

ABSTRACT
Protein post-translational modifications (PTMs) take many shapes, have many effects and are necessary for cellular homeostasis. One of these PTMs, Nε-lysine acetylation, was thought to occur only in the mitochondria, cytosol and nucleus, but this paradigm was challenged in the past decade with the discovery of lysine acetylation in the lumen of the endoplasmic reticulum (ER). This process is governed by the ER acetylation machinery: the cytosol:ER-lumen acetyl-CoA transporter AT-1 (also known as SLC33A1), and the ER-resident lysine acetyltransferases ATase1 and ATase2 (also known as NAT8B and NAT8, respectively). This Review summarizes the more recent biochemical, cellular and mouse model studies that underscore the importance of the ER acetylation process in maintaining protein homeostasis and autophagy within the secretory pathway, and its impact on developmental and age-associated diseases.
KEY WORDS: Lysine acetylation, Endoplasmic reticulum, Secretory pathway, Autophagy
Summary: This Review summarizes recent studies that underscore the importance of the endoplasmic reticulum acetylation process in maintaining protein homeostasis and autophagy within the secretory pathway.
Introduction
Proteins carry out a multitude of critical functions for an organism; for example, they provide structural stability to cells and tissues, give motility to individual cells, carry messages within and between cells, and regulate gene expression and metabolism. In order for a cell to respond to changes in internal and external environmental factors, a broad range of protein co- and post-translational modifications have evolved to expand upon the relatively static properties encoded in protein side-chains. Some consequences of these modifications are alterations in subcellular localization (Cui et al., 2016), protein–protein interactions (Nishi et al., 2011), and protein stability (Amm et al., 2014) and activity (Kapoor and Lozano, 1998). These effects stem from a broad range of reversible and irreversible chemical adornments, including ubiquitylation (Amm et al., 2014; Yu et al., 2014), methylation (Murn and Shi, 2017; Wesche et al., 2017; Zhang et al., 2015) and SUMOylation (Andreou and Tavernarakis, 2009) of lysine and arginine residues, and phosphorylation of tyrosine, serine and threonine residues (Humphrey et al., 2015), as well as Nε acetylation of lysine residues (Choudhary et al., 2014; Menzies et al., 2016), which is the focus of this Review.
The discovery of protein acetylation occurred over 50 years ago with studies on lysine-rich regions of nuclear histones in somatic and testicular cells (Gershey et al., 1968; Polgar, 1964; Vidali et al., 1968). Since then, different groups have identified a large number of nuclear, mitochondrial and cytosolic proteins of varied functions that have conserved lysine acetylation sites. It is an evolutionarily conserved modification, identified in both prokaryotes (Ouidir et al., 2016) and eukaryotes (Finkemeier et al., 2011; Jeffers and Sullivan, 2012; Lundby et al., 2012), which relies on a central metabolite, acetyl-coenzyme A (CoA) (Box 1), to act as a donor of the acetyl group (Drazic et al., 2016) (Fig. 1). This allows for a direct crosstalk between the cellular metabolic state, through the level of acetyl-CoA, and cellular effectors, for instance in the form of modified histone lysine residues, which can fine-tune gene expression, or modified peptidyl-lysine residues, which can modulate protein function (Freiman and Tjian, 2003). The aim of this Review is to introduce the components of the endoplasmic reticulum (ER) acetylation machinery and provide an update on the molecular mechanisms underlying this modification. We will also highlight the effects of dysregulation of acetyl-CoA import into the ER in the context of human diseases and various mouse models.
Box 1. Acetyl-CoA localization and biosynthesis.
The cornerstone metabolite acetyl-CoA is synthesized in the mitochondria (Fujino et al., 2001), the peroxisome (Wanders et al., 2015), the nucleus and the cytoplasm (Chypre et al., 2012) (see Fig. 1). Free CoA is generated from pantothenic acid (vitamin B5) through the sequential action of pantothenate kinase (PANK), phosphopantothenoylcysteine synthetase (PPCS), phosphopantothenoylcysteine decarboxylase (PPCDC), and coenzyme A synthase (COASY) (see Fig. 2). COASY is a bifunctional protein with phosphopantetheine adenylyltransferase (PPAT) and dephospho-CoA kinase (DPCK) activity, which in mammals is encoded by one gene and in bacteria by two genes (Leonardi and Jackowski, 2007). The precise location where CoA is synthesized within the cell is still controversial as the different enzymes have been found in the cytosol, nucleus and mitochondria (Leonardi and Jackowski, 2007).
Generation of acetyl-CoA can be induced by a number of different metabolic inputs (primarily glycolysis and β-oxidation), and the final product is used in a variety of different ways (Fig. 1). Acetate, acetaldehyde, free fatty acids, pyruvate, β-hydroxybutyrate and amino acids are transported into the mitochondria for enzymatic condensation with CoA via a high-energy thioester linkage (Pietrocola et al., 2015). This mitochondrial pool can be used subsequently in ketone body biosynthesis during periods of carbohydrate starvation, by the citric acid cycle for energy purposes (Shi and Tu, 2015) or for mitochondrial protein acetylation (Hosp et al., 2017). Mitochondrial acetyl-CoA can also be sent to the cytosol following its condensation with oxaloacetate through the activity of citrate synthase to yield citrate and free CoA, the former being shuttled across the mitochondrial membrane by solute carrier family 25 member A1 (SLC25A1). Extracellular citrate can also be imported by the plasma membrane citrate/Na+ importer (SLC13A5). The peroxisome can similarly uptake fatty acids to generate acetyl-CoA through α- and β-oxidation, exporting it to the cytoplasm as acetyl-carnitine (Wanders et al., 2015). Under normal conditions, cytosolic acetyl-CoA synthesis mainly originates from the conversion of citrate through the activity of ATP-citrate lyase (ACLY). Under certain stress events, such as hypoxia (Metallo et al., 2011), glutamine can be shunted toward acetyl-CoA production via the action of several enzymes. Some cell types possess alternative cytosolic acetyl-CoA synthetic pathways, such as dehydrogenation of ethanol to form acetaldehyde and subsequent synthesis of acyl-CoA in hepatocytes (You et al., 2002). Acetyl-CoA can travel freely between the cytosol and nucleus, but the nucleus also has some capacity to generate acetyl-CoA from citrate through ACLY (Sivanand et al., 2017), pyruvate through pyruvate dehydrogenase complex (PDC) (Sutendra et al., 2014), and acetaldehyde through acyl-CoA synthetase short-chain family member 2 (ACSS2) (Bulusu et al., 2017). Acetyl-CoA synthesis in the ER has yet to be identified, with the main identified source being cytoplasmic acetyl-CoA that is transported into the ER lumen by AT-1/SLC33A1 (Jonas et al., 2010). Here, it is utilized to post-translationally modify lysine via acetylation, which regulates proteostasis and the induction of reticulophagy (see main text and Pehar and Puglielli, 2013).
Fig. 1.

Metabolic pathways that maintain acetyl-coenzyme A levels in the cell. See also Box 1. Abbreviations: ACAA1, 3-ketoacyl-CoA thiolase; ACAA2, mitochondrial 3-ketoacyl-CoA thiolase; ACDH, 3-hydroxy acyl-CoA dehydrogenase; AceAld, acetaldehyde; AceCS1, cytoplasmic/nuclear acetyl-coenzyme A synthetase; AceCS2, mitochondrial acetyl-coenzyme A synthetase; ACLY, ATP-citrate lyase; ACOT, acyl-CoA thioesterase; ACOX, peroxisomal acyl-CoA oxidase 1; ADH1B, alcohol dehydrogenase 1B; ALDH1, acetaldehyde dehydrogenase; ALDH2, aldehyde dehydrogenase 2; BCAA, branched-chain amino acids; BCAT2, mitochondrial branched-chain amino acid aminotransferase; BCKDC, branched-chain α-keto acid dehydrogenase complex; BDH1, beta-hydroxybutyrate dehydrogenase; B-OH, beta-hydroxybutyric acid; Cit, citrate; CoA, coenzyme A; CPT1, carnitine palmitoyltransferase 1; CPT2 carnitine palmitoyltransferase 2; CS, citrate synthase; DPCK, dephosphocoenzyme A kinase; ECH, 2,3 enoyl-CoA hydrase; ECHD, enoyl-CoA hydratase; ER, endoplasmic reticulum; EtOH, ethanol; FFA, medium/short chain free fatty acid; GDH, glutamate dehydrogenase; Gln, glutamine; GLS, glutaminase; Glu, glutamate; HADH, mitochondrial hydroxyacyl-coenzyme A dehydrogenase; HADH, 3-hydroxyacyl-CoA dehydrogenase; HMG-CoA lyase, 3-hydroxy-3-methylylglutaryl-CoA lyase; HMG-CoA synthase, hydroxymethyl glutaryl-CoA synthase; LC-FACS, long chain fatty acyl-CoA synthetase; LC-FFA, long chain free fatty acid; Mal, malate; MC-CoA, medium chain coenzyme A; MDH, malate dehydrogenase; OAA, oxaloacetate; PANK, pantothenic acid kinase; PDH, pyruvate dehydrogenase; PPAT, phosphopantetheine adenylyl transferase; PPCDC, phosphopantothenoylcysteine decarboxylase; PPCS, phosphopantothenoylcysteine synthetase; Pyr, pyruvate; SLC13A5, solute carrier family 13A5; SLC25A1, solute carrier family 25A1; AT-1 (SLC33A1), solute carrier family 33A1/acetyl-CoA transporter protein 1; TCA, tricarboxylic acid cycle; VLC-FACS, very long chain fatty acyl-CoA synthetase; VLC-FFA, very long chain free fatty acid.
