

Abstract
Asymmetric hydrogenation of sterically hindered substrates still constitutes a long-standing challenge in the area of asymmetric catalysis. Herein, an efficient palladium acetate (an inexpensive Pd salt with low toxicity) catalyzed asymmetric hydrogenation of sterically hindered N-tosylimines is realized with high catalytic activities (S/C up to 5000) and excellent enantioselectivities (ee up to 99.9%). Quantum chemical calculations suggest that uniformly high enantioselectivities are observed due to the structurally different S- and R-reaction pathways.
Sterically hindered unsaturated compounds are challenging substrates for asymmetric hydrogenation. Here, the authors report a palladium/chiral (bis)phosphine catalytic system that hydrogenates a number of hindered N-tosylimines with excellent enantioselectivitites.
Introduction
Sterically hindered chiral amines have found widespread application for the asymmetric syntheses of bioactive substances, drugs, and ligands (Fig. 1a)1–15. Over the past half-century, significant progress has been made in the catalytic asymmetric hydrogenation of various imines for the synthesis of chiral amines16–23. Imines bearing relatively small substituents, such as methyl or ethyl groups directly connected to the carbon atom of a C = N group, have been widely used as substrates in asymmetric hydrogenation for the preparation of chiral amines, providing excellent stereoselectivities16–27. In sharp contrast, the asymmetric hydrogenation of imines bearing bulky substituents (such as the t-butyl group) has proved to be far more challenging, even though this methodology provides a straightforward approach for the preparation of chiral bulky amines, important structural elements found in pharmaceuticals, ligands, and other functional molecules. As evidenced by previous studies, the increased bulk of the alkyl substituents results in reduced yields and enantioselectivities of the chiral amine products (Fig. 1b). In 1996, a ruthenium-catalyzed asymmetric hydrogenation of N-tosylimines was described by Charette and Giroux giving the corresponding products in yields of 82%, 80%, <5%, and ees of 62%, 84%, 17%, with methyl, ethyl, and isopropyl substituents, respectively (R’ = Ph)24. In 2006 and 2007, the groups of Zhang28 and Zhou29 developed a palladium trifluoroacetate-catalyzed asymmetric hydrogenation of N-tosylimines. Enantioselectivities decreased from 99% to 93% and from 96% to 88%, respectively, when a methyl group was replaced by an ethyl group (R’ = Ph).
Fig. 1.

Asymmetric hydrogenation of imines for preparation of chiral amines. a Representative chiral ligands and bioactive compounds bearing sterically hindered chiral amine skeletons. b Previous work about asymmetric hydrogenation of imines. c This work: Pd-catalyzed asymmetric hydrogenation of sterically hindered imines
Previous methods of choice for the preparation of these chiral bulky amines (only one or two examples) have relied on chiral substrates, auxiliaries, or resolving agents, and suffered from low enantioselectivities and/or yields30–38. For example, in 2007, the catalytic enantioselective addition of HN3 to ketenes was disclosed by Fu and co-workers to give chiral methyl(2,2-dimethyl-1-phenylpropyl)carbamate with 76% ee33. In 2009, Zhang and co-workers developed the iridium-catalyzed asymmetric hydrogenation of 2,2-dimethyl-1-phenylpropan-1-imine hydrochloride and 3,3-dimethylbutan-2-imine hydrochloride with 80% ee and 17% ee, respectively34. Thus, an efficient methodology for the synthesis of chiral bulky amines by asymmetric hydrogenation remains elusive and highly desired.
Although palladium-catalyzed homogeneous asymmetric hydrogenations of C = C, C = O, and C = N bonds have been extensively investigated, Pd is less commonly used compared to other transition-metals such as Ru, Rh, and Ir, because Pd catalysts are usually less efficient (S/C ratio of no more than 1000)21,39–43. In addition, almost all these hydrogenations use Pd(OCOCF3)2 as a catalytic precursor, whereas inexpensive Pd salts, such as Pd(OAc)2, have not exhibited high catalytic activities in the previously reported asymmetric hydrogenation reactions28,29,44.
Our group have searched for novel approaches for the preparation of important chiral substances using metal-catalyzed asymmetric hydrogenation45–52. Recently, we developed a palladium-catalyzed asymmetric hydrogenation and hydrogenolysis of α-acyloxy ketones with excellent yields and high S/C (up to 5000–6000) using the bulky ligand DTBM-SegPhos51,52. In continuation of the work, we herein report a high yielding and highly enantioselective palladium acetate-catalyzed asymmetric hydrogenation of sterically hindered N-tosylimines (Fig. 1c). A possible reaction mechanism has been proposed via quantum chemical calculations.
Results
Investigation of reaction conditions
Initially, a model asymmetric hydrogenation of (Z)-t-butyl phenyl N-tosylimines (1a) was carried out under 40 atm H2 pressure at room temperature in TFE using Pd(TFA)2.
