

Summary
Molecular imaging of metastatic “potential” is an unvanquished challenge. To engineer biosensors that can detect and measure the metastatic “potential” of single living cancer cells, we carried out a comprehensive analysis of the pan-cancer phosphoproteome to search for actin remodelers required for cell migration, which are enriched in cancers but excluded in normal cells. Only one phosphoprotein emerged, tyr-phosphorylated CCDC88A (GIV/Girdin), a bona fide metastasis-related protein across a variety of solid tumors. We designed multi-modular biosensors that are partly derived from GIV, and because GIV integrates prometastatic signaling by multiple oncogenic receptors, we named them “‘integrators of metastatic potential (IMP).” IMPs captured the heterogeneity of metastatic potential within primary lung and breast tumors at steady state, detected those few cells that have acquired the highest metastatic potential, and tracked their enrichment during metastasis. These findings provide proof of concept that IMPs can measure the diversity and plasticity of metastatic potential of tumor cells in a sensitive and unbiased way.
Subject Areas: Optical Imaging, Protein Physics, Cancer
Graphical Abstract
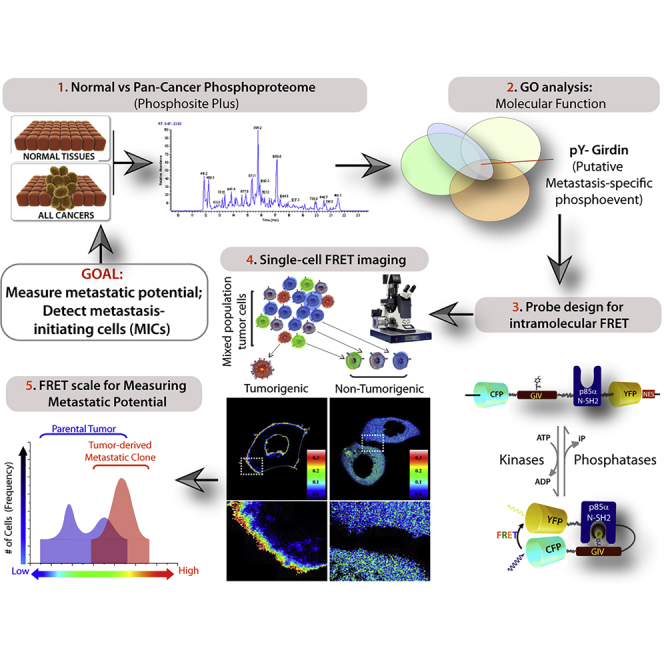
Highlights
-
•
Phosphoproteomes of cancers predicted a putative metastasis-specific phosphoevent
-
•
FRET-based biosensor designed to assess this phosphoevent in living cells
-
•
Biosensor tracks the diversity and plasticity of metastatic potential of cancer cells
-
•
These sensors could direct drug efficacy testing against the most sinister cancer cells
Optical Imaging; Protein Physics; Cancer
Introduction
Metastasis is the dissemination of highly invasive cancer cells from the primary tumor to distant vital organs and the principal cause of cancer-related deaths (McAllister and Weinberg, 2014). However, not all tumors are metastatic, and determining the metastatic proclivity of single tumor cells remains a major challenge due to several reasons. First, and arguably the biggest, hurdle is tumor molecular heterogeneity because finding the few cells (0.02% [McAllister and Weinberg, 2014]) that have the potential to metastasize, interspersed within a mass of tumor cells that will not metastasize, requires precise tools that can pick up a few “signals” amid the thunderous “noise.” Second, our understanding of what imparts metastatic potential remains incomplete despite increased comprehension of a more detailed signaling network with central control nodes (McAllister and Weinberg, 2014) and biomarkers that can detect such potential across different carcinomas are still lacking. Third, the genome of the cancer cell is rewired and signaling networks are reprogrammed during this process of metastatic progression, either to adapt to the changing tumor microenvironment or to overcome the cytotoxicity of drugs (Hanahan and Weinberg, 2011), and biomarkers/tools that monitor any given pathway may lose significance due to the changing/evolving tumor dependency from that pathway to unknown pathway(s) (the so-called addiction switch) (Weinstein, 2002). Consequently, pathway-specific biomarkers that monitor the “known” in the setting of the so-called unknown-unknowns of tumor biology introduce bias in the short run and prove ineffective in the long run. Last, but not least, single-cell studies have shown that when stochastic, invasive, and proliferative events are triggered by perturbations (e.g., mechanical or chemical signals), each individual cell adapts on its own, i.e., cancer cells go solo (Chung et al., 2017, Ellsworth et al., 2017, Ferronika et al., 2017, Kimura et al., 2010, Kubota et al., 2017, Lee et al., 2014, Lorentzen et al., 2018, Ramapathiran et al., 2014, Su et al., 2017, Wu et al., 2018). Hence, any effective biomarker/assay must be sensitive enough to detect the plasticity of metastatic programming within a few sparsely distributed tumorigenic cells, i.e., cells that can metastasize and initiate new tumors at distant sites, within a large population of non-tumorigenic cells.
Despite these shortfalls, experts agree that estimating the metastatic “potential” is a problem that only molecular imaging can resolve, rather than conventional techniques, e.g., immunohistochemistry (IHC) (Winnard et al., 2008). So far, improved imaging platforms have helped detect established metastases (Frangioni, 2008) and assessed tumor cell properties as surrogate markers of metastatic potential (e.g., glucose consumption, hypoxia, angiogenesis, integrin expression patterns, or matrix metalloproteinase activities; reviewed in Winnard et al., 2008) and even visualized the metastatic process in real time in vivo (Sahai, 2007). However, single-cell-based assays to measure the dynamic prometastatic signaling programs that contribute to the “potential” for metastasis remains a Holy Grail. This is largely because conventional approaches (immunofluorescence and IHC) employed to study most biomarkers on fixed tissues are fraught with technical limitations (Sato et al., 2002, Veeriah et al., 2014). Even when live cells or tissues are used in the above-mentioned approaches, one serious flaw remains, i.e., the loss of vital information pertinent to individual cells due to averaging over an ensemble of readouts (Ray, 2013). The concept of single-cell studies has gained traction in the fields of metabolomics (Zenobi, 2013) and signal transduction (Clister et al., 2015, Philips, 2005) and among cell biologists who seek to explore how cellular processes are organized and regulated in vivo (Midde et al., 2013), but has not been applied to study metastatic potential.
Here we report the development of fluorescence resonance energy transfer (FRET) biosensors that measure the metastatic potential of single living cancer cells by overcoming both aforementioned limitations, i.e., they account for the unknown-unknowns of an evolving tumor biology and eliminate averaging losses during conventional molecular imaging.
