroscopy on Full-Length PKA-Phosphorylated Tau (fl-pTau)
15N–1H HSQC spectra were acquired in 3 mm tubes (sample volume 200 μL) using a 900 MHz Bruker Avance spectrometer, equipped with a cryoprobe. Spectra were recorded at 25 °C in a buffer containing 50 mM Tris pH 6.7, 30 mM NaCl, 2.5 mM EDTA, 1 mM DTT, EDTA-free protease inhibitor cocktail (Roche, Switzerland) and 10% (v/v) D2O. The experiments were performed with samples containing 60 μM 15N labeled PKA-phosphorylated Tau, 120 μM 14-3-3ζ and either 360 μM or 1200 μM of modified Tau peptide (3 and 10-fold excess to 14-3-3ζ concentration). The experiments were performed on compounds 3.2e, 4.2c-I, and 4.2e-I. The backbone resonance assignments of the Tau protein, including the phosphorylated residues, were previously reported in the literature.42,45 The reference for the 1H chemical shift was relative to Trimethylsilyl propionate. Spectra were collected and processed with Topspin 3.5 (Bruker Biospin, Karlsruhe, Germany) and analyzed with Sparky 3.12 (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco).
1H NMR Spectroscopy on 4.2e-I
1H spectra containing 4.2e-I were acquired in 3 mm tubes (sample volume 200 μL) using a 600 MHz Bruker Avance I spectrometer equipped with a CPQCI cryoprobe, at 25 °C in a buffer containing 50 mM Tris pH 6.7, 30 mM NaCl, 2.5 mM EDTA and 10% (v/v) D2O. The experiments were performed with 150 μM 4.2e-I alone or in the presence of either 10 μM, 30 μM, 75 μM or 150 μM 14-3-3ζ or 150 μM PKA-phosphorylated Tau. The reference for the 1H chemical shift was relative to Trimethylsilyl propionate. Spectra were collected and processed with Topspin 3.5 (Bruker Biospin, Karlsruhe, Germany).
Acknowledgments
Parts of this research were carried out at PETRA III at DESY, a member of the Helmholtz Association (HGF). We would like to thank Anja Burkhardt for assistance in using the P11 beamline and François-Xavier Cantrelle for NMR data acquisition. Some other crystallographic experiments were performed on the X06SA beamline at the Swiss Light Source, Paul Scherrer Institut, Villigen, Switzerland.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.8b00118.
Synthesis details and characterization data for 1-bromo-2-(2-methoxyethoxy)benzene and 1-bromo-3-(2-methoxyethoxy)benzene; 1H NMR, 13C NMR, and LC-MS or GC-MS spectra for intermediates 2.2, 2.3, 2,4, pyrrolooxazolones 2.5a–g, and benzhydryl pyrrolidine derivatives 2.6a–g; LC-MS data of purified final modified Tau peptides, 3.2a–f and 4.2a–g; thermograms of duplicate ITC measurements of 3.2a–f and 4.2a–g and a summary of the corresponding thermodynamic parameters in table form; data collection and refinement statistics and an in-depth discussion of all crystal structures reported in the paper; supplementary 15N–1H HSQC spectra and intensity plots (PDF)
Accession Codes
Atomic coordinates and structure factors for the crystal structures of 14-3-3σ with ligands 3.2d, 3.2e, 4.2b, 4.2c-I, 4.2e-I, 4.2f-I, and 4.2f-II can be accessed using PDB codes 6FI5, 6FI4, 6FBY, 6FAW, 6FAW, 6FAV, and 6FBW, respectively.
Author Contributions
# S.A.A. and F.A.M. contributed equally to this work. The manuscript was written through contributions of all authors. F.A.M. performed synthesis and biochemical studies; S.A.A. and C.O. performed structural characterization and analysis; J.F.N. performed the NMR experiments; I.L., L.B., C.O., and L.-G.M. designed the studies. All authors have given approval to the final version of the manuscript.
