

Abstract
We report here on long-time all-atomistic molecular dynamics simulations of functional supramolecular nanotubes composed by the self-assembly of peptide-drug amphiphiles (DAs). These DAs have been shown to possess an inherently high drug loading of the hydrophobic anticancer drug camptothecin. We probe the self-assembly mechanism from random with ~ 0.4 microsecond molecular dynamics simulations. Furthermore, we also computationally characterize the interfacial structure, directionality of π-π stacking, and water dynamics within several peptide-drug nanotubes with diameters consistent with the reported experimental nanotube diameter. Insight gained should inform the future design of these novel anticancer drug delivery systems.
Graphical Abstract
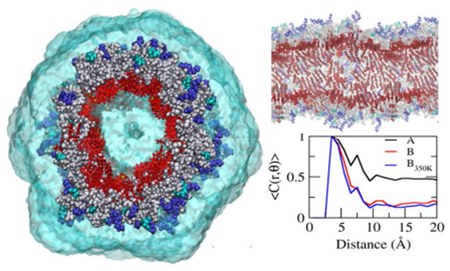
Peptide amphiphiles self-assemble into nanofilamentous morphologies that possess diverse applications as functional materials1–2. These dynamic self-assemblies are found in biomedicine as three dimensional tissue scaffolds3 as well as drug delivery devices4–6. Computer simulations, in particular molecular dynamics (MD), have shown to be a useful methodology to probe the 1D structures of peptide-amphiphiles7–12. Previously, we reported the significant role of π-π stacking on the nucleation and 1D growth of the self-assembled nanofilament of a peptide-based drug amphiphile (DA)13, which consists of a short hexapeptide sequence with a disulfide linker bonded to the hydrophobic anticancer drug camptothecin (CPT). Depending on the number of anticancer drugs in the molecular design, the resulting nanostructures can form either nanofilaments or nanotubes4.
Strongly directional forces such as hydrogen-bonding and/or π–π stacking can reinforce the uni-directional growth of molecular self-assembly, while adding to the complexity of self-assembly9. For example, statistical mechanical models of the self-assembly of chiral rod-like molecules, such as polypeptides, demonstrate the formation of tapes, ribbons, fibrils, and fibers, as a function of molecular concentration14. However, particularly interesting is the self-assembly of molecular components into hollow tubular structures or nanotubes15–19. These results demonstrate that, in addition to molecular topology, the strength of the electrostatic interactions is an important design consideration to the design of nanotubes. Furthermore, these results suggest that this approach can be used to design confined channels of water within these nanoscale assemblies. Experimentally, it was found that increasing the number of drugs attached to the peptide from one to four results in the conversion from a nanofilament to a wider nanotube4, as characterized by TEM. Furthermore, circular dichroism (CD) signals of both ‘mCPT-buSS-Tau’ and ‘qCPT-buSS-Tau’, peptide conjugates with one and four attached CPTs respectively, indicated the strong interactions among CPT units, which was speculated to play a crucial role for formation of the observed nanotube morphology, but the exact mechanism was not clear.4 Within this communication, we characterize in detail the strength of the π-π stacking, as well as the degree to which the π-π stacking propagates throughout the nanotube core.
Herein, we characterize the balance of intermolecular forces stabilizing the shape of molecularly engineered nanotubes composed by DAs with long-time MD simulations. We probe the interfacial density, degree of π–π stacking, and hydrogen bonding in the nanotube structure. We next compare these results with our previously reported computational results of the nanofilament structure13 and find that π-π stacking between CPTs remains strong within the nanotube core, but the contribution of electrostatics to the nanotube structure decreases compared to the nanofilament structure. These results have broad implications to the future design of peptide-based nanotubes as well as drug delivery devices.
