

Abstract
Therapeutics for arachidonic acid pathways began with the development of non-steroidal antiinflammatory drugs that inhibit cyclooxygenase (COX). The enzymatic pathways and arachidonic acid metabolites and respective receptors have been successfully targeted and therapeutics developed for pain, inflammation, pulmonary and cardiovascular diseases. These drugs target the COX and lipoxygenase pathways but not the third branch for arachidonic acid metabolism, the cytochrome P450 (CYP) pathway. Small molecule compounds targeting enzymes and CYP epoxy-fatty acid metabolites have evolved rapidly over the last two decades. These therapeutics have primarily focused on inhibiting soluble epoxide hydrolase (sEH) or agonist mimetics for epoxyeicosatrienoic acids (EET). Based on preclinical animal model studies and human studies, major therapeutic indications for these sEH inhibitors and EET mimics/analogs are renal and cardiovascular diseases. Novel small molecules that inhibit sEH have advanced to human clinical trials and demonstrate promise for cardiovascular diseases. Challenges remain for sEH inhibitor and EET analog drug development; however, there is a high likelihood that a drug that acts on this third branch of arachidonic acid metabolism will be utilized to treat a cardiovascular or kidney disease in the next decade.
Keywords: soluble epoxide hydrolase, epoxyeicosatrienoic acids, hypertension, myocardial infarction, fibrosis, inflammation, nephropathy
Introduction
The development of therapeutics for kidney and cardiovascular diseases has included targeting various lipids and fatty acids. Arachidonic acid is a fatty acid that is metabolized into several metabolites known as eicosanoids. Pharmacological agents have targeted arachidonic acid metabolizing enzymes, eicosanoid metabolites, and eicosanoid receptors (Capra et al., 2007; Grosser et al., 2006; Riberio et al., 2006). There are three primary enzymatic pathways that produce eicosanoid metabolites; the cyclooxygenase (COX) pathway, the lipoxygenase (LOX), pathway, and the cytochrome P450 (CYP) pathway. Drugs have been successfully developed that target the COX and LOX pathways for indications such as pain, inflammation, asthma, and blood clotting (Capra et al., 2007; Grosser et al., 2006; Riberio et al., 2006). More recently, pharmacological agents directed at the CYP pathway have made their way to clinical trials but none have yet to be approved for human use (Bellien & Joannides, 2013; Imig & Hammock, 2009). This review will focus on the prospective for CYP epoxygenase eicosanoid therapeutics for renal and cardiovascular diseases.
CYP Enzymes
Arachidonic acid metabolism by CYP enzymes results in epoxygenase and hydroxylase metabolites. Epoxygenase metabolites are generated by CYP2C or CYP2J enzymes and hydroxylase metabolites are generated by CYP4A or CYP4F enzymes (Capdevila et al., 2007; Karara et al., 1993; Roman, 2002; Zeldin, 2001). Renal and cardiovascular actions for these CYP eicosanoid metabolites have been extensively investigated. Epoxyeicosatrienoic acids (EETs) have acute and chronic actions that are beneficial to kidney and cardiovascular health. Soluble epoxide hydrolase (sEH) is the enzyme responsible for conversion of EETs to diols that lack the overall beneficial biological activities ascribed to EETs (Imig, 2012; Imig & Hammock, 2009). On the other hand, the hydroxylase metabolite, 20-hydroxyeicsatraenoic acid (20-HETE), has acute and chronic actions that could contribute to renal and cardiovascular diseases (Roman, 2002). Although receptors for EETs have yet to be identified, GPR75 has recently been identified as a 20-HETE receptor that is responsible for 20-HETE’s cardiovascular actions (Garcia et al., 2017). Pharmacological agents have been developed that inhibit CYP epoxygenase, CYP hydroxylase, and sEH enzymes (Imig & Hammock, 2009; Miyata et al., 2001; Wang et al., 1998). Novel compounds that act as EET and 20-HETE mimetics and antagonists have also been synthesized and extensively examined (Alonso-Galcia et al., 1999; Campbell et al., 2017; Sudhahar et al., 2010). EET mimetics and sEH inhibitors have been demonstrated to oppose renal and cardiovascular diseases and their potential as a therapeutic will be detailed. For detailed information on the development of hydroxylase and 20-HETE targeted therapeutics please see these excellent references (Kroetz & Xu, 2005; Roman, 2002).
The primary CYP epoxygenase enzymes can regulate EET generation in an organ and cell specific manner (Capdevila et al., 2007; Imig, 2012; Zeldin, 2001). Metabolism of epoxy fatty acids to the corresponding diols is mediated by sEH (Imig, 2012; Imig & Hammock, 2009). Protein expression for sEH is more homogeneous among organs and cell types but can be regulated in an organ or cell selective manner (Capdevila et al., 2007; Morisseau & Hammock, 2013). CYP epoxygenase expression in the renal and cardiovascular systems determines the specific epoxy fatty acids generated (Capdevila et al., 2007; Imig, 2012; Zeldin., 2001). One generalization that can be made is that 11,12-EET and 14,15-EET are the major EETs generated from arachidonic acid by CYP epoxygenase enzymes (Capdevila et al., 2007; Zeldin, 2001). CYP2J enzymes are more prevalent in the heart and cardiomyocytes and convert arachidonic acid to EETs (Oni-Orisan et al., 2014; Zeldin, 2001). Kidney EET levels are regulated by CYP2C enzymes located in tubular epithelial cells (Capdevila et al., 2007; Imig 2012). CYP2C enzymes are also located in endothelial cells to produce EETs that act as endothelial-derived hyperpolarizing factors (EDHFs) (Archer et al., 2003; Campbell & Fleming, 2010; Campbell et al., 1996; Fissthaler et al., 1999; Fleming, 2001). EETs are metabolized by sEH that has a rank order affinity for EETs with 14,15-EET > 11,12-EET > 8,9-EET > 5,6-EET (Imig & Hammock, 2009; Morisseau & Hammock, 2013). In addition, 8,9-EET and 5,6-EET can be metabolized by COX and this metabolic pathway has been clearly demonstrated in the glomerulus of the kidney (Carroll et al., 1992; Imig, 2000; Spector et al., 2004). Regulation of enzymatic activity and the specific epoxy fatty acid metabolites formed will impact renal and cardiovascular function in health and disease.
It is well recognized that arachidonic acid metabolites have important renal and cardiovascular biological activities. Generation of epoxy fatty acid EETs and metabolism by sEH are altered in renal and cardiovascular disease states (Bellien & Joannides, 2013; Imig & Hammock, 2009). In most cases, decreased EETs are associated and contribute to renal and cardiovascular diseases. On the other hand, there is increasing evidence that CYP epoxygenase and sEH generation and regulation of omega-3 and omega-6 fatty acids can impact cardiovascular and renal function (Arnold et al., 2010a). Although the primary focus will be on the therapeutic potential for sEH inhibitors and EET analogs, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) based therapies are an area that is actively being pursued.
Evidence Renal and Cardiovascular Diseases: Animal Models & Human Studies
Extensive experimental studies in renal and cardiovascular disease animal models as well as human studies provides substantial evidence for EETs as a therapeutic target (Figure 1). Experimental animal studies have evaluated CYP2C, CYP2J, and sEH enzymatic expression, epoxygenase metabolite levels, and EET actions (Imig, 2012; Morisseau & Hammock, 2013; Tacconelli & Patrignani, 2015). Genetic and pharmacological manipulation to decrease or increase EETs has also been done to evaluate their contribution to heart, vascular, and kidney function (Campbell et al., 2017; Imig, 2012; Kroetz & Xu, 2005; Morisseau & Hammock, 2013; Oni-Orisan et al., 2014) (Figure 2). Vascular, kidney, and heart EET actions include vasodilation, anti-inflammation, anti-fibrosis, and anti-apoptosis (Campbell et al., 2017; Fleming, 2001; Node et al., 1999; Oni-Orisan et al., 2014). EETs act as EHHFs that are released from endothelial cells to activate vascular smooth muscle cell large conductance KᐩCa channels and vasodilation (Campbell et al., 1996; Fissthaler et al.). Heart specific EET actions on potassium KATP channels contributes to electrical conductivity (Batchu et al., 2012a; Bodiga et al., 2009; Oni-Orisan et al., 2014; Seubert et al., 2004). A renal specific action for EETs is inhibition of epithelial sodium channel (ENaC) to cause sodium excretion or natriuresis (Pidovaka et al., 2013; Sun et al., 2010; Wei et al., 2004). EETs also have anti-platelet and angiogenic actions that are beneficial for cardiovascular and kidney health (Campbell & Fleming, 2010; Imig & Hammock, 2009). These findings that EETs have renal and cardiovascular actions to oppose acute and chronic diseases has been extended to animal disease models and human studies.
Figure 1. Cardiovascular and renal disease pathological mechanisms associated with increased soluble epoxide hydrolase (sEH) activity or decreased epoxyeicosatrienoic acids (EETs).
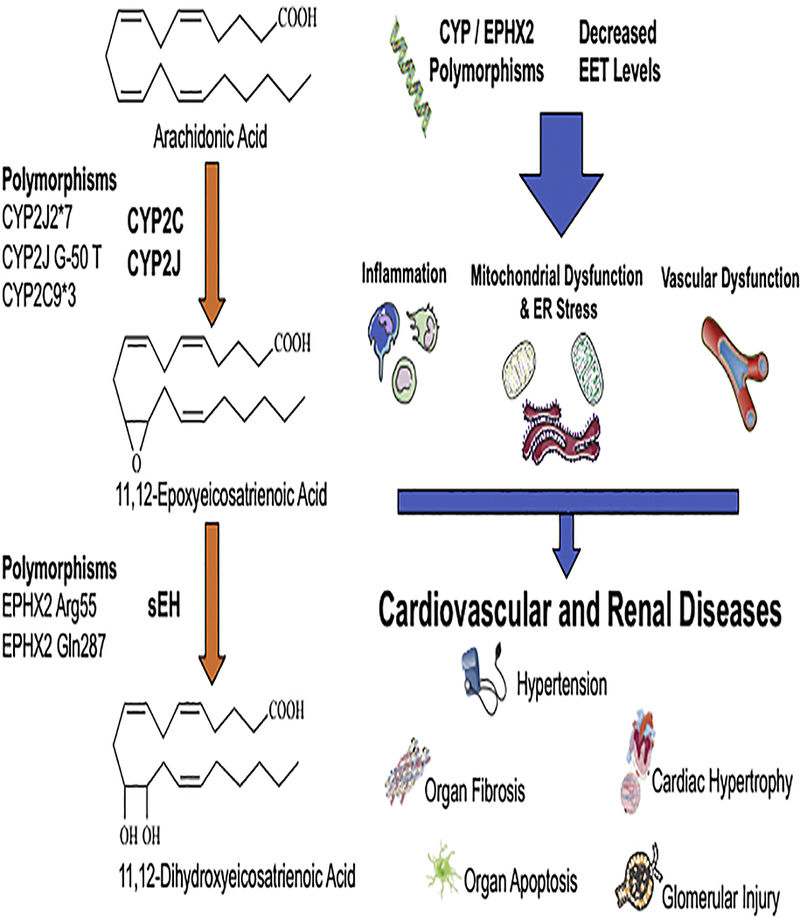
Left panel illustrates the arachidonic acid metabolism by epoxygenase and sEH enzymes. Polymorphisms leading to decreased EET levels or increased sEH activity are described. Right panel illustrates decreased EET levels resulting in inflammation, vascular dysfunction, and endoplasmic reticulum stress that leads to progressive renal and cardiovascular disease.
Figure 2. Epoxyeicosatrienoic acids (EETs) cardiac, vascular, and renal mechanisms of action.
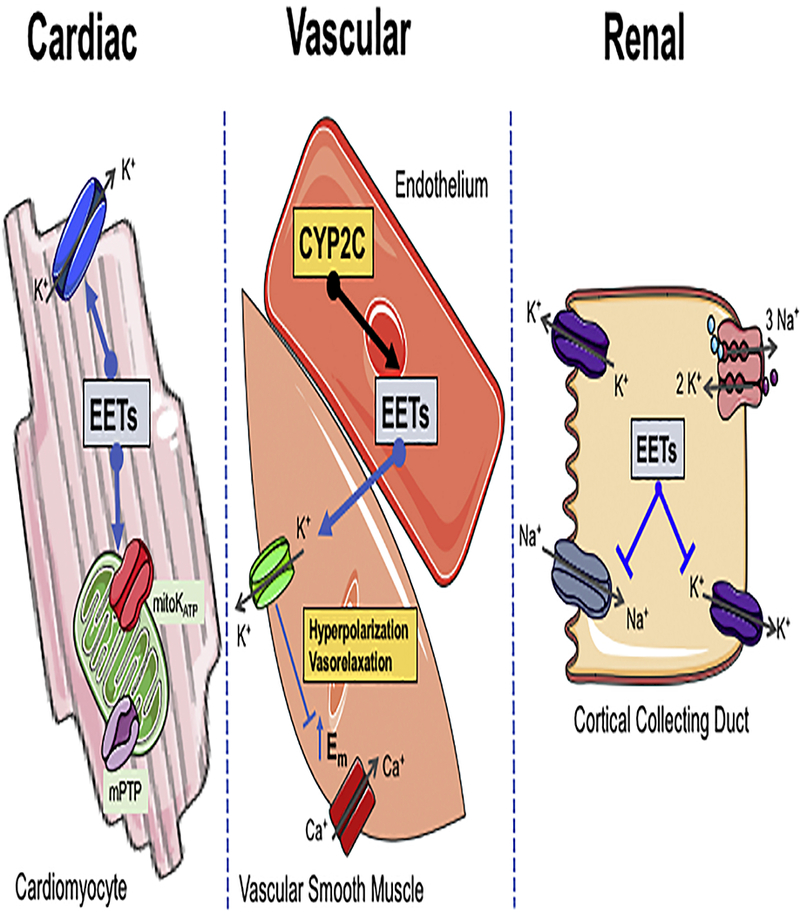
Left panel illustrates EETs actions on cardiomyocyte to activate cell membrane KATP and mitochondrial KATP (mitoKATP) to reduce opening of the mitochondrial permeability transition pore (mPTP) resulting in decreased apoptosis. Middle panel illustrates CYP2C EETs generation in endothelial cells that activate large conductance Ca2+-activated K+ channels (BKCa) resulting in K+ efflux from the smooth muscle cell, membrane hyperpolarization, closing L-type Ca2+ channels to result in vasorelaxation. Right panel illustrates EETs inhibiting cortical collecting duct apical Na+ channels (ENaC) and basolateral K+ channels to decrease sodium reabsorption and cause natriuresis.
Cardiovascular Disease
Cardiovascular disease animal models have provided significant evidence that decreased EETs or increased sEH enzymatic activity contribute to the disease process. Hypertension has been extensively studied and decreased vascular and kidney EET levels contribute to changes in water and electrolyte balance and the elevation in blood pressure (Imig et al., 2002; Lee et al., 2010; Makita et al., 1994; Nakagawa et al., 2006; Zhao et al., 2003). Angiotensin dependent hypertension results in an increase in kidney sEH expression and activity that increases renal vascular resistance and decreases sodium excretion (Imig et al., 2002; Zhao et al., 2004). On the other hand, salt-sensitive hypertension is associated with an inability to properly upregulate CYP2C epoxygenase enzymes (Zhao et al., 2003). As further evidence for epoxygenase enzymes contributing to salt-sensitive hypertension, Cyp2c44−/− mice develop hypertension when fed a high salt diet (Capdevila et al., 2014). Additionally, there is pervasive evidence for decreased EETs contributing to heart injury following myocardial infarction (Oni-Orisan et al., 2014). Increasing EETs in cardiac myocytes in vitro or in acute myocardial infarction rodent models protects the myocardium following ischemia via several interrelated signaling mechanisms (Batchu et al., 2009; Bodiga et al., 2012; Nithipatikom et al., 2006; Seubert et al., 2004; Seubert et al., 2006). These interrelated signaling mechanisms include the PI3K/Akt, MAPK, STAT3, oxidative stress signaling pathways, as well as, KATP channel activation, anti-inflammatory actions, and anti-fibrotic actions (Batchu et al., 2012a; Bodiga et al., 2009; Gross et al., 2007; Seubert et al., 2004). A decrease in EETs has also been demonstrated to contribute to cardiac hypertrophy and heart failure in several rodent diabetic and cardiovascular disease models (Imig & Hammock, 2009; Morisseau & Hammock, 2013; Oni-Orisan et al., 2014). Angiotensin II can induce sEH upregulation to drive cardiac hypertrophy and result in devastating consequences that include left ventricular dysfunction and arrhythmias (Ai et al., 2007; Ai et al., 2009). Taken together, these findings established the important contribution for sEH and EETs in hypertension and heart diseases.
Renal Disease
Extensive evidence in renal disease animal models have implicated an important involvement for EETs and sEH in kidney vascular and epithelial cell injury (Imig, 2002; Imig, 2015; Morisseau & Hammock, 2013). Initial evidence for increased sEH activity and decreased EETs contributing to kidney injury was found in rat angiotensin-dependent hypertension (Imig et al., 2002; Zhao et al., 2003). These studies also discovered that increasing EETs with sEH inhibitors decreased renal inflammation and injury independent of blood pressure lowering (Imig et al., 2002; Olearczyk et al., 2009; Zhao et al., 2003). Diabetic nephropathy has also been determined to involve increased sEH expression and decreased EET levels as contributors to the progressive renal damage (Chen et al., 2012; Elmarakby et al., 2011; Olearczyk et al., 2009). Decreased EETs participate in renal diseases that do not have an associated systemic symptom or disease such as hypertension and diabetes. Acute kidney injury, drug-induced nephrotoxicity, and obstructive kidney fibrotic disease have detrimental changes in sEH or EETs that impact kidney injury (Bettaieb et al., 2017; Hashimoto et al., 2015; Kim et al., 2014; Lee et al., 2012; Liu et al., 2012; Parrish et al., 2009; Zhu et al., 2016). The decrease in EET levels in renal ischemia reperfusion acute kidney injury contribute to endothelial dysfunction and epithelial apoptosis (Lee et al., 2012; Zhu et al., 2016). Endoplasmic reticulum stress is a key signaling mechanism in renal tubular cells that is increased by lower level EET levels in cisplatin drug-induced nephrotoxicity (Khan et al., 2013; Liu et al., 2012). Increased sEH activity and decreased EET levels in unilateral ureteral obstruction allows for inflammation and epithelial to mesenchymal transition mediated kidney fibrosis (Kim et al., 2014; Kim et al., 2015; Skibba et al., 2017). Thus, evidence for decreased EETs levels or increased sEH activity contribut ing to renal and cardiovascular disease in animal models provided substantial data to support the development of therapeutics directed at increasing EET levels.
Human Disease Studies
Human studies demonstrating that increased sEH activity or decreased EETs could contribute to cardiovascular diseases like hypertension has been investigated (Table 1). Reduced plasma EET levels have been demonstrated in humans with pregnancy-induced or renovascular hypertension (Bellien & Joannides, 2013; Elijovich et al., 2017; Jiang et al., 2013). The changes in EET levels in these human studies is postulated to be due to a decrease in CYP production and increased sEH degradation of EETs. Genetic variations in the human CYP2C8, CYP2C9, and CYP2J2 result in decreased arachidonic acid epoxidation rates (Bellien & Joannides, 2013; Morisseau & Hammock, 2013). CYP2J2*7 allele is a genetic variant that results in reduced CYP2J2 transcription and decreased plasma EET levels (King et al., 2005; Polonikov et al., 2008). This CYP2J2 genetic variant associates with essential hypertension in a Russian cohort (Polinikov et al., 2008); however, other human studies have found that the CYP2J2*7 allele decreased risk or resulted in no change for developing hypertension (King et al., 2005). Studies have found that the CYP2C human genetic variants do not associate with hypertension in Caucasian or African American cohorts (Dreisbach et al., 2005; Spiecker et al., 2004). On the other hand, in hypertensive Asian women the frequency for the CYP2C9*3 allele was lower (Yu et al., 2004). Regarding sEH, the evidence for polymorphisms in the sEH gene, EPHX2, that associate with hypertension have not been found (Bellien & Joannides, 2013). Taken together, these findings provide varied evidence for increased sEH activity or decreased EETs in humans contributing to hypertension.
Table 1.