Funded by The Netherlands Organization for Scientific Resarch via ECHO Grant 717014001, VICI Grant 016150366, Gravity Program 024.001.035, by the Initial Training Network TASPPI, funded by the H2020 Marie Curie Actions of the European Commission under Grant Agreement 675179 and by the LabEx (Laboratory of Excellence) DISTALZ (ANR, ANR-11-LABX-009). The NMR facilities were funded by the Nord Region Council, CNRS, Institut Pasteur de Lille, the European Community (ERDF), the French Ministry of Research and the University of Lille and by the CTRL CPER cofunded by the European Union with the European Regional Development Fund (ERDF), by the Hauts de France Regional Council (Contrat No. 17003781), Métropole Européenne de Lille (Contract No. 2016_ESR_05), and French State (Contract No. 2017-R3-CTRL-Phase 1).We acknowledge support for the NMR facilities from TGE RMN THC (CNRS, FR-3050) and FRABio (Univ. Lille, CNRS, FR-3688).
The authors declare no competing financial interest.
Supplementary Material
References
- Cummings J.; Lee G.; Mortsdorf T.; Ritter A.; Zhong K. (2017) Alzheimer’s Disease Drug Development Pipeline: 2017. Alzheimers Dement. Transl. Res. Clin. Interv. 3 (3), 367–384. 10.1016/j.trci.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson P. T.; Alafuzoff I.; Bigio E. H.; Bouras C.; Braak H.; Cairns N. J.; Castellani R. J.; Crain B. J.; Davies P.; Del Tredici K.; et al. (2012) Correlation of Alzheimer Disease Neuropathologic Changes with Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 71 (5), 362–381. 10.1097/NEN.0b013e31825018f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeur E.; Guéret S. M.; Adihou H.; Gopalakrishnan R.; Lemurell M.; Waldmann H.; Grossmann T. N.; Plowright A. T. (2017) New Modalities for Challenging Targets in Drug Discovery. Angew. Chem., Int. Ed. 56 (35), 10294–10323. 10.1002/anie.201611914. [DOI] [PubMed] [Google Scholar]
- Arkin M. R.; Wells J. A. (2004) Small-Molecule Inhibitors of Protein–protein Interactions: Progressing towards the Dream. Nat. Rev. Drug Discovery 3 (4), 301–317. 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- Wells J. A.; McClendon C. L. (2007) Reaching for High-Hanging Fruit in Drug Discovery at Protein–protein Interfaces. Nature 450 (7172), 1001–1009. 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- Arkin M. R.; Tang Y.; Wells J. A. (2014) Small-Molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem. Biol. 21 (9), 1102–1114. 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milroy L.-G.; Grossmann T. N.; Hennig S.; Brunsveld L.; Ottmann C. (2014) Modulators of Protein–Protein Interactions. Chem. Rev. 114 (9), 4695–4748. 10.1021/cr400698c. [DOI] [PubMed] [Google Scholar]
- Pelay-Gimeno M.; Glas A.; Koch O.; Grossmann T. N. (2015) Structure-Based Design of Inhibitors of Protein-Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem., Int. Ed. 54 (31), 8896–8927. 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petta I.; Lievens S.; Libert C.; Tavernier J.; De Bosscher K. (2016) Modulation of Protein–Protein Interactions for the Development of Novel Therapeutics. Mol. Ther. 24 (4), 707–718. 10.1038/mt.2015.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C.; Brunden K. R.; Trojanowski J. Q.; Lee V. M.-Y.; Smith A. B.; Huryn D. M. (2011) Modulation of Protein-Protein Interactions as a Therapeutic Strategy for the Treatment of Neurodegenerative Tauopathies. Curr. Top. Med. Chem. 11 (3), 317–330. 10.2174/156802611794072605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo Y.; Schumacher B.; Landrieu I.; Bartel M.; Smet-Nocca C.; Jang A.; Choi H. S.; Jeon N. L.; Chang K.-A.; Kim H.-S.; et al. (2015) Involvement of 14–3-3 in Tubulin Instability and Impaired Axon Development Is Mediated by Tau. FASEB J. 29 (10), 4133–4144. 10.1096/fj.14-265009. [DOI] [PubMed] [Google Scholar]