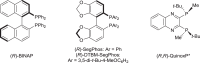
The commonly used ligand, (R)-BINAP, could catalyze this reaction but only gave the desired hydrogenation product with 24% conversion (Table 1, entry 1). The reaction was sluggish when another commonly used ligand, (R)-SegPhos, was used (Table 1, entry 2). However, the bulky ligand (R)-DTBM-SegPhos gave the desired product with almost quantitative conversion and high enantioselectivity (92.2% ee, Table 1, entry 3). To our delight, the hydrogenation proceeded smoothly and the product was obtained with up to 99.9% ee using an electron-rich P-stereogenic diphosphine ligand (R,R)-QuinoxP* (Table 1, entry 4), which has been found to be an efficient chiral diphosphine for Rh- and Ru-catalyzed asymmetric hydrogenations since first being reported in 200553–57. Different Pd precursors were also screened in combination with (R,R)-QuinoxP*. Pd(OAc)2, which is inexpensive, low toxic, and is not commonly used in asymmetric hydrogenations, provided excellent catalytic activity and enantioselectivity (over 99% conversion and 99.9% ee, Table 1, entry 5). Some by-products were produced when using other PdX2-type salts such as PdBr2 and PdCl2 (Table 1, entries 6, 7). The reaction temperature and hydrogenation pressure were also examined. Lowering the temperature to 0 °C had no effect on the reaction conversion and enantioselectivity (Table 1, entry 8). To our surprise, under 1 atm H2 pressure, the reaction proceeded smoothly and gave the product in quantitative conversion and 99.9% ee (Table 1, entry 9). When the S/C ratio was increased to 1000, the product was obtained with no loss in enantioselectivity and full conversion (Table 1, entry 10).
Table 1.
Reaction optimization
|
| ||||
|---|---|---|---|---|
| Entrya | Ligand | Pd source | Yield %b | ee %c |
| 1 | (R)-BINAP | Pd(TFA)2 | 24 | - |
| 2 | (R)-SegPhos | Pd(TFA)2 | <5 | - |
| 3 | (R)-DTBM-SegPhos | Pd(TFA)2 | >99 | 92.2 |
| 4 | (R,R)-QuinoxP* | Pd(TFA)2 | >99 | 99.9 |
| 5 | (R,R)-QuinoxP* | Pd(OAc)2 | >99 | 99.9 |
| 6 | (R,R)-QuinoxP* | PdBr2 | -d | - |
| 7 | (R,R)-QuinoxP* | PdCl2 | -d | - |
| 8e | (R,R)-QuinoxP* | Pd(OAc)2 | >99 | 99.9 |
| 9f | (R,R)-QuinoxP* | Pd(OAc)2 | >99 | 99.9 |
| 10g | (R,R)-QuinoxP* | Pd(OAc)2 | >99 | 99.9 |
| ||||
aConditions: 1a (0.2 mmol), PdX2 (2.0 mol %), ligand (2.1 mol %), TFE (2.0 mL), H2 (40 atm), RT, 24 h, unless otherwise noted
bDetermined by 1H NMR analysis
cThe ee values were determined by HPLC using chiral columns
dA mixture of complex by-products was generated
e0 °C.
fH2 (1 atm)
g1a (0.70 g), S/C = 1000. Pd(TFA)2 = Pd(OCOCF3)2
The chiral (bis)phosphine we used was abbreviated as (R,R)-QuinoxP* by the inventor. Generally speaking, the significance of '*' is chirality.
The influence of solvents on this reaction was also examined (Table 2). Several solvents were studied in order to try and avoid the use of TFE. However, TFE was proved to be superior to these solvents. Alcohols such as MeOH and EtOH showed different reactivities (Table 2, entries 1–3). Compared to the excellent results obtained with TFE, MeOH gave the product in excellent enantioselectivity but only 47% yield. Just a trace amount of product was obtained in EtOH. The low polar solvents THF, toluene, and DCM provided low activities (Table 2, entries 4–6).
Table 2.
Influence of reaction solvent
|
| |||
|---|---|---|---|
| Entrya | Solvent | Yield %b | ee %c |
| 1 | TFE | > 99 | 99.9 |
| 2 | MeOH | 47 | 99.9 |
| 3 | EtOH | Trace | - |
| 4 | THF | Trace | - |
| 5 | toluene | Trace | - |
| 6 | DCM | Trace | - |
aConditions: 1a (0.2 mmol), Pd(OAc)2 (2.0 mol%), (R,R)-QuinoxP* (2.1 mol%), H2 (40 atm), solvents (2.0 mL), RT, 24 h, unless otherwise noted
bDetermined by 1H NMR analysis
cThe ee values were determined by HPLC using chiral columns
Scope of asymmetric catalysis of hindered N-tosylimines
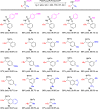
Substrate scope of the catalytic system was explored using the optimized reaction conditions and with a relatively low catalyst loading (S/C = 200, Table 3). All the tested t-Bu-N-tosylimine substrates were converted to their corresponding products with almost quantitative conversions. The six substrates bearing different R and RL were hydrogenated in 95–99% yields and with excellent enantioselectivities (2a, b, 2d–g, 99.0–99.9% ee), while a substrate bearing a 4-fluoro-substituent gave its corresponding product with 96.0% ee (2c). In addition, when the R group was changed to a t-Bu group, none of the corresponding product was detected under the standard conditions. Electron-donating and withdrawing substituents at the 3- or 4-position of the benzene ring of RS did not influence the stereoselectivities (2h-–p, 99.3–99.9% ee). Similarly, excellent enantioselectivities were obtained for disubstituted substrates, including a substrate bearing a 2-naphthyl group (2q–u, 99.4–99.9% ee). It should be noted that even alkyl imine substrates underwent smoothly asymmetric hydrogenation (2v, w, both 99.8% ee).