To begin with, we build seven preassembled nanotubes composed by ‘qCPT-buSS-Tau’, a DA which consists of four CPT drugs conjugated to a β-sheet forming peptide (CGVQIVYKK or Tau), via a biodegradable disulfide linker (buSS) (Figure S1)4. In each pre-assembled nanotube we vary either the number of DAs per layer, conformation of the hydrophobic drugs within the DA, or else the temperature, as summarized in Table S1. The nanotubes are continuous along the z-axis with full periodic boundary conditions. For comparison, a random system is simulated with concentration of 22.1 mM. All systems are solvated with TIP3P water20, and the electrostatic charge is neutralized with Cl− ions. ‘qCPT-buSS-Tau’ is parametrized using the General AMBER force field (GAFF)21. Partial charges are assigned by Vcharge v1.0122. Atomistic MD simulations are carried out using NAMD2.1123. All systems use the NPT ensemble at a selected temperature of 310 K or 350 K and at a pressure of 1 atm. The simulations are performed 400 ns. (see Supporting Information for more details)
Among the seven nanotubes, two pre-assembled nanotubes (nanotube A and nanotube B as shown in Figure 1 A-D) are analyzed in more detail based on the agreement of the relaxed nanotube diameter with the experimentally reported width (Figure S3). TEM images of the nanotube structure show the width to be 9.5 ± 1 nm, with a hollow channel of a width between 2-4 nm (Figure 1F). The relaxed widths of nanotube A and nanotube B are 10.43 ± 0.37 nm and 9.33±0.32 nm respectively (Figure 1, Table S1 and Figure S3). Both starting structures of nanotube A and nanotube B consist of six layers with twelve DAs per layer. The only difference between nanotube A and B is the conformation of the initial DAs used as a building block. (Figure S2A and B) Both nanotube A and nanotube B relax to stable nanotubes, in which the CPTs form the inner wall and the peptides wrap them around with the polar and charged residues exposed to the outside water, as shown in the Figure 1A to D, and quantified in the radial density profile (Figure 1E). In comparison, the width of the simulated nanofilament of ‘mCPT-buSS-Tau’ was much thinner 7.80±0.01 nm13 which agreed well with the experimental reported width of 6.7±1 nm24
Figure 1.
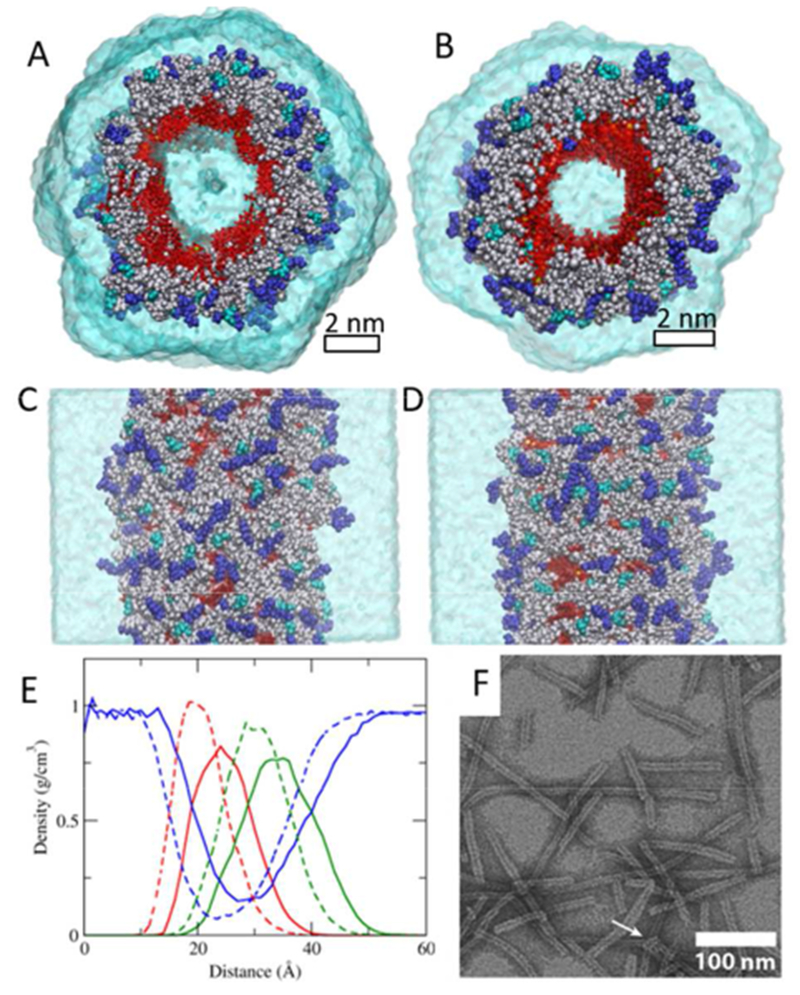
Structure of pre-assembled ‘qCPT-buSS-Tau’. Top (A) and side view (C) of nanotube A. Top (B) and side view (D) nanotube B. CPT and water are displayed in red, and transparent cyan, respectively. Peptides with hydrophobic, polar, and charged side chains are displayed in grey, cyan, and blue, respectively. The scale bar represents 2 nm. E. Radial density of CPT (red), peptide (green) and water (blue) in nanotube A (solid lines) and nanotube B (dotted lines). F. Transmission electron microscope (TEM) image. The scale bar represents 100 nm. The TEM image is adapted with permission from ref. 4, Copyright 2013 American Chemical Society.
