

Abstract
Thrombin-activatable fibrinolysis inhibitor (TAFI) plays a central role in haemostasis, and plasma TAFI concentrations are heritable. Candidate gene studies have identified several variants within the gene encoding TAFI, CPB2 , that explain part of the estimated heritability. Here, we describe an exploratory genome-wide association study to identify novel variants within and outside of the CPB2 locus that influence plasma concentrations of intact TAFI and/or the extent of TAFI activation (measured by released TAFI activation peptide, TAFI-AP) amongst 3,260 subjects from Southern Sweden. We also explored the role of rare variants on the HumanExome BeadChip. We confirmed the association with previously reported common variants in CPB2 for both intact TAFI and TAFI-AP, and discovered novel associations with variants in putative CPB2 enhancers. We identified a gene-based association with intact TAFI at CPB2 ( P SKAT-O = 2.8 × 10 −8 ), driven by two novel rare nonsynonymous single nucleotide polymorphisms (SNPs; I420N and D177G). Carriers of the rare variant of D177G (rs140446990; MAF 0.2%) had lower intact TAFI and TAFI-AP concentrations compared with non-carriers (intact TAFI, geometric mean 53 vs. 78%, P T-test = 5 × 10 −7 ; TAFI-AP 63 vs. 99%, P T-test = 7.2 × 10 −4 ). For TAFI-AP, we identified a genome-wide significant association at an intergenic region of chromosome 3p14.1 and five gene-based associations (all P SKAT-O < 5 × 10 −6 ). Using well-characterized assays together with a genome-wide association study and a rare-variant approach, we verified CPB2 to be the primary determinant of TAFI concentrations and identified putative secondary loci (candidate variants and genes) associated with intact TAFI and TAFI-AP that require independent validation.
Keywords: thrombin-activatable fibrinolysis inhibitor, fibrinolysis inhibitors, plasma levels, genome-wide association study
Introduction
Thrombin-activatable fibrinolysis inhibitor (TAFI), also known as procarboxypeptidase U and procarboxypeptidase B, plays a central role in haemostasis where it acts as a potent inhibitor of fibrinolysis. 1 More recently, a role for TAFI in regulation of inflammation, cell migration and wound healing has also been described. 2 The TAFI zymogen is secreted by the liver into the circulation. A single cleavage at Arg92 by thrombin, plasmin, or the thrombin–thrombomodulin complex removes the activation peptide (TAFI-AP; Phe1-Arg92; 20 kDa) and makes the active site accessible to substrates (active TAFI or TAFIa; Ala93-Val401; 36 kDa 3 4 ). TAFIa can then inhibit fibrinolysis by removing carboxy-terminal lysine residues from partially degraded fibrin, which results in a decreased plasminogen binding and decreased fibrinolytic activity. 5 TAFIa is inactivated through a conformational change to an inactive form (TAFIai) which is subsequently cleaved resulting in two smaller fragments of 25 and 11 kDa. 6 Plasma TAFI levels have been reported to show association with deep-vein thrombosis, 7 8 coronary artery disease, 9 10 and ischaemic stroke. 11 12 13
Most quantitative studies of TAFI have focused on ‘total’ antigen levels (TAFI-Ag) and used assays which recognize the intact, nonactivated TAFI (i.e. zymogen) as well as the various cleavage products. In healthy individuals, plasma levels of TAFI have been shown to be stable over time but exhibit a large interindividual variability poorly explained by lifestyle and environmental factors. 14 Family studies have demonstrated that plasma TAFI levels are heritable (heritability estimates ranging from 40 to 70% for TAFI-Ag and 16% for TAFIa activity 15 16 17 18 19 20 ), suggesting a strong genetic component. Several common single nucleotide polymorphisms (SNPs) located within the gene encoding TAFI ( CPB2 ) have been described (including two that result in amino acid substitutions: Thr147Ala [rs3742264] and Thr325Ile [rs1926447]), and some are reported to be associated with plasma TAFI-Ag, TAFI-AP and/or TAFIa concentrations and risk of arterial and venous thrombotic disease. 7 8 9 10 11 12 13 21 22 23 It should be stressed, however, that the immunological assays used in most of these studies were not validated with respect to putative differential reactivities toward different isoforms of TAFI and cleavage product fragments of TAFI. In particular, Thr325Ile can have a large impact on immunoreactivity. 24 25 Thus, reported clinical effects associated with TAFI SNPs and protein levels as well as heritability estimates should be treated with caution (see Supplementary Table S1 for overview). Genome-wide linkage studies confirmed the CPB2 locus to be a strong determinant of TAFI-Ag 18 20 and TAFI activity 18 levels and found weak evidence for additional linkage on other autosomal loci. 18 20 However, no high-resolution genome-wide association study (GWAS) has been reported; therefore, the exact genetic basis for the heritability remains unknown.
We hypothesized that there are cis - and trans -acting loci in addition to the previously identified CPB2 variants that contribute to the variation in circulating TAFI levels. Furthermore, we hypothesize that rare or low-frequency variants, which are not well covered by GWAS and not easily imputed, in addition to common variants are associated with TAFI levels. Here, we used two well-characterized immunological assays, validated to be isoform independent, which react to the intact nonactivated TAFI, or to the activation peptide (TAFI-AP, a marker of the extent of TAFI activation 26 ). In this explorative study, we tested our hypothesis using a genotyping platform that is enriched with exome content (HumanExome BeadChip) and we searched for both common and rare genetic variants associated with intact TAFI and TAFI-AP levels in data from 3,260 Swedish adults.
Materials and Methods
Study Design
We performed a GWAS and gene-based analyses of plasma TAFI (intact and AP) in a subset of participants from the southern Swedish study on ischaemic stroke. 27 The present study included European ancestry participants with genetic data and in whom plasma levels of TAFI had been measured or citrate plasma was available for analyses. These participants included 600 cases with ischaemic stroke at ages 18 to 69 years and 600 controls matched for age and sex from the Sahlgrenska Academy Study on Ischemic Stroke (SAHLSIS 28 ) and 2,060 population-based participants from the prospective Malmö Diet and Cancer Study (MDC 29 ) of whom 1,812 have not had a cardiovascular event and 248 had an ischaemic stroke during follow-up. The designs of these studies are described in the Supplementary Material . Sample sizes, mean age and sex distribution of study participants in each cohort at the time of the intact TAFI and TAFI-AP determination are summarized in Table 1 .
Table 1. Subject characteristics at the time of blood sampling and genotyping chip used.
| MDC ( n = 2,060) | SAHLSIS ( n = 756) | SAHLSIS a ( n = 444) | Total (3,260) | |
|---|---|---|---|---|
| Cases, n (%) | 0 (0) | 166 (22) | 444 (100) | 600 (18.4) |
| Age, median (IQR) | 58 (53–63) | 59 (52–64) | 58 (50–63) | 58 (52–64) |
| Male gender, n (%) | 818 (39.7) | 477 (63.1) | 293 (66) | 1,588 (48.7) |
| Hypertension, n (%) | 1,304 (63) | 333 (44.2) | 245 (56.1) | 1,882 (57.9) |
| Diabetes mellitus, n (%) | 192 (9.3) | 66 (8.8) | 81 (18.2) | 339 (10.4) |
| Current smoking, n (%) | 405 (22.4) | 174 (23) | 168 (38) | 792 (24.7) |
| Hyperlipidaemia, n (%) | 1,847 (90.2) | 512 (69.3) | 304 (75.1) | 2,663 (83.4) |
| Intact TAFI, n (%) | 2,052 (99.6) | 748 (98.9) | 404 (91.0) | 3,204 (98.3) |
| Median (IQR) | 72 (61–85) | 90 (77–101) | 95 (83–114) | 79 (65–95) |
| TAFI-AP, n (%) | 2,060 (100) | 748 (98.9) | 404 (91.0) | 3,212 (98.5) |
| Median (IQR) | 96 (67–132) | 105 (93–122) | 124 (105–148) | 103 (80–131) |
| Genotyping platform | Human OmniExpress Exome v1.0 | Human OmniExpress Exome v1.0 | Human Omni 5M Exome v1.0 | Imputed to the UK10K + 1000 Genomes Ph3 |
Abbreviations: MDC, Malmö Diet and Cancer Study; SAHLSIS, Sahlgrenska Academy Study on Ischemic Stroke; TAFI-AP, thrombin-activatable fibrinolysis inhibitor activation peptide.
