

Abstract
Histone variants are known to play a central role in genome regulation and maintenance. However, many variants are inaccessible by antibody-based methods or bottom-up tandem mass spectrometry due to their highly similar sequences. For many, the only tractable approach is with intact protein top-down tandem mass spectrometry. Here, ultrahigh-resolution FT-ICR MS and MS/MS yield quantitative relative abundances of all detected HeLa H2A and H2B isobaric and isomeric variants with a label-free approach. We extend the analysis to identify and relatively quantitate 16 proteoforms from 12 sequence variants of histone H2A and 10 proteoforms of histone H2B from three other cell lines: human embryonic stem cells (WA09), U937, and a prostate cancer cell line LaZ. The top-down MS/MS approach provides a path forward for more extensive elucidation of the biological role of many previously unstudied histone variants and post-translational modifications.
Keywords: FT-ICR, FTMS, Proteoform, Histone, Post-translational Modification, Sequence Variants
FOR TOC ONLY
Introduction
The physiological state of the eukaryotic genome is chromatin, a protein-DNA complex. The nucleosome, the primary repeating unit of chromatin, consists of approximately 146 base pairs of DNA wrapped twice around a histone octomer core. The octomer contains 2 copies each of histones H2A, H2B, H3, and H4, also known as core histones. Histones play an essential role in dynamic gene regulation (i.e., transient changes in gene expression)[1–3], epigenetic inheritance of cellular phenotype (i.e., cellular differentiation)[4–5], genomic stability [6], and other genetic activities through post-translational modifications (PTMs) and variations in primary sequence. It is useful to define a “proteoform” as “a chemically distinct combination of post-translational modifications of a protein molecule”. A proteoform thus provides the most accurate description of a protein, including primary amino acid sequence variants as well as the identities and sites of post-translational modifications [7].
Histones H2A and H2B differ from histones H3 and H4 by their greater diversity in sequence and highly dynamic exchange to and from nucleosomes [8]. In humans, among at least 17 members in the H2A gene family and 19 in the H2B family (Figure 1), some have very similar primary sequences, whereas others, such as H2A.Z, H2A.X, and H2BFS exhibit significantly different sequences. Unlike histones H3 and H4, whose functions are mainly determined by PTMs, histones H2A and H2B affect cellular events through both PTMs and sequence variation [9–13].
Figure 1.

Sequence variants of human HeLa histone H2A (top) and H2B (bottom). (www.uniprot.org) Variants in bold have been previously identified in HeLa cells. (www.uniprot.org)
The most-studied histone H2A variants are H2A.V, H2A.Z, H2A.X, H2Abdb, and Macro H2A [14–15]. However, most other H2A variants, the so-called canonical H2A histones, are not well characterized, partially due to high similarity in primary amino acid sequence. As for canonical histone H2A, the variants of the histone H2B family are even less studied because those variants only differ by one or a few amino acids over an average span of 125 AA. Furthermore, their sequence differences frequently occur near both the N- and C-termini. Thus, the complete sequence of the intact histone is required to distinguish the various proteoforms, limiting the success of bottom-up and middle-down MS/MS to separate, identify, and quantitate these variants. The high sequence homology also makes specific antibodies, that do not cross react with other variants, difficult to produce. Most traditional physical separations are incapable of resolving these variants as well [16–18]. Therefore, top-down MS/MS is the best way to characterize these previously unstudied variants.
A top-down MS/MS experiment begins by isolating precursor ions of each (or very few) intact proteoforms. Fragmentation of a given proteoform potentially serves to distinguish and identify each of the variants; however, this requires that there is at least one cleavage between the amino acid positions that differ between the variants [19]. Previous top-down MS/MS studies in the Kelleher lab identified 10 proteoforms of H2A and 7 proteoforms of H2B in HeLa cells based on 8.5 T Fourier transform ion cyclotron resonance (FT-ICR) and combined ECD and IRMPD fragmentation with limited sequence coverage [20–21]. More recent results from the Garcia lab reported more histone H2B proteoforms with limited quantitative information achieved by bottom-up mass spectrometry with an Orbitrap Fusion with front-end electron transfer dissociation (fETD) [22]. The most recent work by the Hunt group identified 3 histone H2A variants with an LC-coupled Orbitrap Velos Pro with fETD [23].