Notably, the stacking of the planar CPTs in the core of the supramolecular nanotube is strong and propagates throughout the nanotubes, as shown in the Figure 2. The molecular structure of camptothecin (CPT) is shown in Figure 2A. Nanotube A and nanotube B display the CPT planes titled 30~40° from the radial vector. To expedite the relaxation of CPT orientations in the tube, nanotube B is also simulated at 350 K for 420 ns after 120 ns-simulation at 310 K. This system is denoted nanotube B350K. The CPT tilting in nanotube B350K falls between nanotube A and B (Figure 2D). While the CPT planes are mostly perpendicular to the axis of the tube in the nanotube A, the corresponding angle is ~120° in nanotube B (Figure 2E). The population of this angle in nanotube B350K is shifted slightly left towards that of the nanotube A. Strong π- π stackings of the planar CPT planes are observed. CPTs are stacked near-parallel within 6 Å in all 3 systems (Figures 2F, S6, and S7). As the distance gets longer, the portion of intermolecular stacking increases. The stackings propagate the system and three-fold stackings are observed in nanotube A. A fraction of the stacking as a function of distance between CPTs. <C(r,θ)>, is calculated with C(r, θ)=1 when |θ|≤ θ cut, and C(r, θ)=0 when |θ|> θ cut, where θ cut is the cutoff angle of 30°. <C(r,θ)> displays three peaks, indicating three-fold stacking: the first (d < 6 Å), the second (6 Å < d < 10 Å), and the third (10 Å < d < 13 Å) stackings (Figure 2F). While the first stackings mainly consist of the intramolecular stackings, more than half of the second and the third stackings are intermolecular (Figure S7). The stacking angles of CPT planes display a wider distribution at the longer distance. In nanotube B, only the first stacking is clear with mostly intramolecular stackings. The distance-dependence of the stacking gets stronger in nanotube B350K, compared to the original nanotube B. The orientation and stacking of the CPT planes suggest that the nanotube B350K is evolving toward nanotube A.
Figure 2.
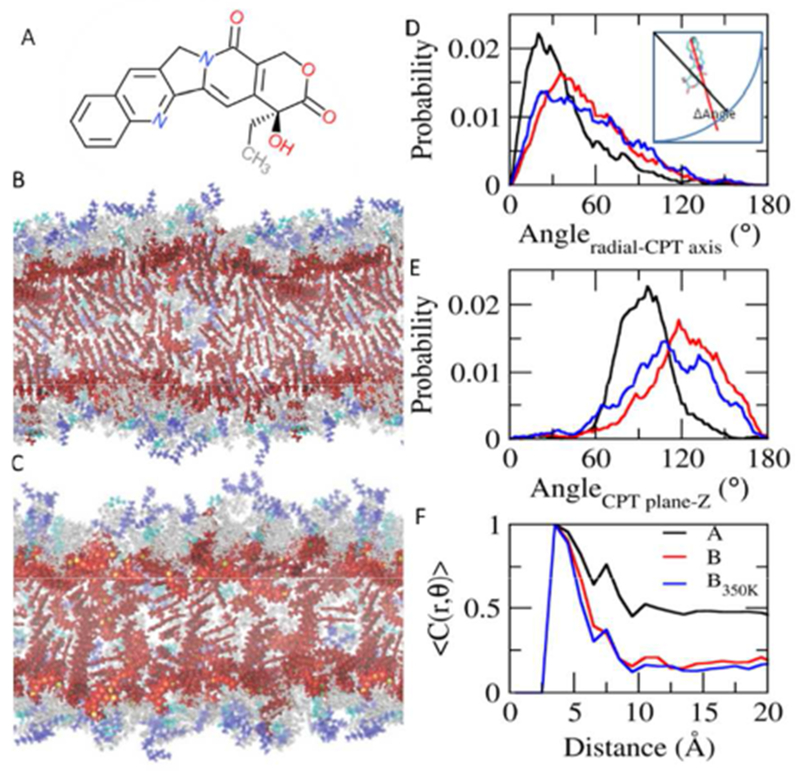
Packing and π-π stacking. A. Structure of camptothecin (CPT). B. Side view of nanotube A. C. Side view of nanotube B. CPTs are highlighted in red and the rest of the molecule are in transparent grey, cyan and blue. D. Distribution of angles between the radial vector and the long CPT axis. E. Distribution of angles between the axis of the tube and the CPT planes. F. Fraction of stacking over distance between CPTs.