Lysine acetylation enters the ER – the components of the ER acetylation machinery
In 2007, while studying the metabolism of the β-site amyloid precursor protein (APP) cleaving enzyme (BACE1), a type I membrane protein that inserts into the secretory pathway, we discovered that both the stability and trafficking of the nascent polypeptide depended on efficient enzymatic Nε-lysine acetylation within the lumen of the ER (Costantini et al., 2007). From the biochemical perspective, enzymatic Nε-lysine acetylation requires three essential components: a molecule to act as the donor of the acetyl group, in the form of acetyl-CoA, an acetyl acceptor, in the form of the lysine side-chain, and an acetyl-CoA:lysine acetyltransferase to facilitate the exchange. Acetyl-CoA is a large, membrane-impermeable compound and, as such, requires a membrane transporter to pass from the cytosol into organelles such as the mitochondria (Kaplan et al., 1995). The sequential identification and enzymatic characterization of the protein product of the mammalian gene SLC33A1 (encoding AT-1) as an ER-localized acetyl-CoA transporter (Jonas et al., 2010), two ER and ER-Golgi intermediate compartment (ERGIC) acetyltransferases (Ko and Puglielli, 2009), and Nε-acetylated lysine residues on ER-resident and -transiting proteins (Pehar et al., 2012b) gave strong evidence that lysine acetylation also occurs in the lumen of the ER (Fig. 2). Below, we present the basic features of each component of the ER acetylation machinery, and discuss the effect of genetic and chemical disruption of the process.
Fig. 2.
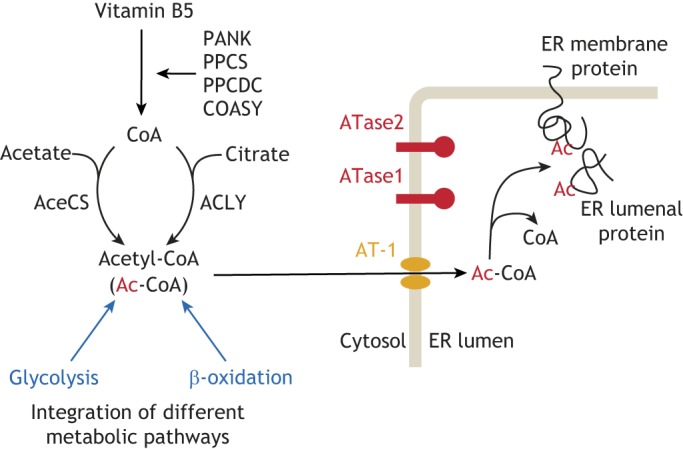
The ER acetylation machinery. Acetyl-CoA is synthesized in the cytosol through a multi-step process and is then transported into the ER lumen by the ER membrane transporter AT-1. In the ER lumen, acetyl-CoA is used by two ER-based acetyltransferases, ATase1 and ATase2, to acetylate ER-cargo and -resident proteins. Both ATase1 and ATase2 are type-II membrane proteins with the catalytic domain facing the lumen of the organelle.
Acetyl-CoA transporter 1 – an essential membrane protein that translocates acetyl-CoA from the cytosol to the ER
In 1997, Kanamori et al. reported the existence of a putative cytosol:ER acetyl-CoA transporter (Kanamori et al., 1997). This 61-kDa protein, termed acetyl-coenzyme A transporter 1 or simply AT-1 (gene name SLC33A1), was predicted to contain six to 12 transmembrane regions as well as a leucine zipper motif, as is observed in other transporter proteins (Eckhardt et al., 1996). Studies using subcellular fractionation approaches showed that this protein localized to the ER, and isolation and reconstitution of recombinant AT-1 into artificial liposomes revealed its ability to transport acetyl-CoA across a lipid bilayer, with free CoA-SH acting as an inhibitor to the process (Jonas et al., 2010). Additionally, we showed that highly purified, native and intact ER vesicles possess the ability to uptake acetyl-CoA with a Km of ∼10–14 μM (Costantini et al., 2007), which is approximately in line with the available concentration of acetyl-CoA in the cytosol (Lee et al., 2014) and the Km of other enzymes that require acetyl-CoA (Mackall and Lane, 1977; Snoswell and Koundakjian, 1972). Finally, overexpression of AT-1 in cellular systems increased acetyl-CoA transport into the ER lumen (Jonas et al., 2010). Therefore, the ER localization of AT-1, its ability to transport acetyl-CoA across both artificial and native lipid bilayers, and the ability of purified ER vesicles to uptake acetyl-CoA point to AT-1 as an ER-localized acetyl-CoA transporter protein (Fig. 2).
The importance of this protein is highlighted by the identification of SLC33A1 mutations associated with human diseases. The first mutation to be identified was the mutation of serine 113 to arginine (S113R) in patients with an autosomal-dominant form of spastic paraplegia 42 (SPG42) (Lin et al., 2008). SPGs are a rather heterogeneous group of diseases characterized by bilateral spasticity, peripheral neuropathy, urinary dysfunction and mild cognitive decline (Salinas et al., 2008). A progressive degeneration of motor axons of the corticospinal tract is a hallmark of the disease, along with defects in the central nervous system that are localized to the memory-forming regions of the brain and disseminated lesions of the cerebral white matter (Depienne et al., 2007). The SPG42 patients were all heterozygous for the S113R mutation and the disease displayed incomplete penetrance with an age of onset ranging between 4 and 42 years (Lin et al., 2008). In later studies, children with additional mutations were also reported; they displayed psychomotor retardation, severe developmental delay, brain atrophy, cerebellar hypoplasia, hearing deficiencies and multi-organ failure, with a life expectancy of 1 to 6 years (Chiplunkar et al., 2016; Huppke et al., 2012). The SLC33A1 mutations in these cases were homozygous and included three large deletions, two frame shifts with premature STOP codons, and one missense mutation (A110P).
The two missense mutations identified so far (A110P and S113R) are both located in the first intraluminal loop of AT-1, suggesting related mechanistic features. So far, in depth studies have only focused on the S113R variant of AT-1. Overexpression of wild-type AT-1 or AT-1 S113R in H4 neuroglioma cells revealed no adverse effects on protein production or stability, but when either the wild-type or mutant protein were separately reconstituted on artificial liposomes, the acetyl-CoA transport ability was completely ablated in the case of AT-1 S113R (Peng et al., 2014). Analytical ultracentrifugation and co-immunoprecipitation experiments showed that AT-1 functions as a homodimer within the membrane of the ER; as the S113R mutation impedes homodimerization of the transporter, it consequentially abolishes the transport activity (Peng et al., 2014). AT-1S113R/S113R mouse embryos are non-viable owing to deficiencies in neural tube closure (Liu et al., 2017; Peng et al., 2014). A similar embryonic requirement was observed in the zebrafish (Lin et al., 2008; Mao et al., 2015). In contrast, AT-1S113R/+ knock-in mice are born with Mendelian ratio and are viable (Peng et al., 2014). When housed in the presence of normal mouse pathogens, they display a short lifespan with an increased propensity for pathogenic infections and reactive inflammatory diseases. Necropsy studies also showed an increased occurrence of malignancies. Housing the mice in a pathogen-free facility eliminates the propensity to infections, inflammation and malignancies (Peng et al., 2014). Haploinsufficiency of AT-1 in the mouse also presents with defects of the peripheral and central nervous system at 10–12 months of age, with intermittent hind-leg clasping, decreased response to pain stimulus, abnormal body rotation and reduced grip strength observed with no concomitant loss of muscle fiber. This is likely attributable to the noted axonal degeneration of the proximal and distal sections of peripheral nerves. Indeed, histology and positron emission tomography (PET) analysis of these mice gave evidence for neuronal loss, axonal degeneration, myelinopathy and reactive inflammation within both the central and the peripheral nervous system. Importantly, the neurodegenerative phenotype was observed in knock-in mice housed in either pathogen-free or open facilities (Peng et al., 2014). Therefore, the propensity to infections, systemic inflammation and malignancies depends on the presence of pathogens within the colony, whereas the neurodegenerative phenotype does not.