As described below, we employed a 9.4T FT-ICR instrument with SWIFT isolation and electron capture dissocition (ECD) MS/MS, and a 14.5 T FT-ICR with fETD MS/MS to provide a more comprehensive, relative quantitative analysis for histones H2A and H2B from a range of cell lines. With our new developed proteoform assignment method and 3-level relative quantitation, we identify 16 proteoforms from 12 sequence variants of histone H2A and 10 proteoforms of histone H2B, with an average ~70% sequence coverage and their relative ion abundances. We also provide the first convincing evidence for the co-existence of the isobaric or near isobaric unmodified histone variants H2B1C, H2B1J, and H2B1O, via ultrahigh resolution splitting of deep z-ions.
Materials and Methods
Cell Culture and Histone Sample Preparation
HeLa S3 cells (American Type Culture Collection, Manassas, VA, USA) were maintained in Joklik’s modified MEM media and harvested on reaching a density of 107 cells/mL. ARCaPE cells (LacZ) were maintained at 37C with 5% CO2 in T-media (Invitrogen) supplemented with 5% heat-inactivated FBS and 1% Pen/Strep. They were subcultured to approximately 80% confluency prior to harvest for nuclei isolation. [33–35] WA09 hESCs were grown in media containing HEREGULIN β1 (10 ng/ml; Peprotech), ACTIVIN A (10 ng/ml; R&D Systems), and human LONGR3 insulin-like growth factor 1 (IGF-1; 200 ng/ml; Sigma) and harvested at confluency. [36–37] The U937 (ATCC® CRL-1593.2™) and U1 (NIH AIDS Reagent Program #165) pleural effusion, lymphocyte cell line was grown in a humidified incubator at 37ºC and 5% CO2. Cells were grown in RPMI1640 supplemented with 10% fetal bovine serum and 30 ng/mL Gentamicin.
Cell nuclei were prepared by use of nuclei isolation buffer (15 mM tris HCl, 60 mM KCl, 15 mM NaCl, 5 mm MgCl2, 1 mM CaCl2, 250 mM sucrose and 0.3% NP-40, to which 1 mM dithiothreitol, 10 nM microcystin, 0.5 mM 4-(2-aminoethyl) benzenesulfonyl fluoride, and 10 mM sodium butyrate were added as inhibitors.) Histones were then obtained by standard acid extraction with 0.2 M H2SO4 at 4 ºC for 2 h under rotation. After centrifugation at 3400 g, raw histones were precipitated from the supernatant by addition of 25% trichloroacetic acid (w/v) on ice for 45 min and then air-dried after washing with acetone.
Raw histones were further purified by reversed-phase high-performance liquid chromatography (RP-HPLC, Thermo Ultimate 3000 system, Thermo Fisher Scientific, San Jose, CA) with a Vydac C18 column (218TP54, 250 mm × 4.6 mm, Grace, Deerfield, IL) and a linear gradient (30% B to 60% B gradient with 0.3% B/min increase at 0.8 mL/min flow rate (Buffer A = 5% acetonitrile:water with 0.2% TFA; Buffer B = 95% acetonitrile:water with 0.188% TFA). Histone H2A eluted as 3 fractions at 42.0% B, 43.4%, and 43.6% B. Histone H2B eluted at 40.8% B. All fractions were collected automatically and dried under vacuum.
9.4 T FT-ICR Mass Spectrometry
Samples were dissolved in 50% acetonitrile, 1% formic acid solution at a final concentration of approximately 25 µg/mL and ionized by positive nanoelectrospray at a flow rate of 0.4 nL/min. A custom-built 9.4 T Fourier transform ion cyclotron resonance mass spectrometer provided both broadband and tandem mass spectra. ECD precursor ions were isolated externally with a quadrupole mass filter and internally by stored waveform inverse Fourier transform (SWIFT) excitation in the ICR cell to within a nominal mass-to-charge ratio window of width, m/z ≈ 1 [24–25]. ECD experiments were performed at an electron beam energy of 12 eV, and 200–500 time-domain transients were digitized and signal-averaged. Mass calibration was achieved externally by two-term frequency-to-m/z conversion [26–27] from Pierce LTQ ESI Positive Ion Calibration Mix. (Thermo Fisher Scientific, San Jose, CA)
14.5 T FT-ICR Mass Spectrometry
Samples were dissolved in 50% acetonitrile, 1% formic acid solution at a final concentration of 25 µg /mL and ionized via positive nanoelectrospray at a flow rate of 0.4 nL/min. A custom-built 14.5T FT-ICR mass spectrometer with Velos Pro front end (Thermo Fisher Scientific, San Jose, CA) provided both broadband and tandem mass spectra. For optimal isolation ETD precursor ions at m/z 810.3 were isolated twice progressively, first with a width of m/z 2.0 and then m/z 1.5, each centered at slightly different isolation centers. Front-end electron transfer dissociation (fETD) was performed with 3 ms reaction period and resolving power, m/Δm50% ≈ 1,500,000 [28]. Mass calibration was performed as for the 9.4 T experiments.