Human gene variants and sEH / EET changes that associate with cardiovascular and renal diseases
| Genetic Variant | Disease Association |
References | |
|---|---|---|---|
| CYP2C9 | CYP2C9*3 | Hypertension | Yu et al., 2004 |
| CYP2J2 | Cyp2J2*7 Decreased EET levels G50-T |
Hypertension Coronary artery disease Type 2 Diabetes |
Driesbach et al., 2005 King et al., 2005 Polonikov et al., 2008 Spiecker et al., 2004 Wang et al., 2010 |
| EPHX2 | Arg55 Increased sEH activity Gln297 Decreased sEH activity |
Decreased forearm blood flow Coronary artery disease Acute kidney injury Increased forearm blood flow Coronary artery plaque |
Lee et al., 2006 Lee et al., 2011 Shuey et al., 2017 Fornage et al., 2004 |
| Plasma EETs |
Coronary artery disease Inflammation Ischemic stroke Decreased forearm blood flow COPD Insulin resistance |
Bellien et al., 2012b
Fritscher et al., 2012 Gangadhariah et al., 2017 Wang et al., 2010 Ward et al., 2011 Yang et al., 2017 |
|
| EPHX2 Expression |
Heart failure | Monti et al., 2008 |
Evidence linking increased sEH activity or decreased EETs could contribute to cardiovascular diseases and endothelial dysfunction that increases kidney disease progression has accelerated therapeutic development. Investigations measuring forearm blood flow responses in humans has supplied the most convincing evidence that reduced EETs can have devastating cardiovascular and renal consequences (Bellien et al., 2006; Bellien et al., 2012a; Bellien et al., 2012b; Lee et al., 2011). CYP inhibition to lower plamsa EET levels reduces forearm blood flow in healthy human volunteers (Bellien et al., 2006). This finding demonstrates that EETs contribute to the basal level of forearm blood flow. Likewise, EPHX2 genetic variations alter the magnitude of human forearm blood flow responses to bradykinin (Lee et al., 2011). The Arg55 EPHX2 genetic variant increases sEH activity and reduces bradykinin-induced increases in forearm blood flow (Lee et al., 2011). An EPHX2 genetic variant Gln287 that decreases sEH activity which would increase EET bioavailability augments forearm blood flow responses to bradykinin (Lee et al., 2011). Intriguingly, an impaired ability to induce EET release was observed in essential hypertensives that contributed to endothelial dysfunction assessed by flow mediated dilation (Bellien et al., 2012a; Bellien et al., 2012b). These human studies provide strong support to the notion that increased sEH activity or decreased EETs results in endothelial dysfunction which is a precursor for renal and cardiovascular diseases.
Other studies have linked genetic EPHX2, CYP2C8, and CYP2J2 variants to coronary artery disease risks (Bellien & Joannides, 2013; Campbell et al., 2017; Morisseau & Hammock, 2013; Oni-Orisan et al., 2014). The CYP2J2 gene G-50 T single nucleotide polymorphism is associated with lower plasma EET levels and increased risk for coronary artery disease (Spiecker et al., 2004). Interestingly, the frequency for the CYP2J2 G-50 T polymorphism is higher in type 2 diabetics under the forty years of age (Wang, et al., 2010). The Atherosclerosis Risk in Communities (ARIC) study found that the EPHX2 Arg55 polymorphism that results in increased sEH activity associated with greater risk for coronary heart disease (Lee et al., 2011). Likewise, this Arg55 gain-of-function polymorphism has been demonstrated to be associated with acute kidney injury following cardiac surgery (Shuey et al., 2017). Human studies have also found plasma EET levels to be elevated and the EET:DHETE ratio increased in coronary artery disease patients (Wang et al., 2010). Plasma EET levels are increased in patients following an ischemic stroke event (Ward, et al., 2011). Likewise, biopsies of heart failure patients with a previous myocardial infarction were found to have decreased EPHX2 expression (Monti et al., 2008). These findings suggest that an increase in EET levels in patients with established coronary artery disease could be a compensatory mechanism. Interestingly, obesity and cigarette smoking was associated with lower EET levels and increased inflammation as measured by monocyte chemoattractant protein-1 (MCP-1) in this coronary artery disease population (Wei et al., 2007; Yang et al., 2008). Evaluation of patients with chronic obstructive pulmonary disease (COPD) have found decreased EET levels and impaired forearm blood flow responses that could be responsible for the increased incidence of cardiovascular disease (Barbera et al., 2001; Fritscher et al., 2012; Yang et al., 2017). The compelling human studies point to decreased CYP EET production and increased EET degradation by sEH as a contributor to endothelial dysfunction and cardiovascular disease risk; however, in many cases an increase in EET levels attempt to compensate following a cardiovascular event.
There is compelling animal renal and cardiovascular disease model and human experimental findings to support the concept that increasing EET levels or inhibiting sEH activity would be an excellent therapeutic. Two primary therapeutic approaches and emerging therapeutic approaches are being developed for renal and cardiovascular diseases. Inhibition of sEH is the most advanced therapeutic approach and has reached the human clinical trial level for hypertension, diabetes, and chronic obstructive pulmonary disease (Arete Therapeutics, 2009; GlaxoSmithKline, 2013). EET analog development has gained steam in the past five years and seems poised to be in human clinical trials within five years. An emerging therapeutic area is bifunctional therapeutics that have sEH inhibition as one the drug activities. This area began with dual sEH / COX inhibitors and now includes peroxisome proliferator-activated receptor (PPAR) agonists and farnesoid X receptor (FXR) agonists activities combined with sEH inhibition. The promise for these three therapeutic approaches for renal and cardiovascular disease will be covered in subsequent sections.
Soluble Epoxide Hydrolase Inhibitors
The development of sEH inhibitors occurred rapidly over the course of one decade and resulted in the first human clinical trial in 2008 (Arete Therapeutics, 2009; Imig & Hammock, 2009). This rapid development was largely due to the determination of Dr. Bruce Hammock and colleagues to advance sEH inhibitors from being an experimental tool to being a drug that could be given orally to animals and humans. The renal and cardiovascular positive effects for sEH inhibitors has been attributed to the increase arachidonic acid EET levels or EET:DHETE ratio (Imig & Hammock, 2009). Although this scenario is highly likely and supported by investigations in animal models and humans, the sEH hydrolase activity can convert other epoxy fatty acids to diols (Morrisseau & Hammock, 2013; Imig et al., 2005). Plasma linoleic acid epoxy to diol ratios increase in animals administered sEH inhibitors (Imig et al., 2005). Likewise, long-chain ω−3 polyunsaturated epoxy fatty acids are metabolized by sEH to their corresponding diols (Schunk et al, 2018). These EPA and DHA epoxy fatty acids are thought to be partially responsible for the positive renal and cardiovascular effects of eating a high dietary ω−3 fatty acid fish oil diet (De Caterina et al., 2011; Schunk et al, 2018; Westphal et al., 2011). Experimental studies are exploring the renal and cardiovascular actions for the different types of epoxy fatty acids (Schunk et al., 2018; Sharma et al, 2016; Ulu et al., 2014). Irrespective of the epoxy fatty acid that provides beneficial renal and cardiovascular actions, positive results in animal models of cardiovascular and kidney diseases combined with medicinal chemistry and a knowledge of the sEH protein structure allowed for the design of potent and selective sEH inhibitors with excellent oral bioavailability (Imig & Hammock, 2009).
Major advantages for sEH inhibitor development were that the sEH gene was cloned and the sEH enzyme structure and catalytic mechanism were known (Imig & Hammock, 2009; Marino, 2009; Shen et al., 2012). The enzyme is high conserved with the carboxy-terminal domain containing the epoxide hydrolase domain and the amino-terminal domain containing phosphatase activity (Imig & Hammock, 2009; Newman et al., 2003; Shen et al., 2012). Although initial sEH inhibitors were potent competitive inhibitors that included chalcone oxides and glycidols, these were quickly inactivated by glutathione and glutathione transferases (Imig & Hammock, 2009; Shen et al., 2012). The development of potent amides, ureas, and cabamate sEH inhibitors led to the first animal studies (Kim et al., 2005; Morisseau et al., 1999; Newman et al., 2003; Yu et al., 2000). Thus far, these inhibitors for sEH have been designed to inhibit the C-terminal epoxide hydrolase activity while lacking alteration of the N-terminal phosphatase activity (Imig & Hammock, 2009; Marino, 2009; Shen et al., 2012). Over the past 15 years, multiple generations of sEH inhibitors have been developed with various structures that include urea-based sEH inhibitors, an amide series of sEH inhibitors, piperidyl urea sEH inhibitors, and aminoheteroarlyl sEH inhibitors (Figure 3) (Imig & Hammock, 2009; Marino, 2009; Shen et al., 2012).
Figure 3. Soluble epoxide hydrolase (sEH) inhibitor development to treat renal and cardiovascular diseases.
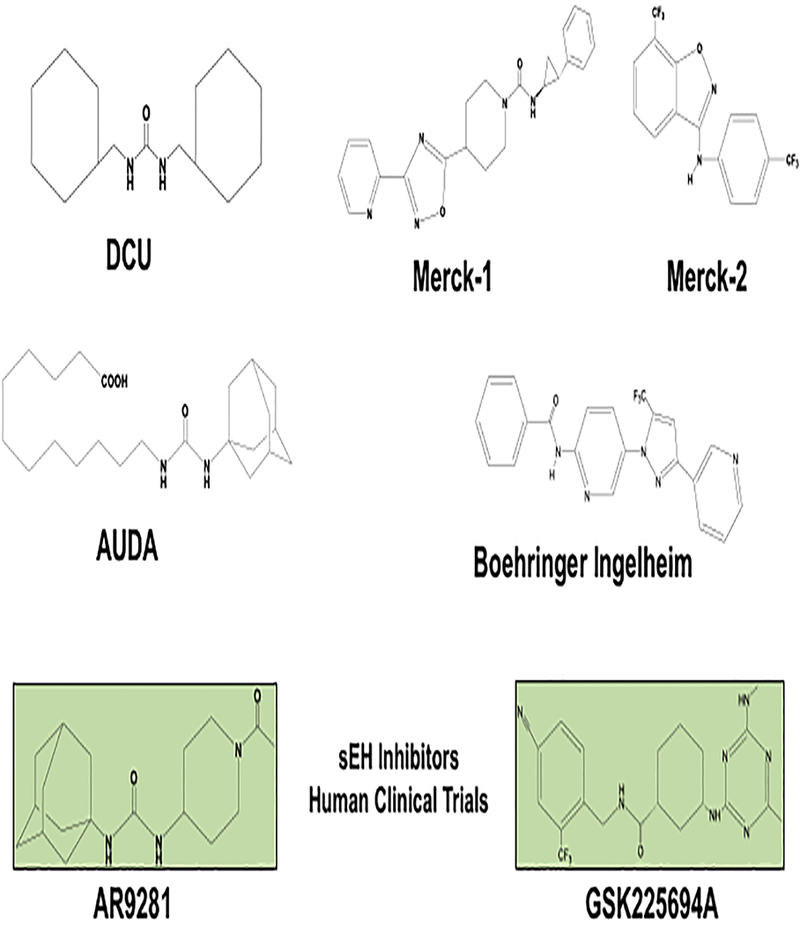
DCU: one of the initial amide, urea, and carbamate classes found to be a potent and stable transition state sEH inhibitor. AUDA: the first sEH inhibitor to be administered orally. Merck-1: a piperidine-derived urea sEH inhibitor. Merck-2: a novel aminohetroaryls that inhibit sEH. Boehringer Ingelheim: a pyrazole amide sEHO inhibitor. Arête Therapeutics AR9281 : the first sEH inhibitor to be tested in humans for hypertension and diabetes. GSK GSK2256294A: second sEH inhibitor to be tested in humans for the indication of chronic obstructive pulmonary disease (COPD).
Early Development for sEH Inhibitors
Small molecule sEH inhibitors of the amide, urea, and carbamate classes changed the landscape because these were found to be potent and stable transition state sEH inhibitors (Imig & Hammock, 2009; Kim et al., 2005; Morisseau et al., 1999; Newman et al., 2003). N-cyclohexyl-N’-decylurea (CDU), a potent inhibitor (human sEH Ki 6 nM), was co-crystalized and demonstrated to be bound in the active site of murine sEH (Argiriadi et al., 2000). Crystal structure studies for sEH and urea inhibitors revealed that the carbonyl urea accepts hydrogen bonds from Tyr-381 and Tyr-465 (Imig & Hammock, 2009; Shen et al., 2012) (Figure 4). Urea and/or -NH groups form a hydrogen bond with sEH Asp-333. Favorable van der Waal interactions occur between the N-cyclohexyl group because of the closeness to Phe-265, Tyr-381, Phe-406, Val-499, and Trp-524 (Imig & Hammock, 2009; Shen et al., 2012). The N’decyl group has favorable van der Waal contacts with hydrophobic residues and is directed towards the opening of the active site (Argiriadi et al., 2000; Imig & Hammock, 2009; Shen et al., 2012). This and other crystallography studies provided information that allowed for attempts to introduce water solubilizing groups on the urea side chain. A transformation small molecule sEH inhibitor, 12-(3-adamantan-l-yl-ureido)dodecanoic acid (AUDA), was developed largely based on this structural data (Imig & Hammock, 2009; Marino, 2009; Shen et al., 2012). AUDA was transformational because it had enough solubility to get into a water solution, could be delivered to animals chronically, and had favorable potency and bioavailability (Jung et al., 2005; Zhao et al.,2003). This led to the development of several sEH inhibitors that have potential to be used in humans to treat renal and cardiovascular diseases.
Figure 4. Bifunctional soluble epoxide hydrolase (sEH) inhibitor structure and binding to the enzymatic pocket.
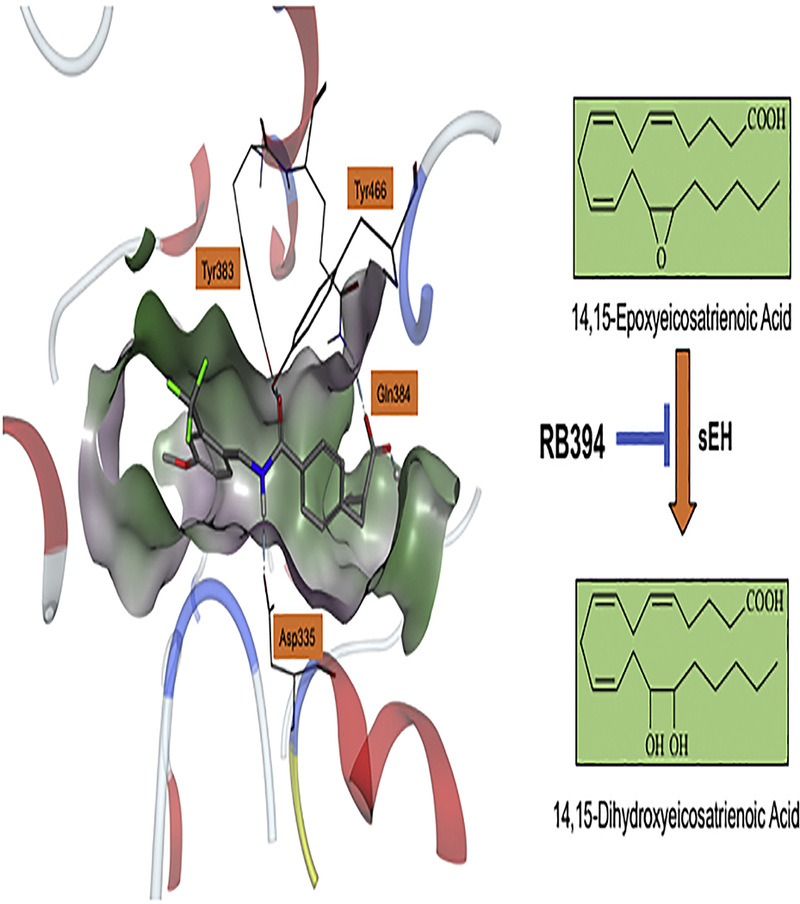
Left panel illustrates the bifunctional sEH inhibitor and peroxisome proliferator-activated receptor-γ (PPAR-γ) agonist, RB394, bound in the sEH (PDB structure 4JNC) enzymatic pocket. Right panel illustrates RB394 sEH inhibition that results in increased epoxyeicosatrienoic acid (EET) levels.
Multiple approaches were used to improve the physical properties and metabolic stability for urea-based sEH inhibitors. One successful approach was to replace the AUDA urea moiety with an amide that resulted in better physical properties at the expense of decreased potency (Marino, 2009; Shen et al., 2012). Eventually, the decreased potency for the amide-based sEH inhibitors was overcome with proper amide substitutions (Marino, 2009; Shen et al., 2012). Peptidyl moieties were also incorporated into sEH inhibitors and required that the peptidyl group be located a proper distance from the primary pharmacophore (Morisseau et al., 2006). Functional groups such as piperazines were added to a series of sEH inhibitors as a secondary pharmacophore that greatly enhanced solubility of the urea-based sEH inhibitors with some loss of potency (Li et al., 2006). Adamantane replacements with other aliphatics or aromatics dramatically increased bioavailability across several species following oral administration (Imig & Hammock, 2009; Tsai et al., 2010). This information was utilized to develop Arête’s AR9281 that exhibited good gut permeability, good potency against sEH, excellent water solubility, and reasonable plasma protein binding (Anandan et al., 2011; Jones et al., 2006). AR92821 was the first sEH inhibitor to be tested in humans (Arete Therapeutics, 2009).
Several sEH inhibitors have been developed to various stages by pharmaceutical companies. Boehringer Ingelheim developed a series of sEH inhibitors based on results from a high throughput screen (Eldrup et al., 2009). Their findings led to hybridization of nicatinamide and urea, piperidylureas, and pyrazole aniline derived amide sEH inhibitors (Eldrup et al., 2009; Shen et al., 2012). The pyrazole amides are good sEH inhibitors with a modest half-life that would be an appealing lead for future optimization (Lo et al., 2010). Merck initially developed 3,3-disubsituted piperidine-derived tri-substituted ureas (Shen et al., 2009b). Another distinct series of potent sEH inhibitors developed by Merck included novel amino-heteroaryl analogues (Shen et al., 2009a). GSK developed the second sEH inhibitor to be tested in humans, GSK2256294 (Lazaar et al., 2015). GSK2256294 is a potent, tight-binding but reversible sEH inhibitor that displays target selectivity and proper physiochemical properties (Lazaar et al., 2015). Like AR92821, GSK2256294 was well-tolerated in humans in Phase I clinical trials and demonstrates excellent potential to advance (Lazaar et al., 2015, Yang et al., 2017).
Hypertension Preclinical Studies
The evolution for sEH inhibitors for use in animals and humans can be traced back to the initial study that an sEH inhibitor, N,N’-dicyclohexylurea (DCU), given i.p. in a corn oil suspension to a spontaneously hypertensive rat (SHR) lowered blood pressure (Yu et al., 2000). Subsequent studies determined that chronic i.p. administration of sEH inhibitors lowered blood pressure in angiotensin dependent hypertension (Imig et al., 2002). This led to a major push to develop sEH inhibitors with pharmacological properties that could eventually be used in humans. As previously mentioned, a key step forward was the development of the orally active sEH inhibitor AUDA (Imig & Hammock, 2009; Zhao et al., 2003). AUDA decreased blood pressure in angiotensin hypertension in rats and mice when used in a therapeutic manner (Jung et al., 2005; Zhao et al., 2003). This provided the launching point to move towards an sEH inhibitor to be used in humans.
Experimental studies in hypertension animal models were the starting point for several studies to determine the benefits for sEH inhibitors to treat cardiovascular diseases (Figure 5). The most consistent finding from these hypertension studies was that sEH inhibitors are very effective in lowering blood pressure in angiotensin dependent hypertension (Imig & Hammock, 2009; Jung et al., 2005; Zhao et al., 2003; Zhao et al., 2004). Angiotensin infusion hypertension results in an upregulation of the sEH enzyme in the kidney (Imig & Hammock, 2009; Zhao et al., 2003). Chronic oral administration of the sEH inhibitor, AUDA, results in an increase in the EET to DHETE ratio, increased sodium excretion, and lowering of blood pressure in angiotensin infusion hypertension in rats and mice (Imig & Hammock, 2009; Jung et al., 2005; Zhao et al., 2003). Blood pressure lowering actions for sEH inhibitors have also been demonstrated in salt-sensitive hypertension (Manhiani et al., 2009; Zhao et al., 2004). Inhibition of sEH in DOCA-salt hypertension and angiotensin salt-sensitive hypertension results in decreases in mean arterial pressure (Manhiani et al., 2009; Zhao et al., 2004). A primary mechanism responsible for sEH inhibitor blood pressure lowering actions in salt-sensitive hypertension is increased urinary sodium excretion, natriuresis (Manhiani et al., 2009; Zhao et al.,2004). Another potential mechanism that contributes to the anti-hypertension for sEH inhibition in salt-sensitive hypertension is a decrease in renal and systemic inflammation (Manhiani et al., 2009; Zhao et al., 2004). Renal vascular actions also contribute to the blood pressure lowering actions and can be ascribed to sEH inhibitor mediated elevation in EETs that oppose the actions of angiotensin II to constrict afferent arterioles (Zhao et al., 2004). These sEH anti-hypertensive actions in angiotensin hypertension provided a strong foundation for their cardiovascular therapeutic potential.
Figure 5. Epoxyeicosatrienoic acid (EET) analogs and soluble epoxide hydrolase (sEH) inhibitors combat cardiovascular diseases.