Table 3.
Substrate scopea
|
aConditions: 1 (0.2 mmol), Pd(OAc)2 (0.5 mol %), (R,R)-QuinoxP* (0.52 mol %), H2 (1 atm), TFE (2.0 mL), RT, 24 h
Scope of asymmetric catalysis of functionalized substrates
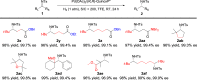
To further extend the substrate scope, many functionalized substrates were designed and synthesized. Under standard reaction conditions with a catalyst loading of 0.5 mol %, the products shown in Table 4 were obtained in almost quantitative yields and excellent stereoselectivities. Substrates with different ester groups and different carbon chain lengths were tested under the hydrogenation conditions, giving their corresponding products with excellent results (2x–z, 99.7%, 99.4% and 99.1% ee). Changing the ester group to an amide group had no negative impact on the reaction (2aa, 97% yield and 99.4% ee). A cyclopentyl-substituted substrate with α-quaternary carbon was also reduced with 99.3% ee (2ab). Substrates in which the carbon atom linked to the tetrasubstituted carbon center was replaced by an oxygen atom were also reduced with high stereoselectivities, irrespective of whether the oxygen atom was exocyclic or within the ring (2ac, ad, 99.8% and 99.4% ee). A substrate bearing an ester group at the side of the quaternary carbon could be hydrogenated to the corresponding chiral amine, albeit with slightly lower ee (2ae, 96% yield and 96.9% ee). Interestingly a diimine could also be reduced completely with excellent de and ee (2af, 98% yield, 99% de and 99.9% ee).
Table 4.
Scope of functionalized substratesa
|
aConditions: 1 (0.2 mmol), Pd(OAc)2 (0.5 mol %), (R,R)-QuinoxP* (0.52 mol %), H2 (1 atm), TFE (2.0 mL), RT, 24 h
Synthetic utility of chiral amines products
The practical utility of this catalytic asymmetric hydrogenation system was evaluated. The catalytic reaction was carried out at a low catalyst loading on a gram scale (1a, 3.60 g, S/C = 5000, Fig. 2). The reaction proceeded smoothly with quantitative conversion and with 99% ee under 60 atm H2 pressure at 60 °C over 48 h. The chiral compounds 2 have the potential to be used for the synthesis of sterically hindered chiral amines commonly found in optically active substances and chiral ligands1–15. Chiral intermediate 4 was obtained in 92% yield and 99% ee by reductive removal of the Ts group of 2a with sodium naphthalide58. This product could be further transformed to the bulky chiral amine structures 5 and 64,9, which can be used as chiral ligands and functional molecular motors. Other ligands, catalysts and bioactive compounds, such as bulky chiral carbene ligands3, amidoiridium complexes10, t-leucine31 and CXCR2/CXCR1 antagonists5, could also be synthesized from compound 4 according to literature procedures.
Fig. 2.

Product derivatization. Reagents and conditions are as follows. a 1a (3.60 g), S/C = 5000, 60 atm H2, 60 °C, 48 h. b 2a (158 mg, 0.5 mmol), naphthalene (5.0 mmol), sodium (5.0 mmol), 1,2-dimethoxyethane (10 mL), –70 °C-RT, 2 h. c 2-pyridinecarboxaldehyde, Et2O, RT, 16 h. d 5H-dibenzo[a,d][7]annulen-5-one, TiCl4, toluene, RT, 24 h
Synthetic utility of functionalized chiral amines products
Furthermore, the ester-functionalized products, 2x and 2y, could be converted to chiral γ- and δ-lactams (8x and 8y), which are useful chiral compounds13–15. Thus, the compounds of 8 were obtained with high yields and stereochemical fidelity via cyclization to form intermediates (7x and 7y) and subsequent removal of the Ts group, respectively (Fig. 3).
Fig. 3.

Functionalized product derivatization. Reagents and conditions are as follows. a 2x, y (0.5 mmol), Me3Al (0.6 mmol), toluene (15 mL), 80 °C, overnight. b 7x, y (0.5 mmol), naphthalene (5.0 mmol), sodium (5.0 mmol), DME (30 mL), –70 °C, 1 min
X-ray crystallographic analysis
The absolute configurations of the products 2a and 7x were determined to be S and R by X-ray crystallographic analysis (Fig. 4). Therefore, substrates with aryl or alkyl groups (including functionalized compounds) are attacked by the hydride on the same favored side.
Fig. 4.

X-ray crystallographic analysis. ORTEP representation of 2a and 7x
Mechanistic considerations
Recently the mechanism of the Pd-catalyzed asymmetric hydrogenation of unprotected indoles has been studied in detail40. There are two possible reaction pathways, inner-sphere hydrogenation with direct coordination of the C = N bond to Pd, and out-of-sphere hydrogenation with direct involvement of a solvent molecules, both employing a Pd–H complex as a catalyst. In the case of cyclic imines (unprotected indoles) the latter mechanism was computed to be favorable. However, for our sulfonated imines, a similar out-of-sphere mechanism is not possible due to the lack of an NH hydrogen atom that is present in indole substrates. Hence, we computed the catalytic cycle for the inner-sphere hydrogenation of 1a using Pd–H complex C1 as a catalyst.