Spectra Analysis and Annotation
Custom software was used to identify variants in the UniProt on-line protein database [29]. Briefly, for each proteoform candidate, the possible fragment ions were enumerated and their natural abundance isotopic distributions calculated. The spectra were then compared to the most abundant peak of the isotopic distribution for each fragment ion within 3 ppm mass error. Neighboring members of the same isotopic distribution were also compared within 1.5 ppm error relative to the most abundant in order to verify the proper charge state and reasonable isotopic distribution. The peak-picking threshold is 10σ of baseline noise. The abundances of identified fragment ions were added together as the Objective Function to determine the quality of the assignment for each proteoform candidate. Assigned fragment ions were later verified manually for the MS/MS product ions.
Results and Discussion
Proteoform Identification.
For each histone fraction collected from the LC-separation (Supplementary Material Figure S1), a high-resolution mass spectrum was collected for accurate intact protein mass measurement (Figure 2A). Each isotopic distribution may contain one or a few histone proteoforms. Because stored waveform inverse Fourier transform (SWIFT) was applied after quadrupole isolation, each MS/MS precursor was limited to only one isotopic distribution and therefore interference was eliminated from neighboring distribution(s) in the MS/MS product ion spectrum (Figure 2B).
Figure 2.

Work flow for the identification and relative quantitation of histone H2B proteoforms from HeLa cells. A) Intact protein mass spectral segment for the 17+ charge state. Each isotopic distribution may present more than one H2B proteoform. B) Precursor ions of interest were isolated by an external quadrupole mass filter followed by in-cell SWIFT mass-selective excitation to within a nominal m/z range of ~1. C) Representative electron capture dissociation (ECD) product ion spectrum. Fragments are assigned based on a custom-input proteoform database by our custom software without prior deconvolution. Proteoform identification was then confirmed manually. D) Relative abundance ratios for corresponding fragments of different proteoforms from a given isolated precursor. E) Partial results of relative quantitation of histone H2B proteoforms.
Figure 1 shows the sequence similarity for canonical histones H2A and H2B. There are 16 possible single amino acid variant sites for histone H2A and 14 sites for histone H2B, providing critical checkpoints for proteoform identification. All MS/MS product ion spectra were searched against the Uniprot histone H2A/H2B sequence database [29] to identify each proteoform, with consideration of possible PTMs and their combinations. A proteoform was identified according to two criteria: A) The intact protein mass was within 3 ppm mass error tolerance window of the calculated value, except if two or more proteoforms with small mass differences overlapped and were unresolved in the broadband mass spectrum. (Supplementary Material Table S1) B) At least two fragment cleavages were observed between any two critical checkpoints that distinguish proteoforms in the MS/MS product ion mass spectrum, except for the adjacent checkpoints or the checkpoints near a terminus. In case three or more proteoforms co-existed in the same MS/MS product ion spectrum, more careful data interpretation is needed as explained below.
Single charge state segments of the broadband spectra for histone fractions H2A-1, H2A-2a, H2A-2b, and H2B are shown in Figure 3. The molecular weight for the highest magnitude isotopic peak from the most abundant isotopic distribution in the mass spectrum of H2A-1 was 14005.88 Da, which matches only proteoform H2A2A-Nαac (i.e., N-terminally acetylated H2A2A) within ±3 Da mass error tolerance when searched against all possible proteoforms. We chose the large ±3 Da window to prevent elimination of possible low-abundance proteoform candidates when they co-exist with more abundant proteoforms but exhibit slightly different mass and thus are unresolved in the broadband mass spectrum. The ECD MS/MS product ion spectrum matched the same proteoform assignment with 80% sequence coverage and thus met the second criterion. (Supplementary Material Figure S2). Also, the calculated mass of proteoform H2A2A-Nαac was within 3 ppm mass tolerance relative to the experimental value and met the first criterion.
