

Abstract
Although a number of studies have recently explored the contribution of the adaptive immunity in interleukin 33 (IL-33)-mediated antitumor effects, innate immune involvement has been poorly characterized. Utilizing Rag1−/− mice (lacking T and B lymphocytes), we show here that either systemic administration of recombinant IL-33 or ectopic expression of IL-33 in melanoma cells is sufficient to inhibit tumor growth independent of adaptive antitumor immunity. We have demonstrated that IL-33-mediated antitumor effects depend on expansion and activation of NK cells. Interestingly, IL-33 also promoted the expansion of active type 2 innate lymphoid cells (ILC2s) via its receptor, ST2, which in turn inhibited NK activation and cytotoxicity. This IL-33-induced ILC2 activity coincided with greater expression of the immunosuppressive ecto-enzyme CD73. Removal of CD73 from ILC2s in culture with NK cells resulted in markedly increased activation levels in NK cells, offering a potential mechanism by which ILC2s might suppress NK cell-mediated tumor killing. Thus, our data reveal an important contribution of IL-33-induced ILC2 to tumor growth by weakening NK cell activation and tumor killing, regardless of adaptive immunity.
Keywords: IL-33, ST2, NK, ILC2, CD73, tumor inhibition
Introduction
Interleukin 33 (IL-33) is a fairly new member of the IL-1 family of cytokines (1). IL-33 has been characterized as being secreted by fibroblasts, epithelial cells, and endothelial cells as an alarmin in response to cell injury (2, 3). Signaling through its receptor, ST2, IL-33 has been shown to act as an early inducer of inflammation, stimulating a variety of immune cells including T helper cells, mast cells, and eosinophils to secrete type 2 cytokines (1, 4).
Though type 2 immune responses have not typically been associated with favorable antitumor effects, recent studies have also indicated IL-33 can stimulate CD8+ T cells and NK cells and thereby inhibit tumor growth and/or metastasis (5–7). For decades, accumulating data have characterized natural killer (NK) cells as powerful innate immune effectors of antitumor responses (8, 9). Complex balancing of stimulatory and inhibitory signaling can drive NK cells to lyse a wide repertoire of target cells (10–12). Through the release of cytotoxic granules and the production of IFN-γ, NK cells possess a number of powerful tools for directly eliminating target cells, while simultaneously stimulating other immune cells (13–16). The exact contribution of NK cells in IL-33 mediated antitumor effects, however, has not been closely examined in the absence of adaptive immunity.
Type 2 innate lymphoid cells (ILC2) are a newly identified population of immune cells expressing ST2 that expand in response to IL-33 (17–19). ILC2s are cytokine-secreting cells of lymphoid morphology like CD4+ and CD8+ T cells; however, they lack rearranged antigen-specific receptors (TCRs) and can be identified as lineage-negative CD25+CD45+CD90+CD127+ST2+ (20). ILC2s were originally observed to be involved in stimulating type 2 immune responses against allergies and helminthes, secreting IL-4, 5, 9, and 13 (18, 20). Recently, ILC2s, that express ST2, are stimulated and expanded upon exposure to IL-33, increasing IL5 and IL13 secretion (19, 21, 22). It is largely unknown whether ILC2s play a role in IL-33-mediated antitumor effects.
In this study, we report that IL-33 is capable of eliciting adequate antitumor responses strictly though the innate immune system, largely dependent on expansion and activation of NK cells. Further, we observed that IL-33-induced ILC2s facilitated tumor growth by suppressing NK cell function, providing the first evidence for the contribution of ILC2s to tumor progression.
Methods
Mice and cancer cell lines
C57BL/6J WT and Rag1−/− mice were purchased from the Jackson Laboratory and bred in-house. All mice used in experiments were between 2–6 months old. Mice were used according to the protocols approved by the Institutional Animal Use Committee at Northwestern University. B16F10 melanoma and EL4 thymoma cells were obtained from Dr. Hans Schreiber (University of Chicago). B16 cell clones designed to express IL-33 (B16-Vec and B16-IL33) were generated and kindly provided by Dr. Binfeng Lu (University of Pittsburgh) (6). A tumor cell line was derived from a spontaneous tumor in Jackson lab’s BrafCA, Tyr::CreER and Ptenlox4−5 mouse model, which we have named BPS1 (23). All cell lines were maintained in complete medium composed of RPMI 1640 containing 5% FBS.