The computed catalytic cycle for the experimentally observed S-product exhibited reasonable activation barriers standing well with the experimentally observed reaction rates (Fig. 5a). Thus, a coplanar orientation of the Pd–H and C = N bonds in the adduct S1 belonging to the S-catalytic cycle enables facile hydride transfer yielding intermediate S2. The latter is further hydrogenated by an additional molecule of H2 providing the product 2a(S) and regenerating the catalyst. Meanwhile, the minor enantiomer R-pathways with higher activation barriers was also computed (Fig. 5b).
Fig. 5.

Computed catalytic cycles for 1a. Relative Gibbs free energies in kcal mol–1 are shown (WB97XD/6-31g(d,p)/SMD(2,2,2-trifluoroethanol, 298.15 K. 1 atm)
The asymmetric center is created during the first hydrogenation step and is conserved throughout the catalytic cycle. The second hydrogenation proceeds through TS2(S), which is lower in free energy compared with TS(S). Therefore, the level of enantioselection is determined by the relative values of TS(S) and TS(R). In Fig. 6, the computed transition states, TS(S) and TS(R), for the hydrogen transfer are shown for substrate 1a. The value of TS(R) was computed to be 3.3 kcal mol–1 higher in energy than TS(S) (Fig. 6, 12.7 vs. 9.4 kcal mol−1). Significant differences in stability between the transition states TS(S) and TS(R) originate from a dissimilarity in the binding modes between the catalyst and the substrate. Thus, the binding of the C = N group coplanar to the Pd–H bond available only in TS(S) leads to the formation of a four-membered ring transition state. Additionally, the numerous weak attractive interactions between the substrate and catalyst are favorable effects in stabilizing the transition state TS(S) (see Fig. 6 and Supplementary Table 1)59–67. Furthermore, the high catalytic activities of this asymmetric hydrogenation may be partly due to the numerous weak attractive interactions between the substrate and catalyst.
Fig. 6.

The structures of TS(S) and TS(R). Computed structures of the transition states for the hydride transfer. (WB97XD/SDD(Pd)/6-31G(d,p)(all others)/SMD(TFE)) (Arrows denote the interactions of two groups)
During the optimization of the TS(R) structure starting from TS(S), the formation of a six-membered transition state takes place via the sulfonyl group of the substrate due to the fixed geometry of the imine, thus removing the migrating hydride from the plane of the catalyst chelate cycle; this can be seen from the values of the corresponding dihedral angles ((Pd–H–C–N) in Fig. 6b, 74.6° vs. 13.4°). As a result, TS(S) is a much “earlier” transition state than TS(R) (compare the Pd–H distances in Fig. 6a, 1.56 vs. 1.69 Å). In addition, the substituents of the substrate are further apart from the substituents of the catalyst in the six-membered TS(R), which decreases the stabilizing effect of the weak intermolecular interactions (see Fig. 6 and Supplementary Table 1).
Discussion
In conclusion, a palladium-catalyzed asymmetric hydrogenation of sterically hindered N-tosylimines has been developed. Chiral N-tosylamines were obtained with excellent enantioselectivities (up to 99.9% ee) as well as high yields under 1 atm hydrogen pressure. Palladium acetate, an inexpensive Pd salt with low toxicity, was found to be a suitable catalyst precursor for the homogeneous asymmetric hydrogenation. High catalytic activities were also observed (up to 5000S/C). The reaction could be conducted on a gram scale and was further applied to the synthesis of useful chiral products and N-ligand. Computations suggested that the enantioselectivity originates from the significant structural differences between the S- and R-pathways. Similarly, excellent enantioselection can be expected for other sulfonated imines possessing a C = N bond with fixed geometry.
Methods
Procedure for asymmetric hydrogenation of N-tosylimines
(R, R)-QuinoxP* (1.4 mg, 2.1 mol %) and Pd(OAc)2 (0.89 mg, 2.0 mol%) were placed in a dried Schlenk tube under nitrogen atmosphere, and degassed anhydrous acetone (1.0 mL) was added. The mixture was stirred at room temperature for 5 min, then the solvent was removed under vacuum to give the dry catalyst. In a glovebox, substrate 1 (0.2 mmol) was stirred in a solvent (0.5 mL) at room temperature for 10 min. Subsequently, the above-prepared catalyst dissolved in solvent (1.5 mL) was added. The hydrogenation was performed at room temperature under H2 in a stainless steel autoclave for 24 h. After releasing hydrogen, the conversion of the product 2 was determined by 1H NMR spectroscopic analysis of the crude reaction mixture. The enantiomeric excesses of the products were determined by HPLC with chiral columns (OD-H, OJ-H, AD-H, or IC-3).
Electronic supplementary material
Acknowledgements
We would like to thank the National Natural Science Foundation of China (Nos. 21620102003 and 21702134), Science and Technology Commission of Shanghai Municipality (Nos. 15JC1402200 and 17ZR1415200), and Shanghai Municipal Education Commission (No. 201701070002E00030) for financial support. We thank the Instrumental Analysis Center of SJTU for characterization. We are grateful to Zhi-Xiang Yu, Peking University, and Yuanyuan Liu, East China Normal University, for helpful discussions concerning our mechanistic studies.