Results
Pan-cancer Phosphoproteome Reveals that Tyrosine Phosphorylation of Girdin Is a Putative Metastasis-Related Phosphoevent
In search of an ideal target, first we carried out a comprehensive analysis of the pan-cancer phosphoproteome using the NIH-supported, continuously curated, and interactive systems biology resource, PhosphoSitePlus (Hornbeck et al., 2004, Hornbeck et al., 2012, Hornbeck et al., 2015) to study experimentally observed post-translational modifications in the regulation of biological processes. Using powerful gene ontology (GO)-analytical tools (PANTHER [Mi et al., 2017], GOrilla [Eden et al., 2009], and REVIGO [Supek et al., 2011]) for mining, interpreting, and visualizing this data, we noted that several pathways and biological processes were overrepresented in the pan-cancer phosphoproteome above the pan-human phosphoproteome set as reference (see Figure S1, Table S1). To identify phosphoproteins that are selectively seen in cancers, we searched for phosphopeptides that are present in cancers, but not in normal tissues. A handful of pathways (Figure S2A) and biological processes (Figures S2B and S2C) were over- or under-represented over the reference set, and only one GO term enriched over the others, i.e., negative regulators of apoptosis (Figure S2D). Because high-resolution imaging of metastasizing cancer cells has underscored the importance of actin cytoskeleton remodeling as a fundamental prerequisite for cancer invasion (Sahai, 2007), next we searched the pan-cancer phosphoproteome using a combination of different criteria for enriching for either “cellular processes” (the largest GO biological process enriched in cancer-specific phosphoproteins; S2b), actin binders/modulators and those involved in cell migration (the most important property of tumor cells that has marked effects on tumor growth, resistance, and spread [Waclaw et al., 2015]), but are excluded from normal tissues (Figure 1A; Tables S2, S3, S4, and S5). Only one phosphopeptide, pY1798, in a protein named GIV/GRDN (Gα-interacting vesicle-associated; a.k.a. Girdin; gene: CCDC88A) fits all the criteria.
Figure 1.

Rationale, Design, and Biochemical Validation of IMPs
(A) The pan-cancer phosphoproteome (Table S1) was analyzed with additional filters (see Transparent Methods and Tables S2, S3, S4, and S5 for list of phosphoproteins returned with each filter/criterion).
(B) Phosphoproteomic map of GIV/Girdin. Tyrosine-phosphorylated peptides are most frequently detected by high-throughput (HTP) mass spectrometry (MS) studies. Tyrosines 1764 and 1798 are known to bind and activate PI3K, whereas the role of Y1743 remains unknown.
(C) Schematic illustrating the modular makeup of IMP probes. A previously validated (Ullman et al., 1997) nuclear export signal (NES) was incorporated to retain most of the probe in the cytosolic compartment.
(D) Immunoblots showing expression and ligand-induced phosphorylation of various IMP probes in Cos7 cells after EGF stimulation.
(E) Immunoblots showing the phosphorylation status of wild-type (WT) IMP-Y1764 and Y1798 and their corresponding YF mutants after EGF stimulation.
(F) Serum-starved Cos7 cells expressing IMP-Y1764 were stimulated with various growth factors before lysis. Lysates were immunoprecipitated with anti-GFP monoclonal antibody, and tyrosine phosphorylation of IMP was analyzed by immunoblotting.
(G and H) Lysates of Cos7 cells co-expressing IMP-Y1798 and WT or kinase-dead (KD) Src-HA (G) or FAK-HA (H) were immunoprecipitated and analyzed for tyrosine phosphorylation of IMP as in (F).
(I) Lysates of Cos7 cells expressing IMP-Y1764 or Y1798 and stimulated with lysophosphatidic acid (LPA) were immunoprecipitated and analyzed for tyrosine phosphorylation of IMP as in (F).
(J) Lysates of Cos7 cells expressing IMP-Y1764 and WT or catalytically dead (CD) SHP1-HA were immunoprecipitated and analyzed for tyrosine phosphorylation of IMP as in (F).
(K) Whole-cell lysates of Cos7 cells expressing various IMP probes were analyzed for phospho(p) and total(t) Akt and ERK, tubulin, and GFP (IMPs) by immunoblotting.
Girdin (a.k.a, GIV, APE, HkRP1) is essentially a large, multi-modular, cytosolic signal transducer that was co-discovered by four groups nearly simultaneously (Anai et al., 2005, Enomoto et al., 2005, Enomoto et al., 2006, Simpson et al., 2005). It has been recognized as a bona fide driver of metastasis because of its ability to enhance phosphatidylinositol 3-kinase (PI3K)-Akt signals and couple them to cytoskeletal remodeling (Takahashi et al., 2015, Weng et al., 2010), an essential feature for tumor cell invasion. Girdin also serves another role, that of a guanine nucleotide exchange modulator, which integrates signals downstream of multiple classes of receptors by coupling ligand-activated receptors to activation of trimeric GTPases (Aznar et al., 2016, DiGiacomo et al., 2018, Ghosh et al., 2017). Consequently, receptor-initiated signals are modulated by G protein intermediates (Garcia-Marcos et al., 2015). Ligand stimulation of a variety of receptors directly (as in the case of growth factor receptor tyrosine kinases [RTKs]) or indirectly (via non-RTKs, as in the case of G-protein coupled receptor [GPCRs], Toll-like receptors, integrins) trigger phosphorylation of GIV on two key tyrosines (Y1764 and Y1798) within its C terminus (Figure 1B) (Dunkel et al., 2016, Kuga et al., 2017, Lin et al., 2011, Lopez-Sanchez et al., 2015, Mizutani et al., 2018, Omori et al., 2015), which directly bind and activate Class 1 PI3Ks, and subsequently, Akt (Lin et al., 2011) (Figure S3A). Multiple groups have independently validated the role of this multi-receotor-GIV-PI3K axis in driving metastasis and/or enhancing prometastatic features of tumor cells across a variety of solid cancers (summarized in Ghosh, 2015, Ghosh et al., 2011, Takahashi et al., 2015, Weng et al., 2010). These studies have established causal links between GIV and several ominous traits that aid in the acquisition of metastatic potential, e.g., collective migration/invasiveness, survival, stemness, and chemoresistance (summarized in Aznar et al., 2016, Takahashi et al., 2015).
Rationale Design and Validation of FRET Probes that Measure Functional Phosphorylation of Girdin
We hypothesized that the measurement of the intensity of functional phosphorylation of GIV within the multi-receptor-GIV-PI3K axis in individual living cells will serve as a surrogate to assess the extent of multi-receptor-driven prometastatic signaling via the GIV-PI3K node. To achieve this goal, we designed intramolecular FRET probes with two modules (Figure 1C): (1) short stretches of GIV flanking either one or both critical tyrosines, Y1764 and Y1798 (Figure S3B) and (2) N-Src homology 2 (SH2) domain of p85α(PI3K); the former serves as a target substrate for multiple tyrosine kinases, and when phosphorylated serves as the ligand for recognition by N-SH2- p85α(PI3K). The two modules are separated by a flexible linker and sandwiched by eCFP (donor) and eYFP (acceptor) proteins, such that binding of the N-SH2- p85α(PI3K) to phosphotyrosine (pY) GIV ligand leads to folding of the probe bringing the donor and acceptor fluorescent proteins into close proximity, with resultant gain in FRET. Because these synthetic probes were built to assess multi-receptor-GIV-PI3K signaling, we named them “integrators of metastatic potential (IMPs).”