Tumor challenge and treatments
B16F10, EL4, B16-Vec or B16-IL33 cells in suspension were injected s.c. (1×106). Treatment with recombinant IL-33 (1μg/mouse; Biolegend) began either 7 days (WT) or 3 days (Rag1−/−) after tumor challenge. In experiments requiring depletion of ILC2s or NK cells, depleting antibodies were first injected i.p. 3 days prior to tumor challenge. Anti-CD90 polyclonal antibodies (T24/31) were purchased from BioXCell and injected at 200 μg per injection. Hybridoma-derived anti-NK1.1 antibodies (PK136) were collected from ascites and routinely tested for NK cell depletion. Groups that did not receive depletion antibodies were administered IgG antibodies as a control. Both antibody depletion and IL-33 treatment were given every other day and continued until completion.
Sample preparation and cell selection
Tumor samples and spleens were harvested and processed into single cell suspension and prepared for flow cytometry as published previously (24). Tumor samples were digested in collagenase D and DNase I. Samples were kept in RPMI 1640 with 10% FBS during processing. Splenic NK cells were isolated using a 2-step labeling process (biotinylated NK1.1 followed by anti-biotin microbeads, BD Biosciences and Stemcell Technologies). ILC2s were purified from spleens of IL-33-treated Rag1−/− mice for in vitro experiments using positive magnetic selection for CD90 microbeads from BD Biosciences and Stemcell Technologies. ILC2 selections first involved selection against NK1.1+ cells prior to CD90 selection to increase purity.
Flow cytometry
The flow antibodies were purchased from Biolegend and eBioscience. Surface staining, annexin V staining, nuclear GATA3 and Ki67 staining, and intracellular cytokine staining were performed as published previously (24). Samples were run on either a MACSQuant Analyzer (Miltenyi Biotec), an LSR II (BD Biosciences), or a FACSCanto II (BD Biosciences). Analysis was done using FlowJo software (Tree Star).
NK cell-mediated cytolytic activity
Splenic NK1.1+ NK cells purified from Rag1−/− or WT mice were incubated with B16F10 cells at a ratio of 20:1 in the presence of recombinant IL-33 (50 ng/ml) for 24h. B16F10 cells with or without IL-33 treatment alone were used as controls. Tumor cell death was measured with annexin V and 7-AAD staining by flow cytometry. To examine the effect of ILC2 on NK cell-mediated tumor cell killing, splenic ILC2 cells were enriched from tumor-bearing Rag1−/− mice as described above. Alternatively, enriched CD90+ cells from naïve Rag1−/− mice were activated and expanded with recombinant IL-33 (1 μg/ml) and IL-2 for 48h in vitro. These ILC2 cells were added to the cocultures of NK and B16F10 cells as descried above at a ratio of 1:20:20 (B16F10:NK:ILC2).
ILC2-mediated suppression assay
Splenic NK1.1+ cells were purified from WT−/− mice and stimulated with IL-15 (10ng/ml and IL-33 (20 ng/ml) for 24 hours. NK cells were then co-cultured with ILC2s prepared using methods described above. Cells were cultured at a 1:1 ratio in the presence of 20 ng/ml IL-33 and 100 μM AMP for 24 hours. NK cell activity was assessed by production of CD107a.
ILC2 generation from bone marrow
Tibias and femurs were removed from WT and CD73−/− C57BL/6 mice using sterile techniques and bone marrow cells (BM) were flushed. Lineage negative CD90+ cells were then purified. ILC2s were generated from purified BM cells using methods similar to those previously described (25). Purified BM cells were cultured in Flt3L (20ng/ml), stem cell factor (20ng/ml), IL-7 (10ng/ml), IL-33 (20ng/ml), and IL-2 (10ng/ml) for 7 days before they were assessed for cytokine production. Generated ILC2s shared similar features with ILC2s isolated from spleens (data not shown).