In addition to the strong aromatic interactions, the balance between hydrophobic interactions, electrostatics interactions, and hydrogen bonding determines the resulting shape of the self-assembly9. The strength of hydrogen bonding along the filament, as well as local salt concentration may govern the mechanical properties of the assembly25. Nanotubes A, B and B350K all display a similar number hydrogen bonds per DA (Figure 3). However, nanotube B350K has the highest ratio of intermolecular hydrogen bonds to the total hydrogen bonds. This suggests a significant rearrangement of the hydrogen bonding network at higher temperatures. Notably, the van der Waals and electrostatic interactions contribute almost equally to the stability of the nanotube (Figure S8), for both nanotube A and B. The contribution of van der Waals interactions is smaller than the electrostatic interaction in the nanofilament of ‘mCPT-buSS-Tau’. The increased contribution of van der Waals interactions in the nanotube stems from the structural change in its building block, ‘qCPT-buSS-Tau’: while keeping the same hydrophilic peptide tail as that of ‘mCPT-buSS-Tau’, the attached hydrophobic drug increases four times with longer linkers, resulting in 2.4-times increase in the volume. This increased hydrophobic volume of the DA leads to the significant decrease in charged/polar fraction (volume ratio of the charged/polar residues, 58 %), density of charged components (primary ammonium ions of the Lysine residues) at the surface (Figure S 9 B) and density of condensed counterions (Figure S 9 A). Interestingly, compared to the nanofilament of ‘mCPT-buSS-Tau’13, the surface charge density around the outer rim of the nanostructure is significantly lower in both of the nanotubes compared to the nanofilament. With more drugs attached, ‘qCPT-buSS-Tau’ molecules get bulkier and assemble into wider nanostructures with a greater surface area. The density of the primary ammonium ion (−NH3+) of the terminal lysine residues is lower in the nanotubes (Figure S9 B). This corresponds to a decreased density of condensed Cl− ions around the nanotubes as compared with the nanofilament (Figure S9 A). Thus, in comparison with the nanofilament structure, electrostatics plays less of a role in stabilizing the nanotube structure. The directional role of electrostatic interactions in the self-assembly of peptide amphiphiles has been suggested in theoretical studies.10
Figure 3.
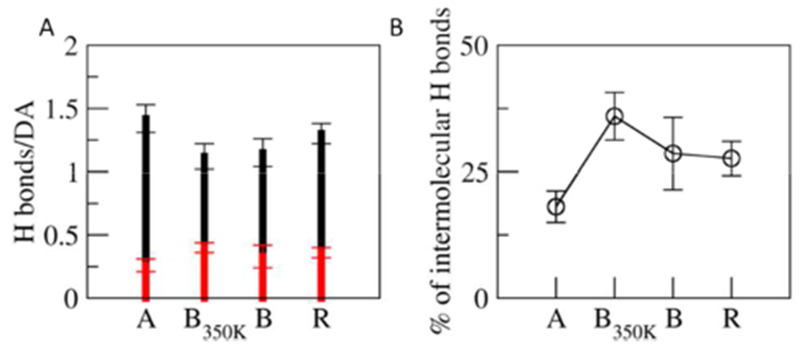
Hydrogen bonds. A. The total number of hydrogen bonds per DA (H bonds, black) and the number of intermolecular hydrogen bonds per DA (red) for the relaxed nanotube A (A), nanotube B (B), nanotube B350K (B350K) and random system (R). B. The percentage of the intermolecular hydrogen bonds.