We first validated the IMP probes biochemically by confirming that they are expressed as fluorescent-tagged proteins of expected size and that they are phosphorylated on tyrosines after ligand (epidermal growth factor [EGF]) stimulation (Figure 1D). Ligand-dependent phosphorylation was virtually abolished when the corresponding phosphorylation-deficient IMPs were expressed in which Tyr (Y) was substituted by Phe (F) (henceforth referred to as YF) (Figures 1E, S4A, and S4B). Consistent with the fact that multiple pathways converge on GIV, IMPs were phosphorylated upon stimulation with multiple growth factors (EGF, insulin, platelet-derived growth factor) (Figures 1F, S4C, and S4D), non-RTKs Src and FAK (Figures 1G, 1H, S4E, and S4F), and ligand for LPAR, a GPCR (Figure 1I). As expected, the expression of catalytically active SHP1, a protein tyrosine phosphatase that dephosphorylates GIV's tyrosines (Mittal et al., 2011), abolished tyrosine phosphorylation of IMPs (Figures 1J and S4G). Finally, because ideal biosensors do not perturb the system under test (Haugh, 2012), and isolated domains of GIV that can bind RTKs or activate G proteins can display a range of biological activity (Midde et al., 2015) and alter the ratio of Akt and ERK signals, we asked which IMP construct is the least perturbing. In the presence of a functional SH2-like or GEF module (Lin et al., 2014, Midde et al., 2015) IMP-SH2 and IMP-GIV-C-terminus (IMP-CT) constructs (shown in Figure S3B) altered the levels of Akt and ERK signals at baseline steady states, whereas under the same conditions the IMP-1764 or IMP-1798, constructs that contain merely 13 amino acids of GIV appeared inert (Figure 1K). Hence, these two minimalist IMPs were deemed appropriate for further evaluation. Consistent with their predicted inertness, overexpression of either IMP-1764 or IMP-1798 or their non-phosphorylatable YF mutants did not affect 2D migration in scratch wound assays (Figure S5). Although both Y1764 and Y1798 cooperatively activate PI3Ks, because Y1764 is a target for both RTKs and non-RTKs, whereas Y1798 is exclusively phosphorylated by non-RTKs (Lin et al., 2011), we focused heavily on IMP-1764.
Next we validated these probes in live cells to determine if ligand-induced tyrosine phosphorylation was also accompanied by intramolecular folding and FRET. FRET signals were analyzed exclusively at the plasma membrane (PM) because Class IA PI3Ks activated by tyrosine-phosphorylated GIV transduce signals primarily by phosphorylating phosphatidylinositol-4,5-bisphosphate at the PM (Engelman, 2009), and because tyrosine phosphorylation of GIV (as determined by immunofluorescence using anti-pYGIV antibody [Lopez-Sanchez et al., 2014]) coincided temporally and spatially with the activation of PI3K (as determined using the fluorescent reporter, YFP-tagged PH-domain of Akt [Lin et al., 2011]) on microdomains at the PM (Figure 2A). These temporal and spatial patterns were reproduced using IMP probes in living cells responding to either EGF (Figures 2B and 2C; Videos S1, S2, S3, and S4) or lysophosphatidic acid (LPA) (Figures 2D and 2E; Videos S5 and S6). Regardless of the pathway activated, FRET was observed at the PM within 2–2.5 min for both IMP-1764 and IMP-1798, albeit with a few notable differences. FRET at the PM was transient in the case of growth factors, whereas a more delayed (peak 4.5 min) and sustained response was seen in the case of LPA. These findings are consistent with the differential kinetics of Akt activation downstream, i.e., Akt phosphorylation typically peaks at 5 min after stimulation with growth factors, but at 15 min after stimulation of GPCRs (Garcia-Marcos et al., 2009). No FRET was observed when each IMP was substituted in the above-mentioned assays with its non-phosphorylatable YF counterpart, indicating that ligand-stimulated tyrosine phosphorylation of GIV is essential for gaining FRET in each instance. Furthermore, compared with starved (0% fetal bovine serum [FBS]) cells, increased ambient signals in serum-fed cells (10% FBS) were accompanied by a higher steady-state probe phosphorylation (Figure 2F) and FRET efficiency (F.E) (Figures 2G and 2H), indicating that IMPs can detect ambient multi-receptor-driven signal flow at a steady state without the need to acutely activate a given receptor/pathway. Furthermore, depletion of GIV in MDA-MB-231 cells, which require GIV for their ability to form lung metastases when xenografted in nude mice (Jiang et al., 2008), was accompanied by a reduction in F.E (Figures 2I–2K). Together, these findings demonstrated that IMPs are the minimal platforms that can measure the timing, intensity, and location of functional phosphorylation of GIV in living cells at steady state.
Figure 2.

Validation of the IMPs using Single-Cell FRET Imaging
(A) Serum-starved Cos7 cells expressing Akt-PH-YFP (pseudocolored green) were stimulated with EGF, fixed, and stained for tyrosine-phosphorylated GIV (anti-pY1764GIV; red) and analyzed by confocal microscopy.
(B) Serum-starved Cos7 cells expressing IMP-Y1764, Y1798, or their corresponding YF mutants were stimulated with EGF and analyzed by confocal live-cell FRET imaging. Representative freeze-frame images from Videos S1, S2, S3, and S4 are shown.
(C) Time traces display the dynamic changes in FRET efficiency in (B); 12–15 cells were analyzed in 3 independent assays.
(D) Serum-starved Cos7 cells expressing IMP-Y1764 or the YF mutant were stimulated with LPA and analyzed by confocal live-cell FRET imaging. Representative freeze-frame images from Videos S5 and S6 are shown.
(E) Time traces display the dynamic changes in FRET efficiency in 12–15 cells that were analyzed in 3 independent assays.
(F) Immunoblots showing the contribution of serum (10%) on the phosphorylation status of wild-type (WT)-IMP-1764 and WT-IMP-1798 probes expressed in Cos7 cells at steady state. Phosphorylation of the IMP peptides and Akt was exclusively observed in fed state (10% FBS), but not in starved (0% FBS) conditions.
(G) Representative steady-state FRET images of Cos7 cells expressing IMP-1764 probe and its phosphorylation triggered by 10% serum. Higher FRET efficiency signals were detected in cells with 10% serum, but not in cells without serum.
(H) Scatterplots comparing the FRET efficiency at the PM in (G). Results are expressed as mean ± SD.
(I) Lysates of control and GIV-depleted MDA-MB-231 cells were analyzed for GIV depletion by immunoblotting (IB). The efficiency of depletion was estimated as ∼85% by band densitometry.
(J) Representative steady-state FRET images of MDA-MB-231 cells in (I) expressing the IMP-1764 probe.
(K) Scatterplots comparing the FRET efficiency at the PM in (J). Results are expressed as mean ± SD.
Video shows ligand-dependent signaling via the GIV-PI3K axis as visualized through FRET imaging in living Cos7 cells expressing the wild-type IMP-Y1764 sensor.
Video shows absence of significant FRET upon EGF stim in Cos7 cells expressing the non-phosphorylatable IMP-pY1764F mutant probe, indicating that phosphorylation and phosphorylation-induced intramolecular rearrangement of the probe are required for the observed changes in FRET in Video S1.
Video shows ligand-dependent signaling via the GIV-PI3K axis as visualized through FRET imaging in living Cos7 cells expressing the wild-type IMP-Y1798 sensor.
This video indicates that phosphorylation and phosphorylation-induced intramolecular rearrangement of the probe are required for the observed changes in FRET in Video S3.
Video shows ligand-dependent signaling via the GIV-PI3K axis as visualized through FRET imaging in living Cos7 cells expressing the wild-type IMP-Y1764 sensor.
This video indicates that phosphorylation and phosphorylation-induced intramolecular rearrangement of the probe are required for the observed dynamic changes in FRET in Video S5.