ELISA
IL-33 was detected by ELISA performed using eBioscience’s kit according to the manufacturer’s protocol. Serum was isolated from whole blood of tumor-bearing mice. Tumor lysates were similarly analyzed following homogenization in RIPA buffer. Fluorescence was measured using a GloMax-Multi Detection System by Promega and IL-33 was quantified using a standard curve derived from the manufacture’s IL-33 standard.
CD73 enzymatic activity assay
AMP consumption was measured using AMP-Glo Assay (Promega) using manufacturer’s protocol. Relative AMP levels were determined by luminescence measured by a GloMax-Multi Detection System by Promega.
Statistical analysis
Mean values were compared using an unpaired two-tailed Student’s t test. P values >0.05 were not considered significant.
Results
IL-33 inhibits tumor growth in Rag1−/− mice.
Consistent with other studies (5–7), our previously reported data suggest an important role of the adaptive immune system in eliciting IL-33-mediated antitumor responses (26). To explore whether IL-33 can inhibit tumor growth independent of an adaptive immune system, B16F10 melanoma cells were s.c. injected into WT mice versus Rag1−/− mice that are deficient in T and B cells followed by systemic administration of recombinant IL-33. As expected, IL-33 treatment significantly delayed the growth of the tumors in WT mice (Figure 1A). Surprisingly, tumor growth was also greatly impaired with IL-33 treatment in Rag1−/− mice (Figure 1B), indicating systemic administration of IL-33 can inhibit melanoma growth solely through the innate immune system. Similar results were observed in Rag1−/− mice challenged with EL4 lymphoma cells (Figure 1C), suggesting that such IL-33-mediated antitumor effect is not restricted to the specific types of tumors. To further verify our findings, we then utilized B16F10 cells engineered to express IL-33 (B16-IL33) (6). B16-IL33 tumors grew significantly slower than control tumors (B16-Vec) in Rag1−/− mice (Figure 1D), indicating local expression of IL-33 is sufficient to inhibit tumor growth independent of an adaptive immune system. Analysis of tumor lysates confirmed significantly high levels of IL-33 in B16-IL33 tumors while IL-33 was undetectable in B16-Vec tumors (Supplementary Figure 1A). Serum levels of IL-33 were detectable in two out of four B16-IL33 tumor-bearing mice while no IL-33 was observable in B16-Vec-bearing mice (Supplementary Figure 1B). We found that IL-33 failed to affect viability of B16F10 melanoma cells cultured in vitro using MTT assay (data not shown), indicating IL-33 does not illicit a direct cytotoxic effect on melanoma cells but presumably recruits other innate immune cells to attenuate tumor growth.
Figure 1. IL-33 reduces tumor growth in Rag1−/− mice.

(A) WT C57BL/6J mice were given s.c. injections of B16F10 tumor cells. After seven days, 1μg recombinant IL-33 was administered every other day and tumor growth was measured every two days (n=5 mice per group). (B) Rag1−/− mice began recombinant IL-33 treatment as described above 3 days after s.c. injection of B16F10 cells (n=5 mice per group) or (C) EL4 cells (n=4 mice per group). (D) Rag1−/− mice (n=5 mice per group) were s.c. injected with either control B16 cells (B16-Vec) or IL-33-expressing B16 cells (B16-IL33). Data are shown as mean ± SEM. ** P< 0.01, *** P<0.001 as determined using a Student’s t-test.
NK cells are implicated in IL-33-mediated tumor suppression.
NK cells have long been implicated in antitumor immune responses and have recently been shown to be stimulated in response to IL-33 exposure in tumor-bearing conditions (5, 6). Indeed, we observed an increase in the number and activation state of intratumoral NK cells in B16-IL33 tumor-bearing Rag1−/− mice (Figure 2A, B). The B16-IL33 tumor-bearing mice also exhibited elevated numbers of splenic NK cells (Figure 2C). To further examine the importance for NK cells in IL-33-mediated antitumor effect, NK cells were depleted by i.p. administration of anti-NK1.1 antibodies prior to B16F10 tumor challenge. Tumors in NK cell-depleted mice grew faster than in those receiving IL-33 alone, though not as fast as control mice receiving only PBS (Figure 2D), indicating NK cells are required for complete IL-33-mediated tumor suppression. In vitro, NK cells treated by IL-33 displayed a greater capacity to kill B16F10 cells than IL-33-untreated NK cells (Figure 2E). As expected, IL-33 alone did not affect the survival status of cultured B16F10 cells. These data suggest that expansion of activated NK cells contributes to IL-33-mediated antitumor effect in the absence of adaptive immunity.