Note: Intact TAFI expressed as relative levels; TAFI-AP, TAFI activation peptide, expressed as relative levels.
Included in the NINDS Stroke Genetic Network (SiGN) study.
Both participating cohorts were granted approval by the appropriate research ethics committees for the research (MDC: LU51/90, 166/2007 and 633/2009; and SAHLSIS: University of Gothenburg Ö-469–99, T-553–03, 873–11 and 823–13). All participants or their next of kin provided written informed consent.
TAFI Antigen and Activity Measurement
Venous blood samples were collected after an overnight fast in tubes containing 10% volume of 0.13 mol/L sodium citrate. For the MDC study, samples were drawn 2 to 4 months after enrolment. For SAHLSIS, blood sampling was performed once for controls and during the convalescent phase at 3-month follow-up for stroke patients. 28 Aliquots of plasma were stored at −80°C. Plasma levels of TAFI were measured by an ELISA which recognizes intact TAFI and equally recognizes the common TAFI isoforms and an ELISA specifically measuring the activation peptide (amino acids residues 1–92, TAFI-AP 26 ). All analyses were performed in the same laboratory. Values are expressed relative to pooled human plasma (%). Intra- and inter-assay coefficients of variation were 6.2 and 8.3%, respectively, for intact TAFI, and 3.1 and 7.3%, respectively, for TAFI-AP. Intact TAFI and TAFI-AP data were naturally log transformed for analysis.
Genotyping, Imputation and Quality Control
DNA was extracted from whole blood and genotyping was performed with either the HumanOmniExpress Exome BeadChip v1.0 at the Broad Institute (SAHLSIS: n = 756; MDC: n = 2,060) or the HumanOmni 5M Exome v1.0 as part of the Stroke Genetics Network (SiGN) study (SAHLSIS: n = 444; Illumina, San Diego, California, United States) as described. 27 30 These arrays have an overlap the Omni content for coverage of common genome-wide variation and both contain an overlapping exome content set of 240k probes (HumanExome BeadChip). Nonoverlapping sites were ignored. Genotypes for the GWAS were auto-called with the Illumina GenomeStudio v2011.1 software and the GenTrain2.0 clustering algorithm. Genotypes for the gene-based tests on rare variants were called using a combination of auto-call and Z-call. 31 The quality control criteria performed by each study for filtering of poorly genotyped individuals and low-quality SNPs included a call rate of <0.95, gender mismatch, identity by descent sharing (>0.375, second-degree relatives), population outliers, excess autosomal heterozygosity and deviation from Hardy-Weinberg equilibrium ( P < 10 −3 ). After separate quality check, the overlapping 731k variants from each dataset were merged and rechecked for quality using the aforementioned criteria. Imputation was performed on autosomal chromosomes, after excluding monomorphic and tri-allelic SNPs, in the merged dataset using the UK10K + 1,000 Genomes Phase 3 reference panel at the Sanger Imputation Service. 32 Prior to imputation, each chromosome was phased against the hg19/GRCh37 reference panel using SHAPEIT2 (v2.r790 33 ). Imputed variants with information score greater than 0.3 were included resulting in approximately 10 million autosomal variants with MAF greater than 1%. The total numbers of subjects included in the analysis were 3,116 for intact TAFI and 3,124 for TAFI-AP.
Genome-wide Association Analyses
Genotype–phenotype association analyses were performed according to a prespecified analysis plan on autosomal variants with a minor allele frequency (MAF) >1%. The association of SNP with natural logarithm-transformed intact TAFI or TAFI-AP values was assessed by a linear model, assuming an additive effect of each risk allele, using PLINK version 1.9. All analyses were adjusted for age, sex, and case–control status. The β value represents the per-allele effect on the natural log-transformed intact TAFI or TAFI-AP concentration. The prespecified threshold of genome-wide significance was set at the conventional level of P = 5 × 10 −8 . Suggestive associations were defined as P < 10 −6 . Based on our sample size and assuming an additive model, the minimal effect size (β-coefficient, β, expressed as standard deviation increase in log-transformed plasma concentration) per allele that is detectable with 80% power at P = 5 × 10 −8 is 0.175 for MAF 30%, 0.26 for MAF 10%, 0.37 for MAF 5% and 0.79 for MAF 1%.
We performed conditional analyses to control for the effects of the lead SNP at the CPB2 locus for intact TAFI as well as for TAFI-AP. The allele dosage (0–2 copies based on imputed genotypes) of the lead variant was used as a covariate in these analyses. For intact TAFI, a significant signal remained; therefore, an additional model was also computed that corrected for the lead SNP from the initial analysis as well as the lead SNP from the conditional analysis (i.e. corrected for two independent SNPs).
Gene-Based Analyses
Gene-based tests are designed to gain power, especially for analyses of variants with low MAF, by aggregating the association signals from variants within the same gene. Thus, we used gene-based tests to investigate the role of rare (MAF < 0.5%) and low-frequency (MAF 0.5% to 5%) variants on intact TAFI and TAFI-AP concentrations. All directly genotyped functional variants (missense, nonsense and splice variants) that overlapped between the datasets with MAF <5% and which passed quality check were included in the gene-based tests (48,898 variants in total). All genes were required to contain at least two variants to be included in the analysis and to have a cumulative minor allele count of ≥ 4 (resulting in 9,325 genes in total). Gene-based tests for association with natural log-transformed intact TAFI and TAFI-AP concentrations were performed using the optimal combination of the sequence kernel association test and the burden test (SKAT-O 34 ), in R package ‘SKAT’ version 1.0.7 ( https://cran.r-project.org/web/packages/SKAT ), using the internal settings kernel = linear, weighted and method = optimal adj, adjusting for age, sex and case–control status. A Bonferroni-corrected, gene-based P -value threshold of 5.36 × 10 −6 was used for gene-based tests (0.05/9,325 genes). Suggestive associations for the gene-based analyses were defined as P < 10 −4 . For significant findings, genotype intensity cluster plots were visually inspected and rare variants were annotated with the most current MAF data from the Exome Aggregation Consortium (ExAc).
Annotation and Functional Prediction of Variants
As gene enhancer elements play a central role in regulating transcription, we speculated that many of the variants remaining in the conditional analysis of intact TAFI may be in high linkage disequilibrium (LD) with or located within putative enhancer elements. The GeneHancer database, embedded in GeneCards ( www.genecards.org ), was used to identify 14 putative enhancer elements (see Supplementary Table S2 for regions). We then used SNAP (SNP Annotation and Proxy Search; Broad Institute), to find proxy SNPs for the 69 variants with MAF greater than 1% located within these regions, based on LD information from the 1000 Genomes Project CEU population and an R 2 threshold of >0.8. The Supplementary Tables are annotated with this information.