Single-Cell Imaging of Signaling through the GIV-PI3K Axis Reveals the Heterogeneity in Metastatic Potential and Tracks the Increasing Potential during Metastasis
Next we asked if the IMPs can measure the steady-state ambient multi-receptor signaling via the GIV-PI3K axis. If F.E. were to be measured at steady state, we assumed such measurement to reflect the equilibrium between the activity of multiple tyrosine kinases (i.e., forward reaction), the antagonistic dephosphorylating action of PTP and SHP1 (i.e., reverse rate reactions), and any upstream triggers to both enzymes. Hence, a high F.E observed would imply the activation of multiple pathways that trigger the prometastatic tyrosine kinase signaling pathways as well as those that suppress SHP1's protein phosphatase activity and would indicate overall hypersignaling via the TK-GIV axis. We specifically asked if such steady-state measurements can serve as a readout of the aberrant TK-GIV prometastatic signaling during the acquisition of metastatic potential. If so, we predicted that such measurement should come with three key advantages: (1) eliminate the need to know to which upstream pathways (drivers versus hitchhikers) a given tumor is addicted; (2) avoid perturbing the intrinsic pathologic pathways with extrinsic ligands that introduce undesired bias, and (3) ensure continued usefulness despite changes in tumor dependency on a given pathway, as during metastatic progression or development of drug resistance. To test these, we first validated the IMPs using three well-characterized metastatic breast and lung adenocarcinoma cell lines, MDA-MB-231, PC-9, and H2030, and their corresponding highly metastatic subclones (BrM) that were selected in mice and display ∼10-fold enhanced ability to metastasize to the bone and brain (Nguyen et al., 2009, Valiente et al., 2014) (Figure S6A). Despite their diverse genetic backgrounds (Figure S6B), all BrM clones consistently displayed one common feature compared with their parental counterparts: higher Akt activity (Figures 3A–3C), whereas the patterns of immunofluorescence staining for tyrosine-phosphorylated GIV (pY1764GIV) showed only minor differences (Figure S7). Using the IMP-Y1764 probe, we found that the steady-state F.E. in individual cells varied over a range in these cells (Figures 3D–3F); the variance is in keeping with the expected heterogeneity in metastatic potential within a given population of cells (Hanahan and Weinberg, 2011). However, the mean F.E. of the BrM cells was consistently ∼2-fold higher than that of their parental counterparts (Figures 3D–3F). Gaussian fits of the histograms of the FRET efficiencies of individual cells from these paired cell lines revealed that parental cells were more heterogeneous (wider spread of F.E with two peaks), whereas their BrM counterparts were less heterogeneous (a single narrow peak), and that a subset of parental cells behaved just like BrM cells (partial overlap between second peak of parental cells and the BrM cells). Because a feature of quantitative single-cell imaging is to allow the measured entity to be quantified by choosing a cutoff value for the histogram of overlap integrals, the data in Figure 3G suggest that 0.14 is a reasonable cutoff; values below or above 0.14 would indicate that the potential for metastasis is low or high, respectively. Because some cells in the parental population displayed F.E. > 0.14 and overlapped with BrM cells, these findings validate that single-cell steady-state F.E. measured using IMP-Y1764 can detect those subpopulations of parental cells that have acquired the metastatic potential, but risk being obscured by averaging (mean F.E. of parental cells = 0.125, which is below the 0.14 cutoff).
Figure 3.

Steady-State FRET Imaging using IMP-Y1764 in Isogenic Cancer Cells with Variable Metastatic Potentials
(A–C) Lysates of parental MDA-MB-231(A), PC-9(B), and H2030(C) cancer cells and their brain metastatic (met) counterparts were analyzed for phospho(p) and total(t) Akt, ERK, tubulin, and GIV by immunoblotting.
(D–F) Parental and brain metastatic clones of MDA-MB-231 (D), PC-9 (E), and H2030 (F) cells expressing IMP-Y1764 were analyzed for steady-state FRET by confocal live-cell imaging. Representative FRET images (left) and scatterplots (right) comparing the FRET efficiency in parental versus brain metastatic clones are displayed. Results are expressed as mean ± SD.
(G) Gaussian fits of composite histograms comparing F.E in parental (blue) versus brain metastatic (met; red) clones of cancer cells in (D–F) are displayed.
(H) Schematic summarizing the clinical course of patient #21, the source of 21T series of normal (NT-ci), primary (PT-ci), and metastatic (MT-1) breast cancer cells (Band et al., 1990).
(I) Graphs display the average FRET efficiency (left y axis) and invasive properties (right y axis; [Souter et al., 2010]) of the 21T cell lines. Results are expressed as mean ± SD.
We also confirmed that the high F.E. observed in BrM cells required a phosphorylatable tyrosine within the IMP-Y1764 probe (Figures S8A and S8B) and yet did not merely reflect a generalized hyperactivation of tyrosine-based signaling (Figure S8C). Furthermore, replacement of the GIV sequence in IMP-Y1764 with a stretch of residues that flank Y941 on IRS1 (Figure S8D), which is phosphorylated by multiple RTKs and binds p85α(PI3K) (Sato et al., 2002), abolished the probe's ability to distinguish parental from their BrM counterparts (Figures S8E and S8F). These results demonstrate the functional specificity of tyrosine-phosphorylated GIV as a portal for the integration of prometastatic PI3K signaling downstream of multiple receptors; findings were not reproducible by other phosphotyrosine substrate that also serve as ligand for p85α(PI3K).
Next we interrogated the ability of the IMP-Y1764 probe to distinguish between the MCF7 and MDA-MB-231 cells, two lines frequently studied for their contrasting metastatic potential (Winnard et al., 2008). MDA-MB-231 cells express full-length GIV (Figure S9A) and metastasize at a frequency approaching ∼100% (100% to LN, 40%–70% to lungs), whereas the MCF7 cells do not metastasize (Garcia-Marcos et al., 2011, Ghosh et al., 2010, Jenkins et al., 2005). We found that that the extent of phosphorylation of IMP-Y1764 (Figure S9B) and the mean F.E. in MCF7 (0.06 ± 0.01) and MDA-MB-231 cells (0.24 ± 0.05) (Figures S9C and S9D) were in keeping with their contrasting metastatic potentials. These results suggest that the IMP probe may be able to distinguish tumor cells with high metastatic potential from the others. Despite the fact that one major outcome of tyrosine phosphorylation of GIV and its ability to bind PI3K is the enhancement of Akt signals, the IMP-Y1764 probe did not capture all forms of ambient Akt signals in cells because we noted that the F.E. in MCF7 cells was low, despite high levels of Akt phosphorylation (Figure S9A). These findings suggest that F.E of IMP-Y1764 can somehow distinguish prometastatic GIV-PI3K-Akt signals from other Akt signals.