Figure 2. NK cells contribute to IL-33-mediated antitumor responses.
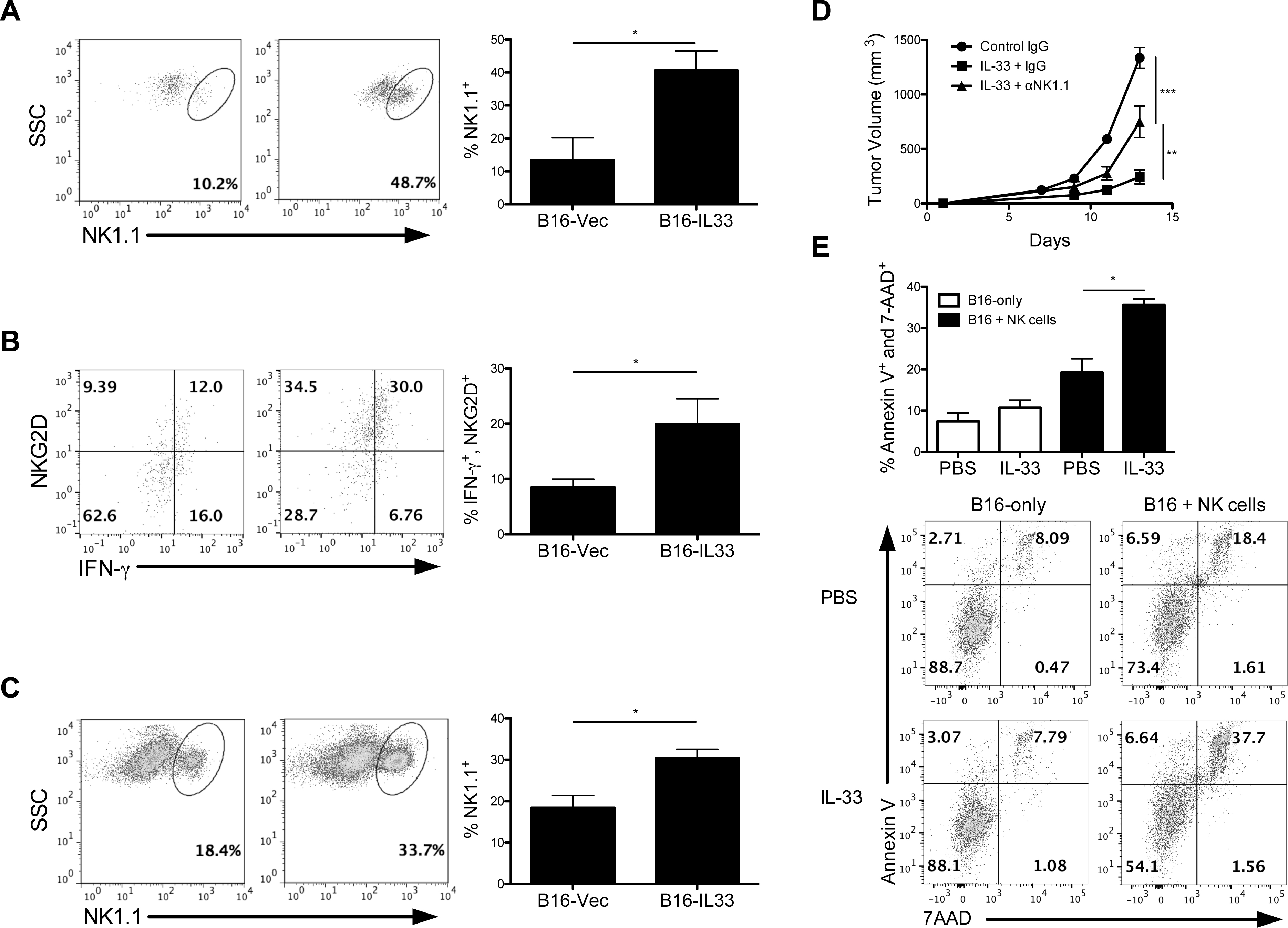
(A) Representative flow cytometry dot plots showed the frequencies of intratumoral NK1.1+ NK cells in Rag1−/− mice bearing B16-Vec versus B16-IL-33 tumors (B) and expression levels of NKG2D and IFN-γ that were summarized in the bar graph (n=5 mice per group). (C) Representative flow cytometry analysis displaying frequencies of splenic NK cells in B16-Vec and B16-IL33 tumor-bearing Rag1−/− mice with corresponding bar graphs (n=5 mice per group). (D) Rag1−/− mice were injected with either IgG or anti-NK1.1 depletion antibodies 3 days prior to B16F10 tumor challenge. 1μg IL-33 was administered every other day starting 3 days after tumor challenge (n=5 mice per group). All NK cells were pregated to exclude dead cells based on forward and side scatter. Intratumoral cells were further pregated on CD45+ cells. (E) B16F10 cells were cultured without or with NK cells at 1:20 ratio treated by recombinant IL-33 or PBS for 24h. B16F10 cell death was determined by flow cytometric analysis measuring annexin V and 7-AAD. Data are shown as mean ± SEM. * P< 0.05, ** P< 0.01, *** P<0.001 as determined using a Student’s t-test.
IL-33 induces expansion of activated ILC2s in tumor-bearing mice
Type 2 innate lymphoid cells (ILC2s) are a subset of the most recently identified constituents of the innate immune system (17–19). ILC2s in particular are known to express ST2, the receptor for IL-33, and respond to IL-33 with the secretion of type 2 immune factors like IL-5 and IL-13 (18, 