The confined water channel within the center of the nanotube results in interesting water dynamics and solvation structure. The hydrophobic CPTs form the inner wall of the nanotube of ‘qCPT-buSS-Tau’ with a diameter of 20~25 Å. In-tube water diffuses faster than the surface water, but slower than the bulk water (Figure 4A). The diffusion coefficients for the bulk water, in-tube water, and surface water (within 4 A from the outer surface of the nanotube) are 3.2×10−9, 2.2×10−9 and 0.9×10−9 m2/s, respectively. The slower lateral diffusion of water on the surface of proteins is well-known.26 Changes of water diffusion in a confined space like cyclic peptide nanotubes have also been observed with MD simulation15. Moreover, recently-reported water dynamics in supramolecular nanostructures27 are consistent with our results. The radial distribution function reveals that the in-tube water is slightly more ordered than the bulk water. The water in the first solvation shell prefers an orientation where more hydrogens face the CPTs and significant less oxygens face on the inner-surface of the tube (Figure 4C and D). This kind of reinforcement of water-to-water interaction due to the hydrophobic environment has been reported as hydrophobic hydration28. Here we demonstrate that confined water channels in self-assembled peptide structures may display complex and interesting water structure and dynamics.
Figure 4.
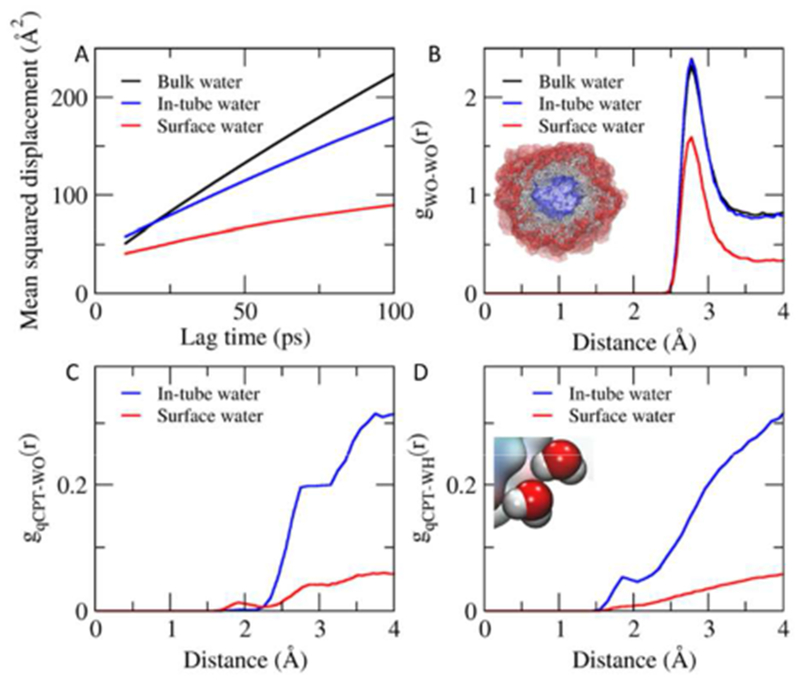
Water in the nanotube. A. Mean squared displacement vs. lag time. B. Radial distribution function of water oxygen-water oxygen. C. Radial distribution function of CPT-water oxygen. D. Radial distribution function of CPT- water hydrogen.
The π-π stacking between the planar CPTs contributes to the 1D growth of the clusters of ‘qCPT-buSS-Tau’ from the early stages of the assembly. Our 400 ns simulation of the random system of ‘qCPT-buSS-Tau’ displays significant CPT stacking that is mostly perpendicular to the longer axis of the central cluster that emerges (Figure 5B). This is consistent with the molecular self-assembly pathway of the ‘mCPT-buSS-Tau’. When the contact distance between atoms in CPT of ‘qCPT-buSS-Tau’ molecules is less than 4.5 Å, it is considered as a CPT cluster. The average size of the CPT clusters continues to grow, indicating the rearrangement of CPTs within the clusters (Figure 5C). The final distribution of angles between CPT planes within 6 Å shows a significant increase, compared to the initial state, and displays two clear peaks near 0° and 180°, indicating the formation of strong stacking in the early stage (< 400 ns) of the self-assembly.