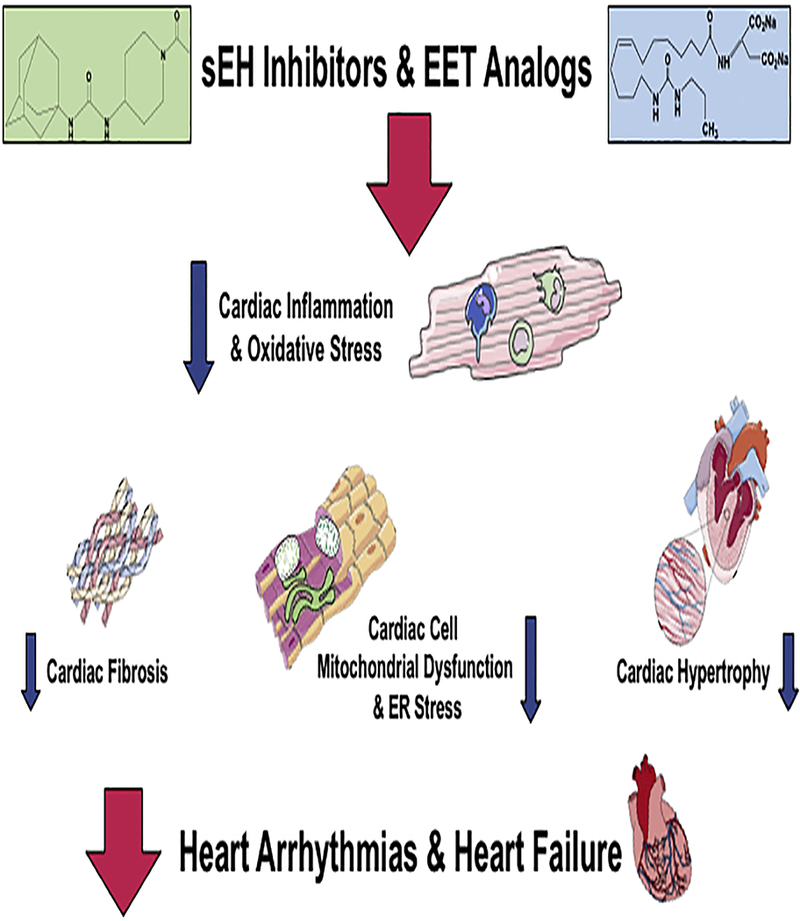
EET analogs and sEH inhibitors decrease cardiac cell inflammation and oxidative stress. Cardiac cell signaling mechanisms leading to fibrosis, endoplasmic reticulum stress and hypertrophy are combated by EET analogs and sEH inhibitors to opposed heart diseases.
On the other hand, a major consideration for sEH inhibitor clinical development was the fact that there were three classes of renin-angiotensin system inhibitors that effectively lowered blood pressure in human hypertension. Unfortunately, the ability for sEH inhibitors to lower blood pressure in other hypertension animal models was variable (Dorrance et al., 2005; Fornage et al., 2002; Olearcyk et al., 2009). The ability for sEH inhibitors to lower blood pressure in SHR has been based on the rat strains (Fornage et al., 2002). Several SHR strains have been shown to have polymorphisms in the Ephx2 gene that could partially explain the lack of ability for sEH to lower blood pressure in these SHR strains (Fornage et al., 2002). Chronic administration of sEH inhibitors have failed to lower blood pressure in Goto-Kakizaki diabetic rats with salt-sensitive hypertension and stroke-prone SHR (Dorrance et al., 2005; Olearczyk et al., 2009). Although blood pressure was not lowered in these hypertensive strains, sEH inhibition did protect from diabetic nephropathy in Goto-Kakizaki diabetic rats and ischemia-induced brain injury in stroke-prone SHR (Dorrance et al., 2005; Olearczyk et al., 2009). A major mechanism responsible for decreased kidney damage in the Goto-Kakizaki diabetic rats with salt-sensitive hypertension by sEH inhibition was the decrease in renal inflammation (Olearczyk et al., 2009). The cerebral protective actions for sEH inhibition in stroke-prone SHR included actions on vascular remodeling and anti-apoptotic actions on neural tissue (Dorrance et al.,2005). These outcomes provided convincing evidence that sEH inhibitors could protect organs in hypertension and other cardiovascular diseases. Thus, the therapeutic potential for sEH inhibitors was significantly expanded.
Cardiac Disease Preclinical Studies
Cardioprotective actions for sEH inhibitors have been extensively studied and remains a viable area for therapeutic development. The potential for sEH inhibitors has been extensively evaluated for left ventricular hypertrophy and ischemia reperfusion injury whereas atrial fibrillation and cardiac arrhythmias have been less well evaluated (Nithikapatkon & Gross, 2010; Oni-Orisan et al., 2014; Seubert et al., 2007). Experimental studies have compared sEH inhibitors to sEH genetic deletion in mice and found comparable results (Ai et al., 2009; Oni-Orisan et al., 2014; Xu et al, 2006). The cardioprotective actions for sEH inhibitors include anti-inflammatory and anti-apoptotic mechanisms (Imig, 2012; Oni-Orisan et al., 2014; Fleming, 2007). There is also significant evidence for sEH inhibitors resulting in cardiomyocyte KATP and mitoKATP activation (Batchu et al., 2011; Batchu et al., 2012b). In addition, the cardiac protective actions for sEH inhibitors are dependent on actions on several cell types within the heart (Nithikapatkon & Gross, 2010; Oni-Orisan et al., 2014; Seubert et al., 2007).
The therapeutic promise for sEH inhibitors to combat cardiac hypertrophy is well documented in animal disease models. Left ventricular hypertrophy in rodent models is associated with decrease EET generation and increased sEH activity (Ai et al., 2009; Xu et al; 2006). Pressure overload left ventricular hypertrophy in mice is prevented and reversed by sEH inhibitors (Xu et al; 2006). Decreased left ventricular hypertrophy in this model in response to sEH inhibition is associated with decreased cardiac myocyte NFkB mediated inflammation (Xu et al; 2006). Angiotensin II mediated cardiac hypertrophy is also attenuated by sEH inhibition (Ai et al., 2009). Likewise, sEH inhibitors decreased cardiac hypertrophy in angiotensin II hypertension; however, the improved heart function could be partially due to the anti-hypertensive effect (Cervenka et al., 2015; Zhao et al., 2004). Cardiac hypertrophy, fibrosis, and inflammation in mice fed a high fat diet was decreased by chronic administration of the sEH inhibitor, t-AUCB (Roche et al., 2015a). Collectively, these studies have supplied strong evidence for sEH inhibitors to oppose cardiac hypertrophy in several animal models.
Improved cardiac function following an ischemic event has been determined with sEH inhibitors. These experimental studies have been conducted in the Langendorff-perfused heart and in vivo in rodents and canines (Gross et al., 2007; Oni-Orisan et al., 2014; Seubert et al., 2006). Acute sEH inhibitor administration to the heart before ischemia or at the start of reperfusion results in decreased cardiac infarct size (Batchu et al., 2009; Oni-Orisan et al., 2014; Seubert et al., 2006; Seubert et al., 2007). Coronary artery administration of AUDA reduced ischemia reperfusion injury in dogs and enhanced the cardioprotective actions of 14,15-EET (Gross et al., 2008). Other studies in rodents have demonstrated that sEH inhibition reduces infarct size and improves left ventricular developed pressure following ischemia reperfusion (Batchu et al., 2011; Seubert et al., 2004). Administration of sEH inhibitors improves cardiac ischemic tolerance in normotensive and hypertensive animals (Merkel et al., 2010; Motoki et al., 2008; Neckar et al., 2012). Anti-apoptotic mechanisms contributing to sEH inhibitor mediated reduction in infarct size include the ERK/MAPK and JAK/STAT signaling pathways (Batchu et al., 2011; Merkel et al., 2010; Motoki et al., 2008). Experimental studies have determined that KATP channel activation is a crucial component for sEH inhibition cardiac protection in ischemia reperfusion injury (Gross et al., 2007; Oni-Orisan et al., 2014; Seubert et al., 2006). Cardiac KATP channel activation acts via downstream PI3 kinase signaling resulting in Akt phosphorylation and increases in phospho-glycogen kinase 3B (p-GSK3B) (Chaudhary et al., 2009; Imig, 2012; Katragadda et al., 2009). This Katp signaling cascade ultimately delays or inhibits mitochondrial permeability transition pore (mPTP) opening and shifts cardiac cells from apoptosis and necrosis toward survival (Chaudhary et al., 2009; Katragadda et al., 2009). Further research has demonstrated that in addition to the short-term cardiac protective actions afforded by sEH inhibitors that this therapeutic approach can decrease the chronic consequences following ischemia reperfusion injury.
Long-term treatment with sEH inhibitors reduces myocardial hypertrophy and fibrosis progression and improves cardiac function following a myocardial infarction (Imig, 2012; Morisseau & Hammock, 2013; Merabet et al., 2012; Oni-Orisan et al., 2014). A major contributor to these longer term sEH inhibitor cardioprotective actions is a reduction in myocardial inflammation. A mouse model of myocardial infarction heart failure where the left anterior descending artery was acutely occluded demonstrated that administration of an sEH inhibitor for three weeks reduced cardiac fibrosis, decreased arrhythmias, and improved left ventricular shortening (Li et al., 2009). Treatment with a sEH inhibitor for five weeks in a rat with permanent coronary artery occlusion reduced collagen deposition in the infarct zone (Kompa et al., 2013). The timing for the sEH inhibitor at various points following the permanent coronary artery occlusion in rats elicits cardioprotective actions (Sirish et al., 2013; Sirish et al., 2016). Administration of sEH inhibitors at 8 days or 47 days post coronary artery ligation improved left ventricular ejection fraction at 50 days whereas the long-term sEH inhibitor treatment elicited an improvement in left ventricular end diastolic pressure (Sirish et al., 2013). Cardiac fibrosis at one month can be reduced when sEH inhibition is started a week following acute coronary occlusion in mice (Sirish et al., 2016). These experimental outcomes establish the therapeutic potential for sEH inhibitors to combat the acute and chronic heart problems associated with myocardial infarction.
Atrial fibrillation and cardiac arrhythmias are other cardiac disorders where sEH inhibitors have been evaluated. Evidence for protective sEH inhibitor actions against atrial fibrillation and cardiac arrhythmias come from studies with ischemia reperfusion and pressure overload animal models (Monti et al., 2008; Sirish et al., 2013; Sirish et al., 2016; Shrestha et al., 2014). Cardiac atrial and ventricular arrhythmias in cardiac hypertrophy models of high dose chronic angiotensin II or transverse aortic constriction were reduced by sEH inhibition (Monti et al., 2008). More recently, the molecular mechanisms responsible for sEH inhibitors to prevent atrial arrhythmias in a thoracic aortic constriction cardiac hypertrophy model has been investigated (Sirish et al., 2013). The anti-inflammatory sEH inhibitor actions is a key mechanism responsible for preventing atrial arrhythmias in cardiac hypertrophy (Sirish et al., 2013; Sirish et al., 2016). Treatment with sEH inhibitors significantly lowers systemic levels of inflammatory cytokines and inhibits NFkB activation in mice following thoracic aortic constriction (Sirish et al., 2016). This sEH inhibitor anti-inflammatory action in this cardiac hypertrophy model led to decreased MAPK and endoplasmic reticulum stress activation in atrial fibroblasts and atrial myocytes to decrease structural remodeling (fibrosis) and electrical remodeling (hypertrophy) to ultimately decrease atrial fibrillation (Sirish et al., 2016). These findings demonstrate that sEH inhibitors have broad cardioprotective actions including reducing atrial fibrillation and cardiac arrhythmias.
Vascular Disease Preclinical Studies
Vascular beneficial therapeutic actions for sEH inhibitors extends to atherosclerosis and vascular remodeling (Fleming, 2001; Imig, 2012; Imig & Hammock, 2009). Vascular smooth muscle cell culture experiments have ascertained that sEH inhibitors decrease proliferation and migration (Davis et al., 2002; Fleming, 2001; Kim et al., 2017). Acute vascular inflammation in mice is attenuated by increased endothelial cell EET levels or sEH inhibition (Deng et al., 2011). Vascular hypertrophic remodeling and collagen deposition in the middle cerebral artery of stroke-prone SHR was attenuated by sEH inhibition (Simpkins et al., 2009). Chronic sEH inhibitor administration attenuated renal arteriolar hypertrophy in angiotensin dependent hypertension (Zhao et al., 2003; Zhao et al., 2004). Likewise, arteriosclerosis in rats and atherosclerosis in apolipoprotein e genetic deficient (Apo-e −/−) mice are reduced by chronic sEH inhibition (Ula et al., 2008; Zhang et al., 2009). Vascular remodeling effects of sEH inhibitors have been assessed in carotid ligation and femoral wire injury model (Simpkins et al., 2010). Even though sEH inhibition decreased the hyperplastic carotid artery ligation response, wire induced femoral artery vascular remodeling was unaltered by sEH inhibition (Simpkins et al., 2010). These findings suggest that an intact endothelium is required for sEH inhibition to prevent vascular neointimal formation. The sEH inhibitor, AR9276, was evaluated in Apo-e −/− mice infused with angiotensin II to induce aneurysm development (Zhang et al., 2009). Aneurysm and atherosclerosis development was attenuated by AR9276 treatment (Zhang et al., 2009). Vascular remodeling and atherosclerosis studies have consistently found that sEH inhibitors decrease pro-inflammatory mediators including cytokines, chemokines and adhesion molecules (Simpkins et al., 2009; Simpkins et al., 2010; Ula et al., 2008; Zhang et al., 2009; Zhang et al., 2015). Lipid and cholesterol levels have been determined to be decreased by sEH inhibition in many but not all studies (Li et al., 2009; Li et al., 2014; Li et al., 2016; Shen et al., 2015; Zhang et al., 2009; Zhang et al., 2015). Thus, anti-inflammatory actions for sEH inhibitors is a major mechanism providing cardiovascular protection.
Ischemic Stroke Preclinical Studies
Stroke is another cardiovascular disease that is reduced by sEH inhibition. Treatment with sEH inhibitors has been time and again demonstrated to decrease cerebral ischemia in rats and mice (Imig, 2012; Morisseau & Hammock, 2013). The sEH inhibitor AUDA reduced cerebral infarct size following ischemia in stroke-prone SHR and normotensive rats (Dorrance et al., 2005; Simpkins et al., 2009). Cerebral ischemia reperfusion injury in mice is also decreased by sEH inhibition (Koerner et al., 2008; Zhang et al., 2008). These cerebral protective actions have been demonstrated with chronic sEH inhibitor administration, sEH inhibitor administration a couple of hours prior to ischemia, and sEH inhibitor treatment at the time of reperfusion (Dorrance et al., 2005; Koerner et al., 2008; Simpkins et al., 2009; Zhang et al., 2008). Several studies have demonstrated that sEH inhibition results in increased EET and decreased DHETE levels to improve stroke outcomes (Jouihan et al., 2013; Kujal et al., 2014; Shaik et al., 2013; Zuloaga et al., 2014). Additional evidence has demonstrated blockade of CYP epoxygenase EET generation abolishes the reduction in cerebral injury afforded by sEH inhibition (Koerner et al., 2008; Zhang et al., 2008). Cerebral protection provided by sEH inhibition is due largely to changes in vascular structure, cerebral blood flow, and neuronal cell apoptotic pathways (Koerner et al., 2008; Shaik et al., 2013; Simpkins et al., 2009; Zhang et al., 2015). Neuronal cell signaling pathways altered by sEH inhibition include increasing several anti-apoptotic genes encoding inhibitors of FAS-induced apoptosis, increasing Mapk8ip that sequesters MAPKs in the cytoplasm to prevent apoptosis, and PI3 kinase-AKT activation to provide neuronal cytoprotection (Simpkins et al., 2009; Zhang et al., 2008). The combination of vascular and neuronal cell protective actions for sEH inhibitors provide support for this as a therapeutic approach for stroke and other cerebral vascular diseases.
Renal Disease Preclinical Studies
Renal diseases are another disease area where there is mounting experimental data in animal models demonstrating therapeutic potential for sEH inhibitors (Figure 6). The therapeutic benefits for sEH inhibitors is evident in progressive kidney disease in hypertension, diabetic nephropathy, drug-induced nephrotoxicity, and renal fibrotic disease (Imig, 2012; Imig, 2015). Initial studies in hypertension demonstrated decreased renal glomerular injury and fibrosis in angiotensin hypertension and salt-sensitive hypertension with sEH inhibitor treatments (Manhiani et al., 2009; Zhao et al., 2003; Zhao et al., 2004). It was difficult to determine if this was due to a direct effect because sEH inhibitors lowered blood pressure. These experimental studies found the sEH inhibition decreased macrophage infiltration and renal inflammation (Manhiani et al., 2009; Zhao et al., 2003; Zhao et al., 2004). Additional studies found that there was a renal protective action for sEH inhibitors that was independent of blood pressure lowering in hypertension (Olearczyk et al, 2009; Kujal et al., 2014; Roche et al., 2015b). Inhibition of sEH also provides significant renal protection in Ren-2 transgenic hypertensive rats that have had 5/6 nephrectomy to induce chronic kidney disease without lowering blood pressure (Kujal et al., 2014). Diabetic nephropathy is reduced by sEH inhibitors that have antidiabetic and anti-inflammatory effects (Elmarakby et al., 2011; Olearczyk et al., 2009). These studies clearly established that sEH inhibitors attenuate hypertensive and diabetic kidney disease.
Figure 6. Epoxyeicosatrienoic acid (EET) analogs and soluble epoxide hydrolase (sEH) inhibitors combat renal diseases.
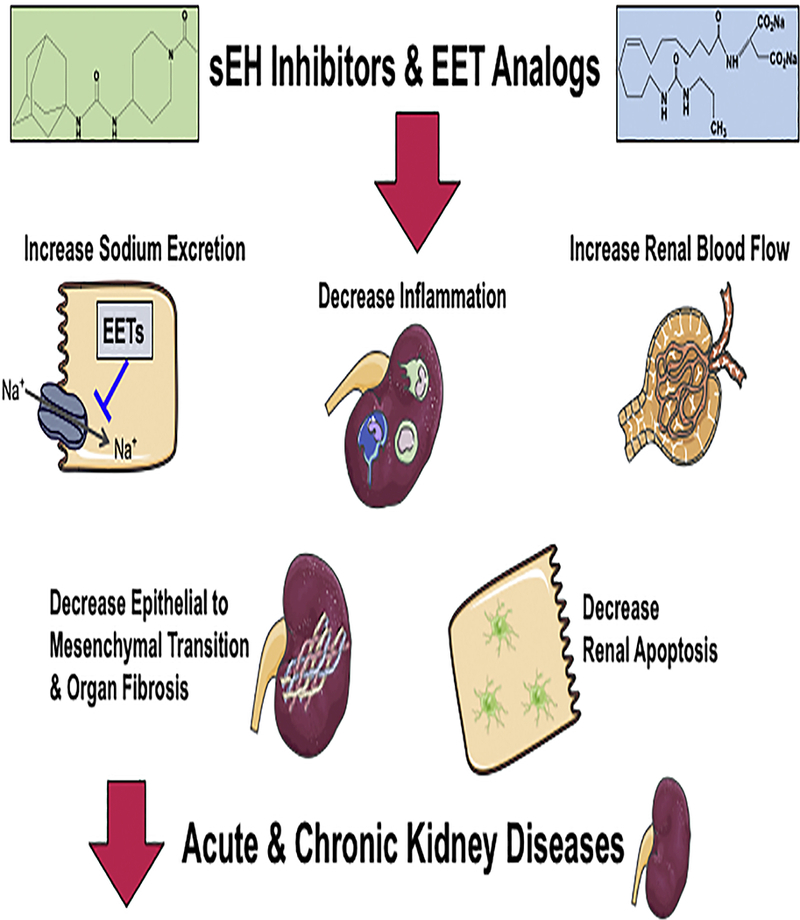
Renal hemodynamic actions, epithelial cell actions, and anti-inflammatory are responsible for EET analogs and sEH inhibitors ability to oppose kidney diseases. EET analogs and sEH inhibitors improve endothelial responses, enhance sodium excretion, decrease apoptosis, and oppose epithelial to mesenchymal transition mediated fibrosis.