While insufficient levels of AT-1 present a myriad of neurological and immunological problems as outlined above, excess of AT-1 has its own consequences. Chromosomal duplications of the 3q25.31 locus, which harbors SLC33A1, are associated with autism spectrum disorder (ASD), intellectual disability, propensity to seizures and facial dysmorphism (Prasad et al., 2012; Sanders et al., 2011; see also SFARI Autism Database, URL: https://gene.sfari.org/database/cnv/3q25.31). To determine the pathogenic role of increased SLC33A1 gene dosage, we generated two different mouse models: one overexpressing AT-1 in forebrain neurons (referred to as AT-1 Tg) and one overexpressing AT-1 systemically (referred to as AT-1 sTg). AT-1 Tg mice displayed reduced cognitive function and modified social behavior (Hullinger et al., 2016). This was observed in-tandem with an increased number of spines and dendritic branches in both cultured neurons and hippocampal tissue, and with defects in synaptic plasticity. Further assessment revealed changes in the acetylation status of ER cargo proteins as well as widespread proteomic changes. By contrast, AT-1 sTg mice displayed a segmental progeria phenotype with a maximum lifespan of only ∼5 months that mimicked an accelerated form of pathogenic aging (Peng et al., 2018). The accelerated aging phenotype was accompanied by tissue inflammation and accumulation of senescent cells.
At the mechanistic level, the reduced AT-1 activity in AT-1S113R/+ mice resulted in a reduced efficiency of the secretory pathway and hyperactivation of ER autophagy (also referred to as reticulophagy) (Peng et al., 2014). In contrast, overexpression of AT-1 in AT-1 Tg and AT-1 sTg mice resulted in increased efficiency of the secretory pathway and reduced activation of reticulophagy (discussed later) (Hullinger et al., 2016; Peng et al., 2018). The fact that homozygous inactivation of AT-1 is lethal and that hemizygous inactivation reduced the import of acetyl-CoA into the ER by ∼50% suggests that AT-1 is the only ER membrane acetyl-CoA transporter (Peng et al., 2014).
The ER acetyltransferases ATase1 and ATase2 acetylate proteins that insert within the secretory pathway
Knowledge that ER-transiting and -resident proteins are acetylated on specific lysine residues in the lumen of the ER spurred efforts to identify potential ER-resident lysine acetyltransferases. These efforts bore fruit in 2009 with the identification and characterization of two ER/ERGIC acetyltransferases (Fig. 2), termed ATase1 (gene name NAT8B) and ATase2 (gene name NAT8) (Ko and Puglielli, 2009). Subcellular fractionation and confocal microscopy studies confirmed that both proteins are localized at the ER and ERGIC (Ding et al., 2012; Veiga-da-Cunha et al., 2010). ATase1 and ATase2 show a molecular mass of ∼25 kDa in reducing gel electrophoresis, share 88% identity between each other, and are members of the camello-like family of proteins under the GCN5-related N-acetyltransferase (GNAT) superfamily of N-acetyltransferases (Dyda et al., 2000; Neuwald and Landsman, 1997), with both retaining the highly conserved R/Q-x-x-G-x-G/A acetyl-CoA-binding motif (Roth et al., 2001). ATase1 appears to be the result of a gene duplication event; however, the promoter region of the two ATases display several differences, suggesting different modes of transcriptional regulation (Hahn and Lee, 2006). Initial analysis of the proteins determined that they both have a short cytosol-exposed N-terminus, a single helical transmembrane anchor spanning residues 43–63, and a large globular, ER-facing catalytic domain (Ko and Puglielli, 2009). ER vesicles from Chinese hamster ovary (CHO) cells overexpressing either ATase1 or ATase2 showed increased acetyltransferase activity compared to non-transfected cells in the presence of detergent, highlighting enzymatic activity within the ER, which was eliminated with increased temperature. Finally, both enzymes also display acetyltransferase activity in vitro (Ko and Puglielli, 2009).
More recent studies have provided additional information regarding their mechanism of action and role within the ER. Specifically, in vitro and ex vivo approaches have revealed that both ATases act as dimers of ∼50-kDa and that the dimerization requires a short stretch of amino acids (amino acids 195–205) near the C-terminus (Ding et al., 2014). Accordingly, deletion of the dimerization domain abolishes the enzymatic activity of the enzymes (Ding et al., 2014). Co-immunoprecipitation (IP) studies of full-length ATase1 and ATase2 showed that they co-elute with components of the oligosaccharyltransferase (OST) complex (see below), a multi-protein complex involved in the N-glycosylation of nascent glycoproteins (Dempski and Imperiali, 2002; Gavel and von Heijne, 1990). This interaction is partially lost when the C-terminus of either ATase is truncated (Ding et al., 2014).
Analysis of the human single nucleotide polymorphism (SNP) database shows multiple SNPs on both NAT8B (https://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?locusId=51471) and NAT8 (https://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?locusId=9027&chooseRs=all). In the case of NAT8B, three nonsense changes are notable. The first is at position R41 of the amino acid sequence (rs757878515), the second at position Y101 (rs780729191), and the third at position Q168 (rs4852974). Amino acid 168 is also reported with two missense changes (glutamine to glutamic acid, rs4852974; and glutamine to histidine, rs369247762). In the case of NAT8 there are two nonsense changes that are notable: the first is at position Y101 (rs757668615) and the second is at position Q172 (rs745439157). The above nonsense and missense mutations do not appear to be associated with human diseases.
The ER acetylation machinery regulates proteostasis and autophagy within the secretory pathway
Important functions of the ER include protein synthesis as well as endolumenal co- and post-translational modifications that help with the folding, and thus stability and activity of newly synthesized membrane and secreted polypeptides (Buchberger et al., 2010; Labbadia and Morimoto, 2015; Pehar and Puglielli, 2013; Trombetta and Parodi, 2003). Although the essential information for folding is present in the primary amino acid sequence, co-translational events (such as N-glycosylation and disulfide bonds) help to preserve the fidelity of the process. Correctly folded and unfolded (or misfolded) polypeptides must also be sorted with the former being allowed to move through the secretory pathway, and the latter being disposed of. For this purpose, transient post-translational events must be in place to distinguish correctly folded from unfolded and/or misfolded polypeptides within the ER.
As we mentioned, both ATase1 and ATase2 act as homodimers of ∼50 kDa; however, when analyzed under native and non-reducing gel conditions, they were found to migrate with a molecular mass of ∼350 kDa (Ding et al., 2014). Mass spectrometry revealed that, in addition to the ATases, this high-molecular-mass complex contained five members of the OST complex, thus suggesting a functional association between the N-glycosylation and Nε-lysine acetylation machineries of the ER (Ding et al., 2014). Studies with truncated versions of BACE1 and CD133, two established substrates of the ATases, revealed that N-glycosylation and Nε-lysine acetylation occur sequentially (Ding et al., 2014). Specifically, full-length and correctly folded polypeptides can be glycosylated and acetylated, while truncated and incorrectly folded polypeptides can be N-glycosylated but not acetylated (Ding et al., 2014). Furthermore, when normally acetylated lysine residues were mutated to alanine or arginine, to generate loss-of-acetylation mutants without affecting the overall tertiary structure of the protein, BACE1 and CD133 failed to exit the ER (Costantini et al., 2007; Mak et al., 2014). When the same lysine residues were mutated to glutamine to generate constitutively acetylated (also referred to as gain-of-acetylation) mutants, BACE1 and CD133 variants were able to exit the ER and reach the cell surface more efficiently (Costantini et al., 2007; Mak et al., 2014). In essence, only correctly folded polypeptides are recognized by the ATases.
It is worth emphasizing that N-glycosylation occurs while the nascent polypeptide is still unfolded (see Fig. 3); the fidelity of the biochemical modification is ensured by the presence of the N-x-(T/S) consensus motif, which is recognized by the OST complex (Dempski and Imperiali, 2002; Gavel and von Heijne, 1990). In contrast, no consensus motif for Nε-lysine acetylation has been identified and only surface-exposed lysine residues are acetylated. Therefore, it is currently accepted that the fidelity of Nε-lysine acetylation is ensured by the tertiary structure of protein (Kouzarides, 2000; Yang and Grégoire, 2007). The standing hypothesis is that the ATases associate with the OST complex during the translocation of nascent polypeptides across the ER membrane; they only recognize and acetylate correctly folded polypeptides. Acetylated polypeptides are able to move through the secretory pathway and complete maturation, while non-acetylated polypeptides are prevented from reaching the Golgi and are disposed of (see Fig. 3). Although we still need to resolve the mechanistic aspects of this novel regulatory function, the above hypothesis is supported by recent studies performed in mice with increased or reduced AT-1 activity where ER-to-Golgi trafficking of mature glycoproteins and levels of cell surface proteins were directly measured (Hullinger et al., 2016; Peng et al., 2018).
Fig. 3.

The ER acetylation machinery is an integral component of ER quality control and regulates the efficiency of the secretory pathway. Secretory membrane or lumenal proteins are synthesized close to the ER and enter the organelle through the translocon (SEC61–SEC62–SEC63) complex (1). Proteins with an N-x-S/T consensus sequence (yellow circles) for N-glycosylation that is 14 or 15 amino acids (∼40 Å) away from the inner membrane, are recognized by the oligosaccharyltransferase (OST) complex, which interacts with the translocon and transfers a preassembled GlcNAc2Man9Glc3 oligosaccharide (branched lines) to the asparagine residue of the nascent glycoprotein (2). Correctly folded glycoproteins are then recognized by the ATases, which interact with the OST to acetylate specific lysine residues (3). The acetylated lysine residues act as a positive marker that allows correctly folded glycoproteins to advance toward the Golgi (4). Unfolded or misfolded glycoproteins, although N-glycosylated, are not recognized by the ATases (5), and, as a result, are prevented from reaching the Golgi and are disposed of (6). Although supported by a number of publications (see text), the above is only a working model for how the ER acetylation machinery might regulate the secretory pathway.