Figure 5.
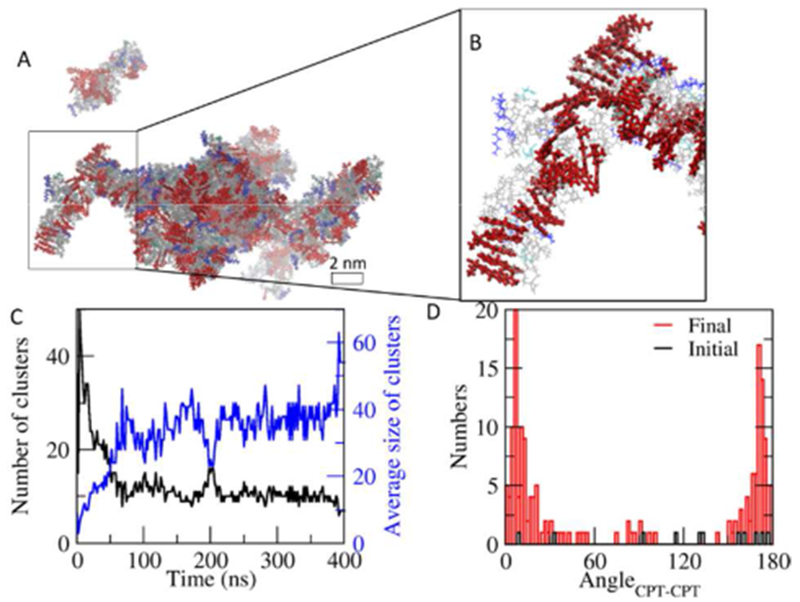
Aggregation in the random system. A. Snapshot at 400 ns. Colors of each component are consistent with those in Figure 1. B. Stacking of CPTs in a cluster. C. The number and the average size of CPT clusters over time. D. Distribution of angles between CPTs within 6 Å at the initial and the final frame.
Herein we report on long-time MD simulations to probe the balance of intermolecular forces governing the stability of supramolecular nanotubes formed by drug amphiphiles. π-π stacking between hydrophobic cancer drugs (CPTs) remains strong within the nanotube core, but the contribution of electrostatics to the stability of the nanotube structure decreases compared to the nanofilament structure, mainly due to the increase in volume of the hydrophobic block. These results provide useful insights into the rational design of functional supramolecular assemblies. In particular, these results suggest that future designs of peptide amphiphiles should consider the balance of hydrophilic charged components with the volume of the hydrophobic block as an important parameter when considering molecular design, in particular with respect to the design of nanotubes. Particularly challenging would be to characterize the stability and mechanical properties of the nanotube vs. filament morphology at the micron scale using specialized coarse-grain molecular dynamics force fields29–30. However, when using simulation techniques with lower resolution, the complex hydration structure of the assembly is lost. These results suggest that the exposure of water to the hydrophobic inner wall of the nanotube induces provocative changes in the local solvation structure, as well as the water dynamics. Shifts in the local solvation structure and water dynamics cannot be captured with more mesoscale simulation techniques. Thus, a multi-scale simulation approach must be used to more fully characterize these unique assemblies.
Furthermore, these results suggest that supramolecular peptide assemblies can be further explored as engineered water channels. Tubular structures such as microtubules31 or the Tobacco Mosaic Virus32 are commonly found in nature. Self-assembly into chiral tubular structures has been suggested to be driven by a balance of both lateral and longitudinal contacts at the monomer level33. An in-depth understanding of the specific intermolecular forces that drive the self-assembly of tubular self-assemblies will provide insight into the molecular design of supramolecular nanotubes.