Author contributions
J.C. conducted most of the synthetic experiments. B.L., F.L., and Y.W. conducted part of the synthetic experiments. I.D.G. conducted the DFT computational study. J.C., Z.Z., I.D.G., and W.Z. wrote the manuscript. Z.Z., M.Z., and T.I. took part in the discussion. W.Z. directed the project.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information file. For the experimental procedures, data of NMR and HPLC analysis and Cartesian coordinates of the optimized structures, see Supplementary Methods and Charts in Supplementary Information file. The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Center (2a: CCDC 1585399, 7x: CCDC 1585398). These data could be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41467-018-07462-w.
References
- 1.Cox PJ, Simpkins NS. Asymmetric synthesis using homochiral lithium amide bases. Tetrahedron.: Asymmetry. 1991;1:1–26. [Google Scholar]
- 2.Kim YH, Choi JY. Diastereoselective addition of organolithiums to new chiral hydrazones. Tetrahedron Lett. 1996;37:5543–5546. doi: 10.1016/0040-4039(96)01151-3. [DOI] [Google Scholar]
- 3.Kündig EP, Seidel TM, Jia YX, Bernardinelli G. Bulky chiral carbene ligands and their application in the palladium-catalyzed asymmetric intramolecular α-arylation of amides. Angew. Chem. Int. Ed. 2007;46:8484–8487. doi: 10.1002/anie.200703408. [DOI] [PubMed] [Google Scholar]
- 4.Constant S, Tortoioli S, Müller J, Lacour J. An enantioselective CpRu-catalyzed carroll rearrangement. Angew. Chem. Int. Ed. 2007;46:2082–2085. doi: 10.1002/anie.200604573. [DOI] [PubMed] [Google Scholar]
- 5.Biju P, et al. Bioorg. med. 3,4-diamino-2,5-thiadiazole-1-oxides as potent CXCR2/CXCR1 antagonists. Chem. Lett. 2008;18:228–231. doi: 10.1016/j.bmcl.2007.10.094. [DOI] [PubMed] [Google Scholar]
- 6.Jia YX, Katayev D, Bernardinelli G, Seidel TM, Kündig EP. New chiral N-heterocyclic carbene ligands in palladium-catalyzed α-arylations of amides: Conformational locking through allylic strain as a device for stereocontrol. Chem. Eur. J. 2010;16:6300–6309. doi: 10.1002/chem.201000031. [DOI] [PubMed] [Google Scholar]
- 7.Huang L, Zhang Y, Staples RJ, Huang RH, Wulff WD. Double stereodifferentiation in the catalytic asymmetric aziridination of imines prepared from a-chiral amines. Chem. Eur. J. 2012;18:5302–5313. doi: 10.1002/chem.201102520. [DOI] [PubMed] [Google Scholar]
- 8.Donets PA, Cramer N. Diaminophosphine oxide ligand enabled asymmetric nickel-catalyzed hydrocarbamoylations of alkenes. J. Am. Chem. Soc. 2013;135:11772–11775. doi: 10.1021/ja406730t. [DOI] [PubMed] [Google Scholar]
- 9.Greb L, Lehn JM. Light-driven molecular motors: imines as four-step or two-step unidirectional rotors. J. Am. Chem. Soc. 2014;136:13114–13114. doi: 10.1021/ja506034n. [DOI] [PubMed] [Google Scholar]
- 10.Sato Y, Kayaki Y, Ikariya T. Comparative study of bifunctional mononuclear and dinuclear amidoiridium complexes with chiral C-N chelating ligands for the asymmetric transfer hydrogenation of ketones. Chem. Asian J. 2016;11:2924–2931. doi: 10.1002/asia.201600955. [DOI] [PubMed] [Google Scholar]
- 11.Ramalingan C, Park YT. Mercury-catalyzed rearrangement of ketoximes into amides and lactams in acetonitrile. J. Org. Chem. 2007;72:4536–4538. doi: 10.1021/jo070297k. [DOI] [PubMed] [Google Scholar]
- 12.DiRocco DA, Oberg KM, Dalton DM, Rovis T. Catalytic asymmetric intermolecular stetter reaction of heterocyclic aldehydes with nitroalkenes: backbone fluorination improves selectivity. J. Am. Chem. Soc. 2009;131:10872–10874. doi: 10.1021/ja904375q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DiRocco DA, Noey EL, Houk KN, Rovis T. Catalytic asymmetric intermolecular stetter reactions of enolizable aldehydes with nitrostyrenes: Computational study provides insight into the success of the catalyst. Angew. Chem. Int. Ed. 2012;51:2391–2394. doi: 10.1002/anie.201107597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Candish L, Forsyth CM, Lupton DW. N-tert-butyl triazolylidenes: catalysts for the enantioselective (3+2) annulation of α,β-unsaturated acyl azoliums. Angew. Chem. Int. Ed. 2013;52:9149–9152. doi: 10.1002/anie.201304081. [DOI] [PubMed] [Google Scholar]