20). Interestingly, ILC2s were enriched among tumor infiltrates from B16F10 tumor-bearing Rag1−/− mice in response to systemic administration of recombinant IL-33 (Figure 3A). ILC2s in Rag1−/− mice were identified as CD11b-CD11c-NK1.1-FcεRI-CD25+CD45+CD90.2+ (Supplementary Figure 1C). Similarly, EL4 tumor-bearing Rag1−/− mice showed significantly elevated levels of ILC2s following IL-33 treatment compared to PBS-treated tumor-bearing Rag1−/− mice (Supplementary Figure 1D). Immunofluorescence verified an elevated presence of tumor-infiltrating CD90+ cells in IL-33-treated Rag1−/− mice (Figure 3B). Given the significant expansion of ILC2s in tumor-bearing mice following IL-33 treatment, we suspected ILC2s might contribute to an IL-33-mediated antitumor response. Surprisingly, depletion of CD90.2+ cells further augmented the IL-33-mediated tumor inhibitory effect (Figure 3C), indicating a tumor-promoting role for CD90+ ILC2s following IL-33 treatment. CD90.2 antibody treatment was confirmed to deplete mice of ILC2s in both tumors and spleens (Supplementary Figure 1E). In a separate experiment, depletion of CD90.2+ cells in Rag1−/− mice had no significant effect on tumor growth in the absence of IL-33 treatment (Figure 3D), suggesting IL-33 elicits pro-tumor behavior in ILCs. In line with these results, subcutaneous co-injection of B16F10 cells with ILC2s isolated from IL-33-treated Rag1−/− mice accelerated the time to tumor occurrence and the time until tumors reached their end stage (Figure 3E, F).
Figure 3. IL-33 treatment promotes expansion of tumor-promoting ILC2s during tumor growth.