Single-Cell Imaging of Signaling through the GIV-PI3K Axis Maintains Its Usefulness in Tracking Metastatic Potential Despite Tumor Evolution and Onset of Chemoresistance
Next we asked if the IMP-Y1764 probe retains its ability to detect the metastatic potential of tumor cells despite evolving signaling programs and tumor biology. Such evolution is encountered under two circumstances:(1) during metastatic progression in humans and (2) during the development of drug resistance. To study the first, we used the 21T lines that were derived from the same patient during breast cancer progression (Band et al., 1990) (Figure 3H) and display an array of changing epigenetic, proteomic, and signaling programs during progressive acquisition of metastatic potential (Figure S10A). Enhanced Akt signaling during metastatic progression in 21T cells (Qiao et al., 2007) has previously been shown to track well with increased expression of GIV mRNA and protein (Garcia-Marcos et al., 2011) as well as enhanced tyrosine phosphorylation of GIV at Y1764/98 (Lin et al., 2011). Here we found that F.E using the IMP-Y1764 probe was low in normal (PT-ci; 0.08 ± 0.01) but high in primary (NT-ci; 0.28 ± 0.07) and metastasized (MT-1; 0.38 ± 0.03) cells (Figures 3I and S10B), indicating that the probe could effectively categorize both NT-ci and MT-1 as cells with “high” potential for metastasis regardless of the changes in tumor biology incurred during disease progression over 1 year and cytotoxic chemotherapy received by the patient (Band et al., 1990). Furthermore, as in the case of MCF7 and MDA-MB-231 cells, the chosen cutoff F.E. of 0.14 was effective in accurately detecting the metastatic potential of 21T cells (Figure S11).
To study the impact of changing tumor biology during the acquisition of drug resistance, we used three breast and lung cancer lines (Figures S12A and 4A–4C) with variable resistance to cytotoxic chemotherapy (docetaxel) or inhibitors of RTKs (erlotinib and lapatinib). Compared with the sensitive clones, both pY1764GIV (Figure S12B) and Akt phosphorylations (Figures 4A–4C) were consistently enhanced and their expressions were upregulated in their resistant counterparts. This is in keeping with the fact that invasiveness/epithelial-mesenchymal transition (EMT) and stemness, two features of drug resistance, are both modulated by GIV (Singh and Settleman, 2010). The F.E measured using IMP-Y1764 was similarly elevated ∼2- to 3-fold in the resistant clones (Figures 4D–4F), indicating that the probe accurately detected the acquisition of higher metastatic potential that coincides with drug resistance. Such detection was possible regardless of the unknown-unknowns of evolving tumor biology during the acquisition of resistance (Figure S12A).
Figure 4.

Steady-State FRET Imaging Using IMP-Y1764 before and after the Emergence of Drug Resistance
(A–C) Lysates of Hs578T(A) and HCC827 (B and C) cancer cells, sensitive or resistant to docetaxel (DTX), lapatinib (Lap), or erlotinib were analyzed by immunoblotting (IB).
(D–F) Sensitive and resistant Hs578T (A) and HCC827 (B and C) cancer cells expressing IMP-Y1764 were analyzed for steady-state FRET by confocal live-cell imaging. Representative FRET images and scatterplots comparing the F.E in sensitive versus resistant cells is shown. Results are expressed as mean ± SD.
(G and H) Summary of findings. Schematic (G; modified and adapted from [Wang and Dick, 2005]) showing that few tumorigenic cells are chemoresistant and account for tumor recurrence and metastasis, whereas most tumor cells are non-tumorigenic and are typically sensitive to chemotherapy. Schematic in (H) summarizes how single-cell FRET-based imaging with IMPs can detect those few cells with tumorigenic potential within the primary tumor (blue; right peak within the bimodal distribution) that are enriched later during the process of metastasis (single red peak).
In conclusion, IMP-Y1764 features three properties that make it ideal for molecular imaging: (1) a unique target, i.e., GIV, whose functional phosphorylation broadly reflects the intensity of convergent signaling from multiple upstream pathways via the prometastatic PI3K-Akt pathway; (2) usefulness in a FRET-based approach that retains single-cell information; and (3) ability to fulfill an urgent and unmet need, i.e., measure metastatic potential in a sensitive, specific, objective, and unbiased way by overcoming the limitations of the unknown (Figures 4G and 4H). That IMP-Y1764 detected the “high” potential for metastasis of 21T-NT-ci cells at diagnosis and 1 year before metastasis to the lungs/pleura (Band et al., 1990) suggests that F.E readouts using the IMP-Y1764 probe and the cutoff of 0.14 may be further developed for use ex vivo or in vitro in assays investigating personalized cancer therapeutic response, such that the sensitivity of any tumor to any small molecule will be studied specifically against those cells with the highest metastatic proclivity in any given tumor.
Discussion
One key goal of the National Cancer Institute's Precision Medicine Initiative focuses on the development of new tools to tailor cancer therapy to the disease status and risk of metastasis—patients at high risk for metastatic disease would receive aggressive, frequently molecularly targeted therapy, whereas those with low risk for metastatic disease would be treated with appropriate local therapies, sparing them toxic side effects of therapy while maintaining high likelihood for cure. One of the main challenges preventing the implementation of precision medicine for metastasis is the limited understanding of signaling molecules and pathways that confer high metastatic potential to a small subset of cancer cells, the so-called unique subset of cancer cells, designated as metastasis-initiating cells (MICs) within a larger, heterogeneous tumor. The identity of MICs remains largely unknown, which poses two critical challenges for cancer therapy. (1) Development of drugs to block metastasis: The ability of disseminated cancer cells to produce clinically evident metastases defines a central bottleneck in disease progression. Targeting key drivers of this process represents an ideal opportunity to stop metastatic disease, even for malignant cells that disseminated before the detection of a primary tumor. (2) Stratification of patients for treatment: Only a subpopulation of patients with cancer develop metastases. Lack of reliable markers to identify MICs and patients at high risk for metastasis leads to over-treatment, causing toxic side effects without benefit to a patient. Identifying MICs remains daunting because of the molecular heterogeneity of cancer cells, uncertainty about what molecules and signaling pathways confer metastatic potential, and dynamic re-wiring of signaling during metastasis. Detection of MICs as a biomarker for risk of metastasis offers the potential to advance cancer research, drug discovery and development, and patient care.
Our major accomplishment here is that we have engineered IMP biosensors that can detect and measure the metastatic “potential” of single living cancer cells (presumably any type of cancer) at a steady state. However, perhaps more importantly, we provided evidence that these biosensors can do so while remaining agnostic to the numerous unknown-unknowns of the tumor's biology, such as any genetic aberration or the epigenetic aberrations of any specific ligand/pathway. Consequently, these sensors maintain their effectiveness despite the evolving changes in the tumor cells during the natural progression of disease, or during progression under the selection pressure of anti-cancer therapies. Thus, our findings provide proof of concept that the IMP biosensors can measure the diversity and plasticity of metastatic potential of tumor cells in a sensitive and unbiased way, and in doing so, may serve as effective tools to visualize, track, and enrich (by sorting) the MICs.