- 15.Liu ZJ, et al. Directing group in decarboxylative cross-coupling: copper-catalyzed site-selective C-N bond formation from nonactivated aliphatic carboxylic acids. J. Am. Chem. Soc. 2016;138:9714–9719. doi: 10.1021/jacs.6b05788. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi S, Ishitani H. Catalytic enantioselective addition to imines. Chem. Rev. 1999;99:1069–1094. doi: 10.1021/cr980414z. [DOI] [PubMed] [Google Scholar]
- 17.Tang W, Zhang X. New chiral phosphorus ligands for enantioselective hydrogenation. Chem. Rev. 2003;103:3029–3069. doi: 10.1021/cr020049i. [DOI] [PubMed] [Google Scholar]
- 18.Zhou YG. Asymmetric hydrogenation of heteroaromatic compounds. Acc. Chem. Res. 2007;40:1357–1366. doi: 10.1021/ar700094b. [DOI] [PubMed] [Google Scholar]
- 19.Xie JH, Zhu SF, Zhou QL. Transition metal-catalyzed enantioselective hydrogenation of enamines and imines. Chem. Rev. 2011;111:1713–1760. doi: 10.1021/cr100218m. [DOI] [PubMed] [Google Scholar]
- 20.Ager DJ, de Vries AHM, de Vries JG. Asymmetric homogeneous hydrogenations at scale. Chem. Soc. Rev. 2012;41:3340–3380. doi: 10.1039/c2cs15312b. [DOI] [PubMed] [Google Scholar]
- 21.Chen QA, Ye ZS, Duan Y, Zhou YG. Homogeneous palladium-catalyzed asymmetric hydrogenation. Chem. Soc. Rev. 2013;42:497–511. doi: 10.1039/C2CS35333D. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z, Butt N, Zhang W. Asymmetric hydrogenation of non-aromatic cyclic substrates. Chem. Rev. 2016;116:14769–14827. doi: 10.1021/acs.chemrev.6b00564. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Z, Butt N, Zhou M, Liu D, Zhang W. Asymmetric transfer and pressure hydrogenation with earth-abundant transition metal catalysts. Chin. J. Chem. 2018;36:443–454. doi: 10.1002/cjoc.201800053. [DOI] [Google Scholar]
- 24.Charette AB, Giroux A. Asymmetric hydrogenation of N-tosylimines catalyzed by BINAP-Ruthenium(II) complexes. Tetrahedron Lett. 1996;37:6669–6672. doi: 10.1016/S0040-4039(96)01471-2. [DOI] [Google Scholar]
- 25.Xiao X, et al. Selective diethylzinc reduction of imines in the presence of ketones catalyzed by Ni(acac)2. Org. Lett. 2006;2006:139–142. doi: 10.1021/ol052628q. [DOI] [PubMed] [Google Scholar]
- 26.Kwak SH, Lee SA, Lee KI. Highly enantioselective Rh-catalyzed transfer hydrogenation of N-sulfonyl ketimines. Tetrahedron Asymmetry. 2010;21:800–804. doi: 10.1016/j.tetasy.2010.04.047. [DOI] [Google Scholar]
- 27.Wang L, et al. Efficient asymmetric transfer hydrogenation of N-sulfonylimines on water. Tetrahedron. 2013;69:6500–6506. doi: 10.1016/j.tet.2013.05.064. [DOI] [Google Scholar]
- 28.Yang Q, Shang G, Gao W, Deng J, Zhang X. A highly enantioselective, Pd-TangPhos-catalyzed hydrogenation of N-tosylimines. Angew. Chem. Int. Ed. 2006;45:3832–3835. doi: 10.1002/anie.200600263. [DOI] [PubMed] [Google Scholar]
- 29.Wang YQ, Lu SM, Zhou YG. Highly enantioselective Pd-catalyzed asymmetric hydrogenation of activated imines. J. Org. Chem. 2007;72:3729–3734. doi: 10.1021/jo0700878. [DOI] [PubMed] [Google Scholar]
- 30.Brunner H, Becker R, Gauder S. Asymmetric catalysis. 291. Optically active primary amines by enantioselective catalytic hydrosilylation of ketoximes. Organometallics. 1986;5:739–746. doi: 10.1021/om00135a020. [DOI] [Google Scholar]
- 31.Boezio AA, Solberghe G, Lauzon CA, Charette B. Orthoacylimines: A new class of chiral auxiliaries for nucleophilic addition of organolithium reagents to imines. J. Org. Chem. 2003;68:3241–3245. doi: 10.1021/jo026571m. [DOI] [PubMed] [Google Scholar]
- 32.Sugimoto H, et al. Enantioselective nucleophilic addition to N-(2-pyridylsulfonyl)imines by use of dynamically induced chirality. Tetrahedron Lett. 2005;46:8941–8944. doi: 10.1016/j.tetlet.2005.10.085. [DOI] [Google Scholar]
- 33.Dai X, Nakai T, Romero JAC, Fu GC. Enantioselective synthesis of protected amines by the catalytic asymmetric addition of hydrazoic acid to ketenes. Angew. Chem. Int. Ed. 2007;46:4367–4369. doi: 10.1002/anie.200700697. [DOI] [PubMed] [Google Scholar]