As mentioned above, another important function of the ER is to dispose of unfolded and/or misfolded polypeptides. Monomeric proteins are preferentially degraded by the proteasome through the ER-associated protein degradation (ERAD) pathway (McCracken and Brodsky, 1996; Trombetta and Parodi, 2003; Werner et al., 1996). In contrast, large protein aggregates are mostly degraded by the autophagy pathway following encapsulation into autophagosomes and subsequent fusion with the lysosome (Axe et al., 2008; Bernales et al., 2006; Ding et al., 2007; Ogata et al., 2006). As noted earlier, AT-1S113R/+ mice display hyperactivation of autophagy (Peng et al., 2014), whereas AT-1 sTg mice show hypoactivation, thus supporting an immediate role of the ER acetylation machinery in the regulation of autophagy at the ER level (hence, the term reticulophagy) (Pehar et al., 2012a; Peng et al., 2018). Interestingly, AT-1 is transcriptionally regulated by X-box-binding-1 protein (XBP-1), which acts immediately downstream of inositol requiring protein-1α (IRE1α; also known as ERN1), one of the three branches of the mammalian unfolded protein response (UPR) system and an upstream initiator of ERAD (Ron and Walter, 2007; Trombetta and Parodi, 2003; Wang and Kaufman, 2016). Downregulation of XBP-1 results in hyperactivation of autophagy and consequent autophagic cell death (Hetz et al., 2009; Matus et al., 2009; Pehar et al., 2012a); however, this can be prevented by overexpressing AT-1 (Pehar et al., 2012a). Furthermore, AT-1 is upregulated under conditions of physiological ER stress, such as during periods of enhanced protein production demands associated with cellular differentiation and activation of B cells (Shaffer et al., 2004). This points toward a possible link between acetyl-CoA levels in the ER, lysine acetylation and protein quality control. Indeed, many proteins involved in protein quality control and ERAD (such as calnexin, calreticulin and ERAD E3 ligase subunits), as well as sensors for protein misfolding [such as GRP76, GRP94 and GRP170 (also known as BiP, HSP90B1 and HYOU1, respectively)] and regulators for autophagy (such as ATG9A), were found to be acetylated in the ER (Pehar et al., 2012b). In essence, both cellular and animal studies suggest that sufficient import and utilization of acetyl-CoA in the ER is required for proper protein folding and passage through the ER, and for tight regulation of reticulophagy (see Figs 3 and 4).
Fig. 4.

The ER acetylation machinery regulates the induction of reticulophagy. ATG9A is acetylated on two lysine residues (K359 and K363) that face the lumen of the ER. The acetylation status of ATG9A regulates its ability to interact with SEC62 and FAM134B. Both SEC62 and FAM134B appear to act as reticulophagy receptors by engaging with cytosolic LC3β, whereas ATG9A appears to act as an ER acetylation sensor. In its acetylated form, ATG9A is unable to interact with either SEC62 or FAM134B, thus impeding further engagement of LC3β and inhibiting the induction of reticulophagy (upper half). In contrast, when non-acetylated, ATG9A is able to interact with SEC62 and FAM134B, which engage LC3β and induce reticulophagy (lower half). Although supported by a number of publications (see text), the above is only a working model for how the ER acetylation machinery might regulate the induction of reticulophagy.
It is also worth noting that ER acetylation occurs within the lumen of the ER, whereas the core autophagic machinery is located in the cytosol. Therefore, the above notion of a possible link between lysine acetylation and reticulophagy would require the existence of a sensor for the levels of acetyl-CoA within the ER that is able to communicate the acetylation status of the ER lumen to the autophagy machinery (Fig. 4). Research efforts in this direction led to identification of ATG9A. The acetylation status of several members of the autophagy-related (ATG) family of proteins has been linked to induction of autophagy, with an increased acetylation status acting as an inhibitory signal and a decreased one as an activation signal (Lee et al., 2008; Lee and Finkel, 2009). ATG9A is an ER-localized integral membrane protein with several ER-facing loops and is crucial for autophagosome formation (Pehar et al., 2012a; Yen and Klionsky, 2007). ATG9A is expressed in several tissues, but the highest levels are found in the brain, spinal cord and liver (Saitoh et al., 2009). Although synthesized and found in the ER, ATG9A can traffic through the trans-Golgi network, endosomes and the autophagosome; this transition is stimulated during periods of cellular starvation and increased autophagic flux (Imai et al., 2016; Lee and Finkel, 2009; Nishimura et al., 2017; Ohashi and Munro, 2010; Tamura et al., 2010; Webber et al., 2007; Young et al., 2006). While investigating the potential role of ATG9A as an acetyl-CoA sensor in the ER, we found that it is acetylated at two lysine positions (K359, K363) on the second ER-facing loop (Pehar et al., 2012a). Expression of a loss-of-acetylation mutant form of ATG9A (K359R/K363R) in H4 neuroglioma cells resulted in widespread autophagic cell death, whereas expression of a gain-of-acetylation mutant (K359Q/K363Q) had no such effect. Furthermore, expression of the K359Q/K363Q mutant provided significant protection against the autophagic cell death caused by knockdown of either AT-1 or XBP1 (Pehar et al., 2012a).
Taken together, these findings provided ample evidence that ATG9A indeed acts as an acetyl-CoA sensor within the ER, although the exact mechanistic relationship between ATG9A acetylation and autophagy remains to be elucidated. Nevertheless, by studying AT-1S113R/+ and AT-1 sTg mice, we were able to show that reduced influx of acetyl-CoA into the ER leads to reduced acetylation of ATG9A and induction of reticulophagy, while increased influx of acetyl-CoA leads to increased ATG9A acetylation and a block in the induction of reticulophagy (Peng et al., 2014, 2018). Furthermore, detailed analysis of AT-1 sTg mice revealed that the acetylation status of ATG9A regulates its ability to interact with FAM134B (also known as RETREG1) and SEC62 (Fig. 4) (Peng et al., 2018), two ER membrane proteins that act as receptors for LC3β (also known as MAP1LC3B), an essential component of the core autophagic machinery (Fumagalli et al., 2016; Khaminets et al., 2015; Mochida et al., 2015; Rubinsztein, 2015; Schuck, 2016). Interestingly, both FAM134B and SEC62 are found on the rough ER, where the bulk of protein biosynthesis occurs (Fumagalli et al., 2016; Khaminets et al., 2015; Mochida et al., 2015; Rubinsztein, 2015; Schuck, 2016). Furthermore, SEC62 is an integral member of the ER translocon machinery and its ability to engage with LC3β requires it to dissociate from the translocon (Fumagalli et al., 2016; Schuck, 2016). It has been proposed that both FAM134B and SEC62 might couple the machinery that allows biosynthesis and insertion of newly synthesized proteins into the ER with the machinery that controls the disposal of unfolded and/or misfolded polypeptides and maintains the size of the ER (Nakatogawa and Mochida, 2015; Schuck, 2016). We discussed above the possible role of the ER acetylation machinery in quality control, efficiency of the secretory pathway and autophagy-mediated disposal of toxic protein aggregates. Therefore, we can envision a model where functional association of ATG9A–FAM134B and ATG9A–SEC62 within the membrane of the ER is an initial step for the induction of reticulophagy. This association is required to engage cytosolic LC3β and can only occur when ATG9A is not acetylated (Fig. 4).
Therapeutic potential of inhibiting the ER acetylation machinery in proteostatic disorders
Given the importance of the regulation of autophagy in order to maintain cellular homeostasis, it is no surprise that both hypoactive and hyperactive autophagy are deleterious in normal mice (Kuma et al., 2017). However, an elevated autophagic flux appears to be beneficial in mice with genetic disorders that result in the accumulation of cytotoxic protein aggregates (Bhuiyan et al., 2013; Hetz et al., 2009; Madeo et al., 2009; Pickford et al., 2008; van Dellen et al., 2000).
By using a combination of cell- and animal-based studies, we discovered that inhibition of the ER acetylation machinery can stimulate autophagy-mediated disposal of toxic protein aggregates that form within the ER and secretory pathway but not those that form in the cytosol (Peng et al., 2016). These findings are in line with the fact that the ER acetylation machinery controls proteostasis within the ER and secretory pathway (discussed above). Importantly, an ATase specific inhibitor (6-chloro-5H-benzo[a]phenoxazin-5-one; commonly referred to as Compound 9), was able to rescue the Alzheimer's disease-like phenotype of APP695/swe mice (Peng et al., 2016) as well as the segmental progeria phenotype of AT-1 sTg mice (Peng et al., 2018).