Furthermore, we used publicly available expression quantitative trait locus (eQTL) browsers to see whether any of the identified variants were eQTLs for genes of interest. For each lead variant within a significant or suggestive locus (listed in Table 2 ), we investigated associations between genotype and expression levels of all genes located within ± 200 kb of the variant, in all available tissues. The browsers and available tissue types used were as follows: the eQTL browser of the Genotype-Tissue Expression (GTEx, accessed December 2016) project includes more than 70 tissue types including liver and whole blood, 35 and the eQTL resources from the Gilad/ Pritchard group includes liver tissue as well as several other tissue types 36 and the blood eQTL browser based on peripheral blood. 37
Table 2. Genome-wide and suggestive loci ( P < 10 − 6 ) for intact TAFI and the TAFI activation peptide (TAFI-AP, a marker of the extent of TAFI activation) .
| Region | Most significant | Effect allele | Alternate allele | MAF | P -Value | Beta | SE | Closest genes |
|---|---|---|---|---|---|---|---|---|
| Intact TAFI | ||||||||
| 13q14.13 | rs56734909 | A | C | 0.343 | 5.3E-30 | 0.09 | 0.008 | CPB2 |
| 5q22.1 | rs11743286 | T | A | 0.022 | 3.0E-07 | −0.15 | 0.030 | TMEM232/SLC25A46 |
| 12q24.21 | rs78402106 | C | T | 0.017 | 4.9E-07 | −0.15 | 0.030 | TBX5/RBM19 |
| 2p25.3 | rs570398477 | C | T | 0.011 | 5.1E-07 | −0.18 | 0.037 | SNTG2/TPO |
| 12q24.31 | rs117742602 | G | A | 0.011 | 7.0E-07 | −0.19 | 0.038 | DENR/HCAR1/HCAR3 |
| 5p15.31 | rs189859825 | C | T | 0.033 | 9.8E-07 | 0.11 | 0.022 | SEMA5A/MIR4636 |
| TAFI-AP | ||||||||
| 13q14.13 | rs940 | G | C | 0.235 | 1.0E-27 | 0.16 | 0.014 | CPB2 |
| 3p14.1 | rs140102162 | G | A | 0.011 | 1.3E-08 | −0.39 | 0.069 | MIR4272/KBTBD8/SUCLG2 |
| 11p11.2 | rs745770348 | A | ACC | 0.016 | 2.8E-07 | −0.27 | 0.053 | CD82/TSPAN18/ALX4 |
| 16q12.2 | rs117122953 | T | C | 0.012 | 3.8E-07 | −0.33 | 0.064 | IRX5/CRNDE/IRX6 |
| 9q33.3 | rs79740971 | A | G | 0.017 | 4.0E-07 | −0.28 | 0.055 | DENND1A/LHX2 |
| 3p26.1 | rs80062757 | T | G | 0.013 | 4.4E-07 | −0.31 | 0.062 | BHLHE40/ITPR1/EGOT |
| 16p12.2 | rs78970023 | A | T | 0.015 | 4.7E-07 | −0.27 | 0.053 | OTOA/METTL9/IGSF6 |
| 15q25.3 | rs111511310 | T | C | 0.025 | 5.5E-07 | −0.22 | 0.045 | LOC101929701/AGBL1/KLHL25 |
| 2q36.2 | rs4674940 | G | A | 0.032 | 6.0E-07 | −0.17 | 0.035 | DOCK10/MIR4439/CUL3 |
| 3p22.3 | rs150338049 | A | AGCAAG | 0.092 | 8.8E-07 | 0.11 | 0.023 | ARPP21/MIR128–2/LOC101928135 |
| 11p14.2 | rs113021326 | T | C | 0.016 | 9.0E-07 | −0.27 | 0.054 | FIBIN/SLC5A12/ANO3 |
Abbreviations: TAFI-AP, thrombin-activatable fibrinolysis inhibitor activation peptide.
Note: Intact TAFI, expressed as relative levels; TAFI-AP, expressed as relative levels.
Statistical Analysis
We used a linear regression model to determine the variance in intact TAFI and TAFI-AP in healthy controls, explained by cardiovascular risk factors (smoking, hypertension, hyperlipidaemia and diabetes mellitus), anthropometric measures (body mass index and waist hip ratio) and high sensitivity C-reactive protein (hsCRP). Likewise, to determine the variance in intact TAFI and TAFI-AP explained by the three covariates included in the GWAS model (age, sex and case–control status), we used linear regression. We then performed a model conditioned on these covariates to determine the remaining variance explained by a particular variant or variant combination.
Results
We conducted a GWAS and additionally evaluated rare and low-frequency functional variants using gene-based tests in 3,260 individuals of European ancestry to identify genetic loci influencing concentrations of circulating intact TAFI and TAFI-AP (a measure of the extent of TAFI activation). Characteristics of participants, genotyping arrays used, and median and interquartile range for each TAFI measure per cohort are summarized in Table 1 . In our study population, cardiovascular risk factors and anthropometric measures explained very little of the variance in intact TAFI or TAFI-AP concentrations, in line with previous reports (1.7 and 3.6% in controls, respectively). The two TAFI traits were moderately correlated (Spearman's Rho = 0.33, P < 0.001) in our study population, also in line with previous studies. 26
Genome-wide Association Study
Intact Thrombin-Activatable Fibrinolysis Inhibitor
In total, 10M imputed autosomal variants with MAF greater than 1% were included in the GWAS. The genomic control coefficient was close to 1 (λ = 1.02), which suggests negligible test-statistic inflation. For intact TAFI, 325 SNPs in a region of chromosome 13 passed the genome-wide significance threshold ( Fig. 1 and Supplementary Table S3 ). All SNPs were within or close to the CPB2 gene, which encodes TAFI ( Fig. 1C ) and the overlapping antisense long noncoding RNA (lncRNA) CPB2-AS1 . As gene enhancer elements play a central role in regulating transcription, we annotated variants listed in Supplementary Table S3 to indicate SNPs located within the 14 putative enhancer elements of the CPB2 gene and SNPs in high LD with SNPs in putative enhancers. Rs56734909 had the smallest P -value (5.3 × 10 −30 ) and is located within a predicted CPB2 enhancer (GeneHancer ID GH130046105). It is also in high LD ( R 2 = 0.96; D′ = 1) with a CPB2 promoter SNP, previously shown to associate with TAFI-Ag concentrations (-2599C > G, rs34813434). 22 38 39 The A allele (frequency, 66%) of rs56734909 was associated with higher intact TAFI levels. Over a third of the remaining SNPs were within ( n = 33) or in high LD ( n = 87) with putative CPB2 enhancers ( Supplementary Table S3 ).
Fig. 1.
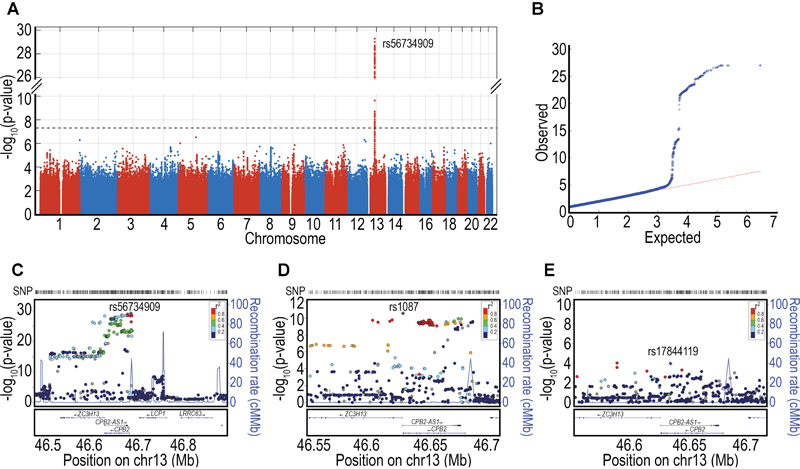
Genome-wide association analyses of intact TAFI. ( A ) Manhattan plot of associations. Dotted line shows genome-wide significance (5 × 10 −8 ). ( B ) QQ-Plot. ( C ) Regional association plot of chromosome 13, CPB2 locus. ( D ) Regional association plot of CPB2 locus in a conditional analysis adjusted for the lead SNP, rs56734909. ( E ) Regional association plot in a further conditional analysis adjusted for rs56734909 and rs1087.