- 34.Hou G, et al. Enantioselective hydrogenation of N-H Imines. J. Am. Chem. Soc. 2009;131:9882–9883. doi: 10.1021/ja903319r. [DOI] [PubMed] [Google Scholar]
- 35.Martjuga M, Belakov S, Liepinsh E, Suna E. Asymmetric synthesis of 1,3-diamines. II: Diastereoselective reduction of atropisomeric n-tert-butanesulfinylketimines. J. Org. Chem. 2011;76:2635–2647. doi: 10.1021/jo1025767. [DOI] [PubMed] [Google Scholar]
- 36.Schramm Y, Barrios-Landeros F, Pfaltz A. Discovery of an iridacycle catalyst with improved reactivity and enantioselectivity in the hydrogenation of dialkyl ketimines. Chem. Sci. 2013;4:2760–2766. doi: 10.1039/c3sc50587a. [DOI] [Google Scholar]
- 37.Fernández-Salas JA, Rodríguez-Fernández MM, Maestro MC, García-Ruano JL. Synthesis of enantiomerically pure (α-phenylalkyl)amines with substituents at the ortho position through diastereoselective radical alkylation reaction of sulfinimines. Eur. J. Org. Chem. 2014;24:5265–5272. doi: 10.1002/ejoc.201402355. [DOI] [Google Scholar]
- 38.Beisel T, Manolikakes G. Palladium-catalyzed enantioselective three-component synthesis of α-substituted amines. Org. Lett. 2015;17:3162–3166. doi: 10.1021/acs.orglett.5b01502. [DOI] [PubMed] [Google Scholar]
- 39.Yu CB, et al. Asymmetric hydrogenation via capture of active intermediates generated from Aza-pinacol rearrangement. J. Am. Chem. Soc. 2014;136:15837–15840. doi: 10.1021/ja5075745. [DOI] [PubMed] [Google Scholar]
- 40.Duan Y, et al. Homogenous Pd-catalyzed asymmetric hydrogenation of unprotected indoles: Scope and mechanistic studies. J. Am. Chem. Soc. 2014;136:7688–7700. doi: 10.1021/ja502020b. [DOI] [PubMed] [Google Scholar]
- 41.Chen ZP, Chen MW, Shi L, Yu CB, Zhou YG. Pd-catalyzed asymmetric hydrogenation of fluorinated aromatic pyrazol-5-ols via capture of active tautomers. Chem. Sci. 2015;6:3415–3419. doi: 10.1039/C5SC00835B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen ZP, Hu SB, Zhou J, Zhou YG. Synthesis of chiral trifluoromethyl-substituted hydrazines via Pd-catalyzed asymmetric hydrogenation and reductive amination. ACS Catal. 2015;5:6086–6089. doi: 10.1021/acscatal.5b01641. [DOI] [Google Scholar]
- 43.Yan Z, Wu B, Gao X, Chen MW, Zhou YG. Enantioselective synthesis of α-amino phosphonates via Pd-catalyzed asymmetric hydrogenation. Org. Lett. 2016;18:692–695. doi: 10.1021/acs.orglett.5b03664. [DOI] [PubMed] [Google Scholar]
- 44.Abe H, Amii H, Uneyama K. Pd-catalyzed asymmetric hydrogenation of α-fluorinated iminoesters in fluorinated alcohol: A new and catalytic enantioselective synthesis of fluoro α-amino acid derivatives. Org. Lett. 2001;3:313–315. doi: 10.1021/ol0002471. [DOI] [PubMed] [Google Scholar]
- 45.Tian F, Yao D, Liu Y, Xie F, Zhang W. Iridium-catalyzed highly enantioselective hydrogenation of exocyclic α,β-unsaturated carbonyl compounds. Adv. Synth. Catal. 2010;352:1841–1845. doi: 10.1002/adsc.201000185. [DOI] [Google Scholar]
- 46.Liu Y, Zhang W. Ir-Catalyzed asymmetric hydrogenation of α-alkylidene succinimides. Angew. Chem. Int. Ed. 2013;52:2203–2206. doi: 10.1002/anie.201209126. [DOI] [PubMed] [Google Scholar]
- 47.Liu Y, Gridnev ID, Zhang W. Mechanism of asymmetric hydrogenation of functionalized olefins with Ir/iPr-BiphPHOX catalyst: NMR and DFT study. Angew. Chem. Int. Ed. 2014;53:1901–1905. doi: 10.1002/anie.201309677. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Wang Y, Liu D, Zhang W. Asymmetric hydrogenation of β-secondary amino ketones catalyzed by a ruthenocenyl phosphino-oxazoline-ruthenium complex (RuPHOX-Ru): the synthesis of γ-secondary amino alcohols. Adv. Synth. Catal. 2015;357:3262–3272. doi: 10.1002/adsc.201500653. [DOI] [Google Scholar]
- 49.Li J, et al. Asymmetric hydrogenation of α-substituted acrylic acids catalyzed by a ruthenocenyl phosphino-oxazoline-ruthenium complex. Org. Lett. 2016;18:2122–2125. doi: 10.1021/acs.orglett.6b00748. [DOI] [PubMed] [Google Scholar]
- 50.Hu Q, et al. Rh-catalyzed chemo- and enantioselective hydrogenation of allylic hydrazones. Chem. Eur. J. 2017;23:1040–1043. doi: 10.1002/chem.201605579. [DOI] [PubMed] [Google Scholar]
- 51.Chen J, et al. Palladium-catalyzed asymmetric hydrogenation of α-Acyloxy-1-aryletheanones. Angew. Chem. Int. Ed. 2013;52:11632–11616. doi: 10.1002/anie.201306231. [DOI] [PubMed] [Google Scholar]