- Milroy L.-G.; Bartel M.; Henen M. A.; Leysen S.; Adriaans J. M. C.; Brunsveld L.; Landrieu I.; Ottmann C. (2015) Stabilizer-Guided Inhibition of Protein-Protein Interactions. Angew. Chem., Int. Ed. 54 (52), 15720–15724. 10.1002/anie.201507976. [DOI] [PubMed] [Google Scholar]
- Kaplan A.; Ottmann C.; Fournier A. E. (2017) 14–3-3 Adaptor Protein-Protein Interactions as Therapeutic Targets for CNS Diseases. Pharmacol. Res. 125 (Pt B), 114–121. 10.1016/j.phrs.2017.09.007. [DOI] [PubMed] [Google Scholar]
- Fu H.; Subramanian R. R.; Masters S. C. (2000) 14–3-3 Proteins: Structure, Function, and Regulation. Annu. Rev. Pharmacol. Toxicol. 40 (1), 617–647. 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- Yaffe M. B. (2002) How Do 14–3-3 Proteins Work? – Gatekeeper Phosphorylation and the Molecular Anvil Hypothesis. FEBS Lett. 513 (1), 53–57. 10.1016/S0014-5793(01)03288-4. [DOI] [PubMed] [Google Scholar]
- Aitken A. (2006) 14–3-3 Proteins: A Historic Overview. Semin. Cancer Biol. 16 (3), 162–172. 10.1016/j.semcancer.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Yaffe M. B.; Rittinger K.; Volinia S.; Caron P. R.; Aitken A.; Leffers H.; Gamblin S. J.; Smerdon S. J.; Cantley L. C. (1997) The Structural Basis for 14–3-3:Phosphopeptide Binding Specificity. Cell 91 (7), 961–971. 10.1016/S0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- Aghazadeh Y.; Papadopoulos V. (2016) The Role of the 14–3-3 Protein Family in Health, Disease, and Drug Development. Drug Discovery Today 21 (2), 278–287. 10.1016/j.drudis.2015.09.012. [DOI] [PubMed] [Google Scholar]
- Freeman A. K.; Morrison D. K. (2011) 14–3-3 Proteins: Diverse Functions in Cell Proliferation and Cancer Progression. Semin. Cell Dev. Biol. 22 (7), 681–687. 10.1016/j.semcdb.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleppe R.; Martinez A.; Døskeland S. O.; Haavik J. (2011) The 14–3-3 Proteins in Regulation of Cellular Metabolism. Semin. Cell Dev. Biol. 22 (7), 713–719. 10.1016/j.semcdb.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Shimada T.; Fournier A. E.; Yamagata K. (2013) Neuroprotective Function of 14-3-3 Proteins in Neurodegeneration. BioMed Res. Int. 564534. 10.1155/2013/564534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries-van Leeuwen I. J.; da Costa Pereira D.; Flach K. D.; Piersma S. R.; Haase C.; Bier D.; Yalcin Z.; Michalides R.; Feenstra K. A.; Jiménez C. R.; et al. (2013) Interaction of 14–3-3 Proteins with the Estrogen Receptor Alpha F Domain Provides a Drug Target Interface. Proc. Natl. Acad. Sci. U. S. A. 110 (22), 8894–8899. 10.1073/pnas.1220809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders C.; Higuchi Y.; Koschinsky K.; Bartel M.; Schumacher B.; Thiel P.; Nitta H.; Preisig-Müller R.; Schlichthörl G.; Renigunta V.; et al. (2013) A Semisynthetic Fusicoccane Stabilizes a Protein-Protein Interaction and Enhances the Expression of K+ Channels at the Cell Surface. Chem. Biol. 20 (4), 583–593. 10.1016/j.chembiol.2013.03.015. [DOI] [PubMed] [Google Scholar]
- Andrei S. A.; Sijbesma E.; Hann M.; Davis J.; O’Mahony G.; Perry M. W. D.; Karawajczyk A.; Eickhoff J.; Brunsveld L.; Doveston R. G.; et al. (2017) Stabilization of Protein-Protein Interactions in Drug Discovery. Expert Opin. Drug Discovery 12 (9), 925–940. 10.1080/17460441.2017.1346608. [DOI] [PubMed] [Google Scholar]