An additional six SNPs outside of the CPB2 locus were suggestively associated ( P < 10 −6 ) with intact TAFI concentrations, and marked five regions on three chromosomes: 5q22.1, 12q24.21, 2p25.3, 12q24.31 and 5p15.31. Each of these regions had solitary suggestive SNP associations, with the exception of 2p25.3 which had two suggestive SNP associations. It is of note that chromosome region 5p15 was previously identified as a suggestive locus in a linkage analysis of TAFI-Ag; however, no obvious candidate genes are present at that locus. 18 Table 2 lists the results for the lead SNP at these loci and regional plots can be found in Supplementary Fig. S1 .
To determine whether multiple independent signals were present in the CPB2 locus (chr13: pos 46.5–46.7Mb), a secondary analysis was conducted conditioned on the lead SNP. After adjustment for rs56734909, 93 variants in the 13q14.11 region retained genome-wide significance ( Fig. 1D ; Supplementary Table S4 ). This suggests there are at least two independent SNPs in the CPB2 locus that regulate intact TAFI concentrations. We hypothesized that the remaining signal may be due to SNPs in high LD with or located within the 14 putative enhancer elements of the CPB2 gene and annotated in Supplementary Table S4 with this information. Of the 93 associated SNPs, the majority were indeed either in predicted CPB2 enhancers ( n = 13) or in high LD ( R 2 < 0.8; n = 44) with predicted CPB2 enhancer variants ( Supplementary Table S4 ). Rs1087 had the smallest P conditional value (2.63 × 10 −11 ), but this variant is not located in any putative enhancers nor is it in LD with any SNPs located within enhancers. Rather, rs1087 is located in the CPB2 3′ UTR and has previously been shown to associate with TAFI-Ag levels (+1583T > A), 22 38 39 40 through affecting mRNA stability. 41 42 The A allele (frequency, 35%) of rs1087 was associated with higher intact TAFI levels. The variance in intact TAFI explained by age, sex and case–control status is 8.7%. The lead SNP (rs56734909) on its own explained 4% of the remaining variance and together with rs1087, 5.4% of the remaining variance was explained.
When conditioning the remaining SNPs in the CPB2 locus on both rs56734909 and rs1087, we found an independent SNP that did not reach genome-wide significance but suggests complex genomic structure at this locus (rs17844119, P conditional value = 0.0001, Fig. 1E ).
In an effort to identify putative candidate genes from the genome-wide significant and suggestive lead variants, we interrogated public regulatory annotation datasets using the lead SNP from each locus. None of our lead variants were eQTLs for any gene in any of the available tissues (data not shown), with the exception of rs1087 in the conditioned analysis which was an eQTL for the lncRNA CPB2-AS1 in four tissues (cerebellum, P GTEX = 1.6 × 10 −6 , β GTEX = − 0.59; basal ganglia, P GTEX = 2.0 × 10 −7 , β GTEX = −0.63; pituitary, P GTEX = 9.1 × 10 −6 , β GTEX = − 0.68; and thyroid, P GTEX = 1.8 × 10 −5 , β GTEX = −0.33).
Thrombin Activatable Fibrinolysis Inhibitor Activation Peptide
The genomic control coefficient for the TAFI-AP GWAS was close to 1 (λ = 0.996). For TAFI-AP, two loci reached the genome-wide significance threshold of 5 × 10 −8 ( Fig. 2 ). Table 2 lists the top variants for each chromosomal region. For the first locus, we identified numerous SNPs ( n = 316) of genome-wide significance in the13q14.11 region that harbours the CPB2 gene and antisense transcript CPB2-AS1 . The lead SNP (rs940, P = 1 × 10 −27 ) resides within the CPB2 3′ UTR. This SNP has previously been shown to associate with TAFI-Ag levels (+1542C > G) 22 and was significantly associated with intact TAFI levels in our dataset as well ( P = 6.62 × 10 −16 ). A regional plot for rs940 can be found in Fig. 2C . This SNP is in perfect LD with several SNPs predicted to be located within two CPB2 enhancer elements (rs2897027, GH130046061; rs2296641, rs3783202 and rs1409435, all in GH130046056), but is not itself located in an enhancer. It is also in high LD with the 3′ UTR SNP identified in the conditional analysis of intact TAFI (rs1087; D′ = 1.0, R 2 = 0.13, based on the 1000 Genomes Project CEU population). The G allele (frequency, 76.5%) of rs940 was associated with higher TAFI-AP levels. When the data were reanalysed conditioning on rs940, we found an independent SNP that did not reach genome-wide significance ( Fig. 2D ; smallest P conditional value = 1.7 × 10 −5 , rs7491577). The variance in TAFI-AP explained by age, sex and case–control status is 4.2%. The proportion of variance in natural log-transformed TAFI-AP (above age, sex and case–control status) explained by rs940 is 3.8%.
Fig. 2.
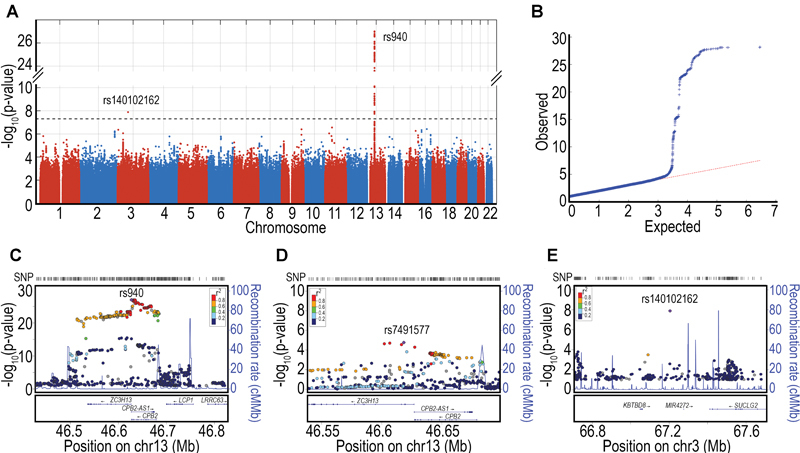
Genome-wide association analyses of TAFI activation peptide. ( A ) Manhattan plot of associations. Dotted line shows genome-wide significance (5 × 10 −8 ). ( B ) QQ-Plot. ( C ) Regional association plot of chromosome 13, CPB2 locus. ( D ) Regional association plot of CPB2 locus in a conditional analysis adjusted for the lead SNP, rs940. ( E ) Regional association plot of rs140102162 on chromosome 3.
The second genome-wide significant locus for TAFI-AP was at chromosomal position 3p14.1 (rs140102162, P -value 1.310 −8 , Table 2 ). This was a solitaire SNP association in an intergenic region between the KBTBD8 (kelch repeat and BTB domain containing 8) and the SUCLG2 (Succinate-CoA Ligase GDP-Forming Beta Subunit) genes ( Fig. 2E ).