It is worth keeping in mind that defects in autophagy, with the consequent disruption of proteostasis, contribute to the progression of many chronic human diseases, including neurodegenerative disorders, such as Alzheimer's disease, Parkinson's disease and Huntington disease, cancer and nephropathies, as well as immune and cardiovascular diseases (Frake et al., 2015; Levine et al., 2015; Nixon, 2013). Defective autophagy has also been implicated in aging (Frake et al., 2015; Levine et al., 2015; Nixon, 2013). Conversely, increased levels of autophagy have been associated with more efficient protein and organelle homeostasis, cytoprotection, lifespan extension and rescue from proteotoxicity (Madeo et al., 2015). With the realization that autophagy can be triggered in a rather specific and well-organized fashion, there has been an effort to focus on specific targets to tightly control its therapeutic potential (Kroemer, 2015; Levine et al., 2015; Vakifahmetoglu-Norberg et al., 2015). With the observation that inhibition of the ER acetylation machinery can induce reticulophagy and rescue proteostatic defects of the ER and secretory pathway in Alzheimer's disease and segmental progeria mouse models, we can easily envision exciting translational implications for different diseases where the primary pathological event resides in the aberrant accumulation of toxic protein aggregates within the ER (and secretory pathway).
Conclusions and outstanding questions
Since its discovery in 2007, the ER acetylation machinery has emerged as a novel biological process that maintains proteostasis within the ER and the overall efficiency of the secretory pathway. The identification of human diseases caused by genetic mutations and gene duplication events that disrupt the ER acetylation machinery, as well as the generation of relevant mouse models, have further shown the biological impact of this previously unknown machinery. Finally, the identification and characterization of compounds that can modulate ER acetylation in vivo has revealed an unexpected translational potential for a broad class of developmental and degenerative diseases. Although much has been learned, there are still several outstanding questions that remain to be addressed, which include those discussed below, as well as many others that we do not discuss owing to space limitations.
Structural biochemistry
Ongoing and future structural biochemistry-based approaches will help define the intrinsic features of AT-1, ATase1 an ATase2 activity. Specifically, it remains unclear where the acetyl-CoA binds or docks within the AT-1 and ATase dimers, where the peptidyl-lysine group of the polypeptide inserts within the ATases, and how AT-1 assembles within the ER membrane. Answering these questions will expand our understanding of how the entire machinery works and help in designing new ATase inhibitors for potential use in medical therapies.
Adaptation of the secretory pathway
Ex vivo and in vivo studies indicate that the secretory pathway is able to adapt to changes in AT-1 activity; increased ER acetylation leads to more proteins being transported through the secretory pathway, whereas reduced ER acetylation leads to the opposite (Hullinger et al., 2016; Peng et al., 2018). However, we still need to discover how this adaptation is ensured. In mammalian cells, the budding of COPII-coated transport vesicles occurs at specific sites that are referred to as transitional ER (tER); these vesicles subsequently evolve into full COPII vesicles and post-ER membrane structures (reviewed in Budnik and Stephens, 2009; D'Arcangelo et al., 2013). The ER could theoretically adapt by regulating (1) the number of ER exit sites (ERES) and, therefore, the number of COPII vesicles; (2) the size of COPII vesicles; or (3) both size and number of COPII vesicles (Aridor et al., 1999; Forster et al., 2006; Guo and Linstedt, 2006). Similarly, the arrival of more or less cargo material at the Golgi will require functional adaptation of the organelle. We specifically measured incorporation of sialic acid into mature glycoproteins and showed that AT-1 sTg mice incorporate more sialic acid whereas AT-1S113R/+ mice incorporated less (Hullinger et al., 2016; Peng et al., 2018). It is worth remembering that fucosylation, galactosylation and sialylation of transiting glycoproteins occurs in the Golgi, with sialic acid being the very last sugar to be added at the reducing end of galactose within the trans-Golgi network (reviewed in Hirschberg et al., 1998). However, it is unclear whether the Golgi adapts to the increased trafficking of glycoproteins through the secretory pathway of AT-1 sTg mice by increasing the expression and/or the catalytic properties of the glycosylation machinery, and whether the Golgi itself must undergo morphological changes.
Regulation of reticulophagy
We have already discussed that the ATG9A–FAM134B and ATG9A–SEC62 interaction is regulated by the acetylation status of ATG9A. However, the specific molecular mechanisms that allow the acetylation of two lysine residues on ATG9A to reduce its ability to interact with FAM134B and SEC62 are still unclear. The intraluminal loop of ATG9A that undergoes acetylation has four small helical segments, one of which contains both acetylated lysine residues (K359 and K363). The acetylated helix is flanked by two coiled regions, which are predicted to create conditions that are favorable for protein–protein interaction. Therefore, it is theoretically possible that the acetylation of this intraluminal loop regulates the ability of ATG9A to interact with FAM134B and SEC62 through alterations in tertiary structure. However, it is currently unclear whether FAM134B has any segment facing the lumen of the ER (Fregno et al., 2018; Khaminets et al., 2015). Therefore, it is also possible that the interaction is regulated by an additional protein that is able to recognize acetylated versus non-acetylated ATG9A and influence the formation of the ATG9A–FAM134B and ATG9A–SEC62 complex by sequestering ATG9A when acetylated.
The Golgi-based deacetylase
Finally, one of the most pressing gaps in our understanding of Nε-lysine acetylation within the secretory pathway is the identity of the deacetylase(s) involved in the removal of the acetyl group. This has been a standing question since the observation that BACE1 is acetylated in the ER and deacetylated in the Golgi as it transits through the secretory pathway (Costantini et al., 2007). Interestingly, an isoform of the Bombesin receptor-activated protein (the protein product of C6orf89), which localizes to the cis-Golgi and Golgi cisternae and possess deacetylase enhancer activity has been identified (Lalioti et al., 2013). Whether C6orf89 is the actual deacetylase or just an enhancer, as suggested, remains to be fully determined. However, it is clear that the identification of the Golgi-based deacetylase would help us further understand how Nε-lysine acetylation regulates the efficiency of the secretory pathway.
In conclusion, the past ten years have witnessed the discovery of the ER acetylation machinery. With it, we have gained a deeper understanding of the intrinsic mechanisms that maintain proteostasis within the ER, and ensure proper sorting and processing of newly synthesized glycoproteins. We have also gained new tools and new hopes to prevent and/or treat different debilitating diseases. Further work in this area will expand our knowledge on important aspects of cell biology and open new avenues of translational research.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Funding
The authors are funded by the National Institutes of Health (grants AG057408, AG053937 and NS094154 to L.P., and T32 AG000213 to M.A.F.). Deposited in PMC for release after 12 months.