- Stevers L. M.; Sijbesma E.; Botta M.; MacKintosh C.; Obsil T.; Landrieu I.; Cau Y.; Wilson A. J.; Karawajczyk A.; Eickhoff J.; et al. (2017) Modulators of 14-3-3 Protein-Protein Interactions. J. Med. Chem. 10.1021/acs.jmedchem.7b00574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland D. W.; Hwo S. Y.; Kirschner M. W. (1977) Purification of Tau, a Microtubule-Associated Protein That Induces Assembly of Microtubules from Purified Tubulin. J. Mol. Biol. 116 (2), 207–225. 10.1016/0022-2836(77)90213-3. [DOI] [PubMed] [Google Scholar]
- Himmler A.; Drechsel D.; Kirschner M. W.; Martin D. W. (1989) Tau Consists of a Set of Proteins with Repeated C-Terminal Microtubule-Binding Domains and Variable N-Terminal Domains. Mol. Cell. Biol. 9 (4), 1381–1388. 10.1128/MCB.9.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindwall G.; Cole R. D. (1984) Phosphorylation Affects the Ability of Tau Protein to Promote Microtubule Assembly. J. Biol. Chem. 259 (8), 5301–5305. [PubMed] [Google Scholar]
- Grundke-Iqbal I.; Iqbal K.; Tung Y. C.; Quinlan M.; Wisniewski H. M.; Binder L. I. (1986) Abnormal Phosphorylation of the Microtubule-Associated Protein Tau (Tau) in Alzheimer Cytoskeletal Pathology. Proc. Natl. Acad. Sci. U. S. A. 83 (13), 4913–4917. 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiguchi M.; Sobue K.; Paudel H. K. (2000) 14–3-3zeta Is an Effector of Tau Protein Phosphorylation. J. Biol. Chem. 275 (33), 25247–25254. 10.1074/jbc.M003738200. [DOI] [PubMed] [Google Scholar]
- Yuan Z.; Agarwal-Mawal A.; Paudel H. K. (2004) 14–3-3 Binds to and Mediates Phosphorylation of Microtubule-Associated Tau Protein by Ser9-Phosphorylated Glycogen Synthase Kinase 3beta in the Brain. J. Biol. Chem. 279 (25), 26105–26114. 10.1074/jbc.M308298200. [DOI] [PubMed] [Google Scholar]
- Li T.; Paudel H. K. (2007) 14–3-3zeta Facilitates GSK3beta-Catalyzed Tau Phosphorylation in HEK-293 Cells by a Mechanism That Requires Phosphorylation of GSK3beta on Ser9. Neurosci. Lett. 414 (3), 203–208. 10.1016/j.neulet.2006.11.073. [DOI] [PubMed] [Google Scholar]
- Qureshi H. Y.; Li T.; MacDonald R.; Cho C. M.; Leclerc N.; Paudel H. K. (2013) Interaction of 14–3-3ζ with Microtubule-Associated Protein Tau within Alzheimer’s Disease Neurofibrillary Tangles. Biochemistry 52 (37), 6445–6455. 10.1021/bi400442d. [DOI] [PubMed] [Google Scholar]
- Li T.; Paudel H. K. (2016) 14-3-3ζ Mediates Tau Aggregation in Human Neuroblastoma M17 Cells. PLoS One 11 (8), e0160635. 10.1371/journal.pone.0160635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillen A.; Barbier P.; Landrieu I.; Lefebvre S.; Wieruszeski J.-M.; Leroy A.; Peyrot V.; Lippens G. (2007) NMR Investigation of the Interaction between the Neuronal Protein Tau and the Microtubules. Biochemistry 46 (11), 3055–3064. 10.1021/bi061920i. [DOI] [PubMed] [Google Scholar]
- Lou K.; Yao Y.; Hoye A. T.; James M. J.; Cornec A.-S.; Hyde E.; Gay B.; Lee V. M.-Y.; Trojanowski J. Q.; Smith A. B.; et al. (2014) Brain-Penetrant, Orally Bioavailable Microtubule-Stabilizing Small Molecules Are Potential Candidate Therapeutics for Alzheimer’s Disease and Related Tauopathies. J. Med. Chem. 57 (14), 6116–6127. 10.1021/jm5005623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grüninger F. (2015) Invited Review: Drug Development for Tauopathies. Neuropathol. Appl. Neurobiol. 41 (1), 81–96. 10.1111/nan.12192. [DOI] [PubMed] [Google Scholar]
- Dammers C.; Yolcu D.; Kukuk L.; Willbold D.; Pickhardt M.; Mandelkow E.; Horn A. H. C.; Sticht H.; Malhis M. N.; Will N.; et al. (2016) Selection and Characterization of Tau Binding d-Enantiomeric Peptides with Potential for Therapy of Alzheimer Disease. PLoS One 11 (12), e0167432. 10.1371/journal.pone.0167432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molzan M.; Schumacher B.; Ottmann C.; Baljuls A.; Polzien L.; Weyand M.; Thiel P.; Rose R.; Rose M.; Kuhenne P.; et al. (2010) Impaired Binding of 14–3-3 to C-RAF in Noonan Syndrome Suggests New Approaches in Diseases with Increased Ras Signaling. Mol. Cell. Biol. 30 (19), 4698–4711. 10.1128/MCB.01636-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey D. J.; O’Hagan D.; Tavasli M. (1997) A Short Synthesis of (S)-2-(Diphenylmethyl)Pyrrolidine, a Chiral Solvating Agent for NMR Analysis. Tetrahedron: Asymmetry 8 (1), 149–153. 10.1016/S0957-4166(96)00495-8. [DOI] [Google Scholar]