Figure 3.

ESI 9.4 T FT-ICR mass spectral segments from histone fractions H2A1, H2A2a, H2A2b, and H2B from asynchronous HeLa cells. Proteoforms identified by ECD MS/MS are labeled. Because H2A fraction 2a (containing 2 major proteoforms) and H2A fraction 2b (containing 5 major proteoforms) were not baseline-resolved by RP-HPLC, proteoform H2A1D Nα-ac and H2A2B Nα-ac are present in both fractions.
If two or more proteoforms were present in the same isotopic distribution, (e.g., the most abundant isotopic distribution from H2B), the 3 ppm error rule was not applied to the minor proteoforms because accurate mass measurement is compromised by interference from the more abundant proteoforms. However, the second criterion assures the confidence of the assignment using this strategy. We identified 10 proteoforms: H2A.V, H2A.Z, H2A2A-Nα-ac, H2A2C-Nα-ac, H2A1-Nα-ac, H2A1B-Nα-ac, H2A1C-Nα-ac, H2A1D-Nα-ac, H2A1J-Nα-ac, and H2A1H-Nα-ac for histone H2A from HeLa. H2A1B-Nα-ac and H2A1D-Nα-ac were found in both histone H2A fractions 2a and 2b, because those fractions were not baseline-resolved. Nine proteoforms were identified: H2B1K, H2B1C, H2B1J, H2B1O, H2B1N, H2B2E, H2B2F, H2B1D, and H2B1B for histone H2B from HeLa. No PTMs were detected in any of the H2B variants. This is in concordance with a previous study by the Kelleher lab of histone H2B in HeLa [21].
MS/MS sequence coverage is typically reported either (A) as the percentage of cleavage sites, and (B) the percentage of cleavage fragments. A cleavage site is the chemical bond between any two adjacent amino acids that can break during MS/MS experiments. The number of possible cleavage sites of a protein is one less than the number of amino acid in the sequence. Most software calculates the type A coverage, which focuses more on how many sites are missing in the MS/MS experiments. However, type B coverage better represents top-down proteomics, because a protein is most reliably identified if both complementary fragment ions are confirmed. For example, when 2 proteoforms that differ only at one amino acid residue (H2A1-Nαac and H2A1D-Nαac) are assigned to the same MS/MS product ion spectrum (the m/z 876 isotopic distribution from fraction H2A2b), the type A coverage for both proteoform is the same (80%). However, H2A1-Nαac yielded a much higher type B coverage (63% compared to H2B1D’s 43%). Also, only H2A1-Nα-ac was within the 3 ppm mass error limit (see Supplementary Material Figure S3).
3-level Relative Quantitation.
Label-free relative quantitation for histones H2A and H2B was carried out at three levels (Figure 4). The first level was based on the RP-HPLC chromatogram (not shown in Figure 4). Histone H2A eluted as 3 fractions from the RP-HPLC: H2A-1, H2A-2a, and H2A-2b, whereas H2A-2a and H2A-2b were not baseline-resolved. We used normalized UV absorption peak areas to represent the relative amount of protein in each peak under the assumption that each sequence variant exhibits the same molar absorptivity--a valid approximation due to their high sequence similarity. Histone H2B appeared in only one fraction; therefore, no LC-level quantitation was applied.
Figure 4.

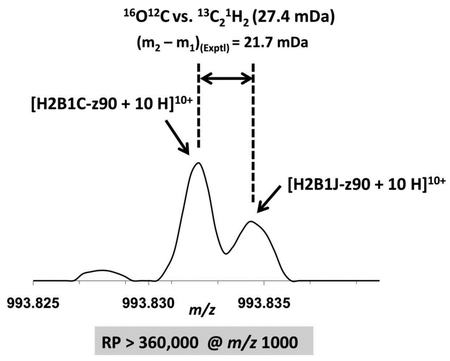
A) Sequences and putative ECD fragments for H2B1C, H2B1J, and H2B1O. Because of very small mass differences (~2 Da), these species will be isolated and fragmented together. Segments in orange or purple represent regions for which ECD fragments are identical in mass for 2 proteoforms. These three species yield unique fragments (segments in light blue, dark blue, and gray) only in the region z87-z124.