- 52.Chen J, Zhang Z, Liu D, Zhang W. Palladium-catalyzed chemo- and enantioselective C-O bond cleavage of α-acyloxy ketones via hydrogenolysis. Angew. Chem. Int. Ed. 2016;55:8444–8447. doi: 10.1002/anie.201603590. [DOI] [PubMed] [Google Scholar]
- 53.Imamoto T, Sugita K, Yoshida K. An air-stable P-chiral phosphine ligand for highly enantioselective transition-metal-catalyzed reactions. J. Am. Chem. Soc. 2005;127:11934–11935. doi: 10.1021/ja053458f. [DOI] [PubMed] [Google Scholar]
- 54.Imamoto T, Nishimura M, Koide A, Yoshida K. t-Bu-QuinoxP* Ligand: Applications in asymmetric Pd-catalyzed allylic substitution and ru-catalyzed hydrogenation. J. Org. Chem. 2007;72:7413–7416. doi: 10.1021/jo071192k. [DOI] [PubMed] [Google Scholar]
- 55.Imamoto T, et al. Rigid P-chiral phosphine ligands with tert-butylmethylphosphino groups for rhodium-catalyzed asymmetric hydrogenation of functionalized alkenes. J. Am. Chem. Soc. 2012;134:1754–1769. doi: 10.1021/ja209700j. [DOI] [PubMed] [Google Scholar]
- 56.Yu K, et al. Synthesis of D-(R)-tyrosine by catalytic asymmetric hydrogenation and its practical application. Chin. J. Org. Chem. 2013;33:1932–1938. [Google Scholar]
- 57.Hu Q, Zhang Z, Liu Y, Imamoto T, Zhang W. ZnCl2-promoted asymmetric hydrogenation of β-secondary-amino ketones catalyzed by a P-chiral bisphosphine-Rh complex. Angew. Chem. Int. Ed. 2015;54:2260–2264. doi: 10.1002/anie.201411384. [DOI] [PubMed] [Google Scholar]
- 58.Yang G, Zhang W. A palladium-catalyzed enantioselective addition of arylboronic acids to cyclic ketimines. Angew. Chem. Int. Ed. 2013;52:7540–7544. doi: 10.1002/anie.201302861. [DOI] [PubMed] [Google Scholar]
- 59.Wagner JP, Schreiner PR. London dispersion in molecular chemistry—reconsidering steric effects. Angew. Chem. Int. Ed. 2015;54:12274–12276. doi: 10.1002/anie.201503476. [DOI] [PubMed] [Google Scholar]
- 60.Cheong PHY, Lagault CY, Um JM, ÇeIebi-Ölçum N, Houk KN. Quantum mechanical investigations of organocatalysis: mechanisms, reactivities, and selectivities. Chem. Rev. 2011;111:5042–5137. doi: 10.1021/cr100212h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gridnev ID, Dub PA. Enantioselection in Asymmetric Catalysis. Boca Raton, London, New York: CRC Press; 2017. [Google Scholar]
- 62.Wang T, et al. Highly enantioselective hydrogenation of quinolines using phosphine-free chiral cationic ruthenium catalysts: Scope, mechanism, and origin of enantioselectivity. J. Am. Chem. Soc. 2011;133:9878–9891. doi: 10.1021/ja2023042. [DOI] [PubMed] [Google Scholar]
- 63.Ding ZY, Chen F, Qin J, He YM, Fan QH. Asymmetric hydrogenation of 2,4-disubstituted 1,5-benzodiazepines using cationic ruthenium diamine catalysts: An unusual achiral counteranion induced reversal of enantioselectivity. Angew. Chem. Int. Ed. 2012;51:5706–5710. doi: 10.1002/anie.201200309. [DOI] [PubMed] [Google Scholar]
- 64.Gridnev ID, Imamoto T. Challenging the major/minor concept in rh-catalyzed asymmetric hydrogenation. ACS Catal. 2015;5:2911–2915. doi: 10.1021/acscatal.5b00424. [DOI] [Google Scholar]
- 65.Gridnev ID, Imamoto T. Enantioselection mechanism in Rh-catalyzed asymmetric hydrogenation. Russ. Chem. Bull. 2016;65:1514–1534. doi: 10.1007/s11172-016-1478-9. [DOI] [Google Scholar]
- 66.Gridnev ID. Attraction versus repulsion in rhodium-catalyzed asymmetric hydrogenation. ChemCatChem. 2016;8:3463–3465. doi: 10.1002/cctc.201600759. [DOI] [Google Scholar]
- 67.Lu G, et al. Ligand-substrate dispersion facilitates the copper-catalyzed hydroamination of unactivated olefins. J. Am. Chem. Soc. 2017;139:16548–16555. doi: 10.1021/jacs.7b07373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information file. For the experimental procedures, data of NMR and HPLC analysis and Cartesian coordinates of the optimized structures, see Supplementary Methods and Charts in Supplementary Information file. The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Center (2a: CCDC 1585399, 7x: CCDC 1585398). These data could be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.