An additional 9 loci, composed of 14 SNPs, were suggestively associated ( P < 10 −6 ) with TAFI-AP ( Table 2 , Supplementary Table S5 , Supplementary Fig. S2 ). Chromosomal position 16p12.2 contained three suggestive SNPs in the OTOA gene, which encodes the otoancorin protein and is expressed in the inner ear and in T-lymphocytes. Rs78970023, the lead SNP ( P = 4.7 × 10 −7 ), is a missense SNP that leads to a phenylalanine-tyrosine (F15Y) substitution. Chromosomal position 2q36.2 contained four SNPs within the DOCK10 (dedicator of cytokinesis 10) gene. Rs4674940, the lead SNP with ( P -value 6.0 × 10 −7 ), is located within an exon and leads to a synonymous substitution. The other loci contained only one suggestive SNP each.
When we interrogated public regulatory annotation datasets using the lead SNP from each genome-wide significant and suggestive locus for TAFI-AP to identify putative candidate genes, none of our lead variants were found to be eQTLs for any gene in any of the available tissues (data not shown), with the exception of rs940 which was an eQTL for CPB2-AS1 in EBV-transformed lymphocytes ( P GTEX = 1.4 × 10 −5 , β GTEX = 0.44).
Gene-Based Analysis
Gene-based tests were used to test for aggregation of low-frequency and rare variants in genes that may have importance for intact TAFI or TAFI-AP. The only significant association that contributed to plasma-level variation in intact TAFI was the CPB2 gene itself ( P SKAT-O =2.75 × 10 −8 , Table 3 ). CPB2 remained associated with intact TAFI after conditioning on the lead SNP from GWAS (rs56734909; P conditional = 1.86 × 10 −6 ). Two markers comprised the signal: rs145067962 (Ile420Asn; MAF, 0.15%; MAF ExAC 0.14%) and rs140446990 (Asp177Gly; MAF, 0.20%; MAF ExAC 0.13%). Carriers of the rs140446990 mutation had significantly lower intact TAFI levels than noncarriers ( n = 14 carriers vs. n = 3,190 noncarriers; geometric mean 52.7 vs. 78.5%; P T-test 5.08 × 10 −7 ). This was true for both ischaemic stroke cases ( n = 4 carriers vs. n = 550 noncarriers; geometric mean 68.4 vs. 94.8%; P T-test 0.008) and controls ( n = 10 carriers vs. n = 2,640 noncarriers; geometric mean 47.4 vs. 75.4%; P T-test 4.78 × 10 −7 ). No difference in intact TAFI values was observed for rs145067962 carriers and noncarriers (data not shown). To determine whether this variant in CPB2 was independent of the two more common amino acid substitutions, we instead conditioned on Thr147Ala (rs3742264) and Thr325Ile (rs1926447) and found that the variant remained significant ( P conditional = 1.66 × 10 −6 ), suggesting that the observed rare variant associations are distinct from known SNPs.
Table 3. Intact TAFI and TAFI activation peptide (TAFI-AP): significant and suggestive loci from gene-based tests (SKAT-O).
| Gene | P -Value | Nr SNPs | Region |
|---|---|---|---|
| Intact TAFI | |||
| CPB2 | 2.8E-08 | 2 | 13q14.13 |
| PHLDB2 | 1.1E-05 | 5 | 3q13.2 |
| SEC14L4 | 2.9E-05 | 2 | 22q12.2 |
| GDPGP1 | 6.2E-05 | 2 | 15q26.1 |
| TAFI-AP | |||
| SGPL1 | 8.3E-07 | 3 | 10q22.1 |
| OTOA | 2.5E-06 | 2 | 16p12.2 |
| ADAL | 3.3E-06 | 2 | 15q15.3 |
| GAST | 3.5E-06 | 2 | 17q21.2 |
| TOR1B | 4.1E-06 | 3 | 9q34.11 |
| CPB2 | 7.2E-06 | 2 | 13q14.13 |
| MLN | 6.6E-05 | 2 | 6p21.31 |
Abbreviations: TAFI-AP, thrombin-activatable fibrinolysis inhibitor activation peptide.
Note: Intact TAFI, expressed as relative levels; TAFI-AP, TAFI activation peptide expressed as relative levels.
In contrast to intact TAFI, for TAFI-AP SKAT-O yielded significant gene-level associations with five genes ( Table 3 ) and only near significance with CPB2 ( P SKAT-O =7.18 × 10 −6 ). The strongest associated gene was SGPL1 (sphingosine-1-phosphate lyase 1), followed by OTOA (described earlier), ADAL (adenosine deaminase-like), GAST (gastrin) and TOR1B (torsin family 1 member B). Of note, the variant driving the association at OTOA in SKAT-O is the same as the lead GWAS SNP at this locus. SGPL1 ( P SKAT-O = 8.33 × 10 −7 ) encodes the enzyme sphingosine phosphate lyase (SPL) which plays a critical role in the regulation of sphingosine-1-phosphate (S1P), a versatile lipid-signaling molecule. Three variants within SGPL1 comprised the signal: K228N, rs191033522, MAF 0.02%, MAF ExAC 0.03%; Y356C, rs147860288, MAF 0.03%, MAF ExAC 0.03%; and Y374C, rs139751906, MAF 0.03%, MAF ExAC 0.02%. Only five individuals were carriers of any of these variants; carriers of the minor allele of rs191033522 and rs139751906 had lower TAFI-AP concentrations compared with noncarriers, while carriers of rs147860288 had elevated TAFI-AP concentrations. With regard to CPB2 , similar to intact TAFI, carriers of the rs140446990 (Asp177Gly) mutation had significantly lower TAFI-AP levels than noncarriers ( n = 14 vs. n = 3,198 noncarriers: geometric mean 63 vs. 99%, P T-test 7.2 × 10 −4 ).
Discussion
This is the first GWAS to examine genetic determinants of intact TAFI and the extent of TAFI activation (measured by TAFI-AP) in plasma. Using a TAFI assay that reacts exclusively to the nonactivated intact TAFI and equally recognizes the common TAFI isoforms, we verified CPB2 at the 13q14.11 locus to be the strongest determinant of intact TAFI concentrations. However, less of the variation was explained for both intact TAFI and TAFI-AP than previously reported for ‘total’ TAFI, denoted TAFI-Ag. Furthermore, we identified an additional genome-wide significant locus for TAFI-AP. We then performed gene-based tests to investigate the role of rare and low-frequency variants and identified novel low frequency coding variants within CPB2 associated with intact TAFI concentrations as well as completely novel associations for TAFI-AP to five genes. As these results are explorative in nature, the novel associations outside the 13q14.11 locus require independent validation.
With regards to the GWAS, the strongest associations for intact TAFI and TAFI-AP concentrations were for variants located in the 13q14.11 locus and both analyses indicate a complex genomic architecture at this locus. Common CPB2 SNPs have been known to influence TAFI levels for some time and early linkage studies also implicated this region. 18 20 Thus, in spite of using a larger sample size and modern techniques including dense imputation, we still find this to be the primary locus, and somewhat unexpectedly we did not identify common variants at any other autosomal region with similar estimated effect sizes. For intact TAFI, two independent genome-wide variants in the CPB2 locus were identified: one in the 5′ UTR of the gene (rs56734909) located in a putative enhancer element and one in the 3′ UTR (rs1087). This is consistent with previous studies that suggested that two polymorphisms in CPB2 , one in the promoter region and the other in the coding or 3′ region, likely underlie the variation in TAFI-Ag. 38 43 A trans -ethnic haplotype study revealed rs1087 to be the most likely eQTL in the 3′ region for TAFI-Ag, 39 supported also by mRNA stability experiments. 41 42 The identification of the putative eQTL in the 5′ region of CPB2 has proven more challenging as a consequence of strong LD between promoter polymorphisms. One SNP in the 5′ region (-2599C > G, rs34813434) that was suggested to contribute to the regulation of TAFI-Ag in the trans -ethnic analysis 39 is in high LD with our lead SNP ( R 2 = 0.96, D' = 1; based on the 1000 Genomes Project CEU population). However, promoter deletion experiments in HepG2-transfected cells do not support a functional role for this SNP. 44 Given that our lead SNP rs56734909 is located 490 bp upstream of rs34813434, it was not analysed in the promoter deletion experiments, and as it is within a predicted enhancer element we evaluated its putative effect on regulatory motifs. According to HaploReg, enhancer histone marks were identified in liver tissue where CPB2 is expressed, and eight regulatory motifs are altered by this SNP. Although speculative, these data suggest that rs56734909 may be the functional 5′ SNP affecting levels, a hypothesis that requires validation through functional experiments.