References
- Amm I., Sommer T. and Wolf D. H. (2014). Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim. Biophys. Acta 1843, 182-196. 10.1016/j.bbamcr.2013.06.031 [DOI] [PubMed] [Google Scholar]
- Andreou A. M. and Tavernarakis N. (2009). SUMOylation and cell signalling. Biotechnol. J. 4, 1740-1752. 10.1002/biot.200900219 [DOI] [PubMed] [Google Scholar]
- Aridor M., Bannykh S. I., Rowe T. and Balch W. E. (1999). Cargo can modulate COPII vesicle formation from the endoplasmic reticulum. J. Biol. Chem. 274, 4389-4399. 10.1074/jbc.274.7.4389 [DOI] [PubMed] [Google Scholar]
- Axe E. L., Walker S. A., Manifava M., Chandra P., Roderick H. L., Habermann A., Griffiths G. and Ktistakis N. T. (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685-701. 10.1083/jcb.200803137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S., McDonald K. L. and Walter P. (2006). Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 4, e423 10.1371/journal.pbio.0040423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuiyan M. S., Pattison J. S., Osinska H., James J., Gulick J., McLendon P. M., Hill J. A., Sadoshima J. and Robbins J. (2013). Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Invest. 123, 5284-5297. 10.1172/JCI70877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger A., Bukau B. and Sommer T. (2010). Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol. Cell 40, 238-252. 10.1016/j.molcel.2010.10.001 [DOI] [PubMed] [Google Scholar]
- Budnik A. and Stephens D. J. (2009). ER exit sites--localization and control of COPII vesicle formation. FEBS Lett. 583, 3796-3803. 10.1016/j.febslet.2009.10.038 [DOI] [PubMed] [Google Scholar]
- Bulusu V., Tumanov S., Michalopoulou E., van den Broek N. J., MacKay G., Nixon C., Dhayade S., Schug Z. T., Vande Voorde J., Blyth K. et al. (2017). Acetate recapturing by nuclear acetyl-CoA synthetase 2 prevents loss of histone acetylation during oxygen and serum limitation. Cell Rep. 18, 647-658. 10.1016/j.celrep.2016.12.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiplunkar S., Bindu P. S., Nagappa M., Bineesh C., Govindaraj P., Gayathri N., Bharath M. M. S., Arvinda H. R., Mathuranath P. S., Sinha S. et al. (2016). Huppke-Brendel syndrome in a seven months old boy with a novel 2-bp deletion in SLC33A1. Metab. Brain Dis. 31, 1195-1198. 10.1007/s11011-016-9854-6 [DOI] [PubMed] [Google Scholar]
- Choudhary C., Weinert B. T., Nishida Y., Verdin E. and Mann M. (2014). The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536-550. 10.1038/nrm3841 [DOI] [PubMed] [Google Scholar]
- Chypre M., Zaidi N. and Smans K. (2012). ATP-citrate lyase: a mini-review. Biochem. Biophys. Res. Commun. 422, 1-4. 10.1016/j.bbrc.2012.04.144 [DOI] [PubMed] [Google Scholar]
- Costantini C., Ko M. H., Jonas M. C. and Puglielli L. (2007). A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem. J. 407, 383-395. 10.1042/BJ20070040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W., Sun M., Zhang S., Shen X., Galeva N., Williams T. D. and Staudinger J. L. (2016). A SUMO-acetyl switch in PXR biology. Biochim. Biophys. Acta 1859, 1170-1182. 10.1016/j.bbagrm.2016.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arcangelo J. G., Stahmer K. R. and Miller E. A. (2013). Vesicle-mediated export from the ER: COPII coat function and regulation. Biochim. Biophys. Acta 1833, 2464-2472. 10.1016/j.bbamcr.2013.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempski R. E. Jr and Imperiali B. (2002). Oligosaccharyl transferase: gatekeeper to the secretory pathway. Curr. Opin. Chem. Biol. 6, 844-850. 10.1016/S1367-5931(02)00390-3 [DOI] [PubMed] [Google Scholar]
- Depienne C., Stevanin G., Brice A. and Durr A. (2007). Hereditary spastic paraplegias: an update. Curr. Opin. Neurol. 20, 674-680. 10.1097/WCO.0b013e3282f190ba [DOI] [PubMed] [Google Scholar]
- Ding W.-X., Ni H.-M., Gao W., Hou Y.-F., Melan M. A., Chen X., Stolz D. B., Shao Z.-M. and Yin X.-M. (2007). Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J. Biol. Chem. 282, 4702-4710. 10.1074/jbc.M609267200 [DOI] [PubMed] [Google Scholar]
- Ding Y., Ko M. H., Pehar M., Kotch F., Peters N. R., Luo Y., Salamat S. M. and Puglielli L. (2012). Biochemical inhibition of the acetyltransferases ATase1 and ATase2 reduces beta-secretase (BACE1) levels and Abeta generation. J. Biol. Chem. 287, 8424-8433. 10.1074/jbc.M111.310136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y., Dellisanti C. D., Ko M. H., Czajkowski C. and Puglielli L. (2014). The endoplasmic reticulum-based acetyltransferases, ATase1 and ATase2, associate with the oligosaccharyl-transferase to acetylate correctly folded polypeptides. J. Biol. Chem. 289, 32044-32055. 10.1074/jbc.M114.585547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drazic A., Myklebust L. M., Ree R. and Arnesen T. (2016). The world of protein acetylation. Biochim. Biophys. Acta 1864, 1372-1401. 10.1016/j.bbapap.2016.06.007 [DOI] [PubMed] [Google Scholar]
- Dyda F., Klein D. C. and Hickman A. B. (2000). GCN5-related N-acetyltransferases: a structural overview. Annu. Rev. Biophys. Biomol. Struct. 29, 81-103. 10.1146/annurev.biophys.29.1.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt M., Muhlenhoff M., Bethe A. and Gerardy-Schahn R. (1996). Expression cloning of the Golgi CMP-sialic acid transporter. Proc. Natl. Acad. Sci. USA 93, 7572-7576. 10.1073/pnas.93.15.7572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkemeier I., Laxa M., Miguet L., Howden A. J. M. and Sweetlove L. J. (2011). Proteins of diverse function and subcellular location are lysine acetylated in Arabidopsis. Plant Physiol. 155, 1779-1790. 10.1104/pp.110.171595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster R., Weiss M., Zimmermann T., Reynaud E. G., Verissimo F., Stephens D. J. and Pepperkok R. (2006). Secretory cargo regulates the turnover of COPII subunits at single ER exit sites. Curr. Biol. 16, 173-179. 10.1016/j.cub.2005.11.076 [DOI] [PubMed] [Google Scholar]
- Frake R. A., Ricketts T., Menzies F. M. and Rubinsztein D. C. (2015). Autophagy and neurodegeneration. J. Clin. Invest. 125, 65-74. 10.1172/JCI73944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregno I., Fasana E., Bergmann T. J., Raimondi A., Loi M., Solda T., Galli C., D'Antuono R., Morone D., Danieli A. et al. (2018). ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J. 37, e99259 10.15252/embj.201899259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiman R. N. and Tjian R. (2003). Regulating the regulators: lysine modifications make their mark. Cell 112, 11-17. 10.1016/S0092-8674(02)01278-3 [DOI] [PubMed] [Google Scholar]
- Fujino T., Kondo J., Ishikawa M., Morikawa K. and Yamamoto T. T. (2001). Acetyl-CoA synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J. Biol. Chem. 276, 11420-11426. 10.1074/jbc.M008782200 [DOI] [PubMed] [Google Scholar]
- Fumagalli F., Noack J., Bergmann T. J., Cebollero E., Pisoni G. B., Fasana E., Fregno I., Galli C., Loi M., Soldà T. et al. (2016). Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 18, 1173-1184. 10.1038/ncb3423 [DOI] [PubMed] [Google Scholar]
- Gavel Y. and von Heijne G. (1990). Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: implications for protein engineering. Protein Eng. 3, 433-442. 10.1093/protein/3.5.433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershey E. L., Vidali G. and Allfrey V. G. (1968). Chemical studies of histone acetylation. The occurrence of epsilon-N-acetyllysine in the f2a1 histone. J. Biol. Chem. 243, 5018-5022. [PubMed] [Google Scholar]
- Guo Y. and Linstedt A. D. (2006). COPII-Golgi protein interactions regulate COPII coat assembly and Golgi size. J. Cell Biol. 174, 53-63. 10.1083/jcb.200604058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn Y. and Lee B. (2006). Human-specific nonsense mutations identified by genome sequence comparisons. Hum. Genet. 119, 169-178. 10.1007/s00439-005-0125-6 [DOI] [PubMed] [Google Scholar]
- Hetz C., Thielen P., Matus S., Nassif M., Court F., Kiffin R., Martinez G., Cuervo A. M., Brown R. H. and Glimcher L. H. (2009). XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 23, 2294-2306. 10.1101/gad.1830709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg C. B., Robbins P. W. and Abeijon C. (1998). Transporters of nucleotide sugars, ATP, and nucleotide sulfate in the endoplasmic reticulum and Golgi apparatus. Annu. Rev. Biochem. 67, 49-69. 10.1146/annurev.biochem.67.1.49 [DOI] [PubMed] [Google Scholar]
- Hosp F., Lassowskat I., Santoro V., De Vleesschauwer D., Fliegner D., Redestig H., Mann M., Christian S., Hannah M. A. and Finkemeier I. (2017). Lysine acetylation in mitochondria: from inventory to function. Mitochondrion 33, 58-71. 10.1016/j.mito.2016.07.012 [DOI] [PubMed] [Google Scholar]
- Hullinger R., Li M., Wang J., Peng Y., Dowell J. A., Bomba-Warczak E., Mitchell H. A., Burger C., Chapman E. R., Denu J. M. et al. (2016). Increased expression of AT-1/SLC33A1 causes an autistic-like phenotype in mice by affecting dendritic branching and spine formation. J. Exp. Med. 213, 1267-1284. 10.1084/jem.20151776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey S. J., James D. E. and Mann M. (2015). Protein phosphorylation: a major switch mechanism for metabolic regulation. Trends Endocrinol. Metab. 26, 676-687. 10.1016/j.tem.2015.09.013 [DOI] [PubMed] [Google Scholar]