The therapeutic potential for sEH inhibitors has also been demonstrated in kidney diseases that are not associated with a systemic disease. Drug-induced nephrotoxicity studies provided strong evidence for a direct renal protection by sEH inhibition. Cisplatin induced acute kidney injury was opposed by sEH inhibition (Liu et al., 2012; Parrish et al., 2009). The decrease in renal tubular damage afforded by sEH inhibition was due to decreased NFkB activation and TNFα inflammation (Liu et al., 2012). Renal fibrosis induced by unilateral ureter obstruction is prevented by sEH inhibitor administration. EET levels were increased and tubulointerstitial fibrosis decreased by sEH inhibition in unilateral ureter obstruction mice (Kim et al., 2014). Decreased inflammation was demonstrated by decreased neutrophil influx, decreased MCP-1, TNFα and ICAM-1 levels in unilateral ureter obstruction mice administered an sEH inhibitor (Kim et al., 2014). Another mechanism responsible for the sEH inhibitor anti-fibrotic actions is changes in epithelial to mesenchymal transition. Epithelial to mesenchymal transition induction in cultured NRK-52E renal epithelial cells decreases E-cadherin, increases α-smooth muscle actin (α-SMA) resulting in a myofibroblast phenotype (Liang et al., 2015). AUDA suppressed epithelial to mesenchymal transition by increasing E-cadherin, decreasing α-SMA, and preventing the phenotype transition (Kim et al., 2014; Liang et al., 2015). Epithelial cell signaling GSK-β and PI3-Akt activation were inhibited by sEH inhibition (Liang et al., 2015). Overall, sEH inhibitors provide renal protection through anti-inflammatory and anti-fibrotic mechanisms that are consistent with the cardiovascular protective mechanisms attributed to sEH inhibitors.
Potential Adverse Effects
Although there is great promise for sEH inhibitors to treat renal and cardiovascular diseases, there are potential unwanted effects that need to be considered. Possible adverse effects include angiogenesis, tumorigenesis, and metastasis, (Michaelis et al., 2003; Panigraphy et al., 2012; Pokreisz et al., 2006; Pozzi et al., 2010; Wei et al., 2014; Yang et al., 2009). Angiogenesis and anti-apoptotic actions for sEH inhibitors are likely contributing factors to the potential for tumorigenesis and metastasis (Michaelis et al., 2003; Panigraphy et al., 2012; Pozzi et al., 2010; Yang et al., 2009). Initial data demonstrated that sEH inhibitors enhance tumorigenesis and metastasis in lung small cell carcinomas (Panigraphy et al., 2012); however, subsequent data have demonstrated anti-tumor actions for sEH inhibitors alone or when combined with COX-2 inhibitor (Zhang et al., 2013a; Zhang et al., 2013b; Zhang et al., 2014). Angiogenesis can also be a negative or positive effect depending on the renal or cardiovascular disease being treated (Tanaka et al., 2015; Tanaka & Nangaku, 2013; Khurena et al., 2005; Simons & Ware, 2003). Augmentation of angiogenesis to vascularize ischemic tissue is a potential use for sEH inhibitors. Targeted delivery or incorporation of sEH inhibitors into biomaterials for the ischemic tissue could be done (Deveza et al., 2012).
Vascular actions on the pulmonary circulation to cause hypoxic vasoconstriction could lead to adverse events (Keseru et al., 2010; Pokreisz et al., 2006). Recovery from cardiopulmonary resuscitation is delayed in mice by sEH inhibitors or EPHX2 gene deletion (Hutchens et al., 2008). Increased generation of EETs is associated with pulmonary hypertension and epoxygenase inhibition reduces hypoxic pulmonary vasoconstriction (Keseru et al., 2010; Pokreisz et al., 2006). Likewise, EPHX2 gene deletion in mice resulted in acute hypoxic-induced pulmonary vasoconstriction (Keseru et al., 2010). These findings suggest that sEH inhibitors could have the potential unwanted effect of pulmonary vasoconstriction. In contrast, sEH inhibition reduced pulmonary vascular remodeling and the development of monocrotaline-induced pulmonary hypertension (Revermann et al, 2009). Interestingly, this potential pulmonary effect was not observed in initial clinical trials for sEH inhibitors (Arete Therapeutics, 2009; GlaxoSmithKine, 2013). One human clinical trial gave an sEH inhibitor to obese smokers with no significant consequences on the pulmonary circulation (Yang et al., 2017). Even with these concerns for unwanted side effects with sEH inhibitors, human clinical trials have proceeded primarily for chronic diseases.
Human Clinical Trials
The development of sEH inhibitors has advanced to a point where they have been tested in human clinical trials for several diseases including cardiovascular diseases. The first sEH inhibitor to enter human clinical trials was Arête Therapeutics small molecule drug AR9281 (Arete Therapeutics, 2009). AR9281 was well-tolerated and did not demonstrate major side effects of toxicity in Phase I clinical trials (Chen et al., 2012). A Phase IIa clinical trial with AR2981 for hypertension and diabetes failed to provide sufficient positive results (Arete Therapeutics, 2009). The second sEH inhibitor to reach the human clinical trial level was GSK2256294 developed by GSK to potentially treat chronic obstructive pulmonary disease (GlaxoSmithKline, 2013; Yang et al., 2017). Phase I clinical trial results were positive for GSK2256294 when evaluating safety and pharmacokinetics (Lazaar et al., 2015). GSK2256294 was well-tolerated and there were no serious adverse events when administered to healthy male subjects or obese smokers (Lazaar et al., 2015). The most frequent adverse effects for GSK2256294 were headache and contact dermatitis (Lazaar et al., 2015). A major finding for the human clinical trial was that GSK2256294 improved endothelial dysfunction in obese smokers assessed by forearm blood flow response to the vasodilator bradykinin (Yang et al., 2017). This finding provides the first evidence in humans that sEH inhibitors have beneficial cardiovascular actions. Evaluation for GSK2256294 in another cardiovascular disease has begun (Martini, 2017). Recruitment began in October 2017 for a Phase I clinical trial to evaluate GSK2256294 as a treatment for subarachnoid hemorrhage (Martini, 2017). Taken together, these human clinical trials offer exciting initial results that sEH inhibitors could treat renal or cardiovascular disease in the future.
EET Analogs
The development of EET mimetics or analogs initially occurred to provide pharmacological tools for investigators to evaluate EET biological activities (Campbell et al., 2017; Imig, 2012; Sudhahar et al., 2010). EETs are difficult to use for biological experiments due to limited solubility and storage issues. Modifications were made to the carboxylic acid to prevent β-oxidation and to the epoxide to prevent metabolism by sEH (Gauthier et al., 2003; Imig et al., 1999; Imig et al., 2008; Imig et al., 2010; Sodhi et al., 2009). Another important aspect for the EET analog advancement was to determine structure activity relationships for EETs (Falck et al., 2003; Falck et al., 2009; Falck et al., 2014; Gauthier et al., 2004). The EET structure activity relationship also led to the discovery of EET antagonists (Bukhari et al., 2011; Gauthier et al., 2003; Gauthier et al., 2004;). EET antagonists selective for 11,12-EET and 14,15-EET have established that 14,15-EET selective antagonism attenuated the mesenteric resistance artery flow-mediated relaxation, whereas 11,12-EET selective antagonism did not significantly decrease the response (Bukhari et al., 2012). This finding supports the notion that 11,12-EET and 14,15-EET have distinct biological activities and binding sites/receptors that are responsible for distinct vascular functions. These studies indicate that 14,15-EET, but not 11,12-EET contribute to flow-mediated vasodilation (Bukhari et al., 2012). Progress also eventually led to EET analogs for 11,12-EET and 14,15-EET that could be administered orally and chronically to renal and cardiovascular disease animal models (Imig et al., 2010; Jichova et al., 2016; Khan et al., 2013a; Khan et al., 2013b; Sodhi et al., 2009). One current drawback is that EET antagonists that can be administered to animals chronically are not currently available. Even with this being the case, EET analogs have demonstrated great potential as a therapeutic for renal and cardiovascular diseases.
EET Agonists and Antagonists
The bulk of the initial development for EET analogs concentrated on 14,15-EET analogs and 11,12-EET analogs (Figure 7). First generation EET analogs that resisted β-oxidation were developed by synthesizing methyl esters and sulfonamide carboxylic acids (Gauthier et al., 2003; Imig et al., 1999; Imig et al., 2008). These were followed by a next series of EET analogs that eliminated double carbon bonds to improve solubility (Falck et al., 2003; Falck et al., 2009; Gauthier et al., 2004. Sulphur and ether moieties replaced the 11,12- or 14,15 epoxide oxygen to prevent metabolism of sEH and to mimic the epoxide (Falck et al., 2003; Gauthier et al., 2004; Imig et al., 2010). Carbon chain length modifications were another aspect that provided structure activity information on EETs (Falck et al., 2009; Gauthier et al., 2004; Sudhahar et al., 2010). These EET analogs were extensively tested in vitro for cardiovascular activity (Gauthier et al., 2004; Sudhahar et al., 2010). 14,15-EET-Me, 11,12-EET-Me, 14,15-EET-SI and 11,12-EET-SI all demonstrated full agonistic vasorelaxation when tested on coronary arteries and afferent arterioles (Gauthier et al., 2003; Gauthier et al., 2004; Imig et al., 2008). Evaluation of EET analogs by these three major modifications to 11,12-EET and 14,15-EET revealed a structure activity relationship that included an acidic carboxyl group at carbon 1, a Δ8 carbon double bond, and 20-chain carbon length for full vascular agonistic function (Campbell et al., 2017; Sudhahar et al., 2010). These experimental findings have also provided important insight concerning biological actions that are EET versus DHETE dependent. Overall, experimental findings from EET analogs validate that EETs are the primary contributors to epoxyeicosanoid renal and cardiovascular actions (Campbell et al., 2017; Sudhahar et al., 2010).
Figure 7. Epoxyeicosatrienoic acid (EET) analog development to treat renal and cardiovascular diseases.
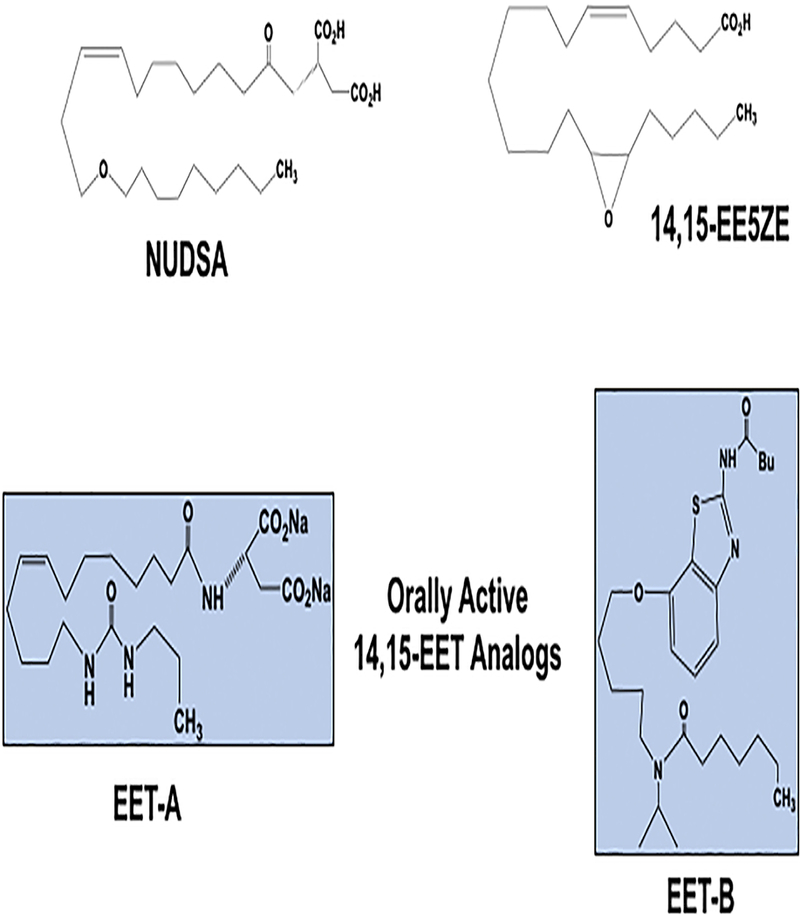
NUDSA: 11,12-EET analog first EET analog to be found to combat cardiovascular disease. 14,15-EE5ZE: non-selective EET antagonists used widely to determine EET biological actions. EET-A: initial 14,15-EET analog to be administered orally to rodents with renal and cardiovascular diseases. EET-B: departure from the carbon backbone to a fused bicyclic maintained renal and cardiovascular protective activity.
Along with determining structure activity relationship, the development of EET analogs led to the identification of EET antagonists (Bukhari et al., 2011; Bukhari et al., 2012; Gauthier et al., 2004). The first major EET antagonist was 14,15-epoxyeicosa-5 (Z)-enoic acid (14,15-EE5ZE) (Gauthier et al., 2003; Gauthier et al., 2004). Vascular studies evaluating 14,15-EE5ZE found that it was a non-selective EET antagonist (Gauthier et al., 2003; Gauthier et al., 2004). 14,15-EE5ZE inhibited bradykinin and methacholine EDHF mediated coronary artery relaxations (Gauthier et al., 2002). EET cardioprotective actions against ischemia reperfusion injury are eliminated by 14,15-EE5ZE administration (Gross et al., 2009; Nithipatikom et al., 2006). Surprisingly when evaluating coronary artery relaxation, it was found that the sulfonamide for 14,15-EE5ZE, 14,15-EE5ZE-SI, was a selective antagonist for 14,15-EET vasodilation (Bukhari et al., 2011; Gauthier et al., 2004). In addition, 14,15-DHE5ZE, the 14,15-EE5ZE hydrolysis product, inhibited vasorelaxations to 14,15-EET but was without effect on the relaxations to other EET regioisomers (Bukhari et al., 2011; Gauthier et al., 2004). Thus, 14,15-DHE5ZE is a selective 14,15-EET antagonist. Subsequent evaluation of novel EET small molecules demonstrated that 11,12,20-THE8ZE is a selective 11,12-EET antagonist (Bukhari et al., 2012). These EET antagonists allow for experimental studies to distinguish the contribution of 11.12-EET and 14,15-EET to renal and cardiovascular functions. These EET selective antagonists await extensive evaluation by investigators and further development for chronic use in animal models of renal and cardiovascular diseases.
Although EET antagonists await further advancement to be used in animal disease models, EET analogs have progressed to a state where they can be evaluated chronically in hypertension, cardiac diseases, and kidney diseases (Campbell et al., 2017; Imig, 2012; Sudhahar et al., 2010). A series of 11,12-EET analogs based on the 11-nonyloxy-undec-8(Z)-enoic acid structure with an acyclic ether were generated to enhance solubility, resist sEH metabolism, and resist β-oxidation (Hye Khan et al, 2014; Imig et al., 2008; Imig et al., 2010). Renal vascular dilation responses were evaluated for nine 11,12-EET analogs and three evaluated for blood pressure lowering in angiotensin hypertension (Imig et al., 2010). One promising 11,12-EET analog, NUDSA, that is the aspartic amide of 11-no nyloxy-undec-8(Z)-enoic acid was found to be active following intraperitoneal administration (Imig et al., 2010; Sodhi et al., 2009). The next generation was focused on more efficacious 11,12-EET and 14,15-EET analogs with sEH resistant bioisosteres or surrogates (Falck et al., 2014). These EET analogs found that replacing the acyclic ether with a 1,3-disubstituted urea gave an EET analog with similar vasorelaxation activity (Falck et al., 2014). An advantage for the EET analog series was that the epoxide bioisosteres are achiral and lack an asymmetric center (Falck et al., 2014). These EET analogs are therefore capable of binding to receptors or recognition binding sites irrespective of enatiomeric preferences. In addition to changes at the epoxide, major modifications were made with various bioisosteres to the carboxylic acid (Falck et al., 2014). These carboxylic acid changes to the EET analogs had the goal of improving water solubility, oral bioavailability, and plasma half-life. Small molecular weight amides were functional whereas phosphonate was less effective despite being more acidic than the carboxylic acid (Campbell et al., 2017; Falck et al., 2014). Other small heterocyclic bioisosteres for the carboxylic acid included tetrazoles and oxathiadiazole-2-oxides (Falck et al., 2014). There appears to be significant latitude for the design for EET analogs because a departure from the carbon backbone to a fused bicyclic maintained vascular activity. These third generation EET analogs provided a pharmacological toolbox with good water solubility, oral bioavailability, and a reasonable plasma half-life to allow for chronic administration in renal and cardiovascular diseases.
Hypertension and EET Analogs
EET analogs were administered by intraperitoneal injection initially in animal models of hypertension (Hye Khan et al., 2014; Imig et al., 2010; Sodhi et al., 2009). NUDSA was the first EET analog tested and decreased blood pressure in angiotensin hypertension after a single injection (Imig et al., 2010). Subsequent evaluation demonstrated that NUDSA decreased blood pressure over a 5-day period in SHR and ameliorated the metabolic syndrome phenotype by decreasing blood pressure and endothelial dysfunction in heme-oxygenase 2 null mice (Hye Khan et al., 2014; Sodhi et al., 2009). A major next step was identification of novel orally active 14,15-EET analogs, EET-A and EET-B (Khan et al., 2013 a). Administering a series of EET analogs to SHR determined that blood pressure lowering required at least three days and the blood pressure returned to hypertensive levels within a week after discontinuing treatment (Hye Khan et al., 2014). Anti-hypertensive actions were demonstrated for EET-A or EET-B when administered for two weeks in SHR and angiotensin hypertension (Hye Khan et al., 2014). These studies also determined that EET analogs lowered blood pressure through vasodilation and inhibition of the renal epithelial sodium channel, ENaC, to promote sodium excretion (Hye Khan et al., 2014). In these hypertension studies blood pressure was lowered to near normal levels and renal injury was prevented by orally administered EET analogs (Hye Khan et al., 2014). Angiotensin-mediated malignant hypertension in Cyp1a1-Ren-2 transgenic rats was prevented by EET-A administration for two weeks (Jichova et al., 2016). Importantly, EET-A was effective in genetic deficient Cyp2c44 −/− mice with decreased EET production and salt-sensitive hypertension. EET-A resulted in afferent arteriolar dilation, reduced ENaC activity and expression, and lowered blood pressure in Cyp2c44 −/− salt-sensitive hypertensive mice (Capdevila et al., 2014; Hye Khan et al.,2014). Experimental studies in hypertension animal models also revealed that EET analogs were antiinflammatory by decreasing MCP-1 levels and renal macrophage infiltration (Hye Khan et al., 2014; Khan et al., 2103b). Endothelial dysfunction associated with hypertension was also prevented with chronic EET analog treatment (Khan et al., 2013b; Sodhi et al., 154). Taken together, these findings are consistent with multiple EET analog activities including vasodilation, anti-inflammation, and natriuresis as contributing to blood pressure lowering in hypertension.
Cardiac Diseases and EET Analogs
Therapeutic potential for EET analogs has also been demonstrated in various heart disease animal models (Figure 3). The EET analog NUDSA provided the starting point for evaluation in cardiac ischmia reperfusion models (Seubert et al., 2007). NUDSA administered to mice after myocardial infarction resulted in improvements in heart function (Cao et al., 2015; Seubert et al., 2007). Treatment with NUDSA was started five days following a left anterior descending coronary artery ligation and continued for one month (Cao et al., 2015). Echocardiography and cardiac histological analysis revealed that NUDSA treatment improved left ventricular end diastolic area and fractional area and significantly decreased diastolic dysfunction (Cao et al., 2015). Cardiac histological analysis showed decreased fibrosis in myocardial infarcted animals administered NUDSA (Cao et al.,2014). Cardio-protective action of the EET analog UA-8 against acute ischemia/reperfusion injury was also demonstrated (Seubert et al., 2007). Indeed, acute administration of UA-8 improved cardiac ischemic tolerance in isolated mice hearts (Seubert et al., 2007). Experimental studies in hypertension demonstrated that EET-B treatment administered for two months in SHR subjected to a 30-minute left coronary artery occlusion attenuated progressive heart failure and decreased cardiac fibrosis (Neckar et al., 2018). EET-B administration was also found to prevent lung edema and reduce ischemic zone fibrosis induced by myocardial infarction (Neckar et al., 2018). The cardioprotective EET analog actions are through multiple mechanisms. EET analogs oppose apoptosis and provide mitochondrial protection (Campbell et al., 2017; Oni-Orisan et al., 2014). Mitochondrial protection via EET analogs includes an increase in OPA1 oligomers and mitochondrial cristae density and activation of mitochondrial KATP channels (El-Shikhry et al., 2016; Oni-Orisan et al., 2014). Pro-survival activation of MAP kinase signaling in the heart occurs with EET analog treatment to prevent ischemic stress induced cardiac cell apoptosis (El-Shikhry et al., 2016; Oni-Orisan et al., 2014). EET analogs can decrease hypertension induced left ventricular hypertrophy and heart fibrosis (Khan et al., 2013b). Overall, the ability for EET analogs to oppose cardiac fibrosis has been demonstrated in hearts exposed to pressure overload, myocardial infarction, and insulin resistance (Cao et al., 2015; Oni-Orisan et al., 2014; Sodhi et al., 2009).