B) Mass spectral segment for the z90 10+ ions of H2B1C and H2B1J to show the resolving power required for performing reliable top-down ECD MS/MS. The mass difference between H2B1C-z90 and H2B1J-z90 is the difference between 16O and 12C1H2 (1.97926 Da) and the split at m/z 993.83 is due to the small mass difference between 16O12C2and 13C212C1H2 (27.4 mDa). Note that the minimum required resolving power at m/z 1,000 is 360,000.
C) Mass scale-expanded segment, showing the narrow split at m/z 993.83 between 16O12C2 and 13C212C1H2 (27.4 mDa).
CD) Abundance ratio for the c ions (orange crosses) or z ions (blue squares) generated from H2B1C, H2B1J, or H2B1O for the m/z 811 precursor from HeLa cells. The ratio for the c-ion and z-ion series spanning AA1 to AA38 represents the ratio between the sum of H2B1C and H2B1J to H2B1O, whereas those in the range of AA39 to AA125 reflect the ratio between H2B1J and the sum of H2B1C and H2B1O. From the expressions at bottom left, the relative abundances of H2B1C, H2B1J, and H2B1O were calculated to be 3.2:1.0:0.82, in agreement with the numbers derived from the resolved peaks in Figure 4B.
The next level was based on the intact protein broadband mass spectra. As shown in Figure 3D, 5 isotopic distributions were above 5% in relative abundance, and each contained one or a few proteoforms. They were weighted based on their normalized mass spectral peak heights (Figure 2E). If more than one proteoforms were observed in a given isotopic distribution, the peak height was then considered as the sum of all unresolved variants and their contributions were further weighted based on MS/MS assignments.
Figure 5.

Relative abundances of histones H2A (top) and H2B (bottom) variants for HeLa cells, human embryonic stem cell line (WA09), U937 cell line, and prostate cancer cell line LaZ.
ECD MS/MS fragment ions were used to quantitate co-isolated variants. For example, the isotopic distribution centered at m/z 812 contained 3 proteoforms (H2B1N, H2B2E, and H2B2F), each with unique c4-c38 ions. The ratios of those c ions thus represent the relative abundances of their precursor proteins (Figure 3D).
Advantages of High Resolving Power.
High mass resolving power and high mass accuracy enable accurate intact protein mass measurement and validate proteoform assignments (Supplementary Material Table S1). Moreover, resolving power is crucial for identifying highly similar proteoforms. Three sequence variants were identified from the isotopic distribution centered at m/z 811 in Figure 3D by the two previously described criteria: H2B1C, H2B1J, and H2B1O. H2B1C and H2B1O are structural isomers (13775 Da) and H2B1J (13773 Da) is 2 Da lighter (O vs. CH2) but could not be resolved from the others in the broadband mass spectrum due to its low abundance. If 3 or more proteoforms co-exist in the same isotopic distribution, we look further for critical fragments (i.e., fragments unique to only one of the proteoforms) to confirm the presence of each proteoform [30]. In this case, H2B1C would constitute a false positive. Fortunately, the ultrahigh resolving power of FT-ICR MS and MS/MS enables confident verification of the co-existence of all 3 proteoforms.
H2B1O, H2B1C, and H2B1J differ at 3 sites: 2, 39, and 124 (Figure 4A). As a result, H2B1C yields c3-c38 ions (shown in orange) identical to H2B1J and z3-z86 ions (also shown in orange) identical to H2B1O. H2B1C also has c39-c129 ions (shown in purple) isomeric with H2B1O and thus indistinguishable by MS/MS. Therefore, only z87-z124 ions are critical fragments for this group and are essential to verify the existence of all 3 proteoforms. In this region, z ions from H2B1O are 14 Da heavier than those from H2B1C and 16 Da heavier than those from H2B1J, and thus easily identified. However, z ions from H2B1C are only 2 Da heavier than those from H2B1J. (16O vs 12C1H2). Because of these are interlaced isotopic distributions, the separation between 2 peaks in the MS/MS product ion spectrum is only 27.4 mDa (16O12C vs. 13C21H2). With the ultrahigh resolving power of our 14.5 T FT-ICR instrument, we successfully resolved z90 ions of H2B1J from H2B1C (Figure 4B) and thereby confirmed the existence of both proteoforms. Z90 ions from H2B1C are 2 Da heavier and three times more abundant. If the ions from the two peaks at m/z 993.83 were equally abundant, the minimum mass resolving power to barely resolve them would be 360,000 at m/z 1000, which is equivalent to ~1M resolving power at m/z 400. However, in this sample, H2B1C was ~3 times as abundant as H2B1J, so that even higher resolving power is required.