- Huppke P., Brendel C., Kalscheuer V., Korenke G. C., Marquardt I., Freisinger P., Christodoulou J., Hillebrand M., Pitelet G., Wilson C. et al. (2012). Mutations in SLC33A1 cause a lethal autosomal-recessive disorder with congenital cataracts, hearing loss, and low serum copper and ceruloplasmin. Am. J. Hum. Genet. 90, 61-68. 10.1016/j.ajhg.2011.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K., Hao F., Fujita N., Tsuji Y., Oe Y., Araki Y., Hamasaki M., Noda T. and Yoshimori T. (2016). Atg9A trafficking through the recycling endosomes is required for autophagosome formation. J. Cell Sci. 129, 3781-3791. 10.1242/jcs.196196 [DOI] [PubMed] [Google Scholar]
- Jeffers V. and Sullivan W. J. Jr. (2012). Lysine acetylation is widespread on proteins of diverse function and localization in the protozoan parasite Toxoplasma gondii. Eukaryot. Cell 11, 735-742. 10.1128/EC.00088-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas M. C., Pehar M. and Puglielli L. (2010). AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability. J. Cell Sci. 123, 3378-3388. 10.1242/jcs.068841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori A., Nakayama J., Fukuda M. N., Stallcup W. B., Sasaki K., Fukuda M. and Hirabayashi Y. (1997). Expression cloning and characterization of a cDNA encoding a novel membrane protein required for the formation of O-acetylated ganglioside: a putative acetyl-CoA transporter. Proc. Natl. Acad. Sci. USA 94, 2897-2902. 10.1073/pnas.94.7.2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan R. S., Mayor J. A., Gremse D. A. and Wood D. O. (1995). High level expression and characterization of the mitochondrial citrate transport protein from the yeast Saccharomyces cerevisiae. J. Biol. Chem. 270, 4108-4114. 10.1074/jbc.270.8.4108 [DOI] [PubMed] [Google Scholar]
- Kapoor M. and Lozano G. (1998). Functional activation of p53 via phosphorylation following DNA damage by UV but not gamma radiation. Proc. Natl. Acad. Sci. USA 95, 2834-2837. 10.1073/pnas.95.6.2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A., Heinrich T., Mari M., Grumati P., Huebner A. K., Akutsu M., Liebmann L., Stolz A., Nietzsche S., Koch N. et al. (2015). Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354-358. 10.1038/nature14498 [DOI] [PubMed] [Google Scholar]
- Ko M. H. and Puglielli L. (2009). Two endoplasmic reticulum (ER)/ER golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J. Biol. Chem. 284, 2482-2492. 10.1074/jbc.M804901200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. (2000). Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 19, 1176-1179. 10.1093/emboj/19.6.1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G. (2015). Autophagy: a druggable process that is deregulated in aging and human disease. J. Clin. Invest. 125, 1-4. 10.1172/JCI78652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A., Komatsu M. and Mizushima N. (2017). Autophagy-monitoring and autophagy-deficient mice. Autophagy 13, 1619-1628. 10.1080/15548627.2017.1343770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J. and Morimoto R. I. (2015). The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435-464. 10.1146/annurev-biochem-060614-033955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalioti V. S., Vergarajauregui S., Villasante A., Pulido D. and Sandoval I. V. (2013). C6orf89 encodes three distinct HDAC enhancers that function in the nucleolus, the golgi and the midbody. J. Cell. Physiol. 228, 1907-1921. 10.1002/jcp.24355 [DOI] [PubMed] [Google Scholar]
- Lee I. H. and Finkel T. (2009). Regulation of autophagy by the p300 acetyltransferase. J. Biol. Chem. 284, 6322-6328. 10.1074/jbc.M807135200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I. H., Cao L., Mostoslavsky R., Lombard D. B., Liu J., Bruns N. E., Tsokos M., Alt F. W. and Finkel T. (2008). A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 105, 3374-3379. 10.1073/pnas.0712145105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. V., Carrer A., Shah S., Snyder N. W., Wei S., Venneti S., Worth A. J., Yuan Z.-F., Lim H.-W., Liu S. et al. (2014). Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 20, 306-319. 10.1016/j.cmet.2014.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi R. and Jackowski S. (2007). Biosynthesis of pantothenic acid and coenzyme A. EcoSal Plus 2, doi: 10.1128/ecosalplus.3.6.3.4 10.1128/ecosalplus.3.6.3.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B., Packer M. and Codogno P. (2015). Development of autophagy inducers in clinical medicine. J. Clin. Invest. 125, 14-24. 10.1172/JCI73938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P., Li J., Liu Q., Mao F., Li J., Qiu R., Hu H., Song Y., Yang Y., Gao G. et al. (2008). A missense mutation in SLC33A1, which encodes the acetyl-CoA transporter, causes autosomal-dominant spastic paraplegia (SPG42). Am. J. Hum. Genet. 83, 752-759. 10.1016/j.ajhg.2008.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P., Jiang B., Ma J., Lin P., Zhang Y., Shao C., Sun W. and Gong Y. (2017). S113R mutation in SLC33A1 leads to neurodegeneration and augmented BMP signaling in a mouse model. Dis. Model. Mech. 10, 53-62. 10.1242/dmm.026880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundby A., Lage K., Weinert B. T., Bekker-Jensen D. B., Secher A., Skovgaard T., Kelstrup C. D., Dmytriyev A., Choudhary C., Lundby C. et al. (2012). Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2, 419-431. 10.1016/j.celrep.2012.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackall J. C. and Lane M. D. (1977). Changes in mammary-gland acetyl-coenzyme A carboxylase associated with lactogenic differentiation. Biochem. J. 162, 635-642. 10.1042/bj1620635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo F., Eisenberg T. and Kroemer G. (2009). Autophagy for the avoidance of neurodegeneration. Genes Dev. 23, 2253-2259. 10.1101/gad.1858009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo F., Zimmermann A., Maiuri M. C. and Kroemer G. (2015). Essential role for autophagy in life span extension. J. Clin. Invest. 125, 85-93. 10.1172/JCI73946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak A. B., Pehar M., Nixon A. M. L., Williams R. A., Uetrecht A. C., Puglielli L. and Moffat J. (2014). Post-translational regulation of CD133 by ATase1/ATase2-mediated lysine acetylation. J. Mol. Biol. 426, 2175-2182. 10.1016/j.jmb.2014.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao F., Li Z., Zhao B., Lin P., Liu P., Zhai M., Liu Q., Shao C., Sun W. and Gong Y. (2015). Identification and functional analysis of a SLC33A1: c.339T>G (p.Ser113Arg) variant in the original SPG42 family. Hum. Mutat. 36, 240-249. 10.1002/humu.22732 [DOI] [PubMed] [Google Scholar]
- Matus S., Nassif M., Glimcher L. H. and Hetz C. (2009). XBP-1 deficiency in the nervous system reveals a homeostatic switch to activate autophagy. Autophagy 5, 1226-1228. 10.4161/auto.5.8.10247 [DOI] [PubMed] [Google Scholar]
- McCracken A. A. and Brodsky J. L. (1996). Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J. Cell Biol. 132, 291-298. 10.1083/jcb.132.3.291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies K. J., Zhang H., Katsyuba E. and Auwerx J. (2016). Protein acetylation in metabolism - metabolites and cofactors. Nat. Rev. Endocrinol. 12, 43-60. 10.1038/nrendo.2015.181 [DOI] [PubMed] [Google Scholar]
- Metallo C. M., Gameiro P. A., Bell E. L., Mattaini K. R., Yang J., Hiller K., Jewell C. M., Johnson Z. R., Irvine D. J., Guarente L. et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380-384. 10.1038/nature10602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida K., Oikawa Y., Kimura Y., Kirisako H., Hirano H., Ohsumi Y. and Nakatogawa H. (2015). Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 522, 359-362. 10.1038/nature14506 [DOI] [PubMed] [Google Scholar]
- Murn J. and Shi Y. (2017). The winding path of protein methylation research: milestones and new frontiers. Nat. Rev. Mol. Cell Biol. 18, 517-527. 10.1038/nrm.2017.35 [DOI] [PubMed] [Google Scholar]
- Nakatogawa H. and Mochida K. (2015). Reticulophagy and nucleophagy: new findings and unsolved issues. Autophagy 11, 2377-2378. 10.1080/15548627.2015.1106665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald A. F. and Landsman D. (1997). GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem. Sci. 22, 154-155. 10.1016/S0968-0004(97)01034-7 [DOI] [PubMed] [Google Scholar]
- Nishi H., Hashimoto K. and Panchenko A. R. (2011). Phosphorylation in protein-protein binding: effect on stability and function. Structure 19, 1807-1815. 10.1016/j.str.2011.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T., Tamura N., Kono N., Shimanaka Y., Arai H., Yamamoto H. and Mizushima N. (2017). Autophagosome formation is initiated at phosphatidylinositol synthase-enriched ER subdomains. EMBO J. 36, 1719-1735. 10.15252/embj.201695189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon R. A. (2013). The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983-997. 10.1038/nm.3232 [DOI] [PubMed] [Google Scholar]
- Ogata M., Hino S., Saito A., Morikawa K., Kondo S., Kanemoto S., Murakami T., Taniguchi M., Tanii I., Yoshinaga K. et al. (2006). Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 26, 9220-9231. 10.1128/MCB.01453-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi Y. and Munro S. (2010). Membrane delivery to the yeast autophagosome from the Golgi-endosomal system. Mol. Biol. Cell 21, 3998-4008. 10.1091/mbc.e10-05-0457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouidir T., Kentache T. and Hardouin J. (2016). Protein lysine acetylation in bacteria: current state of the art. Proteomics 16, 301-309. 10.1002/pmic.201500258 [DOI] [PubMed] [Google Scholar]