- Stevers L. M.; Lam C. V.; Leysen S. F. R.; Meijer F. A.; van Scheppingen D. S.; de Vries R. M. J. M.; Carlile G. W.; Milroy L. G.; Thomas D. Y.; Brunsveld L.; et al. (2016) Characterization and Small-Molecule Stabilization of the Multisite Tandem Binding between 14–3-3 and the R Domain of CFTR. Proc. Natl. Acad. Sci. U. S. A. 113 (9), E1152–1161. 10.1073/pnas.1516631113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrieu I.; Lacosse L.; Leroy A.; Wieruszeski J.-M.; Trivelli X.; Sillen A.; Sibille N.; Schwalbe H.; Saxena K.; Langer T.; et al. (2006) NMR Analysis of a Tau Phosphorylation Pattern. J. Am. Chem. Soc. 128 (11), 3575–3583. 10.1021/ja054656+. [DOI] [PubMed] [Google Scholar]
- Lippens G.; Wieruszeski J.-M.; Leroy A.; Smet C.; Sillen A.; Buée L.; Landrieu I. (2004) Proline-Directed Random-Coil Chemical Shift Values as a Tool for the NMR Assignment of the Tau Phosphorylation Sites. ChemBioChem 5 (1), 73–78. 10.1002/cbic.200300763. [DOI] [PubMed] [Google Scholar]
- Mukrasch M. D.; Bibow S.; Korukottu J.; Jeganathan S.; Biernat J.; Griesinger C.; Mandelkow E.; Zweckstetter M. (2009) Structural Polymorphism of 441-Residue Tau at Single Residue Resolution. PLoS Biol. 7 (2), e1000034. 10.1371/journal.pbio.1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smet C.; Leroy A.; Sillen A.; Wieruszeski J.-M.; Landrieu I.; Lippens G. (2004) Accepting Its Random Coil Nature Allows a Partial NMR Assignment of the Neuronal Tau Protein. ChemBioChem 5 (12), 1639–1646. 10.1002/cbic.200400145. [DOI] [PubMed] [Google Scholar]
- Powell H. R.; Johnson O.; Leslie A. G. W. (2013) Autoindexing Diffraction Images with IMosflm. Acta Crystallogr., Sect. D: Biol. Crystallogr. 69 (7), 1195–1203. 10.1107/S0907444912048524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J. (2007) Solving Structures of Protein Complexes by Molecular Replacement with. Acta Crystallogr., Sect. D: Biol. Crystallogr. 63 (1), 32–41. 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Grosse-Kunstleve R. W.; Hung L. W.; Ioerger T. R.; McCoy A. J.; Moriarty N. W.; Read R. J.; Sacchettini J. C.; Sauter N. K.; Terwilliger T. C. (2002) PHENIX: Building New Software for Automated Crystallographic Structure Determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 58 (11), 1948–1954. 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66 (4), 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danis C.; Despres C.; Bessa L. M.; Malki I.; Merzougui H.; Huvent I.; Qi H.; Lippens G.; Cantrelle F.-X.; Schneider R.; et al. (2016) Nuclear Magnetic Resonance Spectroscopy for the Identification of Multiple Phosphorylations of Intrinsically Disordered Proteins. J. Visualized Exp. 118. 10.3791/55001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H.; Despres C.; Prabakaran S.; Cantrelle F.-X.; Chambraud B.; Gunawardena J.; Lippens G.; Smet-Nocca C.; Landrieu I. (2017) The Study of Posttranslational Modifications of Tau Protein by Nuclear Magnetic Resonance Spectroscopy: Phosphorylation of Tau Protein by ERK2 Recombinant Kinase and Rat Brain Extract, and Acetylation by Recombinant Creb-Binding Protein. Methods Mol. Biol. 1523, 179–213. 10.1007/978-1-4939-6598-4_11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.