However, there were not enough critical peaks to confidently show statistical significance for the quantification of histones H2B1C, H2B1J, and H2B1O. Therefore, we also calculated the ratios of their non-critical fragments (Figure 4D). Note the significant discontinuity at amino acid 39 in the graph, due to the presence of H2B1C: before AA39 the ratio is between the sum of H2B1C and H2B1J to H2B1O and after AA39 the ratio is between H2B1J and the sum of H2B1C and H2B1O. We were thus able to derive relative abundance ratios by solving the system of equations: H2B1J + H2B1C = 5.75 × H2B1O and 0.257 × (H2B1O+H2B1C) = H2B1J. The relative abundances of H2B1C:H2B1J:H2B1O were calculated to be 3.2:1.0:0.72, which match the numbers derived from the resolved critical peaks in Figure 4B.
Relative Quantitation of Histone H2A/H2B Variants from 4 Different Cell Lines.
We proceeded to apply the same method to human embryonic stem cells (WA09), U937 cell line, and a prostate cancer cell line LaZ. The results are summarized in Figure 5. 16 proteoforms from 12 sequence variants of histone H2A and 10 proteoforms of histone H2B were identified and relatively quantitated by a combination of RP-HPLC, high-resolution FT-ICR MS, and top-down FT-ICR MS/MS. Histone H2A2A and H2A1 are the most abundant variants for all four cell lines and contribute to more than 50% of all histone H2A. Some variants, such as H2A2A, H2A1D, H2A2C do not vary much across cell lines whereas others show great diversity, such as H2A.Z, H2A.V, H2A1J, and H2A1H. Note that H2A variant H2A1H (both H2A1H_Nαac and H2A1H_NαacK5ac) is much more abundant in stem cells than in the other three cell lines. Stem cells in general exhibit a higher abundance of lysine 5 (K5) acetylation, which has been previously associated with transcriptional activation [31]. U937 cells are more heavily phosphorylated at serine 1 (S1). S1 phosphorylation in canonical histone H2A has been proven to relate to transcription repression [32]. We identified S1 phosphorylation in both H2A2A and H2A1, and they exhibit a similar percentage of phosphorylation. Our top-down method is capable of studying the sequence-dependence of individual PTMs, a phenomenon we have observed previously; however, in this case these analogous phosphorylation sites behave identically. Histone H2B variant abundances varied markedly across the four cell lines, and only variant H2B2F abundance was conserved across all of the cell lines. There is limited knowledge of the function of different H2B variants generally. For example, the Dulac lab discovered that histone H2B variant H2BE affects olfactory function in mice [10]. However, the functions of H2B variants are not clear in humans. Our top-down method provides a path to understanding the biological function of both canonical H2A and H2B variants.
Supplementary Material
Acknowledgments
This work was supported by NSF Division of Materials Research through DMR-06–54118, NIH General Medical Sciences through P01GM85354, and the State of Florida.
Footnotes
Supporting Information:
The following files are available free of charge at ACS website http://pubs.acs.org: Supplementary Table S1 Table of measured intact protein mass. Supplementary Figures S1-S3 LC Chromatography of off-line histone fractionation, ion map of the assignment, and sequence coverage.