Renal Diseases and EET Analogs
The kidney is another major organ that EET analogs have demonstrated significant therapeutic potential (Figure 5). EET analogs can decrease drug-induced nephrotoxicity, acute kidney disease, and chronic kidney disease (Imig, 2015; Khan et al., 2013a; Khan et al., 2013b; Skibba et al., 2017). Orally active EET analogs were first demonstrated to be effective in combating drug-induced nephrotoxicity (Khan et al., 2013a). EET-A or EET-B treatment protected the kidneys from cisplatin-induced injury through reduced inflammation, oxidative stress, and decreased apoptosis (Khan et al., 2013a). Cyclosporine-induced nephrotoxicity and hypertension were prevented by EET analog therapy (Yeboah et al., 2016). EET-B treatment for one month decreased cyclosporine-mediated oxidative stress, inflammation, fibrosis, and apoptosis (Yeboah et al., 2016). Experimental studies in renal epithelial cells provided evidence that EET-B eliminates cyclosporine-induced apoptosis through decreasing endoplasmic reticulum stress and pro-apoptotic GRP78 levels (Yeboah et al., 2016). Taken together, these findings demonstrate that EET analogs have therapeutic potential to combat drug-induced nephrotoxicity.
Progressive kidney disease associated with hypertension is also prevented by EET analogs (Hye Khan et al., 2014; Khan et al., 2013b). EET-B administration to Dahl salt-sensitive hypertensive rats decreased renal fibrosis, intratubular cast formation, and glomerular injury (Khan et al., 2013b). EET-B dramatically decreased renal inflammation, oxidative stress, and endoplasmic reticulum stress in the kidneys of Dahl salt-sensitive hypertensive rats (Khan et al., 2013b). EET analog treatment decreases renal injury and improves renal hemodynamics in Cyplal-Ren-2 transgenic malignant hypertensive rats (Jichova et al., 2016). Experimental studies in unilateral ureter obstruction mice support the notion that EET analogs can combat progressive chronic kidney disease by direct renal actions (Skibba et al., 2017). EET-A had a strong anti-fibrotic effect in unilateral ureter obstruction mice (Skibba et al., 2017). Renal collagen positive area, kidney hydroxyproline content, and renal α-smooth muscle actin levels were reduced by EET-A treatment (Skibba et al., 2017). Interestingly, EET-A markedly prevented the elevation in the epithelial to mesenchymal transition inducers including renal Snaill and ZEB-1 expression in unilateral ureter obstruction mice (Skibba et al., 2017). Myofibroblast markers FSP-1, fibrotic cytoskeletal protein markers (fibronectin and desmin), E-cadherin, and renal matrix protein collagen type III were reduced by EET-A treatment (Skibba et al., 2017). Therefore, the anti-fibrotic EET analog actions were due to preventing activation of the renal epithelial to mesenchymal signaling pathways.
EET-A has also been demonstrated to reduce renal injury due to radiation exposure (Hye Khan et al, 2016a). Administration of EET-A two days following exposure to 11 GY radiation mitigated the progression of nephropathy (Hye Khan et al., 2016a). Three months following radiation exposure led to increases in blood urea nitrogen, albuminuria, and renal histopathological injury (Hye Khan et al., 2016a). EET-A treatment reduced these renal injury parameters through reductions on the p53/Fas/FasL (Fas ligand) apoptotic pathway (Hye Khan et al., 2016a). Renal afferent arteriolar endothelial dysfunction in radiation nephropathy was also prevented by EET-A treatment (Hye Khan et al., 2016a). Overall, EET analogs can oppose chronic kidney disease through multiple mechanisms that improve epithelial cell and renal hemodynamic function.
Future for EET Analogs
The development of EET analogs have lagged sEH inhibitors for obvious reasons. EET analogs lack defined protein targets, actions could be different for analogs of each EET regioisomer, and synthesis for the EET analogs is more complex. The precise molecular and protein target for EETs has yet to be identified which does not allow for target-based screening drug design approaches; however, this has been overcome by a structure-activity approach that has defined the pharmacophore model for EET analogs (Campbell et al., 2017; Sudhahar et al., 2010). Interestingly, when comparing FDA drugs approved between 1999–2008 it was found that phenotypic screening was the most successful approach for first-in-class drugs whereas target-based screening was most successful for follower drugs (Swinney & Anthony, 2011). Intriguingly, nitro fatty acid analogs that lack a protein target are currently in clinical trials for focal segmental glomerulosclerosis and pulmonary arterial hypertension (Klinke et al., 2014; Liu et al., 2013; Wang et al., 2106). To define the potential differences between EET regioisomeric analogs biological actions and cell-signaling mechanisms have been extensively evaluated for regioisomeric EETs (Fleming, 2001; Imig, 2012; Spector et al., 2004). The recent development of 11,12-EET and 14,15-EET selective antagonists will provide a means to clearly define biological actions for regioisomeric EETs and their corresponding EET analogs (Bukhari et al., 2011; Bukhari et al., 2013). In addition, 8,9-EET analogs have been developed and could have selective actions on the kidney glomerulus barrier function (Sharma et al., 2009). Additional evaluation for the 8,9-EET analogs needs to be conducted. Even though EET analog development has been slower, there is great potential for EET analogs to successfully treat renal and cardiovascular diseases.
New Vistas
There are new vistas that are in the beginning stages and remain largely unexplored for epoxyeicosanoid based drugs. These areas include the expansion of novel small molecules that are bifunctional (Proschak et al., 2017). Development of sEH inhibitors with complementary activities such as COX-2 inhibition are being developed (Hwang et al., 2011; Hye Khan et al., 2016b; Zhang et al., 2014). Expanding the therapeutic indications for sEH inhibitors, EET analogs, EET anatagonists, and bifunctional molecules is another vista worthy of exploration. Diseases such as pre-eclampsia and metabolic disorders including non-alcoholic fatty liver disease could be treated by epoxyeicosanoid based drugs. There is recent evidence that EPHX1 can hydrolyze epoxyeicosanoids and contributes to cardiac ischemia reperfusion injury recovery (Edin et al., 2018). This finding suggest that therapeutics that target EPHX1 could have benefits in cardiovascular diseases. In addition to omega-6 EETs, CYP epoxygenases convert the omega-3 EPA and DHA into novel epoxyeicosatetraenoic acids (EEQs) and epoxydocosapentaenoic acids (EDPs), respectively (Westphal et al., 2011). Targeting and developing drugs for EEQs and EDPs has begun for renal and cardiovascular diseases (Arnold et al., 2010a; Westphal et al., 2011). Lastly, there has also been interest in plant-derived sEH inhibitors as a potential therapeutic.
Bifunctional Small Molecules
The concept that bifunctional small molecules could be developed to treat cardiovascular and renal diseases has been energized by the approval of Entresto, a combined neprilysin and angiotensin type 1 receptors inhibitor, for heart failure (Schiering et al., 2016; von Lueder et al., 2015). Bifunctional molecules that include sEH inhibitor activity are being developed because of the positive cardiovascular effects (Proschak et al., 2017) (Figure 4). These dual-acting sEH inhibitors have focused on including inhibition of another fatty acid enzymatic pathway, kinases, and nuclear receptor agonists (Proschak et al., 2017). The first novel dual-acting COX-2/sEH inhibitor, PTUPB [4-(5-phenyl-3-{3-[3-(4-trifluoromethylphenyl)-ureido]-propyl}-pyrazol-1-yl)-benzenesulfonamide] was developed because selective inhibition of either COX-2 or sEH provides beneficial outcomes in models of hypertension, inflammation, cardiovascular disease and diabetes (Hwang et al., 2011). Interestingly, PTUPB was first tested in a cancer animal model and was shown to suppress tumor growth and metastasis (Zhang et al., 2014). The ability for PTUPB to combat metabolic abnormalities and diabetic kidney injury was demonstrated in the Zucker Diabetic Fatty rat (Hye Khan et al., 2016b). Like sEH inhibitors, PTUPB reduced oxidative stress and inflammation in the Zucker Diabetic Fatty rat kidney (Hye Khan et al., 2016b). Several other dual acting bifunctional small molecules that include sEH inhibitor activity have been developed (Proschak et al., 2017). These dual-acting molecules combine sEH inhibition with c-Raf inhibition, fatty acid amide hydrolase inhibition, 5-LOX inhibition, farnesoid X receptor agonism, or PPAR agonism (Meirer, et al., 2016; Proschak et al., 2017; Schmidt et al., 2017; Temml et al., 2017). In addition, a few sEH inhibitors such as AUDA have EET agonistic activity and EET analogs have sEH inhibitor activity that varies from none to nanomolar IC50 sEH inhibitor activity (Falck et al., 2014; Olearczyk et al., 2006). EET analogs have also been designed that have PPAR agonistic activity (Blocher et al., 2016; Proschak et al., 2017). Although several of these dual-acting small molecules with sEH inhibitor or EET analog activity exists there has been very limited testing in renal and cardiovascular diseases.
Novel Epoxy Fatty Acid Therapeutic Targeting Approaches
Additional means for therapeutically targeting EETs and other epoxy fatty acids include development of EET antagonists, EEQ and EDP analogs, and plant derived sEH inhibitors. There is increasing recognition that sEH inhibition can metabolize other epoxy fatty acids other than EETs (Arnold et al., 2010b; Arnold et al., 2016). EET antagonists that are selective for 11,12-EET and 14,15-EET have been developed based on the structure activity relationship for these EET regioisomers (Bukhari et al., 2012). There are potential therapeutic applications where elevated EET levels could be contributing to disease. Although there has been limited evaluation of pathological conditions where elevated EETs have been described, potential clinical areas are ones with increased angiogenesis or elevated organ blood flow.
Epidemiological and clinical studies demonstrate that long-chain 𝛚-3 polyunsaturated fatty acids protect against cardiovascular diseases (De Caterina et al., 2011, Kris-Etherton et al., 2002; Westphal et al., 2011). Several cardiac beneficial effects have been attributed to increases in dietary 𝛚-3 EPA and DHA levels (Westphal et al., 2011). Clinical studies have increased EPA and DHA levels by formulating concentrated fish oil supplements or giving purified EPA/DHA-ethylesters. Cardiac sudden death, arrhythmias, and heart failure incidences were reduced by consuming approximately 1 gram EPA/DHA supplements (Lavie et al., 2009; Mozaddrian & Rimm, 2006). Dietary EPA and DHA were found to protect against arrhythmias and cardiac sudden death in angiotensin II hypertension (Fischer et al., 2008). These findings led to evaluating specific EPA and DHA metabolites for cardiovascular protective actions and led to the development of metabolite analogs. Experimental studies have found protective effects on the heart for EPA epoxygenase EEQ metabolites (Arnold et al., 2010a; Falck et al., 2011; Westphal et al., 2011). These EPA metabolites are known to increase with increased cold water fish consumption and are strongly associated with decreased incidence in cardiovascular disease (Arnold et al., 2010a; Fischer et al., 2014). Regarding EEQ and EDP analogs, this field is actively being pursued for therapeutic potential based on the biological activities found for EPA and DHA metabolites (Arnold et al., 2010a).
Based on evidence for 17,18-EEQ having cardioprotective actions, EEQ analogs are actively being examined as a treatment for atrial fibrillation and cardiac arrhythmias (Arnold et al., 2010b; Falck et al., 2011; Schunck et al., 2018). Experimental studies in neonatal rat cardiomyocytes EPA and 17,18-EEQ were found to have antiarrhythmic actions (Falck et al, 2011; Kang et al., 1995). DHA metabolites also appear to have beneficial cardiovascular actions to combat disease progression (Arnold et al., 2010a). DHA metabolites have been demonstrated to cause coronary artery relaxation (Arnold et al., 2010a; Falck et al., 2011). 19, 20-EDP was found to reduce cardiomyocyte contractility (Arnold et al., 2010b). To date, 17,18-EEQ analogs have been synthesized and tested for cardioprotective actions (Falck et al., 2011). 17,18-EEQ analogs attenuate ventricular tachyarrhythmia following coronary artery ligation in rats (Schunk et al, 2010). More recently, atrial fibrillation in mice has been demonstrated to be reduced by 17,18-EEQ analogs (Fischer et al., 2017). A clinical phase 1 trial began in February 2017 (EudraCTNo. : 2016–003445-28) by OMEICOS Therapeutics for a lead candidate 17,18-EEQ analog (Schunck etal., 2018).
Kidney and cardiovascular diseases are being explored for dietary DHA and DHA metabolites. Increasing dietary DHA levels combined with sEH inhibitors has anti-hypertensive and kidney protective actions (Ula et al., 2013). This therapeutic combination could be more effective than sEH inhibitors given alone. Other studies have demonstrated that the DHA epoxygenase metabolite 19,20-EDP given intraperitoneally decreases kidney fibrosis in mice with unilateral ureter obstruction (Sharma et al, 2016). The 19,20-EDP renal anti-fibrotic effects were due to inhibition of epithelial to mesenchymal transition (Sharma et al, 2016). Anti-arrhythmic actions have also been described for 19,20-EDP in rat neonatal cardiomyocytes (Arnold et al, 2010b). Newly described co-3 endocannabinoid epoxide metabolites have been demonstrated to have vascular and anti-inflammatory actions (McDougle et al, 2017). Thus, there is an expanding number epoxide metabolites that demonstrate biological activities that could be manipulated to treat renal and cardiovascular diseases.
The search for therapeutics in nature that could alter epoxy-fatty acid levels has recently intensified. Plant-derived sEH inhibitors have recently been identified from Cimicifuga dahurica roots and from plants in the order Brassicales (Kitamura et al., 2017; Thao et al., 2017). These plant-derived sEH inhibitors have not been tested for favorable renal and cardiovascular actions. Interestingly, flaxseed consumption results in sEH inhibition and lowering of blood pressure in patients with hypertension (Caligiuri et al, 2014). Overall, the future for EET anatagonists, EPA metabolites, DHA metabolites, and plant-derived sEH inhibitors will require considerable effort, however; initial data in preclinical renal and cardiovascular disease animal data provide excitement for their therapeutic prospective.
Additional Diseases for Epoxy Fatty Acid Therapies
The utility of sEH inhibitors, EET analogs, and new novel drugs aimed at epoxy fatty acids will expand the potential for treating several diseases. These diseases include non-alcoholic fatty liver disease, cancers, neuropathies, retinopathies, pre-eclampsia, and organ transplantation (Bellien & Joannides, 2013; Morisseau & Hammock, 2013). Recent findings demonstrate that decreased EET levels contribute to insulin sensitivity in mice and humans (Gangadhariah et al., 2017). Exciting data with sEH inhibitors and dual-acting sEH inhibitors in obesity and metabolic diseases have demonstrate positive actions on the pancreas and liver (Blocher et al., 2016; Elmarkby et al., 2011; Hye Khan et al., 2016b; Luria et al., 2011). The anti-inflammatory, anti-apoptotic, and decreases in endoplasmic stress described for EETs and sEH inhibition provide a means for combating complex diseases like metabolic syndrome, diabetes, and non-alcoholic fatty liver disease (Imig, 2012; Imig & Hammock, 2009). Other areas being investigated that demonstrate initial promise for sEH inhibitors, EET analogs, and new novel drugs aimed at epoxy fatty acids include cancer, neuropathies, and retinopathies (Morisseau & Hammock, 2013; Taguchi et al., 2016). Recently, novel epoxy fatty metabolites have been identified in retinopathies that contribute to the disease (Hu et al., 2017). Another area that has long-been associated with alterations in EET metabolites is pre-eclampsia (Herse et al., 2012; Jiang et al., 2013; Vedove et al., 2015). There have been initial studies in pre-clinical animal models of pre-eclampsia that require additional follow up (Huang et al., 2006; Zhou et al., 2005). Lastly, organ transplantation is an area worthy of additional investigation for epoxy fatty acid based drugs. The first indication for EETs was that the addition of EETs to an organ transplantation solution demonstrates positive effects on organ function (Yang et al., 2003). Additionally, EET analogs have been demonstrated to combat cyclosporine hypertensive and renal vasoconstrictive actions (Yeboah et al., 2016). Because cyclosporine is used as an immunosuppressive in greater than ninety percent of the transplants worldwide, a therapeutic that could combat these devastating side effects could ensure better transplantation outcomes. Even though the future for sEH inhibitors, EET analogs, and new novel drugs aimed at epoxy fatty acids to combat these diseases is possible, extensive investigations in these disease indications need to be conducted.
Unexplored Areas - Microsomal Epoxide Hydrolase (mEH) and sEH Phosphatase
Microsomal epoxide hydrolase (EPHX1, mEH) was identified three decades ago as being capable of hydrolyzing EETs to DHETEs (Morisseau & Hammock, 2005; Vaclavikova et al, 2015). The contribution for mEH to EET conversion to DHETEs is thought to be minimal due to the low catalytic turnover (Decker et al, 2012). On the other hand, mEH is known to importantly contribute to the metabolism of environmental contaminants and epoxide drugs like carbamazepine (Morisseau & Hammock, 2005; Newman et al., 2005). Mutations in EPHX1 are associated with the cardiovascular disease condition, preeclampsia (Zusterzeel et al., 2001). More recently, the development of mice with EPHX1 and EPHX2 gene deletion provide evidence that EPHXl(mEH) could contribute to EET metabolism and alter the response to cardiac recovery following ischemia (Edin et al., 2018). In this study EPHX1 was found to contribute to EET hydrolysis in vivo especially when EET formation rates are low and mice with EPHX1 and EPHX2 gene deletion had improved cardiac function after ischemia when compared to wild-type of EPHX2 gene deleted mice (Edin et ah, 2018). Other novel epoxide hydrolases include EPHX3 and EPHX4; however, a significant contribution to EET hydrolysis or renal and cardiovascular diseases has yet to be identified.
Unlike the C-terminal domain that encodes the lipid epoxide hydrolase activity, the sEHN-terminal domain that contains phosphatase activity is less well investigated. There has been insight into the structure and single nucleotide polymorphisms in the sEH phosphatase domain and speculation as to the physiological roles for the domain (Kramer & Proschak, 2017). Three single nucleotide polymorphisms have been identified in the phosphatase domain that result in amino acid substitutions and the Lys55Arg that reduces phosphatase and increases hydrolase activity (Lee et al., 2011; Przbyla-Zawislak et al., 2003). A physiological role for the sEH phosphatase domain is that it can influence cholesterol, lipid, and steroid levels (Luria et al., 2009). Sphingosine-1-phosphatase and lysophosphatidic acid are substrates for the sEH phosphatase domain and could be linked to vascular endothelial growth factor signaling (Kramer & Proschak, 2017). Another potential physiological role would impact endothelial nitric oxide and coronary artery endothelial function (Harris & Hammock, 2013; Hou et al., 2012). Assays for the sEH N-terminal phosphatase domain have been developed and have led to the identification of sEH phosphatase inhibitors (Kramer & Proschak, 2017; Morisseau et al., 2013). SMTP-7 has been identified as a sEH phosphatase inhibitor that demonstrates antiinflammatory actions and enhances thrombolytic activity by plasminogen modulation that could be promising for the treatment of ischemic stroke (Matsumoto et al., 2015). Evaluation of SMTP-7 revealed that it acts as an allosteric inhibitor of the sEH phosphatase domain (Kramer & Proschak, 2017). Future research is required to clearly identify the physiological role for the sEH N-terminal phosphatase domain and the potential for sEH phosphatase inhibitors to treat cardiovascular diseases.
Conclusion
The development of epoxy fatty acid therapeutics to treat renal and cardiovascular diseases has progressed rapidly since the turn of the century. A wealth of information was gathered from biochemical evaluation of the arachidonic acid CYP pathway that led to studies to determine biological activities. EET renal epithelial cell and vascular actions were initially described followed by EET effects to combat inflammation. Pharmacological manipulation of the pathway through sEH inhibition to increase EET levels led the way to evaluate sEH inhibitors in renal and cardiovascular diseases. Another major piece to the development for sEH inhibitors as a therapeutic was experimental studies in humans that linked increased sEH activity or decreased EET levels with cardiovascular diseases. Ultimately, sEH inhibitors have successfully developed to a point that they have made it to the human clinical trial phase for hypertension, diabetes, chronic obstructive pulmonary disease, and subarachnoid hemorrhage. EET analogs have lagged in development; however, EET analogs look poised to advance to human clinical trials.