- Pehar M. and Puglielli L. (2013). Lysine acetylation in the lumen of the ER: a novel and essential function under the control of the UPR. Biochim. Biophys. Acta 1833, 686-697. 10.1016/j.bbamcr.2012.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehar M., Jonas M. C., Hare T. M. and Puglielli L. (2012a). SLC33A1/AT-1 protein regulates the induction of autophagy downstream of IRE1/XBP1 pathway. J. Biol. Chem. 287, 29921-29930. 10.1074/jbc.M112.363911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehar M., Lehnus M., Karst A. and Puglielli L. (2012b). Proteomic assessment shows that many endoplasmic reticulum (ER)-resident proteins are targeted by N(epsilon)-lysine acetylation in the lumen of the organelle and predicts broad biological impact. J. Biol. Chem. 287, 22436-22440. 10.1074/jbc.C112.362871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y., Kim M. J., Hullinger R., O'Riordan K. J., Burger C., Pehar M. and Puglielli L. (2016). Improved proteostasis in the secretory pathway rescues Alzheimer's disease in the mouse. Brain 139, 937-952. 10.1093/brain/awv385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y., Li M., Clarkson B. D., Pehar M., Lao P. J., Hillmer A. T., Barnhart T. E., Christian B. T., Mitchell H. A., Bendlin B. B. et al. (2014). Deficient import of acetyl-CoA into the ER lumen causes neurodegeneration and propensity to infections, inflammation, and cancer. J. Neurosci. 34, 6772-6789. 10.1523/JNEUROSCI.0077-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y., Shapiro S. L., Banduseela V. C., Dieterich I. A., Hewitt K. J., Bresnick E. H., Kong G., Zhang J., Schueler K. L., Keller M. P. et al. (2018). Increased transport of acetyl-CoA into the endoplasmic reticulum causes a progeria-like phenotype. Aging Cell 17, e12820 10.1111/acel.12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickford F., Masliah E., Britschgi M., Lucin K., Narasimhan R., Jaeger P. A., Small S., Spencer B., Rockenstein E., Levine B. et al. (2008). The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Invest. 118, 2190-2199. 10.1172/JCI33585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrocola F., Galluzzi L., Bravo-San Pedro J. M., Madeo F. and Kroemer G. (2015). Acetyl Coenzyme A: a central metabolite and second messenger. Cell Metab. 21, 805-821. 10.1016/j.cmet.2015.05.014 [DOI] [PubMed] [Google Scholar]
- Polgar L. (1964). Specific acetylation of a lysine residue during the hydrolytic action of glyceraldehyde-3-phosphate dehydrogenase. Acta Physiol. Acad. Sci. Hung. 25, 1-4. [PubMed] [Google Scholar]
- Prasad A., Merico D., Thiruvahindrapuram B., Wei J., Lionel A. C., Sato D., Rickaby J., Lu C., Szatmari P., Roberts W. et al. (2012). A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 (Bethesda) 2, 1665-1685. 10.1534/g3.112.004689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D. and Walter P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519-529. 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- Roth S. Y., Denu J. M. and Allis C. D. (2001). Histone acetyltransferases. Annu. Rev. Biochem. 70, 81-120. 10.1146/annurev.biochem.70.1.81 [DOI] [PubMed] [Google Scholar]
- Rubinsztein D. C. (2015). Cell biology: receptors for selective recycling. Nature 522, 291-292. 10.1038/nature14532 [DOI] [PubMed] [Google Scholar]
- Saitoh T., Fujita N., Hayashi T., Takahara K., Satoh T., Lee H., Matsunaga K., Kageyama S., Omori H., Noda T. et al. (2009). Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. USA 106, 20842-20846. 10.1073/pnas.0911267106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas S., Proukakis C., Crosby A. and Warner T. T. (2008). Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 7, 1127-1138. 10.1016/S1474-4422(08)70258-8 [DOI] [PubMed] [Google Scholar]
- Sanders S. J., Ercan-Sencicek A. G., Hus V., Luo R., Murtha M. T., Moreno-De-Luca D., Chu S. H., Moreau M. P., Gupta A. R., Thomson S. A. et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863-885. 10.1016/j.neuron.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck S. (2016). On keeping the right ER size. Nat. Cell Biol. 18, 1118-1119. 10.1038/ncb3430 [DOI] [PubMed] [Google Scholar]
- Shaffer A. L., Shapiro-Shelef M., Iwakoshi N. N., Lee A.-H., Qian S.-B., Zhao H., Yu X., Yang L., Tan B. K., Rosenwald A. et al. (2004). XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 21, 81-93. 10.1016/j.immuni.2004.06.010 [DOI] [PubMed] [Google Scholar]
- Shi L. and Tu B. P. (2015). Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr. Opin. Cell Biol. 33, 125-131. 10.1016/j.ceb.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivanand S., Rhoades S., Jiang Q., Lee J. V., Benci J., Zhang J., Yuan S., Viney I., Zhao S., Carrer A. et al. (2017). Nuclear acetyl-CoA production by ACLY promotes homologous recombination. Mol. Cell 67, 252-265.e6. 10.1016/j.molcel.2017.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoswell A. M. and Koundakjian P. P. (1972). Relationships between carnitine and coenzyme A esters in tissues of normal and alloxan-diabetic sheep. Biochem. J. 127, 133-141. 10.1042/bj1270133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutendra G., Kinnaird A., Dromparis P., Paulin R., Stenson T. H., Haromy A., Hashimoto K., Zhang N., Flaim E. and Michelakis E. D. (2014). A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 158, 84-97. 10.1016/j.cell.2014.04.046 [DOI] [PubMed] [Google Scholar]
- Tamura H., Shibata M., Koike M., Sasaki M. and Uchiyama Y. (2010). Atg9A protein, an autophagy-related membrane protein, is localized in the neurons of mouse brains. J. Histochem. Cytochem. 58, 443-453. 10.1369/jhc.2010.955690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta E. S. and Parodi A. J. (2003). Quality control and protein folding in the secretory pathway. Annu. Rev. Cell Dev. Biol. 19, 649-676. 10.1146/annurev.cellbio.19.110701.153949 [DOI] [PubMed] [Google Scholar]
- Vakifahmetoglu-Norberg H., Xia H.-G. and Yuan J. (2015). Pharmacologic agents targeting autophagy. J. Clin. Invest. 125, 5-13. 10.1172/JCI73937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dellen A., Blakemore C., Deacon R., York D. and Hannan A. J. (2000). Delaying the onset of Huntington's in mice. Nature 404, 721-722. 10.1038/35008142 [DOI] [PubMed] [Google Scholar]
- Veiga-da-Cunha M., Tyteca D., Stroobant V., Courtoy P. J., Opperdoes F. R. and Van Schaftingen E. (2010). Molecular identification of NAT8 as the enzyme that acetylates cysteine S-conjugates to mercapturic acids. J. Biol. Chem. 285, 18888-18898. 10.1074/jbc.M110.110924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidali G., Gershey E. L. and Allfrey V. G. (1968). Chemical studies of histone acetylation. The distribution of epsilon-N-acetyllysine in calf thymus histones. J. Biol. Chem. 243, 6361-6366. [PubMed] [Google Scholar]
- Wanders R. J., Waterham H. R. and Ferdinandusse S. (2015). Metabolic interplay between peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front. Cell Dev. Biol. 3, 83 10.3389/fcell.2015.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. and Kaufman R. J. (2016). Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326-335. 10.1038/nature17041 [DOI] [PubMed] [Google Scholar]
- Webber J. L., Young A. R. J. and Tooze S. A. (2007). Atg9 trafficking in Mammalian cells. Autophagy 3, 54-56. 10.4161/auto.3419 [DOI] [PubMed] [Google Scholar]
- Werner E. D., Brodsky J. L. and McCracken A. A. (1996). Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc. Natl. Acad. Sci. USA 93, 13797-13801. 10.1073/pnas.93.24.13797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesche J., Kühn S., Kessler B. M., Salton M. and Wolf A. (2017). Protein arginine methylation: a prominent modification and its demethylation. Cell. Mol. Life Sci. 74, 3305-3315. 10.1007/s00018-017-2515-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.-J. and Grégoire S. (2007). Metabolism, cytoskeleton and cellular signalling in the grip of protein Nepsilon - and O-acetylation. EMBO Rep. 8, 556-562. 10.1038/sj.embor.7400977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen W.-L. and Klionsky D. J. (2007). Atg27 is a second transmembrane cycling protein. Autophagy 3, 254-256. 10.4161/auto.3823 [DOI] [PubMed] [Google Scholar]
- You M., Fischer M., Deeg M. A. and Crabb D. W. (2002). Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J. Biol. Chem. 277, 29342-29347. 10.1074/jbc.M202411200 [DOI] [PubMed] [Google Scholar]
- Young A. R. J., Chan E. Y., Hu X. W., Kochl R., Crawshaw S. G., High S., Hailey D. W., Lippincott-Schwartz J. and Tooze S. A. (2006). Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 119, 3888-3900. 10.1242/jcs.03172 [DOI] [PubMed] [Google Scholar]
- Yu Y., Carter C. R. J., Youssef N., Dyck J. R. B. and Light P. E. (2014). Intracellular long-chain acyl CoAs activate TRPV1 channels. PLoS ONE 9, e96597 10.1371/journal.pone.0096597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Huang Y. and Shi X. (2015). Emerging roles of lysine methylation on non-histone proteins. Cell. Mol. Life Sci. 72, 4257-4272. 10.1007/s00018-015-2001-4 [DOI] [PMC free article] [PubMed] [Google Scholar]