For the GWAS of TAFI-AP, the lead SNP in the CPB2 locus, rs940, is located within the 3′ UTR and has previously been shown to associate with TAFI-Ag levels (+1542C > G). 19 22 This SNP is in perfect LD with four SNPs predicted to be located within two putative enhancers (rs2296641, rs3783202 and rs1409435 in enhancer GH130046056; and rs2897027 in enhancer GH130046061) and high LD with three additional putative enhancer SNPs (rs4942470 in GH130046061; rs7139571 in GH130046087; and rs7989892 in GH130046056). Two known common SNPs in CPB2 lead to amino acid substitutions in TAFI, Thr147Ala (rs3742264) and Thr325Ile (rs1926447). Individuals with Ile-325 variants have a more stable TAFIa enzyme with an extended half-life, 45 whereas the Ala-147 variant does not affect the stability. Both were significantly associated with TAFI-AP in the unconditioned analysis (rs3742264, P = 1.010 −13 and rs1926447, P = 7.98 × 10 −26 ) but not in the analysis conditioned on rs940 (rs3742264, P = 3.19 × 10 −4 and rs1926447, P = 0.302). Because SNP rs940 is tightly linked with rs1926447 ( R 2 = 0.83, D' = 1; based on the 1000 Genomes Project CEU population) and multiple SNPs in putative enhancers, the question remains open as to which one is functional.
It is of note that the antisense lncRNA CPB2-AS1 is located on the opposite DNA strand and fully overlapping with the CPB2 gene. Antisense lncRNAs can modulate expression of their sense transcripts through various mechanisms (reviewed in the study of Villegas and Zaphiropoulos 46 ). For example, sense RNA and antisense lncRNA transcripts can hybridize and form RNA duplexes which can interfere with splicing, RNA editing and alter mRNA stability. Therefore, it is plausible that some SNPs associated with intact TAFI and/or TAFI-AP levels may exert their affects via the CPB2-AS1 transcript rather than CPB2 gene. In line with this, rs1087 and rs940 associated with intact TAFI and TAFI-AP, respectively, were found to be eQTLs for CPB2-AS1 in the GTEX database. However, to our knowledge, the putative functional effect from CPB2-AS1 on CPB2 transcript level has not been reported upon in the literature and is unknown.
Taken together, polymorphisms within CPB2 were found to explain only a modest amount of the variation in intact TAFI (5.4%) and TAFI-AP (3.8%) levels, (above age, sex and case–control status). This is much lower than observed in older works that used immunological assays that measured ‘total’ TAFI (TAFI-Ag, i.e. intact TAFI as well as the various cleavage products). Many of these assays were later demonstrated to have decreased reactivity toward the TAFI Ile325 isoform, and as a result overestimated the TAFI heritability and effects associated with CPB2 polymorphisms. 16 17 19 Two small studies ( n < 300) that used isoform-independent TAFI assays reported a combined variance estimate for CPB2 SNPs of 15 to 25% in univariate haplotype analyses. 38 39 While higher than our findings, the difference in variance estimates could be due in part to the different methodologies used in the measurement of TAFI, the different populations studied and the fact that different covariates were used in the calculation/models.
Using GWAS, we also identified one novel variant associated with TAFI-AP concentrations on chromosome 3 (rs140102162) and several suggestive loci. However, despite the fact that rs140102162 was genome-wide significant ( P = 1.3 × 10 −8 ), it was a solitary SNP within an intergenic region. Similarly, most suggestive findings were also due to solitary SNPs. We were unable to identify any eQTLs for these variants in any tissue in publically available resources. Thus, apart from CPB2 , we did not identify any strong evidence for additional candidate loci for determining plasma TAFI concentrations. Combined with the fact that rs140102162 and the majority of suggestive variants were low frequency, we did not proceed into replication as this would require very large sample sizes.
CPB2 was also significantly associated with intact TAFI and suggestively associated with TAFI-AP in the gene-based analysis ( P = 2.8 × 10 −8 and 7.2 × 10 −6 , respectively), and this was due to a rare coding variant. This association was independent of the more common variants at this locus. Carriers of the rs140446990 (Asp177Gly) mutation had significantly lower intact TAFI and TAFI-AP levels than noncarriers. Since intact TAFI and TAFI-AP are moderately correlated (Spearman's Rho = 0.33), this indirectly suggests that the SNP has no direct bearing on the extent of activation (i.e.: TAFI-AP, amino acids residues 1–92), and simply reflects this interrelationship.
The gene-based analyses also showed completely novel associations for TAFI-AP with five genes. The strongest signal was for SGPL1 , which encodes the enzyme SPL. SPL has been directly implicated in various physiological and pathological processes, including cell stress responses, cancer, immunity, hematopoietic function, muscle homeostasis, inflammation and development. Furthermore, SPL irreversibly degrades the lipid signaling molecule S1P, which has recently emerged as a critical mediator linking the coagulation factor system to vascular inflammation 47 and has been implicated in various cardiovascular events such as myocardial infarction and stroke. 48 49 50 Thus, it would be of interest to seek replication and also to investigate associations between rare variants in SGPL1 and cardiovascular diseases. However, as the MAF is as low as 0.02 to 0.03%, this will require very large datasets genotyped with rare variant information (e.g. exome chips or exome sequencing) and as far as we are aware it is not feasible at this time.
The strengths of this study include that approximately 10 million markers spread throughout the genome, covering the full allelic spectrum of common variants (assessed via GWAS) and rare variants (assessed via gene-based testing) were genotyped in 3,260 individuals with standardized and well-characterized TAFI assays. Despite these strengths, there are some limitations which deserve mention. Although this is the largest study on the genetic regulation of intact TAFI and TAFI-AP published to date, it is still small for GWAS standards, and in the future an expansion of the number of study subjects is needed to improve the power of detecting genetic variants related to TAFI. Specifically, this study is still underpowered to detect variants with low MAF in single variant analyses. Although we used gene-based tests to increase power for low-frequency and rare variants, one limitation is that the exome array does not provide complete coverage of all functional variants at each locus. Exome or whole-genome sequencing would be required to completely assess variants associated with TAFI levels. As such, it remains possible that a burden of rare mutations could contribute to plasma TAFI levels. Additionally, it should also be emphasized that the results were generated in European ancestry populations, and our findings many not be generalizable to populations of different ethnicity. Finally, the results are explorative in nature and clearly require future replication in an independent cohort.
In conclusion, using genome-wide genotype data from a total of 3,260 subjects, we confirmed the CPB2 locus to be the strongest determinant of plasma TAFI, but showed that the effect was more modest than previously reported. Through gene-based testing, we identified novel putative functional variants within the CPB2 locus and in five other genes that require future independent replication.