Supplementary Material
ACKNOWLEDGMENT
S.M.L. acknowledges support from the NIH (R15EB020343-01A1). In addition, this research was supported, in part, by the NSF through grant 1506937 as well as XSEDE resources under grant number TG-CHE130099 and a grant of computer time from the City University of New York High Performance Computing Center under NSF Grants CNS-0855217, CNS-0958379 and ACI-1126113. S.M.L. also acknowledges start-up funding received from College of Staten Island and City University of New York. M. K. also acknowledges the partial support from the Rosemary O’Halloran scholarship from College of Staten Island.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Detailed simulation methods, summary of the simulated systems, initial conformations of DAs, widths of the nanotubes and decomposition of CPT stackings.
Authors declared no competing financial interest.
REFERENCES
- 1.Aida T; Meijer EW; Stupp SI, Functional Supramolecular Polymers. Science 2012, 335, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kokkoli E; Mardilovich A; Wedekind A; Rexeisen EL; Garg A; Craig JA, Self-Assembly and Applications of Biomimetic and Bioactive Peptide-Amphiphiles. Soft Matter 2006, 2, 1015–1024. [DOI] [PubMed] [Google Scholar]
- 3.Matson JB; Stupp SI, Self-Assembling Peptide Scaffolds for Regenerative Medicine. Chem. Commun 2012, 48, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheetham AG; Zhang PC; Lin YA; Lock LL; Cui HG, Supramolecular Nanostructures Formed by Anticancer Drug Assembly. J. Am. Chem. Soc 2013, 135, 2907–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Webber MJ; Appel EA; Meijer EW; Langer R, Supramolecular Biomaterials. Nat. Mater 2016, 15, 13–26. [DOI] [PubMed] [Google Scholar]
- 6.Webber MJ; Langer R, Drug Delivery by Supramolecular Design. Chem. Soc. Rev 2017, 46, 6600–6620. [DOI] [PubMed] [Google Scholar]
- 7.Manandhar A; Kang M; Chakraborty K; Tang PK; Loverde SM, Molecular Simulations of Peptide Amphiphiles. Org. Biomol. Chem 2017, 15, 7993–8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee OS; Stupp SI; Schatz GC, Atomistic Molecular Dynamics Simulations of Peptide Amphiphile Self-Assembly into Cylindrical Nanofibers. J. Am. Chem. Soc 2011, 133, 3677–3683. [DOI] [PubMed] [Google Scholar]
- 9.Velichko YS; Stupp SI; de la Cruz MO, Molecular Simulation Study of Peptide Amphiphile Self-Assembly. J. Phys. Chem. B 2008, 112, 2326–2334. [DOI] [PubMed] [Google Scholar]
- 10.Tsonchev S; Schatz GC; Ratner MA, Electrostatically-Directed Self-Assembly of Cylindrical Peptide Amphiphile Nanostructures. J. Phys. Chem. B 2004, 108, 8817–8822. [Google Scholar]
- 11.Lee OS; Liu YM; Schatz GC, Molecular Dynamics Simulation of Beta-Sheet Formation in Self-Assembled Peptide Amphiphile Fibers. J. Nanopart. Res 2012, 14. [Google Scholar]
- 12.Lee OS; Cho V; Schatz GC, Modeling the Self-Assembly of Peptide Amphiphiles into Fibers Using Coarse-Grained Molecular Dynamics. Nano Lett. 2012, 12, 4907–4913. [DOI] [PubMed] [Google Scholar]
- 13.Kang M; Zhang P; Cui H; Loverde SM, Π–Π Stacking Mediated Chirality in Functional Supramolecular Filaments. Macromolecules 2016, 49, 994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aggeli A; Nyrkova IA; Bell M; Harding R; Carrick L; McLeish TCB; Semenov AN; Boden N, Hierarchical Self-Assembly of Chiral Rod-Like Molecules as a Model for Peptide Beta-Sheet Tapes, Ribbons, Fibrils, and Fibers. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 11857–11862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engels M; Bashford D; Ghadiri MR, Structure and Dynamics of Self-Assembling Peptide Nanotubes and the Channel-Mediated Water Organization and Self-Diffusion - a Molecular-Dynamics Study. J. Am. Chem. Soc 1995, 117, 9151–9158. [Google Scholar]