(A) Representative flow dot plots showing intratumoral presence of CD90+Lin- ILC2s and corresponding bar graphs illustrating the percentage of ILC2s among infiltrates from B16F10-bearing Rag1−/− mice treated by PBS versus IL-33. ILC2s were pregated on CD45+CD11b-CD11c-NK1.1-FcεRI-. (n=5 mice per group). (B) Immunofluorescence of BPS1 tumors (X20) of Rag1−/− mice illustrate presence of CD90+ cells among IL-33-treated mice. (C) Tumor growth curves of B16F10 tumors demonstrating effects of IL-33 treatment with or without concurrent depletion of ILCs by anti-CD90 antibodies in Rag1−/− mice (n=5 mice per group). (D) Similar tumor experiment examining the impact of anti-CD90 treatment alone on tumor B16F10 tumor growth in Rag1−/− mice (n=4 mice per group). In both experiments involving CD90 depletion, equivalent amounts of rat IgG was injected in control mice as an isotype control. (E) Following the co-injection of B16F10 cells and ILC2s in Rag1−/− mice, mice were monitored until tumor occurrence and (F) until tumors reached their endpoint. 5000 B16F10 cells were co-injected with 1*106 ILC2s (n=5 mice per group). Tumor endpoint was determined when tumors reached 1cm3. Data are shown as mean ± SEM. * P<0.05 ** P< 0.01 as determined using a Student’s t-test.
Characterization of IL-33-induced ILC2s
Due to the novelty of innate lymphoid cells, characterization of the numerous subsets is still in flux. Considering the shifting nature of current literature, it is important to characterize the phenotype and function of our IL-33-induced ILC2s. CD90+ splenocytes were collected from IL-33-treated Rag1−/− mice and cultured with or without IL-33. Ex vivo treatment with IL-33 promoted CD90+ ILC2 activation and proliferation reflected by increased expression of CD25 and Ki-67, respectively (Figure 4A). Increased proliferation was further verified through efluor450 signal dilution and elevated cell numbers among treated ILC2s (Figure 4B). These cells also exhibited elevated levels of typical ILC2 cytokines IL-5 and IL-13, as well as the critical ILC2 transcription factor GATA3 (Figure 4C). Examination of intratumoral ILCs revealed similar characteristics. B16-IL-33 tumors exhibited a dramatic increase in IL-5-secreting ST2+ ILCs compared to B16-Vec, indicative of functional ILC2s (Figure 4D). This ILC2 population was preferentially expanded compared to other ILC subsets based on cytokine production (Supplementary Figure 2A,B), further supporting the fact that IL-33 promotes the accumulation of intratumoral ILC2s. Interestingly, a subset of IL-5-secreting intratumoral ILC2s also expressed produced IL-10 (Supplementary Figure 2C). ILC2s are the only ILC subset known to be stimulated by IL-33 and/or IL-25 (17, 27). ST2 blockade completely abrogated activation of ILC2s induced by IL-33 in vitro, denoted by the loss of CD25 expression (Figure 4E). These data support the notion that IL-33 may directly drive expansion and activation of ILC2s via ST2 during tumor growth.
Figure 4. IL-33 drives ILC2 expansion and activity.

(A) CD25 and Ki-67 expression in ILC2s in vitro measured by flow cytometry. (B) Flow cytometric assessment of ILC2 proliferation indicated by efluor450 signal and cell number after 24 hours. (C) Measurement of IL-5, IL-13, and GATA3 in ILCs in vitro. (D) Evaluation of ST2 and IL-5 expression by intratumoral ILC2s from B16-Vec or B16-IL33 tumor-bearing Rag1−/− mice (n=4 mice per group). Cells were pregated on CD90+ cells. (E) Flow cytometric analysis of ILC2 activation in vitro following blockade of IL-33-ST2 signaling by anti-ST2 (clone DJ8) antibodies. Rat IgG was used as an isotype control. Data are shown as mean ± SEM. * P< 0.05, ** P< 0.01, *** P<0.001 as determined using a Student’s t-test.
Inhibition of NK activity by ILC2.