References
- [1].Jenuwein T; Allis CD Translating the histone code. Science 2001, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]; Berger SL histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev 2002, 12, 142–148. [DOI] [PubMed] [Google Scholar]
- [2].Biel M; Wascholowski V; Giannis A Epigenetics-an epicenter of gene regulation: histones and histone-modifying enzymes. Angew. Chem. Int. Edit 2005, 44, 3186–3216. [DOI] [PubMed] [Google Scholar]
- [3].Bannister A; Kouzarides T Regulation of chromatin by histone modifications. Cell Res 2011, 21, 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Reik W Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [DOI] [PubMed] [Google Scholar]
- [5].Liu C; Liu L; Shan J; Shen J; Xu Y; Zhang Q; Yang Z; Wu L; Xia F; Bie P; Cui Y; Zhang X; Bian X; Quan C Histone deacetylase 3 participates in self-renewal of liver cancer stem cells through histone modification. Cancer Letters 2013, 339, 60–69. [DOI] [PubMed] [Google Scholar]
- [6].Groth A; Rocha W; Verreault A; Almouzni G Chromatin challenges during DNA replication and repair. Cell 2007, 128, 721–733. [DOI] [PubMed] [Google Scholar]
- [7].Smith LM; Kelleher NL, and The Consortium for Top Down Proteomics Proteoform: a single term describing protein complexity. Nature Methods 2013, 10, 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xu M; Long C; Chen X, Huang C; Chen S; Zhu B Partitioning of Histone H3-H4 Tetramers During DNA Replication–Dependent Chromatin. Science, 2010, 328, 94–98. [DOI] [PubMed] [Google Scholar]
- [9].Banaszynski LA; Allis CD; Lewis PW Histone variants in metazoan development. Dev. Cell 2010, 19, 662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Santoro SW; Dulac C The activity-dependent histone variant H2BE modulates the life span of olfactory neurons. eLife 2012, 1: e00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tessarz P; Santos-Rosa H; Robson SC; Sylvestersen KB; Nelson CJ; Nielsen ML; Kouzarides T Glutamine methylation in histone H2A is an RNA-polymerase-I-dedicated modification. Nature 2014, 505, 564–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Maze I; Noh K; Soshnev AA; Allis CD Every amino acid matters: essential contributions of histone variants to mammalian development and disease. Nature Rev. Genet 2014, 15, 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mahajan K; Fang B; Koomen JM; Mahajan NP H2B Tyr37 phosphorylation suppresses expression of replication-dependent core histone genes. Nat. Struct. Mol. Biol 2012, 19, 930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bönisch C; Hake SB Histone H2A variants in nucleosomes and chromatin: more or less stable? Nucleic Acids Res 2012, 40, 10719–10741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kimura H; Cook PR Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol 2001, 153, 1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fischle W; Tseng BS; Dormann HL; Ueberheide BM; Garcia BA; Shabanowitz J; Hunt DF; Funabiki H; Allis CD Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 2015, 438, 1116–1122. [DOI] [PubMed] [Google Scholar]
- [17].Angel TE; Aryal UK; Hengel SM; Baker ES; Kelly RT; Robinson EW; Smith RD Mass spectrometry-based proteomics: existing capabilities and future directions. Chem Soc Rev 2012, 41, 3912–3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kalli A; Sweredoski MJ; Hess S Data-dependent middle-down nano-liquid chromatography-electron capture dissociation-tandem mass spectrometry: an application for the analysis of unfractionated histones. Anal. Chem 2013, 85, 3501–3507. [DOI] [PubMed] [Google Scholar]
- [19].Catherman AD; Skinner OS; Kelleher NL Top Down proteomics: Facts and perspectives. Biochem. Biophys. Res. Commun 2014, 445, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Boyne II MT; Pesavento JJ; Mizzen CA; Kelleher NL Precise characterization of human histone in the H2A gene family by top down mass spectrometry. J. Proteome Res 2006, 5, 248–253. [DOI] [PubMed] [Google Scholar]