Limitations of the Study and Future Directions
Despite our success in validation of the IMP probes in cultured tumor cells, further evolution of the probes is a must before they can be used for in vivo imaging studies (e.g., use of better fluorophores for deep tissue imaging explants in nude mice). Because the signaling event (tyrosine phosphorylation of GIV) specifically localizes to focal adhesions, it may be critical that any sorting-related application does so while maintaining cell-extra-cellular matrix (ECM) interactions. Despite our use of OxyFluor to minimize reactive oxygen species (ROS)-induced photobleaching, it is possible that increased ROS production in tumor cells affect either the signaling pathways monitored with IMPs or the IMP probes themselves, or both. Finally, the commonly used dimerization-preventing mutation (A206K) was not present in IMPs (see details in Methods). Because on and off rate balance in IMPs at steady state critically depends on the intramolecular affinities of fluorescent proteins (FPs), any further optimization of the IMPs (either by design, or change in FPs, or based on the proposed mode of use, i.e., at steady state versus after an acute stimuli) will require considering how these rates may affect readouts. Such optimization and thoughtful considerations are warranted because successful validation of tools to detect MICs will be a transformative advance for cancer cell biology. For example, IMPs could be adapted and evolved for the development of an imaging-based ex vivo screening platform for assessing the effectiveness of anti-cancer drugs in killing the MICs within any tumor type; they could define quantitative metrics for activation of GIV that correlates with metastatic potential, providing the foundation needed to advance GIV as a prognostic biomarker for metastasis and potential drug target to eliminate MICs. Ultimately, these efforts could enable the clinical implementation of precision cancer therapy to improve the treatment and quality of life for patients.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Gordon N. Gill and Marilyn G. Farquhar (UCSD) and Gary D. Luker and Kathryn Luker (University of Michigan) for their critical input during the preparation of the manuscript. We thank Frank Furnari (Ludwig's Cancer Institute) and Joan Massague for cell lines and reagents they shared as generous gifts. This work was supported by National Institutes of Health (NIH) grants CA100768, CA160911, and DK099226 (to P.G.). P.G. was also supported by the American Cancer Society (ACS-IRG 70-002) and by Padres Pedal the Cause/C3 (#PTC, 2017) pilot grant award and Moores Cancer Center intramural funding.
Author Contributions
N.N.S cloned and generated all constructs used in this work and characterized them biochemically. K.M, H.D and L.J carried out all FRET based studies showcased here. C.R and P.G performed all bioinformatic analysis of normal and pan-cancer phosphoproteomic datasets. K.M and P.G conceived the project. C.R and P.G wrote and edited the manuscript. P.G supervised and funded the project.
Declaration of Interests
P.G. has a pending patent application related to the discovery of IMP probes.
Published: December 21, 2018
Footnotes
Supplemental Information includes Transparent Methods, 12 figures, 5 tables, and 6 videos can be found with this article online at https://doi.org/10.1016/j.isci.2018.11.022.
Supplemental Information
The PhosphoSitePlus(PSP) was mined for proteins expressed in all cancers. The search yielded 535 phosphosites in 324 proteins.
The PhosphoSitePlus(PSP) was mined for proteins expressed in all cancers and also excluded if expressed in normal tissue. The search yielded 113 phosphosites in 72 proteins.
The PhosphoSitePlus(PSP) was mined for proteins expressed in all cancers, excluded if expressed in normal tissue, and also involved in cellular processes. Such search yielded 149 phosphosites in 92 proteins.
The PhosphoSitePlus(PSP) was mined for proteins expressed in all cancers, excluded if expressed in normal tissue, involved in cellular processes, and involved in regulating actin. The search yielded 63 phosphosites in 42 proteins.
The PhosphoSitePlus(PSP) was mined for proteins expressed in all cancers, excluded if expressed in normal tissue, involved in cellular processes, involved in regulating actin, and also involved in migration. The search yielded 34 phosphosites in 16 proteins.
References
- Anai M., Shojima N., Katagiri H., Ogihara T., Sakoda H., Onishi Y., Ono H., Fujishiro M., Fukushima Y., Horike N. A novel protein kinase B (PKB)/AKT-binding protein enhances PKB kinase activity and regulates DNA synthesis. J. Biol. Chem. 2005;280:18525–18535. doi: 10.1074/jbc.M500586200. [DOI] [PubMed] [Google Scholar]
- Aznar N., Kalogriopoulos N., Midde K.K., Ghosh P. Heterotrimeric G protein signaling via GIV/Girdin: breaking the rules of engagement, space, and time. Bioessays. 2016;38:379–393. doi: 10.1002/bies.201500133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Band V., Zajchowski D., Swisshelm K., Trask D., Kulesa V., Cohen C., Connolly J., Sager R. Tumor progression in four mammary epithelial cell lines derived from the same patient. Cancer Res. 1990;50:7351–7357. [PubMed] [Google Scholar]
- Chung W., Eum H.H., Lee H.O., Lee K.M., Lee H.B., Kim K.T., Ryu H.S., Kim S., Lee J.E., Park Y.H. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017;8:15081. doi: 10.1038/ncomms15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clister T., Mehta S., Zhang J. Single-cell analysis of G-protein signal transduction. J. Biol. Chem. 2015;290:6681–6688. doi: 10.1074/jbc.R114.616391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiacomo V., Marivin A., Garcia-Marcos M. When Heterotrimeric G proteins are not activated by G protein-coupled receptors: structural insights and evolutionary conservation. Biochemistry. 2018;57:255–257. doi: 10.1021/acs.biochem.7b00845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkel Y., Diao K., Aznar N., Swanson L., Liu L., Zhu W., Mi X.Y., Ghosh P. Prognostic impact of total and tyrosine phosphorylated GIV/Girdin in breast cancers. FASEB J. 2016;30:3702–3713. doi: 10.1096/fj.201600500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth D.L., Blackburn H.L., Shriver C.D., Rabizadeh S., Soon-Shiong P., Ellsworth R.E. Single-cell sequencing and tumorigenesis: improved understanding of tumor evolution and metastasis. Clin. Transl. Med. 2017;6:15. doi: 10.1186/s40169-017-0145-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman J.A. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat. Rev. Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- Enomoto A., Murakami H., Asai N., Morone N., Watanabe T., Kawai K., Murakumo Y., Usukura J., Kaibuchi K., Takahashi M. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev. Cell. 2005;9:389–402. doi: 10.1016/j.devcel.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Enomoto A., Ping J., Takahashi M. Girdin, a novel actin-binding protein, and its family of proteins possess versatile functions in the Akt and Wnt signaling pathways. Ann. N. Y. Acad. Sci. 2006;1086:169–184. doi: 10.1196/annals.1377.016. [DOI] [PubMed] [Google Scholar]
- Ferronika P., van den Bos H., Taudt A., Spierings D.C.J., Saber A., Hiltermann T.J.N., Kok K., Porubsky D., van der Wekken A.J., Timens W. Copy number alterations assessed at the single-cell level revealed mono- and polyclonal seeding patterns of distant metastasis in a small-cell lung cancer patient. Ann. Oncol. 2017;28:1668–1670. doi: 10.1093/annonc/mdx182. [DOI] [PubMed] [Google Scholar]