- 16.Huang Z; Kang SK; Banno M; Yamaguchi T; Lee D; Seok C; Yashima E; Lee M, Pulsating Tubules from Noncovalent Macrocycles. Science 2012, 337, 1521–1526. [DOI] [PubMed] [Google Scholar]
- 17.Pizzey CL; Pomerantz WC; Sung BJ; Yuwono VM; Gellman SH; Hartgerink JD; Yethiraj A; Abbott NL, Characterization of Nanofibers Formed by Self-Assembly of Beta-Peptide Oligomers Using Small Angle X-Ray Scattering. J. Chem. Phys 2008, 129. [DOI] [PubMed] [Google Scholar]
- 18.Ghadiri MR; Granja JR; Milligan RA; McRee DE; Khazanovich N, Self-Assembling Organic Nanotubes Based on a Cyclic Peptide Architecture. Nature 1993, 366, 324–327. [DOI] [PubMed] [Google Scholar]
- 19.Horne WS; Stout CD; Ghadiri MR, A Heterocyclic Peptide Nanotube. J. Am. Chem. Soc 2003, 125, 9372–9376. [DOI] [PubMed] [Google Scholar]
- 20.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- 21.Wang JM; Wolf RM; Caldwell JW; Kollman PA; Case DA, Development and Testing of a General Amber Force Field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- 22.Gilson MK; Gilson HSR; Potter MJ, Fast Assignment of Accurate Partial Atomic Charges: An Electronegativity Equalization Method That Accounts for Alternate Resonance Forms. J. Chem. Inf. Comput. Sci 2003, 43, 1982–1997. [DOI] [PubMed] [Google Scholar]
- 23.Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kale L; Schulten K, Scalable Molecular Dynamics with Namd. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheetham AG; Zhang PC; Lin Y-A; Lock LL; Cui HG, Supramolecular Nanostructures Formed by Anticancer Drug Assembly. J. Am. Chem. Soc 2013, 135, 2907–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tantakitti F; Boekhoven J; Wang X; Kazantsev RV; Yu T; Li JH; Zhuang E; Zandi R; Ortony JH; Newcomb CJ; Palmer LC; Shekhawat GS; de la Cruz MO; Schatz GC; Stupp SI, Energy Landscapes and Functions of Supramolecular Systems. Nat. Mater 2016, 15, 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang M; Roberts C; Cheng Y; Chang C.-e. A., Gating and Intermolecular Interactions in Ligand-Protein Association: Coarse-Grained Modeling of Hiv-1 Protease. J. Chem. Theory Comput. 2011, 7, 3438–3446. [DOI] [PubMed] [Google Scholar]
- 27.Ortony JH; Qiao B; Newcomb CJ; Keller TJ; Palmer LC; Deiss-Yehiely E; Olvera de la Cruz M; Han S; Stupp SI, Water Dynamics from the Surface to the Interior of a Supramolecular Nanostructure. J. Am. Chem. Soc 2017. [DOI] [PubMed] [Google Scholar]
- 28.Garde S; Hummer G; García AE; Paulaitis ME; Pratt LR, Origin of Entropy Convergence in Hydrophobic Hydration and Protein Folding. Phys. Rev. Lett 1996, 77, 4966–4968. [DOI] [PubMed] [Google Scholar]
- 29.Kang M; Cui H; Loverde SM, Coarse-Grained Molecular Dynamics Studies of the Structure and Stability of Peptide-Based Drug Amphiphile Filaments. Soft Matter 2017, 13, 7721–7730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shinoda W; DeVane R; Klein ML, Multi-Property Fitting and Parameterization of a Coarse Grained Model for Aqueous Surfactants Mol. Simul. 2007, 33, 27–36. [Google Scholar]
- 31.Alushin GM; Lander GC; Kellogg EH; Zhang R; Baker D; Nogales E; Ludtke SJ; Pechatnikova E; Wilson-Kubalek EM; Whittaker M; al e., High-Resolution Microtubule Structures Reveal the Structural Transitions In ??-Tubulin Upon Gtp Hydrolysis. Cell 2014, 157, 1117–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klug A, The Tobacco Mosaic Virus Particle: Structure and Assembly. Philos. Trans. R. Soc., B 1999, 354, 531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng S; Stevens MJ, Self-Assembly of Chiral Tubules. Soft Matter 2014, 10, 510–518. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.