A major consideration going forward for sEH inhibitors, EET analogs, dual-acting sEH inhibitors, and other epoxy fatty acid based drugs will be deciding on the appropriate clinical indication and proper design for the human clinical trials. Previous clinical trials have determined that sEH inhibitors appear to lack toxicity and are devoid of major side effects (Arete Therapeutics, 2009; Lazaar et al., 2016). In addition, the GSK clinical trial with GSK2256294A demonstrated improved endothelial function in obese males with chronic obstructive pulmonary disease (Yang et al., 2017). This finding clearly demonstrates potential for sEH inhibitors to combat cardiovascular disease in humans. On the other hand, the renal and cardiovascular diseases that would be viable from a pharmaceutical economic development standpoint will likely require a sophisticated clinical trial design. Drugs to improve organ preservation and transplantation are needed but have been largely avoided due to the very difficult clinical trial design. Cerebral ischemic stroke is another indication where sEH inhibitors and EET analogs have demonstrated great promise. Clinical trials for stroke are extremely difficult to design in a way to truly determine drug effectiveness. Myocardial infarction and acute kidney injury are two other ischemic reperfusion injury problems that are in dire need of a therapeutic. Several clinical trials for these indications have all failed due to difficulties in predictable progression and outcomes, different etiologies, and poor biomarkers to evaluate therapeutic effectiveness. There continue to be improvements in designing these difficult clinical trials to overcome these problems. Even with these barriers, the sEH inhibitors, EET analogs, dual-acting sEH inhibitors, and other epoxy fatty acid based drugs have actions in preclinical animal models and initial human trials that make them worthy of pursuing their therapeutic value in renal and cardiovascular diseases.
ACKNOWLEDGEMENTS
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases DK103616 and Dr. Ralph and Marian Falk Medical Research Trust Bank of America, N.A., Trustee. I thank Dr. Ewgenij Proschak for providing the model for solubule epoxide hydrolase with RB394 binding presented in Figure 4. Servier Medical Art was used to generate Figures 1, 2, 5, 6 and is licensed by Servier under a Creative Commons Attribution 3.0 Unported License.
ABBREVIATIONS
- α-SMA
α-smooth muscle actin
- AUDA
12-(3-adamantan-l-yl-ureido)dodecanoic acid
- ARIC
Atherosclerosis Risk in Communities
- CARDIA
Coronary Artery Risk Development in young adults
- COX
cyclooxygenase
- CYP
cytochrome P450
- DHETEs
dihydroxyeicosatrienoic acids
- DHA
docosahexaenoic acid
- EDHFs
endothelial-derived hyperpolarizing factors
- EPA
eicosapentaenoic acid
- ENaC
epithelial sodium channel
- EDPs
epoxydocosapentaenoic
- EEQs
epoxyeicosatetraenoic
- EETs
epoxyeicosatrienoic acids
- FXR
and farnesoid X receptor
- HETEs
hydroxyeicosatetraenoic acids
- LOX
lipoxygenase
- mPTP
mitochondrial transition pore
- PPAR
peroxisome proliferator-activated receptor
- sEH
soluble epoxide hydrolase
- SHR
spontaneously hypertensive rat
Footnotes
CONFLICT OF INTEREST STATEMENT
Dr. Imig has patents that cover the composition of matter for EET analogs and bifunctional sEH inhibitors. There are no other conflicts of interest, financial or otherwise, are declared by the author.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ai D, Fu Y, Guo D, Tanaka H, Wang N, Tang C, et al. (2007) Angiotensin II up-regulates soluble epoxide hydrolase in vascular endothelium in vitro and in vivo. Proc Natl Acad Sci U S A 104, 9018–9023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai D, Pang W, Li N, Xu M, Jones PD, Yang J, et al. (2009) Soluble epoxide hydrolase plays an essential role in angiotensin II-induced cardiac hypertrophy. Proc Natl Acad Sci U S A 106, 564–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Galicia M, Falck JR, Reddy KM, Roman RJ (1999) 20-HETE agonists and antagonists in the renal circulation. Am J Physiol 277, F790–F796. [DOI] [PubMed] [Google Scholar]
- Anandan SK, Webb HK, Chen D, Wang YX, Aavula BR, Cases S, et al. (2011) 1-(1-acetyl-piperidin-4-yl)-3-adamantan-l-yl-urea (AR9281) as a potent, selective, and orally available soluble epoxide hydrolase inhibitor with efficacy in rodent models of hypertension and dysglycemia. Bioorg Med Chem Lett 21, 983–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therapeutics Arete. (2009) Evaluation of soluble epoxide hydrolase (s-EH) inhibitor in patients with mild to moderate hypertension and impaired glucose tolerance https://clinicaltrials.gov/ct2/show/NCT00847899
- Argiriadi MA, Morisseau C, Goodrow MH, Dowdy DL, Hammock BD, Christianson DW (2000) Binding of alkylurea inhibitors to epoxide hydrolase implicates active site tyrosines in substrate activation. J Biol Chem 275, 15265–15270. [DOI] [PubMed] [Google Scholar]
- Archer SL, Gragasin FS, Wu X, Wang S, McMurtry S, Kim DH, et al. (2003) Endothelium-derived hyperpolarizing factor in human internal mammary artery is 11,12-epoxyeicosatrienoic acid and causes relaxation by activating smooth muscle BK(Ca) channels. Circulation 107, 769–776. [DOI] [PubMed] [Google Scholar]
- Arnold C, Konkel A, Fischer R, Schunck WH (2010a) Cytochrome P450-dependent metabolism of omega-6 and omega-3 long-chain polyunsaturated fatty acids. Pharmacol Rep 62, 536–547. [DOI] [PubMed] [Google Scholar]
- Arnold C, Markovic M, Blossey K, Wallukat G, Fischer R, Dechend R, et al. (2010b) Arachidonic acid-metabolizing cytochrome P450 enzymes are targets of {omega}−3 fatty acids. J Biol Chem 285, 32720–32733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold WR, Baylon JL, Tajkhorshid E, Das A (2016) Asymmetric binding and metabolism of polyunsaturated fatty acids (PUFAs) by CYP2J2 epoxygenase. Biochemistry 55, 6969–6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberà JA, Peinado VI, Santos S, Ramirez J, Roca J, Rodriguez-Roisin R (2001) Reduced expression of endothelial nitric oxide synthase in pulmonary arteries of smokers. Am J Respir Crit Care Med 164, 709–713. [DOI] [PubMed] [Google Scholar]
- Batchu SN, Chaudhary KR, El-Sikhry H, Yang W, Light PE, Oudit GY, et al. (2012a) Role of PI3Kα and sarcolemmal ATP-sensitive potassium channels in epoxyeicosatrienoic acid mediated cardioprotection. J Mol Cell Cardiol 53, 43–52. [DOI] [PubMed] [Google Scholar]
- Batchu SN, Law E, Brocks DR, Falck JR, Seubert JM (2009) Epoxyeicosatrienoic acid prevents postischemic electrocardiogram abnormalities in an isolated heart model. J Mol Cell Cardiol 46, 67–74. [DOI] [PubMed] [Google Scholar]
- Batchu SN, Lee SB, Qadhi RS, Chaudhary KR, El-Shikhry H, Kodela R, et al. (2011) Cardioprotective effect of a dual acting epoxyeicosatrienoic acid analogue towards ischaemia reperfusion injury. British Journal of Pharmacology 162, 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchu SN, Lee SB, Samokhvalov V, Chaudhary KR, El-Sikhry H, Weldon SM, et al. (2012b) Novel soluble epoxide hydrolase inhibitor protects mitochondrial function following stress. Can J Physiol Pharmacol 90, 811–823. [DOI] [PubMed] [Google Scholar]
- Bellien J, Iacob M, Gutierrez L, Isabelle M, Lahary A, Thuillez C, et al. (2006) Crucial role of NO and endothelium-derived hyperpolarizing factor in human sustained conduit artery flow-mediated dilatation. Hypertension 48, 1088–1094. [DOI] [PubMed] [Google Scholar]
- Bellien J, Iacob M, Remy-Jouet I, Lucas D, Monteil C, Gutierrez L, et al. (2012a) Epoxyeicosatrienoic acids contribute with altered nitric oxide and endothelin-1 pathways to conduit artery endothelial dysfunction in essential hypertension. Circulation 125, 1266–1275. [DOI] [PubMed] [Google Scholar]
- Bellien J & Joannides R (2013) Epoxyeicosatrienoic acid pathway in human health and diseases. J Cardiovasc Pharmacol 61, 188–196. [DOI] [PubMed] [Google Scholar]
- Bellien J, Remy-Jouet I, Iacob M, Blot E, Mercier A, Lucas D, et al. (2012b) Impaired role of epoxyeicosatrienoic acids in the regulation of basal conduit artery diameter during essential hypertension. Hypertension 60, 1415–1421. [DOI] [PubMed] [Google Scholar]
- Bettaieb A, Koike S, Chahed S, Zhao Y, Bachaalany S, Hashoush N, et al. (2017) Podocyte-specific soluble epoxide hydrolase deficiency in mice attenuates acute kidney injury. FEBS J 284, 1970–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blöcher R, Lamers C, Wittmann SK, Merk D, Hartmann M, Weizel L, et al. (2016) N-Benzylbenzamides: A novel merged scaffold for orally available dual soluble epoxide hydrolase/peroxisome proliferator-activated receptor γ modulators. J Med Chem 59, 61–81. [DOI] [PubMed] [Google Scholar]
- Bodiga S, Zhang R, Jacobs DE, Larsen BT, Tampo A, Manthati VL, et al. (2009) Protective actions of epoxyeicosatrienoic acid: dual targeting of cardiovascular PI3K and KATP channels. J Mol Cell Cardiol 46, 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukhari IA, Gauthier KM, Jagadeesh SG, Sangras B, Falck JR, Campbell WB (2011) 14,15-Dihydroxy-eicosa-5(Z)-enoic acid selectively inhibits 14,15-epoxyeicosatrienoic acid-induced relaxations in bovine coronary arteries. J Pharmacol Exp Ther 336, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukhari IA, Shah AJ, Gauthier KM, Walsh KA, Konduru SR, Imig JD, et al. (2012) 11,12,20-Trihydroxy-eicosa-8(Z)-enoic acid: a selective inhibitor of 11,12-EET induced relaxations of bovine coronary and rat mesenteric arteries. Am J Physiol Heart Circ Physiol 302, H1574–H1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligiuri SP, Aukema HM, Ravandi A, Guzman R, Dibrov E, Pierce GN (2014) Flaxseed consumption reduces blood pressure in patients with hypertension by altering circulating oxylipins via an α-linolenic acid-induced inhibition of soluble epoxide hydrolase. Hypertension 64, 53–59. [DOI] [PubMed] [Google Scholar]
- Campbell WB & Fleming I (2010) Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch 459, 881–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell WB, Gebremedhin D, Pratt PF, Harder DR (1996) Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res 78, 415–423. [DOI] [PubMed] [Google Scholar]
- Campbell WB, Imig JD, Schmitz JM, Falck JR (2017) Orally active epoxyeicosatrienoic acid analogs. J Cardiovasc Pharmacol 70, 211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila JH, Falck JR, Imig JD (2007) Roles of the cytochrome P450 arachidonic acid monooxygenases in the control of systemic blood pressure and experimental hypertension. Kidney Int 72, 683–689. [DOI] [PubMed] [Google Scholar]
- Capdevila JH, Pidkovka N, Mei S, Gong Y, Falck JR, Imig JD, et al. (2014) The Cyp2c44 epoxygenase regulates epithelial sodium channel activity and the blood pressure responses to increased dietary salt. J Biol Chem 289, 4377–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Tsenovoy PL, Thompson EA, Falck JR, Touchon R, Sodhi K, et al. (2015) Agonists of epoxyeicosatrienoic acids reduce infarct size and ameliorate cardiac dysfunction via activation of HO-1 and Wnt1 canonical pathway. Prostaglandins Other Lipid Mediat. 116-117, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra V, Thompson MD, Sala A, Cole DE, Folco G, Rovati GE (2007) Cysteinyl-leukotrienes and their receptors in asthma and other inflammatory diseases: critical update and emerging trends. Med Res Rev 27, 469–527. [DOI] [PubMed] [Google Scholar]
- Carroll MA, Garcia ΜP, Falck JR, McGiff JC (1992) Cyclooxygenase dependency of the renovascular actions of cytochrome P450-derived arachidonate metabolites. J Pharmacol Exp Ther 260, 104–109. [PubMed] [Google Scholar]
- Červenka L, Melenovský V, Husková Z, Sporková A, Bürgelová M, Škaroupková P, et al. (2015) Inhibition of soluble epoxide hydrolase does not improve the course of congestive heart failure and the development of renal dysfunction in rats with volume overload induced by aorto-caval fistula. Physiol Res 64, 857–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary KR, Batchu SN, Das D, Suresh MR, Falck JR, Graves JP, et al. (2009) Role of B-type natriuretic peptide in epoxyeicosatrienoic acid-mediated improved post-ischaemic recovery of heart contractile function. Cardiovasc Res 83, 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Whitcomb R, MacIntyre E, Tran V, Do ZN, Sabry J, et al. (2012) Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J Clin Pharmacol 52, 319–328. [DOI] [PubMed] [Google Scholar]
- Chen G, Xu R, Wang Y, Wang P, Zhao G, Xu X, et al. (2012) Genetic disruption of soluble epoxide hydrolase is protective against streptozotocin-induced diabetic nephropathy. Am J Physiol Endocrinol Metab 303, E563–E575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BB, Thompson DA, Howard LL, Morisseau C, Hammock BD, Weiss RH (2002) Inhibitors of soluble epoxide hydrolase attenuate vascular smooth muscle cell proliferation. Proc Natl Acad Sci USA 99, 2222–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Caterina R (2011)n-3 fatty acids in cardiovascular disease. N Engl J Med 364, 2439–2450. [DOI] [PubMed] [Google Scholar]
- Decker M, Adamska M, Cronin A, Di Giallonardo F, Burgener J, Marowsky A, et al. (2012) EH3 (ABHD9): the first member of a new epoxide hydrolase family with high activity for fatty acid epoxides. J Lipid Res 53, 2038–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Edin ML, Theken KN, Schuck RN, Flake GP, Kannon MA, et al. (2011) Endothelial CYP epoxygenase overexpression and soluble epoxide hydrolase disruption attenuate acute vascular inflammatory responses in mice. FASEB J 25, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveza L, Choi J, Yang F (2012). Therapeutic Angiogenesis for Treating Cardiovascular Diseases. Theranostics, 2, 801–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrance AM, Rupp N, Pollock DM, Newman JW, Hammock BD, Imig JD (2005) An epoxide hydrolase inhibitor, 12-(3-adamantan-l-yl-ureido)dodecanoic acid (AUDA), reduces ischemic cerebral infarct size in stroke-prone spontaneously hypertensive rats. J Cardiovasc Pharmacol 46, 842–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreisbach AW, Japa S, Sigel A, Parenti MB, Hess AE, Srinouanprachanh SL, et al. (2005) The Prevalence of CYP2C8, 2C9, 2J2, and soluble epoxide hydrolase polymorphisms in African Americans with hypertension. Am J Hypertens 18, 1276–1281. [DOI] [PubMed] [Google Scholar]
- Edin ML, Gholipour Hamedani B, Gruzdev A, Graves JP, Lih FB, Arbes SJ, et al. (2018) Epoxide hydrolase 1 (EPHX1) hydrolyzes epoxyeicosanoids and impairs cardiac recovery after ischemia. J Biol Chem 293, 3281–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldrup AB, Soleymanzadeh F, Taylor SJ, Muegge I, Farrow NA, Joseph D, et al. (2009) Structure-based optimization of arylamides as inhibitors of soluble epoxide hydrolase. J Med Chem 52, 5880–5895. [DOI] [PubMed] [Google Scholar]
- Elijovich F, Milne GL, Brown NJ, Laniado-Schwartzman M, Laffer CL (2018) Two pools of epoxyeicosatrienoic acids in humans: Alterations in salt-sensitive normotensive subjects. Hypertension 71, 346–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmarakby AA, Faulkner J, Al-Shabrawey M, Wang MH, Maddipati KR, Imig JD (2011) Deletion of soluble epoxide hydrolase gene improves renal endothelial function and reduces renal inflammation and injury in streptozotocin-induced type 1 diabetes. Am J Physiol Regul Integr Comp Physiol 301, R1307–R1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sikhry HE, Alsaleh N, Dakarapu R, Falck JR, Seubert JM (2016) Novel Roles of Epoxyeicosanoids in Regulating Cardiac Mitochondria. PLoS One 11, e0160380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck JR, Kodela R, Manne R, Atcha KR, Puli N, Dubasi N, et al. (2009) 14,15-Epoxyeicosa-5,8,ll-trienoic acid (14,15-EET) surrogates containing epoxide bioisosteres: influence upon vascular relaxation and soluble epoxide hydrolase inhibition. J Med Chem 52, 5069–5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck JR, Koduru SR, Mohapatra S, Manne R, Atcha R, Manthati VL, et al. (2014) 14,15-Epoxyeicosa-5,8,ll-trienoic acid (14,15-EET) surrogates: carboxylate modifications. J Med Chem 57, 6965–6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck JR, Reddy LM, Reddy YK, Bondlela M, Krishna UM, Ji Y, et al. (2003) 11,12-epoxyeicosatrienoic acid (11,12-EET): structural determinants for inhibition of TNF-alpha-induced VCAM-1 expression. Bioorg Med Chem Lett 13, 4011–4014. [DOI] [PubMed] [Google Scholar]
- Falck JR, Wallukat G, Puli N, Goli M, Arnold C, Konkel A, et al. (2011) 17(R),18(S)-epoxyeicosatetraenoic acid, a potent eicosapentaenoic acid (EPA) derived regulator of cardiomyocyte contraction: structure-activity relationships and stable analogues. J Med Chem 54, 4109–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer R, Dechend R, Qadri F, Markovic M, Feldt S, Herse F, et al. (2008) Dietary n-3 polyunsaturated fatty acids and direct renin inhibition improve electrical remodeling in a model of high human renin hypertension. Hypertension 51, 540–546. [DOI] [PubMed] [Google Scholar]
- Fischer R, Konkel A, Mehling H, Blossey K, Gapelyuk A, Wessel N, et al. (2014) Dietary omega-3 fatty acids modulate the eicosanoid profile in man primarily via the CYP-epoxygenase pathway. J Lipid Res 55, 1150–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer R, Konkel A, Wesser T, Westphal P, Schunck W-H, Westphal C, Falck JR (2017). Metabolically robust analogs of Cyp-eicosanoids for the treatment of cardiac disease. (WO 2017/013265).
- Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, et al. (1999) Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 401, 493–497. [DOI] [PubMed] [Google Scholar]
- Fleming I (2007a) Epoxyeicosatrienoic acids, cell signaling and angiogenesis. Prostaglandins Other Lipid Mediat 82, 60–67. [DOI] [PubMed] [Google Scholar]
- Fleming I (2001) Cytochrome p450 and vascular homeostasis. Circ Res 89, 753–762. [DOI] [PubMed] [Google Scholar]
- Fleming I (2007b) DiscrEET regulators of homeostasis: epoxyeicosatrienoic acids, cytochrome P450 epoxygenases and vascular inflammation. Trends Pharmacol Sci 28, 448–452. [DOI] [PubMed] [Google Scholar]
- Fornage M, Hinojos CA, Nurowska BW, Boerwinkle E, Hammock BD, Morisseau CH, et al. (2002) Polymorphism in soluble epoxide hydrolase and blood pressure in spontaneously hypertensive rats. Hypertension 40, 485–490. [DOI] [PubMed] [Google Scholar]
- Fritscher LG, Post M, Rodrigues MT, Silverman F, Balter M, Chapman KR, et al. (2012) Profile of eicosanoids in breath condensate in asthma and COPD. J Breath Res 6, 026001. [DOI] [PubMed] [Google Scholar]
- Gangadhariah MH, Dieckmann BW, Lantier L, Kang L, Wasserman DH, Chiusa M, et al. (2017) Cytochrome P450 epoxygenase-derived epoxyeicosatrienoic acids contribute to insulin sensitivity in mice and in humans. Diabetologia 60, 1066–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia V, Gilani A, Shkolnik B, Pandey V, Zhang FF, Dakarapu R, et al. (2017) 20-HETE signals through G-protein-coupled receptor GPR75 (G(q)) to affect vascular function and trigger hypertension. Circ Res 120, 1776–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier KM, Deeter C, Krishna UM, Reddy YK, Bondlela M, Falck JR, et al. (2002) 14,15-Epoxyeicosa-5(Z)-enoic acid: a selective epoxyeicosatrienoic acid antagonist that inhibits endothelium-dependent hyperpolarization and relaxation in coronary arteries. Circ Res 90, 1028–1036. [DOI] [PubMed] [Google Scholar]
- Gauthier KM, Falck JR, Reddy LM, Campbell WB (2004) 14,15-EET analogs: characterization of structural requirements for agonist and antagonist activity in bovine coronary arteries. Pharmacol Res 49, 515–524. [DOI] [PubMed] [Google Scholar]
- Gauthier KM, Jagadeesh SG, Falck JR, Campbell WB (2003) 14,15-epoxyeicosa-5(Z)-enoic-mSI: a 14,15- and 5,6-EET antagonist in bovine coronary arteries. Hypertension 42, 555–561. [DOI] [PubMed] [Google Scholar]
- GlaxoSmithKline (2013) A Study to Assess the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Single Doses of GSK2256294 in Healthy Volunteers, and Single and Repeat Doses of GSK2256294 in Adult Male Moderately Obese Smokers http://clinicaltrials.gov/ct2/show/NCT01762774?term=epoxide+hydrolase&rank=2
- Gross GJ, Gauthier KM, Moore J, Campbell WB, Falck JR, Nithipatikom K (2009) Evidence for role of epoxyeicosatrienoic acids in mediating ischemic preconditioning and postconditioning in dog. Am J Physiol Heart Circ Physiol 297, H47–H52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross GJ, Gauthier KM, Moore J, Falck JR, Hammock BD, Campbell WB, et al. (2008) Effects of the selective EET antagonist, 14,15-EEZE, on cardioprotection produced by exogenous or endogenous EETs in the canine heart. Am J Physiol Heart Circ Physiol 294, H2838–H2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross GJ, Hsu A, Falck JR, Nithipatikom K (2007) Mechanisms by which epoxyeicosatrienoic acids (EETs) elicit cardioprotection in rat hearts. J Mol Cell Cardiol 42, :687–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosser T, Fries S, FitzGerald GA (2006) Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest 116, 4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TR & Hammock BD (2013) Soluble epoxide hydrolase: gene structure, expression and deletion. Gene 526, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Fang YI, Ohata H, Honda K (2015) Change in soluble epoxide hydrolase (sEH) during cisplatin-induced acute renal failure in mice. J Toxicol Sci 40, 451–457. [DOI] [PubMed] [Google Scholar]
- Herse F, Lamarca B, Hubel CA, Kaartokallio T, Lokki AI, Ekholm E, et al. (2012) Cytochrome P450 subfamily 2J polypeptide 2 expression and circulating epoxyeicosatrienoic metabolites in preeclampsia. Circulation 126, 2990–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou HH, Hammock BD, Su KH, Morisseau C, Kou YR, Imaoka S, et al. (2012) N-terminal domain of soluble epoxide hydrolase negatively regulates the VEGF-mediated activation of endothelial nitric oxide synthase. Cardiovasc Res 93, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Dziumbla S, Lin J, Bibli SI, Zukunft S, de Mos J, et al. (2017) Inhibition of soluble epoxide hydrolase prevents diabetic retinopathy. Nature 552, 248–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Chang HH, Xu Y, Reddy DS, Du J, Zhou Y, et al. (2006) Epoxyeicosatrienoic acid inhibition alters renal hemodynamics during pregnancy. Exp Biol Med (Maywood) 231, 1744–1752. [DOI] [PubMed] [Google Scholar]
- Hutchens MP, Nakano T, Dunlap J, Traystman RJ, Hurn PD, Alkayed NJ (2008) Soluble epoxide hydrolase gene deletion reduces survival after cardiac arrest and cardiopulmonary resuscitation. Resuscitation 76, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SH, Wagner KM, Morisseau, Liu JY, Dong H, Wecksler AT, et al. (2011) Synthesis and structure-activity relationship studies of urea-containing pyrazoles as dual inhibitors of cyclooxygenase-2 and soluble epoxide hydrolase. J Med Chem 54, 3037–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hye Khan MA, Fish B, Wahl G, Sharma A, Falck JR, Paudyal MP, et al. (2016a) Epoxyeicosatrienoic acid analogue mitigates kidney injury in a rat model of radiation nephropathy. Clin Sci (Lond) 130, 587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hye Khan MA, Hwang SH, Sharma A, Corbett JA, Hammock BD, Imig JD (2016b) A dual COX-2/sEH inhibitor improves the metabolic profile and reduces kidney injury in Zucker diabetic fatty rat. Prostaglandins Other Lipid Mediat 125, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hye Khan MA, Pavlov TS, Christain SV, Neckář J, Staruschenko A, Gauthier KM, et al. (2014) Epoxyeicosatrienoic acid analogue lowers blood pressure through vasodilation and sodium channel inhibition. Clin Sci (Lond) 127, 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD (2000) Eicosanoid regulation of the renal vasculature. Am J Physiol Renal Physiol 279, F965–F981. [DOI] [PubMed] [Google Scholar]
- Imig JD (2015) Epoxyeicosatrienoic acids, hypertension, and kidney injury. Hypertension 65, 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD (2012) Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiological Reviews 92, 101–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Dimitropoulou C, Reddy DS, White RE, Falck JR (2008) Afferent arteriolar dilation to 11, 12-EET analogs involves PP2A activity and Ca2+-activated K+ Channels. Microcirculation 15, 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Elmarakby A, Nithipatikom K, Wei S, Capdevila JH, Tuniki VR, et al. (2010) Development of epoxyeicosatrienoic acid analogs with in vivo anti-hypertensive actions. Frontiers in Vascular Physiology 1, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD & Hammock BD (2009) Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov 8, 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Inscho EW, Deichmann PC, Reddy KM, Falck JR (1999) Afferent arteriolar vasodilation to the sulfonimide analog of 11, 12-epoxyeicosatrienoic acid involves protein kinase A. Hypertension 33, 408–413. [DOI] [PubMed] [Google Scholar]
- Imig JD, Zhao X, Capdevila JH, Morisseau C, Hammock BD (2002) Soluble epoxide hydrolase inhibition lowers arterial blood pressure in angiotensin II hypertension. Hypertension 39, 690–694. [DOI] [PubMed] [Google Scholar]
- Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, et al. (2005) An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension 46, 975–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, McGiff JC, Fava C, Amen G, Nesta E, Zanconato G, et al. (2013) Maternal and fetal epoxyeicosatrienoic acids in normotensive and preeclamptic pregnancies. Am J Hypertens 26, 271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jíchová Š, Kopkan L, Husková Z, Doleželová Š, Neckář J, Kujal P, et al. (2016) Epoxyeicosatrienoic acid analog attenuates the development of malignant hypertension, but does not reverse it once established: a study in Cyplal-Ren-2 transgenic rats. J Hypertens 34, 2008–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PD, Tsai HJ, Do ZN, Morisseau C, Hammock BD (2006) Synthesis and SAR of conformationally restricted inhibitors of soluble epoxide hydrolase. Bioorg Med Chem Lett 16, 5212–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouihan SA, Zuloaga KL, Zhang W, Shangraw RE, Krasnow SM, Marks DL, et al. (2013) Role of soluble epoxide hydrolase in exacerbation of stroke by streptozotocin-induced type 1 diabetes mellitus. J Cereb Blood Flow Metab 33, 1650–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, et al. (2005) Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension 45, 759–765. [DOI] [PubMed] [Google Scholar]
- Kang JX, Xiao YF, Leaf A (1995) Free, long-chain, polyunsaturated fatty acids reduce membrane electrical excitability in neonatal rat cardiac myocytes. Proc Natl Acad Sci U S A 92, 3997–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karara A, Makita K, Jacobson HR, Falck JR, Guengerich FP, DuBois RN, et al. (1993) Molecular cloning, expression, and enzymatic characterization of the rat kidney cytochrome P-450 arachidonic acid epoxygenase. J Biol Chem 268, 13565–13570. [PubMed] [Google Scholar]
- Katragadda D, Batchu SN, Cho WJ, Chaudhary KR, Falck JR, Seubert JM (2009) Epoxyeicosatrienoic acids limit damage to mitochondrial function following stress in cardiac cells. J Mol Cell Cardiol 46, 867–875. [DOI] [PubMed] [Google Scholar]
- Keserü B, Barbosa-Sicard E, Schermuly RT, Tanaka H, Hammock BD, Weissmann N, et al. (2010) Hypoxia-induced pulmonary hypertension: comparison of soluble epoxide hydrolase deletion vs. inhibition. Cardiovasc Res 85, 232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MAH, Liu J, Kumar G, Skapek SX, Falck JR, Imig JD (2013) Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. FASEB J 27, 2946–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MAH, Manthati V, Errabelli R, Pavlov TS, Staruschenko A, Falck JR, et al. (2013) An orally active epoxyeicosatrienoic acid analog attenuates kidney injury in hypertensive Dahl salt sensitive rat. Hypertension 62, 905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana R, Simons M, Martin JF, Zachary IC (2005) Role of angiogenesis in cardiovascular disease: a critical appraisal. Circulation 112, 1813–1824. [DOI] [PubMed] [Google Scholar]
- Kim HS, Kim SK, Kang KW (2017) Differential Effects of sEH Inhibitors on the Proliferation and Migration of Vascular Smooth Muscle Cells. Int J Mol Sci 18, pii: E2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IH, Heirtzler FR, Morisseau C, Nishi K, Tsai HJ, Hammock BD (2005) Optimization of amide-based inhibitors of soluble epoxide hydrolase with improved water solubility. J Med Chem 48, 3621–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Imig JD, Yang J, Hammock BD, Padanilam BJ (2014) Inhibition of soluble epoxide hydrolase prevents renal interstitial fibrosis and inflammation. Am J Physiol Renal Physiol 307, F971–F980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Yoon SP, Toews ML, Imig JD, Hwang SH, Hammock BD, et al. (2015) Pharmacological inhibition of soluble epoxide hydrolase prevents renal interstitial fibrogenesis in obstructive nephropathy. Am J Physiol Renal Physiol 308, F131–F139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King LM, Gainer JV, David GL, Dai D, Goldstein JA, Brown NJ, et al. (2005) Single nucleotide polymorphisms in the CYP2J2 and CYP2C8 genes and the risk of hypertension. Pharmacogenet Genomics 15, 7–13. [DOI] [PubMed] [Google Scholar]
- Kitamura S, Morisseau C, Harris TR, Inceoglu B, Hammock BD (2017) Occurrence of urea-based soluble epoxide hydrolase inhibitors from the plants in the order Brassicales. PLoS One 12, e0176571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinke A, Möller A, Pekarova M, Ravekes T, Friedrichs K, Berlin M, et al. (2014) Protective effects of 10-nitro-oleic acid in a hypoxia-induced murine model of pulmonary hypertension. Am J Respir Cell Mol Biol 51, 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerner IP, Zhang W, Cheng J, Parker S, Hurn PD, Alkayed NJ (2008) Soluble epoxide hydrolase: regulation by estrogen and role in the inflammatory response to cerebral ischemia. Front Biosci 13, 2833–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kompa AR, Wang BH, Xu G, Zhang Y, Ho PY, Eisennagel S, et al. (2013) Soluble epoxide hydrolase inhibition exerts beneficial anti-remodeling actions post-myocardial infarction. Int J Cardiol 167, 210–219. [DOI] [PubMed] [Google Scholar]
- Kramer J & Proschak E (2017) Phosphatase activity of soluble epoxide hydrolase. Prostaglandins Other Lipid Mediat 133, 88–92. [DOI] [PubMed] [Google Scholar]
- Kris-Etherton PM, Harris WS, Appel LJ (2002) American Heart Association. Nutrition Committee. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation 106, 2747–2757. [DOI] [PubMed] [Google Scholar]
- Kroetz DL & Xu F (2005) Regulation and inhibition of arachidonic acid omega-hydroxylases and 20-HETE formation. Annu Rev Pharmacol Toxicol 45, 413–438. [DOI] [PubMed] [Google Scholar]
- Kujal P, Čertíková Chábová V, Škaroupková P, Husková Z, Vernerová Z, Kramer HJ, et al. (2014) Inhibition of soluble epoxide hydrolase is renoprotective in 5/6 nephrectomized Ren-2 transgenic hypertensive rats. Clin Exp Pharmacol Physiol 41,227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavie CJ, Milani RV, Mehra MR, Ventura HO (2009) Omega-3 polyunsaturated fatty acids and cardiovascular diseases. J Am Coll Cardiol 54, 585–594. [DOI] [PubMed] [Google Scholar]
- Lazaar AL, Yang L, Boardley RL, Goyal NS, Robertson J, Baldwin SJ, et al. (2016) Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br J Clin Pharmacol 81, 971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CR, Pretorius M, Schuck RN, Burch LH, Bartlett J, Williams SM, et al. (2011) Genetic variation in soluble epoxide hydrolase (EPHX2) is associated with forearm vasodilator responses in humans. Hypertension 57, 116–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JP, Yang SH, Lee HY, Kim B, Cho JY, Paik JH, et al. (2012) Soluble epoxide hydrolase activity determines the severity of ischemia-reperfusion injury in kidney. PLoS One 7, e37075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Liu Y, Zhang X, Lv H, Pang W, Sun X, et al. (2016) Inhibition of soluble epoxide hydrolase alleviated atherosclerosis by reducing monocyte infiltration in Ldlr(−/−) mice. J Mol Cell Cardiol 98, 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HY, Jin Y, Morisseau C, Hammock BD, Long YQ (2006) The 5-substituted piperazine as a novel secondary pharmacophore greatly improving the physical properties of urea-based inhibitors of soluble epoxide hydrolase. Bioorg Med Chem 14, 6586–6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Li N, Pang W, Zhang X, Hammock BD, Ai D, et al. (2014) Opposite effects of gene deficiency and pharmacological inhibition of soluble epoxide hydrolase on cardiac fibrosis. PLoS One 9, e94092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Liu JY, Timofeyev V, Qiu H, Hwang SH, Tuteja D, et al. (2009) Beneficial effects of soluble epoxide hydrolase inhibitors in myocardial infarction model: Insight gained using metabolomic approaches. J Mol Cell Cardiol 47, 835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Jing Z, Deng H, Li Z, Zhuang Z, Wang S, et al. (2015) Soluble epoxide hydrolase inhibition ameliorates proteinuria-induced epithelial-mesenchymal transition by regulating the PI3K-Akt-GSK-3β signaling pathway. Biochem Biophys Res Commun 463, 70–75. [DOI] [PubMed] [Google Scholar]
- Liu S, Jia Z, Zhou L, Liu Y, Ling H, Zhou SF, et al. (2013) Nitro-oleic acid protects against adriamycin-induced nephropathy in mice. Am J Physiol Renal Physiol 305, F1533–F1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Webb HK, Fukushima H, Micheli J, Markova S, Olson JL, et al. (2012) Attenuation of cisplatin-induced renal injury by inhibition of soluble epoxide hydrolase involves nuclear factor kB signaling. J Pharmacol Exp Ther 341, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo HY, Man CC, Fleck RW, Farrow NA, Ingraham RH, Kukulka A, et al. (2010) Substituted pyrazoles as novel sEH antagonist: investigation of key binding interactions within the catalytic domain. Bioorg Med Chem Lett 20, 6379–6383. [DOI] [PubMed] [Google Scholar]
- Luria A, Bettaieb A, Xi Y, Shieh GJ, Liu HC, Inoue H, et al. (2011) Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in a model of insulin resistance. Proc Natl Acad Sci USA 108, 9038–9043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luria A, Morisseau C, Tsai HJ, Yang J, Inceoglu B, De Taeye B, et al. (2009) Alteration in plasma testosterone levels in male mice lacking soluble epoxide hydrolase. Am J Physiol Endocrinol Metab 297, E375–E383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manhiani M, Quigley JE, Knight SF, Tasoobshirazi S, Moore T, Brands MW, et al. (2009) Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am J Physiol Renal Physiol 297, F740–F448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makita K, Takahashi K, Karara A, Jacobson HR, Falck JR, Capdevila JH (1994) Experimental and/or genetically controlled alterations of the renal microsomal cytochrome P450 epoxygenase induce hypertension in rats fed a high salt diet. J Clin Invest 94, 2414–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino JP Jr. (2009) Soluble epoxide hydrolase, a target with multiple opportunities for cardiovascular drug discovery. Curr Top Med Chem 9, 452–463. [DOI] [PubMed] [Google Scholar]
- Martini R (2017) Subarachnoid hemorrhage and soluble epoxide hydrolase inhibition trial (SUSHI) https://clinicaltrials.gov/ct2/show/NCT03318783
- Matsumoto N, Suzuki E, Tsujihara K, Nishimura Y, Hasumi K (2015) Structure-activity relationships of the plasminogen modulator SMTP with respect to the inhibition of soluble epoxide hydrolase. J Antibiot (Tokyo) 68, 685–690. [DOI] [PubMed] [Google Scholar]
- McDougle DR, Watson JE, Abdeen AA, Adili R, Caputo MP, Krapf JE, et al. (2017) Anti-inflammatory ω−3 endocannabinoid epoxides. Proc Natl Acad Sci USA 114, E6034–E6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirer K, Glatzel D, Kretschmer S, Wittmann SK, Hartmann M, Blöcher R, et al. (2016) Design, synthesis and cellular characterization of a dual inhibitor of 5-lipoxygenase and soluble epoxide hydrolase. Molecules 22, pii: E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merabet N, Bellien J, Glevarec E, Nicol L, Lucas D, Remy-Jouet I, et al. (2012) Soluble epoxide hydrolase inhibition improves myocardial perfusion and function in experimental heart failure. J Mol Cell Cardiol 52, 660–666. [DOI] [PubMed] [Google Scholar]
- Merkel MJ, Liu L, Cao Z, Packwood W, Young J, Alkayed NJ, et al. (2010) Inhibition of soluble epoxide hydrolase preserves cardiomyocytes: role of STAT3 signaling. Am J Physiol Heart Circ Physiol 298, H679–H687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis UR, Fisslthaler B, Medhora M, Harder D, Fleming I, Busse R (2003) Cytochrome P450 2C9-derived epoxyeicosatrienoic acids induce angiogenesis via cross-talk with the epidermal growth factor receptor (EGFR). FASEB J 17, 770–772. [DOI] [PubMed] [Google Scholar]
- Miyata N, Taniguchi K, Seki T, Ishimoto T, Sato-Watanabe M, Yasuda Y, et al. (2001) HET0016, a potent and selective inhibitor of 20-HETE synthesizing enzyme. Br J Pharmacol 133, 325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti J, Fischer J, Paskas S, Heinig M, Schulz H, Gösele C, et al. (2008) Soluble epoxide hydrolase is a susceptibility factor for heart failure in a rat model of human disease. Nat Genet 40, 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C, Goodrow ΜH, Dowdy D, Zheng J, Greene JF, Sanborn JR, et al. (1999) Potent urea and carbamate inhibitors of soluble epoxide hydrolases. Proc Natl Acad Sci USA 96, 8849–8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C & Hammock BD (2005) Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol 45, 311–333. [DOI] [PubMed] [Google Scholar]
- Morisseau C & Hammock BD (2013) Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol 53, 37–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C, Newman JW, Tsai HJ, Baecker PA, Hammock BD (2006) Peptidyl-urea based inhibitors of soluble epoxide hydrolases. Bioorg Med Chem Lett 16, 5439–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisseau C, Sahdeo S, Cortopassi G, Hammock BD (2013) Development of an HTS assay for EPHX2 phosphatase activity and screening of nontargeted libraries. Anal Biochem 434, 105–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoki A, Merkel ΜJ, Packwood WH, Cao Z, Liu L, Iliff J, et al. (2008) Soluble epoxide hydrolase inhibition and gene deletion are protective against myocardial ischemia-reperfusion injury in vivo. Am J Physiol Heart Circ Physiol 295, H2128–H2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozaffarian D & Rimm EB (2006) Fish intake, contaminants, and human health: evaluating the risks and the benefits. JAMA 296, 1885–1899. [DOI] [PubMed] [Google Scholar]
- Nakagawa K, Holla VJ, Wei Y, Wang WH, Gatica A, Wei S, et al. (2006) Salt-sensitive hypertension is associated with dysfunctional Cyp4a10 gene and kidney epithelial sodium channel. J Clin Invest 116, 1696–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckar J, Khan MAH, Gross GJ, Falck JR, Campbell WB, Kolar F, et al. (2018) Epoxyeicosatrienoic acid analog EET-B attenuates post-myocardial infarction remodeling and renal injury in spontaneously hypertensive rats Scientific Reports (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckář J, Kopkan L, Husková Z, Kolář F, Papoušek F, Kramer HJ, et al. (2012) Inhibition of soluble epoxide hydrolase by cis-4-[4-(3-adamantan-l-ylureido)cyclohexyl-oxy]benzoic acid exhibits antihypertensive and cardioprotective actions in transgenic rats with angiotensin II-dependent hypertension. Clin Sci (Lond) 122, 513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JW, Morisseau C, Hammock BD. (2005) Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog Lipid Res 44, 1–51. [DOI] [PubMed] [Google Scholar]
- Newman JW, Morisseau C, Harris TR, Hammock BD (2003) The soluble epoxide hydrolase encoded by EPXH2 is a bifunctional enzyme with novel lipid phosphate phosphatase activity. Proc Natl Acad Sci USA 100, 1558–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nithipatikom K & Gross GJ (2010) Review article: epoxyeicosatrienoic acids: novel mediators of cardioprotection. J Cardiovasc Pharmacol Ther 15, 112–119. [DOI] [PubMed] [Google Scholar]
- Nithipatikom K, Moore JM, Isbell MA, Falck JR, Gross GJ (2006) Epoxyeicosatrienoic acids in cardioprotection: ischemic versus reperfusion injury. Am J Physiol Heart Circ Physiol 291, H537–H542. [DOI] [PubMed] [Google Scholar]
- Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, et al. (1999) Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 285, 1276–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olearczyk JJ, Field MB, Kim IH, Morisseau C, Hammock BD, Imig JD (2006) Substituted adamantyl-urea inhibitors of the soluble epoxide hydrolase dilate mesenteric resistance vessels. J Pharmacol Exp Ther 318, 1307–1314. [DOI] [PubMed] [Google Scholar]
- Olearczyk JJ, Quigley JE, Mitchell BC, Yamamoto T, Kim IH, Newman JW, et al. (2009) Administration of a substituted adamantyl urea inhibitor of soluble epoxide hydrolase protects the kidney from damage in hypertensive Goto-Kakizaki rats. Clin Sci (Lond) 116, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oni-Orisan A, Alsaleh N, Lee CR, Seubert JM (2014) Epoxyeicosatrienoic acids and cardioprotection: the road to translation. J Mol Cell Cardiol 74, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahy D, Edin ML, Lee CR, Huang S, Bielenberg DR, Butterfield CE, et al. (2012) Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J Clin Invest 122, 178–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish AR, Chen G, Burghardt RC, Watanabe T, Morisseau C, Hammock BD (2009) Attenuation of cisplatin nephrotoxicity by inhibition of soluble epoxide hydrolase. Cell Biol Toxicol 25, 217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister SL, Gauthier KM, Campbell WB (2010) Vascular pharmacology of epoxyeicosatrienoic acids. Adv Pharmacol 60, 27–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidkovka N, Rao R, Mei S, Gong Y, Harris RC, Wang WH, et al. (2013) Epoxyeicosatrienoic acids (EETs) regulate epithelial sodium channel activity by extracellular signal-regulated kinase 1/2 (ERK1/2)-mediated phosphorylation. J Biol Chem 288, 5223–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokreisz P, Fleming I, Kiss L, Barbosa-Sicard E, Fisslthaler B, Falck JR, et al. (2006) Cytochrome P450 epoxygenase gene function in hypoxic pulmonary vasoconstriction and pulmonary vascular remodeling. Hypertension 47, 762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polonikov AV, Ivanov VP, Solodilova MA, Khoroshaya IV, Kozhuhov MA, Ivakin VE, et al. (2008) A common polymorphism G-50T in cytochrome P450 2J2 gene is associated with increased risk of essential hypertension in a Russian population. Dis Markers 24, 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi A, Popescu V, Yang S, Mei S, Shi M, Puolitaival SM, et al. (2010) The anti-tumorigenic properties of peroxisomal proliferator-activated receptor alpha are arachidonic acid epoxygenase-mediated. J Biol Chem 285, 12840–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proschak E, Heitel P, Kalinowsky L, Merk D (2017) Opportunities and challenges for fatty acid mimetics in drug discovery. J Med Chem 60, 5235–5266. [DOI] [PubMed] [Google Scholar]
- Przybyla-Zawislak BD, Srivastava PK, Vazquez-Matias J, Mohrenweiser HW, Maxwell JE, Hammock BD, et al. (2003) Polymorphisms in human soluble epoxide hydrolase. Mol Pharmacol 64, 482–490. [DOI] [PubMed] [Google Scholar]
- Revermann M, Barbosa-Sicard E, Dony E, Schermuly RT, Morisseau C, Geisslinger G, et al. (2009) Inhibition of the soluble epoxide hydrolase attenuates monocrotaline-induced pulmonary hypertension in rats. J Hypertens 27, 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro JD, Toro AA, Baracat EC (2006) Antileukotrienes in the treatment of asthma and allergic rhinitis. J Pediatr (Rio J) 82(5 Suppl), S213–S221. [DOI] [PubMed] [Google Scholar]
- Roche C, Besnier M, Cassel R, Harouki N, Coquerel D, Guerrot D, et al. (2015a) Soluble epoxide hydrolase inhibition improves coronary endothelial function and prevents the development of cardiac alterations in obese insulin-resistant mice. Am J Physiol Heart Circ Physiol 308, H1020–H1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche C, Guerrot D, Harouki N, Duflot T, Besnier M, Rémy-Jouet I, et al. (2015b) Impact of soluble epoxide hydrolase inhibition on early kidney damage in hyperglycemic overweight mice. Prostaglandins Other Lipid Mediat 120, 148–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman RJ (2002) P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82, 131–185. [DOI] [PubMed] [Google Scholar]
- Schiering N, D’Arcy A, Villard F, Ramage P, Logel C, Cumin F, et al. (2016) Structure of neprilysin in complex with the active metabolite of sacubitril. Sci Rep 6, 27909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J, Rotter M, Weiser T, Wittmann S, Weizel L, Kaiser A, et al. (2017) A dual modulator of farnesoid X receptor and soluble epoxide hydrolase to counter nonalcoholic steatohepatitis. J Med Chem 60, 7703–7724. [DOI] [PubMed] [Google Scholar]
- Schunck WH, Konkel A, Fischer R, Weylandt KH (2018) Therapeutic potential of omega-3 fatty acid-derived epoxyeicosanoids in cardiovascular and inflammatory diseases. Pharmacol Ther 183, 177–204. [DOI] [PubMed] [Google Scholar]
- Schunck W-H, Wallukat G, Fischer R, Schmidt C, Müller DN, Puli N, Falck JR (2010). Novel eicosanoid derivatives. (WO 2010/081683).