Acknowledgements
The authors thank research nurse Ingrid Eriksson for her excellent work and assistance with the study patients and Miet Peeters for the analyses of intact TAFI and TAFI-AP.
Conflict of Interest There are no direct or indirect conflicts of interest to declare.
Financial Support
The present study was supported by the Swedish Research Council and grants from the Swedish state (ALF), the Swedish Heart-Lung Foundation and the Swedish Stroke Association. The National Institute of Neurological Disorders and Stroke (NINDS) funded the genotyping of patients included in the SiGN study. PI, Steven Kittner.
What is known about this topic?
Elevated circulating TAFI concentrations have been reported in patients with cardiovascular disease and family studies have demonstrated that plasma TAFI concentrations are heritable.
Several common single nucleotide polymorphisms (SNPs) located within the gene encoding TAFI ( CPB2 ) have been described and some are reported to be associated with plasma TAFI concentrations and risk of arterial and venous thrombotic disease.
What does this paper add?
This is the first genome-wide association study and the first study that used gene-based tests of low-frequency and rare variants to identify the genetic basis for the heritability of circulating TAFI concentrations.
We verified CPB2 to be the primary determinant of TAFI concentrations and identified putative secondary loci (candidate variants and genes) that require independent validation.
Supplementary Material
References
- 1.Swaisgood C M, Schmitt D, Eaton D, Plow E F. In vivo regulation of plasminogen function by plasma carboxypeptidase B. J Clin Invest. 2002;110(09):1275–1282. doi: 10.1172/JCI15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boffa M B, Koschinsky M L. Curiouser and curiouser: recent advances in measurement of thrombin-activatable fibrinolysis inhibitor (TAFI) and in understanding its molecular genetics, gene regulation, and biological roles. Clin Biochem. 2007;40(07):431–442. doi: 10.1016/j.clinbiochem.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 3.Bajzar L, Morser J, Nesheim M. TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem. 1996;271(28):16603–16608. doi: 10.1074/jbc.271.28.16603. [DOI] [PubMed] [Google Scholar]
- 4.Mao S S, Cooper C M, Wood T, Shafer J A, Gardell S J. Characterization of plasmin-mediated activation of plasma procarboxypeptidase B. Modulation by glycosaminoglycans. J Biol Chem. 1999;274(49):35046–35052. doi: 10.1074/jbc.274.49.35046. [DOI] [PubMed] [Google Scholar]
- 5.Wang W, Boffa M B, Bajzar L, Walker J B, Nesheim M E. A study of the mechanism of inhibition of fibrinolysis by activated thrombin-activable fibrinolysis inhibitor. J Biol Chem. 1998;273(42):27176–27181. doi: 10.1074/jbc.273.42.27176. [DOI] [PubMed] [Google Scholar]
- 6.Marx P F, Dawson P E, Bouma B N, Meijers J C. Plasmin-mediated activation and inactivation of thrombin-activatable fibrinolysis inhibitor. Biochemistry. 2002;41(21):6688–6696. doi: 10.1021/bi015982e. [DOI] [PubMed] [Google Scholar]
- 7.Franco R F, Fagundes M G, Meijers J C et al. Identification of polymorphisms in the 5′-untranslated region of the TAFI gene: relationship with plasma TAFI levels and risk of venous thrombosis. Haematologica. 2001;86(05):510–517. [PubMed] [Google Scholar]
- 8.van Tilburg N H, Rosendaal F R, Bertina R M. Thrombin activatable fibrinolysis inhibitor and the risk for deep vein thrombosis. Blood. 2000;95(09):2855–2859. [PubMed] [Google Scholar]
- 9.Santamaría A, Martínez-Rubio A, Borrell M, Mateo J, Ortín R, Fontcuberta J. Risk of acute coronary artery disease associated with functional thrombin activatable fibrinolysis inhibitor plasma level. Haematologica. 2004;89(07):880–881. [PubMed] [Google Scholar]
- 10.Schroeder V, Wilmer M, Buehler B, Kohler H P. TAFI activity in coronary artery disease: a contribution to the current discussion on TAFI assays. Thromb Haemost. 2006;96(02):236–237. [PubMed] [Google Scholar]
- 11.Ladenvall C, Gils A, Jood K, Blomstrand C, Declerck P J, Jern C. Thrombin activatable fibrinolysis inhibitor activation peptide shows association with all major subtypes of ischemic stroke and with TAFI gene variation. Arterioscler Thromb Vasc Biol. 2007;27(04):955–962. doi: 10.1161/01.ATV.0000259354.93789.a6. [DOI] [PubMed] [Google Scholar]
- 12.Leebeek F W, Goor M P, Guimaraes A H et al. High functional levels of thrombin-activatable fibrinolysis inhibitor are associated with an increased risk of first ischemic stroke. J Thromb Haemost. 2005;3(10):2211–2218. doi: 10.1111/j.1538-7836.2005.01484.x. [DOI] [PubMed] [Google Scholar]
- 13.Montaner J, Ribó M, Monasterio J, Molina C A, Alvarez-Sabín J. Thrombin-activable fibrinolysis inhibitor levels in the acute phase of ischemic stroke. Stroke. 2003;34(04):1038–1040. doi: 10.1161/01.STR.0000063139.06585.45. [DOI] [PubMed] [Google Scholar]
- 14.Chetaille P, Alessi M C, Kouassi D, Morange P E, Juhan-Vague I. Plasma TAFI antigen variations in healthy subjects. Thromb Haemost. 2000;83(06):902–905. [PubMed] [Google Scholar]
- 15.Ariëns R AS, de Lange M, Snieder H, Boothby M, Spector T D, Grant P J.Activation markers of coagulation and fibrinolysis in twins: heritability of the prethrombotic state Lancet 2002359(9307):667–671. [DOI] [PubMed] [Google Scholar]
- 16.Bladbjerg E M, de Maat M PM, Christensen K, Bathum L, Jespersen J, Hjelmborg J. Genetic influence on thrombotic risk markers in the elderly--a Danish twin study. J Thromb Haemost. 2006;4(03):599–607. doi: 10.1111/j.1538-7836.2005.01778.x. [DOI] [PubMed] [Google Scholar]
- 17.Peetz D, Victor A, Adams P et al. Genetic and environmental influences on the fibrinolytic system: a twin study. Thromb Haemost. 2004;92(02):344–351. doi: 10.1160/TH04-01-0001. [DOI] [PubMed] [Google Scholar]
- 18.Sabater-Lleal M, Buil A, Souto J C et al. A genome-wide exploration suggests an oligogenic model of inheritance for the TAFI activity and its antigen levels. Hum Genet. 2008;124(01):81–88. doi: 10.1007/s00439-008-0527-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trégouet D A, Aubert H, Henry M et al. Combined segregation-linkage analysis of plasma thrombin activatable fibrinolysis inhibitor (TAFI) antigen levels with TAFI gene polymorphisms. Hum Genet. 2001;109(02):191–197. doi: 10.1007/s004390100558. [DOI] [PubMed] [Google Scholar]
- 20.Warren D M, Cole S A, Dyer T D et al. A locus on chromosome 13 influences levels of TAFI antigen in healthy Mexican Americans. Hum Biol. 2006;78(03):329–339. doi: 10.1353/hub.2006.0049. [DOI] [PubMed] [Google Scholar]
- 21.Brouwers G J, Vos H L, Leebeek F W et al. A novel, possibly functional, single nucleotide polymorphism in the coding region of the thrombin-activatable fibrinolysis inhibitor (TAFI) gene is also associated with TAFI levels. Blood. 2001;98(06):1992–1993. doi: 10.1182/blood.v98.6.1992. [DOI] [PubMed] [Google Scholar]