- Frangioni J.V. New technologies for human cancer imaging. J. Clin. Oncol. 2008;26:4012–4021. doi: 10.1200/JCO.2007.14.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Marcos M., Ghosh P., Farquhar M.G. GIV is a nonreceptor GEF for G alpha i with a unique motif that regulates Akt signaling. Proc. Natl. Acad. Sci. U S A. 2009;106:3178–3183. doi: 10.1073/pnas.0900294106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Marcos M., Ghosh P., Farquhar M.G. GIV/Girdin transmits signals from multiple receptors by triggering trimeric G protein activation. J. Biol. Chem. 2015;290:6697–6704. doi: 10.1074/jbc.R114.613414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Marcos M., Jung B.H., Ear J., Cabrera B., Carethers J.M., Ghosh P. Expression of GIV/Girdin, a metastasis-related protein, predicts patient survival in colon cancer. FASEB J. 2011;25:590–599. doi: 10.1096/fj.10-167304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P. Heterotrimeric G proteins as emerging targets for network based therapy in cancer: end of a long futile campaign striking heads of a Hydra. Aging (Albany NY) 2015;7:469–474. doi: 10.18632/aging.100781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P., Beas A.O., Bornheimer S.J., Garcia-Marcos M., Forry E.P., Johannson C., Ear J., Jung B.H., Cabrera B., Carethers J.M. A G{alpha}i-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate. Mol. Biol. Cell. 2010;21:2338–2354. doi: 10.1091/mbc.E10-01-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P., Garcia-Marcos M., Farquhar M.G. GIV/Girdin is a rheostat that fine-tunes growth factor signals during tumor progression. Cell Adh. Migr. 2011;5:237–248. doi: 10.4161/cam.5.3.15909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P., Rangamani P., Kufareva I. The GAPs, GEFs, GDIs and…now, GEMs: new kids on the heterotrimeric G protein signaling block. Cell Cycle. 2017;16:607–612. doi: 10.1080/15384101.2017.1282584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Haugh J.M. Live-cell fluorescence microscopy with molecular biosensors: what are we really measuring? Biophys. J. 2012;102:2003–2011. doi: 10.1016/j.bpj.2012.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck P.V., Chabra I., Kornhauser J.M., Skrzypek E., Zhang B. PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics. 2004;4:1551–1561. doi: 10.1002/pmic.200300772. [DOI] [PubMed] [Google Scholar]
- Hornbeck P.V., Kornhauser J.M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins D.E., Hornig Y.S., Oei Y., Dusich J., Purchio T. Bioluminescent human breast cancer cell lines that permit rapid and sensitive in vivo detection of mammary tumors and multiple metastases in immune deficient mice. Breast Cancer Res. 2005;7:R444–R454. doi: 10.1186/bcr1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P., Enomoto A., Jijiwa M., Kato T., Hasegawa T., Ishida M., Sato T., Asai N., Murakumo Y., Takahashi M. An actin-binding protein Girdin regulates the motility of breast cancer cells. Cancer Res. 2008;68:1310–1318. doi: 10.1158/0008-5472.CAN-07-5111. [DOI] [PubMed] [Google Scholar]
- Kimura H., Hayashi K., Yamauchi K., Yamamoto N., Tsuchiya H., Tomita K., Kishimoto H., Bouvet M., Hoffman R.M. Real-time imaging of single cancer-cell dynamics of lung metastasis. J. Cell Biochem. 2010;109:58–64. doi: 10.1002/jcb.22379. [DOI] [PubMed] [Google Scholar]
- Kubota S.I., Takahashi K., Nishida J., Morishita Y., Ehata S., Tainaka K., Miyazono K., Ueda H.R. Whole-body profiling of cancer metastasis with single-cell resolution. Cell Rep. 2017;20:236–250. doi: 10.1016/j.celrep.2017.06.010. [DOI] [PubMed] [Google Scholar]
- Kuga D., Ushida K., Mii S., Enomoto A., Asai N., Nagino M., Takahashi M., Asai M. Tyrosine phosphorylation of an actin-binding protein girdin specifically marks tuft cells in human and mouse gut. J. Histochem. Cytochem. 2017;65:347–366. doi: 10.1369/0022155417702586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.C., Lopez-Diaz F.J., Khan S.Y., Tariq M.A., Dayn Y., Vaske C.J., Radenbaugh A.J., Kim H.J., Emerson B.M., Pourmand N. Single-cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proc. Natl. Acad. Sci. U S A. 2014;111:E4726–E4735. doi: 10.1073/pnas.1404656111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., Ear J., Midde K., Lopez-Sanchez I., Aznar N., Garcia-Marcos M., Kufareva I., Abagyan R., Ghosh P. Structural basis for activation of trimeric Gi proteins by multiple growth factor receptors via GIV/Girdin. Mol. Biol. Cell. 2014;25:3654–3671. doi: 10.1091/mbc.E14-05-0978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., Ear J., Pavlova Y., Mittal Y., Kufareva I., Ghassemian M., Abagyan R., Garcia-Marcos M., Ghosh P. Tyrosine phosphorylation of the Galpha-interacting protein GIV promotes activation of phosphoinositide 3-kinase during cell migration. Sci. Signal. 2011;4:ra64. doi: 10.1126/scisignal.2002049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Sanchez I., Dunkel Y., Roh Y.S., Mittal Y., De Minicis S., Muranyi A., Singh S., Shanmugam K., Aroonsakool N., Murray F. GIV/Girdin is a central hub for profibrogenic signalling networks during liver fibrosis. Nat. Commun. 2014;5:4451. doi: 10.1038/ncomms5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Sanchez I., Kalogriopoulos N., Lo I.C., Kabir F., Midde K.K., Wang H., Ghosh P. Focal adhesions are foci for tyrosine-based signal transduction via GIV/Girdin and G proteins. Mol. Biol. Cell. 2015;26:4313–4324. doi: 10.1091/mbc.E15-07-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorentzen A., Becker P.F., Kosla J., Saini M., Weidele K., Ronchi P., Klein C., Wolf M.J., Geist F., Seubert B. Single cell polarity in liquid phase facilitates tumour metastasis. Nat. Commun. 2018;9:887. doi: 10.1038/s41467-018-03139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister S.S., Weinberg R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 2014;16:717–727. doi: 10.1038/ncb3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H., Huang X., Muruganujan A., Tang H., Mills C., Kang D., Thomas P.D. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45:D183–D189. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midde K., Rich R., Marandos P., Fudala R., Li A., Gryczynski I., Borejdo J. Comparison of orientation and rotational motion of skeletal muscle cross-bridges containing phosphorylated and dephosphorylated myosin regulatory light chain. J. Biol. Chem. 2013;288:7012–7023. doi: 10.1074/jbc.M112.434209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midde K.K., Aznar N., Laederich M.B., Ma G.S., Kunkel M.T., Newton A.C., Ghosh P. Multimodular biosensors reveal a novel platform for activation of G proteins by growth factor receptors. Proc. Natl. Acad. Sci. U S A. 2015;112:E937–E946. doi: 10.1073/pnas.1420140112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal Y., Pavlova Y., Garcia-Marcos M., Ghosh P. Src homology domain 2-containing protein-tyrosine phosphatase-1 (SHP-1) binds and dephosphorylates G(alpha)-interacting, vesicle-associated protein (GIV)/Girdin and attenuates the GIV-phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway. J. Biol. Chem. 2011;286:32404–32415. doi: 10.1074/jbc.M111.275685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani Y., Kuga D., Iida M., Ushida K., Takagi T., Tokita Y., Takahashi M., Asai M. Use of anti-phospho-girdin antibodies to visualize intestinal tuft cells in free-floating mouse jejunum cryosections. J. Vis. Exp. 2018:57475–57480. doi: 10.3791/57475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D.X., Chiang A.C., Zhang X.H., Kim J.Y., Kris M.G., Ladanyi M., Gerald W.L., Massague J. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell. 2009;138:51–62. doi: 10.1016/j.cell.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori K., Asai M., Kuga D., Ushida K., Izuchi T., Mii S., Enomoto A., Asai N., Nagino M., Takahashi M. Girdin is phosphorylated on tyrosine 1798 when associated with structures required for migration. Biochem. Biophys. Res. Commun. 2015;458:934–940. doi: 10.1016/j.bbrc.2015.02.065. [DOI] [PubMed] [Google Scholar]