- Seubert JM, Sinal CJ, Graves J, DeGraff LM, Bradbury JA, Lee CR, et al. (2006) Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ Res 99, 442–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seubert J, Yang B, Bradbury JA, Graves J, Degraff LM, Gabel S, et al. (2004) Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res 95, 506–514. [DOI] [PubMed] [Google Scholar]
- Seubert JM, Zeldin DC, Nithipatikom K, Gross GJ (2007) Role of epoxyeicosatrienoic acids in protecting the myocardium following ischemia/reperfusion injury. Prostaglandins Other Lipid Mediat 82, 50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaik JS, Ahmad M, Li W, Rose ΜE, Foley LM, Hitchens TK, et al. (2013) Soluble epoxide hydrolase inhibitor trans-4-[4-(3-adamantan-l-yl-ureido)-cyclohexyloxy]-benzoic acid is neuroprotective in rat model of ischemic stroke. Am J Physiol Heart Circ Physiol 305, H1605–H1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Hye Khan MA, Levick SP, Lee KS, Hammock BD, Imig JD (2016) Novel omega-3 fatty acid epoxygenase metabolite reduces kidney fibrosis. Int J Mol Sci 17, pii: E751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, McCarthy ET, Reddy DS, Patel PK, Savin VJ, Medhora M, et al. (2009) 8,9-Epoxyeicosatrienoic acid protects the glomerular filtration barrier. Prostaglandins Other Lipid Mediat 89, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HC, Ding FX, Deng Q, Xu S, Tong X, Zhang X, et al. (2009a) A strategy of employing aminoheterocycles as amide mimics to identify novel, potent and bioavailable soluble epoxide hydrolase inhibitors. Bioorg Med Chem Lett 19, 5716–5721. [DOI] [PubMed] [Google Scholar]
- Shen HC, Ding FX, Wang S, Xu S, Chen HS, Tong X, et al. (2009b) Discovery of spirocyclic secondary amine-derived tertiary ureas as highly potent, selective and bioavailable soluble epoxide hydrolase inhibitors. Bioorg Med Chem Lett 19, 3398–3404. [DOI] [PubMed] [Google Scholar]
- Shen HC & Hammock BD (2012) Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J Med Chem 55, 1789–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Peng H, Peng R, Fan Q, Zhao S, Xu D, et al. (2015) Inhibition of soluble epoxide hydrolase in mice promotes reverse cholesterol transport and regression of atherosclerosis. Atherosclerosis 239, 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha A, Krishnamurthy PT, Thomas P, Hammock BD, Hwang SH (2014) Soluble epoxide hydrolase inhibitor, t-TUCB, protects against myocardial ischaemic injury in rats. J Pharm Pharmacol 66, 1251–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuey MM, Billings FT 4th, Wei S, Milne GL, Nian H, Yu C, et al. (2017) Association of gain-of-function EPHX2 polymorphism Lys55Arg with acute kidney injury following cardiac surgery. PLoS One 12, e0175292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M & Ware JA (2003) Therapeutic angiogenesis in cardiovascular disease. Nat Rev Drug Discov 2, 863–871. [DOI] [PubMed] [Google Scholar]
- Simpkins AN, Rudic RD, Roy S, Tsai HJ, Hammock BD, Imig JD (2010) Soluble epoxide hydrolase inhibition modulates vascular remodeling. Am J Physiol Heart Circ Physiol 298, H795–H806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins AN, Rudic RD, Schreihofer DA, Roy S, Manhiani M, Tsai HJ, et al. (2009) Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. Am J Pathol 174, 2086–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirish P, Li N, Liu JY, Lee KS, Hwang SH, Qiu H, et al. (2013) Unique mechanistic insights into the beneficial effects of soluble epoxide hydrolase inhibitors in the prevention of cardiac fibrosis. Proc Natl Acad Sci USA 110, 5618–5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirish P, Li N, Timofeyev V, Zhang XD, Wang L, Yang J, et al. (2016) Molecular mechanisms and new treatment paradigm for atrial fibrillation. Circ Arrhythm Electrophysiol 9, pii: e003721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibba M, Hye Khan MA, Kolb LL, Yeboah MM, Falck JR, Amaradhi R, et al. (2017) Epoxyeicosatrienoic acid analog decreases renal fibrosis by reducing epithelial-to-mesenchymal transition. Front Pharmacol 8, 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodhi K, Inoue K, Gotlinger KH, Canestraro M, Vanella L, Kim DH, et al. (2009) Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J Pharmacol Exp Ther 331, 906–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector AA, Fang X, Snyder GD, Weintraub NL (2004) Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res 43, 55–90. [DOI] [PubMed] [Google Scholar]
- Spiecker M, Darius H, Hankeln T, Soufi M, Sattler AM, Schaeferj JR, et al. (2004) Risk of coronary artery disease associated with polymorphism of the cytochrome P450 epoxygenase CYP2J2. Circulation 110, 2132–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhahar V, Shaw S, Imig JD (2010) Epoxyeicosatrienoic acid analogs and vascular function. Curr Med Chem 17, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun P, Lin DH, Yue P, Jiang H, Gotlinger KH, Schwartzman ML, et al. (2010) High potassium intake enhances the inhibitory effect of 11,12-EET on ENaC. J Am Soc Nephrol 21, 1667–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinney DC & Anthony J (2011) How were new medicines discovered? Nat Rev Drug Discov 10, 507–519. [DOI] [PubMed] [Google Scholar]
- Tacconelli S & Patrignani P (2014) Inside epoxyeicosatrienoic acids and cardiovascular disease. Front Pharmacol 5, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi N, Nakayama S, Tanaka M (2016) Single administration of soluble epoxide hydrolase inhibitor suppresses neuroinflammation and improves neuronal damage after cardiac arrest in mice. Neurosci Res 111, 56–63. [DOI] [PubMed] [Google Scholar]
- Tanaka T & Nangaku M (2013) Angiogenesis and hypoxia in the kidney. Nat Rev Nephrol 9, 211–222. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Tanaka T, Nangaku M (2015) Hypoxia and dysregulated angiogenesis in kidney disease. Kidney Dis (Basel) 1, 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temml V, Garscha U, Romp E, Schubert G, Gerstmeier J, Kutil Z, et al. (2017) Discovery of the first dual inhibitor of the 5-lipoxygenase-activating protein and soluble epoxide hydrolase using pharmacophore-based virtual screening. Sci Rep 7, 42751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thao NP, Luyen BT, Lee JS, Kim JH, Kim YH (2017) Soluble epoxide hydrolase inhibitors of indolinone alkaloids and phenolic derivatives from Cimicifuga dahurica (Turcz.) Maxim. Bioorg Med Chem Lett 27, 1874–1879. [DOI] [PubMed] [Google Scholar]
- Tsai HJ, Hwang SH, Morisseau C, Yang J, Jones PD, Kasagami T, et al. (2010) Pharmacokinetic screening of soluble epoxide hydrolase inhibitors in dogs. Eur J Pharm Sci 40, 222–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulu A, Davis BB, Tsai HJ, Kim IH, Morisseau C, Inceoglu B, et al. (2008) Soluble epoxide hydrolase inhibitors reduce the development of atherosclerosis in apolipoprotein e-knockout mouse model. J Cardiovasc Pharmacol 52, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulu A, Harris TR, Morisseau C, Miyabe C, Inoue H, Schuster G, et al. (2013) Anti-inflammatory effects of omega-3 polyunsaturated fatty acids and soluble epoxide hydrolase inhibitors in angiotensin II dependent hypertension. J Cardiovasc Pharmacol 62, 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulu A, Stephen Lee KS, Miyabe C, Yang J, Hammock BG, Dong H, et al. (2014) An omega-3 epoxide of docosahexaenoic acid lowers blood pressure in angiotensin-II-dependent hypertension. J Cardiovasc Pharmacol 64, 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Václavíková R, Hughes DJ, Souček P (2015) Microsomal epoxide hydrolase 1 (EPHX1): Gene, structure, function, and role in human disease. Gene 571, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedove FD, Fava C, Jiang H, Zanconato G, Quilley J, Brunelli M, et al. (2015) 8C.05: Epoxyeicosatrienoic acids are increased in placentas of preeclamptic pregnancies. J Hypertens 33 Suppl 1, e111. [Google Scholar]
- von Lueder TG & Krum H (2015) New medical therapies for heart failure. Nat Rev Cardiol 12, 730–740. [DOI] [PubMed] [Google Scholar]
- Wang CP, Hung WC, Yu TH, Chiu CA, Lu LF, Chung FM, et al. (2010) Genetic variation in the G-50T polymorphism of the cytochrome P450 epoxygenase CYP2J2 gene and the risk of younger onset type 2 diabetes among Chinese population: potential interaction with body mass index and family history. Exp Clin Endocrinol Diabetes 118, 346–352. [DOI] [PubMed] [Google Scholar]
- Wang MH, Brand-Schieber E, Zand BA, Nguyen X, Falck JR, Balu N, et al. (1998) Cytochrome P450-derived arachidonic acid metabolism in the rat kidney: characterization of selective inhibitors. J Pharmacol Exp Ther 284, 966–973. [PubMed] [Google Scholar]
- Wang W, Li C, Yang T (2016) Protection of nitro-fatty acid against kidney diseases. Am J Physiol Renal Physiol 310, F697–F704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward NC, Croft KD, Blacker D, Hankey GJ, Barden A, Mori TA, et al. (2011) Cytochrome P450 metabolites of arachidonic acid are elevated in stroke patients compared with healthy controls. Clin Sci (Lond) 121, 501–507. [DOI] [PubMed] [Google Scholar]
- Wei Q, Doris PA, Pollizotto MV, Boerwinkle E, Jacobs DR Jr, Siscovick DS, et al. (2007) Sequence variation in the soluble epoxide hydrolase gene and subclinical coronary atherosclerosis: interaction with cigarette smoking. Atherosclerosis 190, 26–34. [DOI] [PubMed] [Google Scholar]
- Wei X, Zhang D, Dou X, Niu N, Huang W, Bai J, et al. (2014) Elevated 14,15-epoxyeicosatrienoic acid by increasing of cytochrome P450 2C8, 2C9 and 2J2 and decreasing of soluble epoxide hydrolase associated with aggressiveness of human breast cancer. BMC Cancer 14, 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Lin DH, Kemp R, Yaddanapudi GS, Nasjletti A, Falck JR, et al. (2004) Arachidonic acid inhibits epithelial Na channel via cytochrome P450 (CYP) epoxygenase-dependent metabolic pathways. J Gen Physiol 124, 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal C, Konkel A, Schunck WH (2011) CYP-eicosanoids--a new link between omega-3 fatty acids and cardiac disease? Prostaglandins Other Lipid Mediat 96, 99–108. [DOI] [PubMed] [Google Scholar]
- Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, et al. (2006) Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci USA 103, 18733–18738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Olsson T, Johansson CA, Öster L, Beisel HG, Rohman M, et al. (2016) Fragment screening of soluble epoxide hydrolase for lead generation-structure-based hit evaluation and chemistry exploration. Chem Med Chem 11, 497–508. [DOI] [PubMed] [Google Scholar]
- Yang L, Cheriyan J, Gutterman DD, Mayer RJ, Ament Z, Griffin JL, et al. (2017) Mechanisms of vascular dysfunction in COPD and effects of a novel soluble epoxide hydrolase inhibitor in smokers. Chest 151, 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Underwood MJ, Hsin MK, Liu XC, He GW (2008) Dysfunction of pulmonary vascular endothelium in chronic obstructive pulmonary disease: basic considerations for future drug development. Curr Drug Metab 9, 661–667. [DOI] [PubMed] [Google Scholar]
- Yang Q, Zhang RZ, Yim AP,, He GW (2003) Effect of 11,12-epoxyeicosatrienoic acid as an additive to St. Thomas’ cardioplegia and University of Wisconsin solutions on endothelium-derived hyperpolarizing factor-mediated function in coronary microarteries: influence of temperature and time. Ann Thorac Surg 76, 1623–1630. [DOI] [PubMed] [Google Scholar]
- Yang S, Wei S, Pozzi A, Capdevila JH (2009) The arachidonic acid epoxygenase is a component of the signaling mechanisms responsible for VEGF-stimulated angiogenesis. Arch Biochem Biophys 489, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeboah MM, Hye Khan MA, Chesnik MA, Sharma A, Paudyal MP, Falck JR, et al. (2016) The epoxyeicosatrienoic acid analog PVPA ameliorates cyclosporine-induced hypertension and renal injury in rats. Am J Physiol Renal Physiol 311, F576–F585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu BN, Luo CH, Wang D, Wang A, Li Z, Zhang W, et al. (2004) CYP2C9 allele variants in Chinese hypertension patients and healthy controls. Clin Chim Acta 348, 57–61. [DOI] [PubMed] [Google Scholar]
- Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, et al. (2000) Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res 87, 992–998. [DOI] [PubMed] [Google Scholar]
- Zeldin DC (2011) Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem 276, 36059–36062. [DOI] [PubMed] [Google Scholar]
- Zhao X, Pollock DM, Inscho EW, Zeldin DC, Imig JD (2003) Decreased renal cytochrome P450 2C enzymes and impaired vasodilation are associated with salt-sensitive hypertension. Hypertension 41, 709–714. [DOI] [PubMed] [Google Scholar]
- Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, et al. (2004) Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol 15, 1244–1253. [PubMed] [Google Scholar]
- Zhang G, Panigrahy D, Hwang SH, Yang J, Mahakian LM, Wettersten HI, et al. (2014) Dual inhibition of cyclooxygenase-2 and soluble epoxide hydrolase synergistically suppresses primary tumor growth and metastasis. Proc Natl Acad Sci USA 111, 11127–11132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liu YS, Lu QH (2015) Therapeutic effects of the soluble epoxide hydrolase (sEH) inhibitor AUDA on atherosclerotic diseases. Pharmazie 70, 24–28. [PubMed] [Google Scholar]
- Zhang LN, Vincelette J, Cheng Y, Mehra U, Chen D, Anandan SK, et al. (2009) Inhibition of soluble epoxide hydrolase attenuated atherosclerosis, abdominal aortic aneurysm formation, and dyslipidemia. Arterioscler Thromb Vasc Biol 29, 1265–1270. [DOI] [PubMed] [Google Scholar]
- Zhang W, Li H, Dong H, Liao J, Hammock BD, Yang GY (2013a) Soluble epoxide hydrolase deficiency inhibits dextran sulfate sodium-induced colitis and carcinogenesis in mice. Anticancer Res 33, 5261–5271. [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Liao J, Li H, Dong H, Bai H, Yang A, et al. (2013b) Reduction of inflammatory bowel disease-induced tumor development in IL-10 knockout mice with soluble epoxide hydrolase gene deficiency. Mol Carcinog 52, 726–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Otsuka T, Sugo N, Ardeshiri A, Alhadid YK, Iliff JJ, et al. (2008) Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke 39, 2073–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Chang HH, Du J, Wang CY, Dong Z, Wang MH (2005) Renal epoxyeicosatrienoic acid synthesis during pregnancy. Am J Physiol Renal Physiol 288, F221–F226. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Blum M, Hoff U, Wesser T, Fechner M, Westphal C, et al. (2016) Renal ischemia/reperfusion injury in soluble epoxide hydrolase-deficient mice. PLoS One 11, e0145645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuloaga KL, Krasnow SM, Zhu X, Zhang W, Jouihan SA, Shangraw RE, et al. (2014) Mechanism of protection by soluble epoxide hydrolase inhibition in type 2 diabetic stroke. PLoS One 9, e97529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zusterzeel PL, Peters WH, Visser W, Hermsen KJ, Roelofs HM, Steegers EA. (2001) A polymorphism in the gene for microsomal epoxide hydrolase is associated with pre-eclampsia. J Med Genet 38, 234–237. [DOI] [PMC free article] [PubMed] [Google Scholar]