- 22.Henry M, Aubert H, Morange P E et al. Identification of polymorphisms in the promoter and the 3′ region of the TAFI gene: evidence that plasma TAFI antigen levels are strongly genetically controlled. Blood. 2001;97(07):2053–2058. doi: 10.1182/blood.v97.7.2053. [DOI] [PubMed] [Google Scholar]
- 23.Zhao L, Morser J, Bajzar L, Nesheim M, Nagashima M. Identification and characterization of two thrombin-activatable fibrinolysis inhibitor isoforms. Thromb Haemost. 1998;80(06):949–955. [PubMed] [Google Scholar]
- 24.Gils A, Alessi M C, Brouwers E et al. Development of a genotype 325-specific proCPU/TAFI ELISA. Arterioscler Thromb Vasc Biol. 2003;23(06):1122–1127. doi: 10.1161/01.ATV.0000074145.58172.BD. [DOI] [PubMed] [Google Scholar]
- 25.Guimarães A H, van Tilburg N H, Vos H L, Bertina R M, Rijken D C. Association between thrombin activatable fibrinolysis inhibitor genotype and levels in plasma: comparison of different assays. Br J Haematol. 2004;124(05):659–665. doi: 10.1111/j.1365-2141.2004.04824.x. [DOI] [PubMed] [Google Scholar]
- 26.Ceresa E, Brouwers E, Peeters M, Jern C, Declerck P J, Gils A. Development of ELISAs measuring the extent of TAFI activation. Arterioscler Thromb Vasc Biol. 2006;26(02):423–428. doi: 10.1161/01.ATV.0000199246.08616.98. [DOI] [PubMed] [Google Scholar]
- 27.Söderholm M, Almgren P, Jood K et al. Exome array analysis of ischaemic stroke: results from a southern Swedish study. Eur J Neurol. 2016;23(12):1722–1728. doi: 10.1111/ene.13086. [DOI] [PubMed] [Google Scholar]
- 28.Jood K, Ladenvall C, Rosengren A, Blomstrand C, Jern C. Family history in ischemic stroke before 70 years of age: the Sahlgrenska Academy Study on Ischemic Stroke. Stroke. 2005;36(07):1383–1387. doi: 10.1161/01.STR.0000169944.46025.09. [DOI] [PubMed] [Google Scholar]
- 29.Li C, Engström G, Hedblad B, Berglund G, Janzon L. Risk factors for stroke in subjects with normal blood pressure: a prospective cohort study. Stroke. 2005;36(02):234–238. doi: 10.1161/01.STR.0000152328.66493.0a. [DOI] [PubMed] [Google Scholar]
- 30.NINDS Stroke Genetics Network (SiGN); International Stroke Genetics Consortium (ISGC).Loci associated with ischaemic stroke and its subtypes (SiGN): a genome-wide association study Lancet Neurol 20161502174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldstein J I, Crenshaw A, Carey J et al. zCall: a rare variant caller for array-based genotyping: genetics and population analysis. Bioinformatics. 2012;28(19):2543–2545. doi: 10.1093/bioinformatics/bts479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCarthy S, Das S, Kretzschmar W et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48(10):1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delaneau O, Howie B, Cox A J, Zagury J F, Marchini J. Haplotype estimation using sequencing reads. Am J Hum Genet. 2013;93(04):687–696. doi: 10.1016/j.ajhg.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee S, Emond M J, Bamshad M J et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012;91(02):224–237. doi: 10.1016/j.ajhg.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lonsdale J, Thomas J, Salvatore M et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(06):580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Veyrieras J B, Kudaravalli S, Kim S Y et al. High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet. 2008;4(10):e1000214. doi: 10.1371/journal.pgen.1000214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Westra H-J, Peters M J, Esko T et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45(10):1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frère C, Morange P E, Saut N et al. Quantification of thrombin activatable fibrinolysis inhibitor (TAFI) gene polymorphism effects on plasma levels of TAFI measured with assays insensitive to isoform-dependent artefact. Thromb Haemost. 2005;94(02):373–379. doi: 10.1160/TH04-08-0497. [DOI] [PubMed] [Google Scholar]
- 39.Frère C, Tregouet D A, Morange P E et al. Fine mapping of quantitative trait nucleotides underlying thrombin-activatable fibrinolysis inhibitor antigen levels by a transethnic study. Blood. 2006;108(05):1562–1568. doi: 10.1182/blood-2006-01-008094. [DOI] [PubMed] [Google Scholar]
- 40.Biswas A, Ranjan R, Meena A et al. TAFI antigen level variability in young healthy Asian Indians; first report from Asia. Clin Biochem. 2008;41(09):750–753. doi: 10.1016/j.clinbiochem.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 41.Boffa M B, Maret D, Hamill J D et al. Effect of single nucleotide polymorphisms on expression of the gene encoding thrombin-activatable fibrinolysis inhibitor: a functional analysis. Blood. 2008;111(01):183–189. doi: 10.1182/blood-2007-03-078543. [DOI] [PubMed] [Google Scholar]
- 42.Maret D, Boffa M B, Brien D F, Nesheim M E, Koschinsky M L. Role of mRNA transcript stability in modulation of expression of the gene encoding thrombin activable fibrinolysis inhibitor. J Thromb Haemost. 2004;2(11):1969–1979. doi: 10.1111/j.1538-7836.2004.00971.x. [DOI] [PubMed] [Google Scholar]
- 43.Morange P E, Tregouet D A, Frere C et al. TAFI gene haplotypes, TAFI plasma levels and future risk of coronary heart disease: the PRIME Study. J Thromb Haemost. 2005;3(07):1503–1510. doi: 10.1111/j.1538-7836.2005.01486.x. [DOI] [PubMed] [Google Scholar]
- 44.Boffa M B, Reid T S, Joo E, Nesheim M E, Koschinsky M L. Characterization of the gene encoding human TAFI (thrombin-activable fibrinolysis inhibitor; plasma procarboxypeptidase B) Biochemistry. 1999;38(20):6547–6558. doi: 10.1021/bi990229v. [DOI] [PubMed] [Google Scholar]
- 45.Schneider M, Boffa M, Stewart R, Rahman M, Koschinsky M, Nesheim M. Two naturally occurring variants of TAFI (Thr-325 and Ile-325) differ substantially with respect to thermal stability and antifibrinolytic activity of the enzyme. J Biol Chem. 2002;277(02):1021–1030. doi: 10.1074/jbc.M104444200. [DOI] [PubMed] [Google Scholar]
- 46.Villegas V E, Zaphiropoulos P G. Neighboring gene regulation by antisense long non-coding RNAs. Int J Mol Sci. 2015;16(02):3251–3266. doi: 10.3390/ijms16023251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mahajan-Thakur S, Böhm A, Jedlitschky G, Schrör K, Rauch B H. Sphingosine-1-phosphate and its receptors: a mutual link between blood coagulation and inflammation. Mediators Inflamm. 2015;2015:831059. doi: 10.1155/2015/831059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knapp M. Cardioprotective role of sphingosine-1-phosphate. J Physiol Pharmacol. 2011;62(06):601–607. [PubMed] [Google Scholar]
- 49.Wei Y, Yemisci M, Kim H H et al. Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann Neurol. 2011;69(01):119–129. doi: 10.1002/ana.22186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Z, Wang R, Huo Z, Li C, Wang Z. Characterization of the anticoagulant and antithrombotic properties of the Sphingosine 1-phosphate mimetic FTY720. Acta Haematol. 2017;137(01):1–6. doi: 10.1159/000448837. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.