- Philips M.R. Teaching resources. Imaging signal transduction in living cells with fluorescent proteins. Sci. STKE. 2005;2005:tr28. doi: 10.1126/stke.3142005tr28. [DOI] [PubMed] [Google Scholar]
- Qiao M., Iglehart J.D., Pardee A.B. Metastatic potential of 21T human breast cancer cells depends on Akt/protein kinase B activation. Cancer Res. 2007;67:5293–5299. doi: 10.1158/0008-5472.CAN-07-0877. [DOI] [PubMed] [Google Scholar]
- Ramapathiran L., Bernas T., Walter F., Williams L., Dussmann H., Concannon C.G., Prehn J.H. Single-cell imaging of the heat-shock response in colon cancer cells suggests that magnitude and length rather than time of onset determines resistance to apoptosis. J. Cell Sci. 2014;127:609–619. doi: 10.1242/jcs.137158. [DOI] [PubMed] [Google Scholar]
- Ray L.B. Single-cell biology. Cells go solo. Introduction. Science. 2013;342:1187. doi: 10.1126/science.342.6163.1187. [DOI] [PubMed] [Google Scholar]
- Sahai E. Illuminating the metastatic process. Nat. Rev. Cancer. 2007;7:737–749. doi: 10.1038/nrc2229. [DOI] [PubMed] [Google Scholar]
- Sato M., Ozawa T., Inukai K., Asano T., Umezawa Y. Fluorescent indicators for imaging protein phosphorylation in single living cells. Nat. Biotechnol. 2002;20:287–294. doi: 10.1038/nbt0302-287. [DOI] [PubMed] [Google Scholar]
- Simpson F., Martin S., Evans T.M., Kerr M., James D.E., Parton R.G., Teasdale R.D., Wicking C. A novel hook-related protein family and the characterization of hook-related protein 1. Traffic. 2005;6:442–458. doi: 10.1111/j.1600-0854.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- Singh A., Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souter L.H., Andrews J.D., Zhang G., Cook A.C., Postenka C.O., Al-Katib W., Leong H.S., Rodenhiser D.I., Chambers A.F., Tuck A.B. Human 21T breast epithelial cell lines mimic breast cancer progression in vivo and in vitro and show stage-specific gene expression patterns. Lab. Invest. 2010;90:1247–1258. doi: 10.1038/labinvest.2010.97. [DOI] [PubMed] [Google Scholar]
- Su Y., Wei W., Robert L., Xue M., Tsoi J., Garcia-Diaz A., Homet Moreno B., Kim J., Ng R.H., Lee J.W. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc. Natl. Acad. Sci. U S A. 2017;114:13679–13684. doi: 10.1073/pnas.1712064115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F., Bosnjak M., Skunca N., Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M., Asai N., Enomoto A. Akt-Girdin as oncotarget. Oncoscience. 2015;2:811–812. doi: 10.18632/oncoscience.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman K.S., Powers M.A., Forbes D.J. Nuclear export receptors: from importin to exportin. Cell. 1997;90:967–970. doi: 10.1016/s0092-8674(00)80361-x. [DOI] [PubMed] [Google Scholar]
- Valiente M., Obenauf A.C., Jin X., Chen Q., Zhang X.H., Lee D.J., Chaft J.E., Kris M.G., Huse J.T., Brogi E. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014;156:1002–1016. doi: 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeriah S., Leboucher P., de Naurois J., Jethwa N., Nye E., Bunting T., Stone R., Stamp G., Calleja V., Jeffrey S.S. High-throughput time-resolved FRET reveals Akt/PKB activation as a poor prognostic marker in breast cancer. Cancer Res. 2014;74:4983–4995. doi: 10.1158/0008-5472.CAN-13-3382. [DOI] [PubMed] [Google Scholar]
- Waclaw B., Bozic I., Pittman M.E., Hruban R.H., Vogelstein B., Nowak M.A. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature. 2015;525:261–264. doi: 10.1038/nature14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.C., Dick J.E. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15:494–501. doi: 10.1016/j.tcb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Weinstein I.B. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- Weng L., Enomoto A., Ishida-Takagishi M., Asai N., Takahashi M. Girding for migratory cues: roles of the Akt substrate Girdin in cancer progression and angiogenesis. Cancer Sci. 2010;101:836–842. doi: 10.1111/j.1349-7006.2009.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnard P.T., Jr., Pathak A.P., Dhara S., Cho S.Y., Raman V., Pomper M.G. Molecular imaging of metastatic potential. J. Nucl. Med. 2008;49 Suppl 2:96S–112S. doi: 10.2967/jnumed.107.045948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Chen S., Yu J., Li Y., Zhang X.Y., Yang L., Zhang H., Hou Q., Jiang M., Brunicardi F.C. Single-cell transcriptome analyses reveal molecular signals to intrinsic and acquired paclitaxel resistance in esophageal squamous cancer cells. Cancer Lett. 2018;420:156–167. doi: 10.1016/j.canlet.2018.01.059. [DOI] [PubMed] [Google Scholar]
- Zenobi R. Single-cell metabolomics: analytical and biological perspectives. Science. 2013;342:1243259. doi: 10.1126/science.1243259. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video shows ligand-dependent signaling via the GIV-PI3K axis as visualized through FRET imaging in living Cos7 cells expressing the wild-type IMP-Y1764 sensor.
Video shows absence of significant FRET upon EGF stim in Cos7 cells expressing the non-phosphorylatable IMP-pY1764F mutant probe, indicating that phosphorylation and phosphorylation-induced intramolecular rearrangement of the probe are required for the observed changes in FRET in Video S1.
Video shows ligand-dependent signaling via the GIV-PI3K axis as visualized through FRET imaging in living Cos7 cells expressing the wild-type IMP-Y1798 sensor.
This video indicates that phosphorylation and phosphorylation-induced intramolecular rearrangement of the probe are required for the observed changes in FRET in Video S3.
Video shows ligand-dependent signaling via the GIV-PI3K axis as visualized through FRET imaging in living Cos7 cells expressing the wild-type IMP-Y1764 sensor.
This video indicates that phosphorylation and phosphorylation-induced intramolecular rearrangement of the probe are required for the observed dynamic changes in FRET in Video S5.
The PhosphoSitePlus(PSP) was mined for proteins expressed in all cancers. The search yielded 535 phosphosites in 324 proteins.
The PhosphoSitePlus(PSP) was mined for proteins expressed in all cancers and also excluded if expressed in normal tissue. The search yielded 113 phosphosites in 72 proteins.
The PhosphoSitePlus(PSP) was mined for proteins expressed in all cancers, excluded if expressed in normal tissue, and also involved in cellular processes. Such search yielded 149 phosphosites in 92 proteins.
The PhosphoSitePlus(PSP) was mined for proteins expressed in all cancers, excluded if expressed in normal tissue, involved in cellular processes, and involved in regulating actin. The search yielded 63 phosphosites in 42 proteins.
The PhosphoSitePlus(PSP) was mined for proteins expressed in all cancers, excluded if expressed in normal tissue, involved in cellular processes, involved in regulating actin, and also involved in migration. The search yielded 34 phosphosites in 16 proteins.