- [21].Siuti N; Roth MJ; Mizzen CA; Kellerher NL; Pesavento JJ Gene-specific characterization of human histone H2B by electron capture dissociation. J. Proteome Res 2006, 5, 233–239. [DOI] [PubMed] [Google Scholar]
- [22].Molden RC; Bhanu NV; LeRoy G; Arnaudo AM Garcia BA. Multi-faceted quantitative proteomics analysis of histone H2B isoforms and their modifications. Epigenetics Chromatin 2015, 8:15 DOI 10.1186/s13072-015-0006-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Anderson LC; Karch KR; Ugrin SA; Coradin M; English AM; Sidoli S; Shabanowitz J; Garcia BA; Hunt DF Analysis of histone proteoforms using front-end electron transfer dissociation-enabled Orbitrap instruments. Mol Cell Proteomics 2016, O115.053843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kaiser NK; Quinn JP; Blakney GT; Hendrickson CL; Marshall AG A Novel 9.4 Tesla FTICR Mass Spectrometer with Improved Sensitivity, Mass Resolution, and Mass Range. J. Am. Soc. Mass Spectrom 2011, 22, 1343–1351. [DOI] [PubMed] [Google Scholar]
- [25].Guan S; Marshall AG Stored Waveform Inverse Fourier Transform (SWIFT) Ion Excitation in Trapped-Ion Mass Spectrometry: Theory and Applications, Int. J. Mass Spectrom. Ion Proc 1996, 158, 5–37. [Google Scholar]
- [26].Ledford EB Jr., Rempel DL, and Gross ML Space charge effects in Fourier transform mass spectrometry mass calibration. Anal. Chem 1984, 56, 2744–2748. [DOI] [PubMed] [Google Scholar]
- [27].Shi SDH, Drader JJ, Freitas MA, Hendrickson CL, and Marshall AG Comparison and Interconversion of the Two Most Common Frequency-to-Mass Calibration Functions for Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Int. J. Mass Spectrom 2000, 196, 591–598. [Google Scholar]
- [28].Anderson LC; He L; Marshall AG; Hendrickson CL Analysis of a Monoclonal Antibody by Top-Down LC-MS/MS with a 21 Tesla FT-ICR Mass Spectrometer, Sanibel Conference on Characterization of Protein Therapeutics by Mass Spectrometry, Hilton Clearwater Beach, Florida, 21-24 Jan, 2016. [Google Scholar]
- [29]. http://www.uniprot.org/
- [30].Dang X; Scotcher J; Wu S; Chu RK; Tolic N.; Ntai I.; Thomas PM.; Fellers RT.; Early BP.; Zheng Y.; Durbin KR.; LeDuc RD.; Wolff JJ.; Thompson CJ.; Pan J.; Han J.; Shaw JB.; Salisbury JP.; Easterling M.; Borchers CH.; Brodbelt JS.; Agar JN.; Pasa-Tolic L.; Kelleher NL.; Young NL. The first pilot project of the consortium for top-down proteomics: A status report. Proteomics 2014, 14, 1130–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Tafrova JI; Tafrov ST Human histone acetyltransferase 1 (Hat1) acetylates lysine 5 of histone h2A in vivo. Mol. Cell. Biochem 2014, 392, 259–272. [DOI] [PubMed] [Google Scholar]
- [32].Zhang Y; Griffin K; Mondal N; Parvin JD Phosphorylation of histone H2A inhibits transcription on chromatin templates. J Biol. Chem 2004, 279, 21866–21872. [DOI] [PubMed] [Google Scholar]
- [33].Zhau HY; Chang SM; Chen BQ; Wang Y; Zhang H; Kao C; Sang QA; Pathak SJ; Chung LW Androgen-repressed phenotype in human prostate cancer. Proc. Natl. Acad. Sci 1996, 93, 15152–15157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Hance MW; Dole K; Gopal U; Bohonowych JE; Jezierska-Drutel A; Neumann CA; Liu H; Garraway IP; Isaacs JS Secreted Hsp90 is a novel regulator of the epithelial to mesenchymal transition (EMT) in prostate cancer. J. Biol. Chem 2012, 287, 37732–37744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nolan KD; Franco OE; Hance MW; Hayward SW; Isaacs JS Tumor-secreted Hsp90 subverts polycomb function to drive prostate tumor growth and invasion. J. Biol. Chem 2015, 290, 8271–8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Singh AM; Chappell J; Trost R; Lin L; Wang T; Tang J; Matlock BK; Weller KP; Wu H; Zhao S; Jin P; Dalton S Cell-cycle control of developmentally regulated transcription factors accounts for heterogeneity in human pluripotent cells. Stem Cell Reports 2013, 1, 532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Singh AM; Trost R; Boward B; Dalton S Utilizing FUCCI reporters to understand pluripotent stem cell biology. Methods 2015. pii: S1046–2023(15)30103–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.