Given the tumor-promoting effect of ILC2s following IL-33 therapy, we examined whether IL-33-induced ILC2 cells regulated NK cell activity during tumor growth. As expected, we found that IL-33 treatment increased the proportion of NK cells in the spleens of tumor-bearing Rag1−/− mice (Figure 5A). Further, a larger proportion of these NK cells expressed elevated levels of NKG2D (Figure 5B), one of the best-characterized activating receptors expressed by NK cells (28). Importantly, depletion of ILC2 cells with anti-CD90 further augmented IL-33-mediated NK cell infiltration and NKG2D expression. A similar trend was observed within tumors; depletion of ILC2s resulted in a significant increase in the infiltration of NKG2D+ NK cells (Figure 5C). In vitro, NK cells cocultured with ILC2s demonstrated an impaired ability to eliminate B16 cells (Figure 5D). ST2-blocked ILC2s failed to suppress cytokine production by IL-33-stimulated NK cells (Figure 5E). These results indicate ILC2s may promote tumor growth through suppression of NK cell expansion and activity.
Figure 5. ILC2s suppress NK cell function during IL-33 treatment.

) Flow cytometric analysis of NK cell presence and activation in spleens (A,B) and tumors (C) following depletion of ILC2s using anti-CD90 antibodies in combination with IL-33 treatment. NK cells were identified by CD49b expression and activation was evaluated by NKG2D. The difference between ‘IL-33+IgG’ and ‘IL-33+αCD90’ in figure c is significant only by one-tailed Student’s t-test. Experiment was performed in B16F10 tumor-bearing Rag1−/− mice. Rat IgG was used as an isotype control. (n=4 mice per group). (D) Coculture of B16F10, NK cells, and ILC2s with 10ng/ml recombinant IL-33 were assessed for B16F10 cell death measured by annexin V. Cells were cultured at a ratio of 1:20:20 B16F10:NK:ILC2. (E) IFN-γ production by NK cells following coculture with ILC2s. NK cells were pretreated with or without 20 ng/ml recombinant IL-33 and 10 ng/ml IL-2. ILC2s were pretreated with 20 ng/ml IL-33 and 10 ng/ml IL-2 with or without anti-ST2 blocking antibody (clone: DJ8). NK cells and ILC2s were then washed and co-cultured for 24 hours. Rat IgG was used as an isotype control. Data are shown as mean ± SEM. * P< 0.05, ** P< 0.01, *** P<0.001 as determined using a Student’s t-test.
CD73 involvement in ILC2-mediated NK cell suppression
CD73 is a widely expressed ecto-enzyme involved in regulating both innate and adaptive immune responses (29–31). Together with CD39, CD73 facilitates the dephosphorylation of ATP to adenosine, which has potent immunosuppressive effects and has been linked to increased tumor growth and metastasis (32–34). Notably, CD73 expression was dramatically elevated on tumoral ST2+ ILC2s in B16-IL33-bearing mice compared to B16-Vec tumors (Figure 6A). Further, activated CD25+ splenic ILC2s displayed increased expression of CD73 from tumor-bearing Rag1−/− mice following IL-33 treatment (Figure 6B). Furthermore, we confirmed the preferential expression of CD73 in CD25+ST2+CD90+ ILC2s induced by IL-33 in vitro (Figure 6C). To explore the potential significance of ILC2-derived CD73 in NK cell suppression, NK cells were cultured with either WT ILC2s or CD73-deficient ILC2s collected from CD73 knock-out (CD73−/−) mice. NK cells that were cultured with WT ILC2s displayed reduced expression of IFN-γ and CD107a. Interestingly, CD73−/− ILC2s failed to impair NK cell activity significantly (Figure 6D). WT ILC2s successfully catabolized AMP in vitro, indicative of CD73-mediated adenosine production, whereas CD73−/− ILC2s failed to consume AMP (Supplementary Figure 3A). However, CD73−/− ILC2s generated from bone marrow—using a previously described method (25)—secreted comparable amounts of IL-5 compared to WT (Supplementary Figure 3B), indicating CD73 does not directly affect ILC2 development or activity. Additionally, NK cells did not express CD73 (confirmed by comparison with CD73−/− mice) (Supplementary Figure 4A), nor did CD73−/− NK cells demonstrate any defect in tumor-killing capacity (Supplementary Figure 4B), further substantiating the importance of ILC2-derived CD73 in suppressing NK cell activity. Thus, these findings provide a possible mechanism by which ILC2s inhibit NK cell-mediated antitumor effect.
Figure 6. ILC2 expression of CD73 is involved in ILC2-mediated suppression of NK cells.
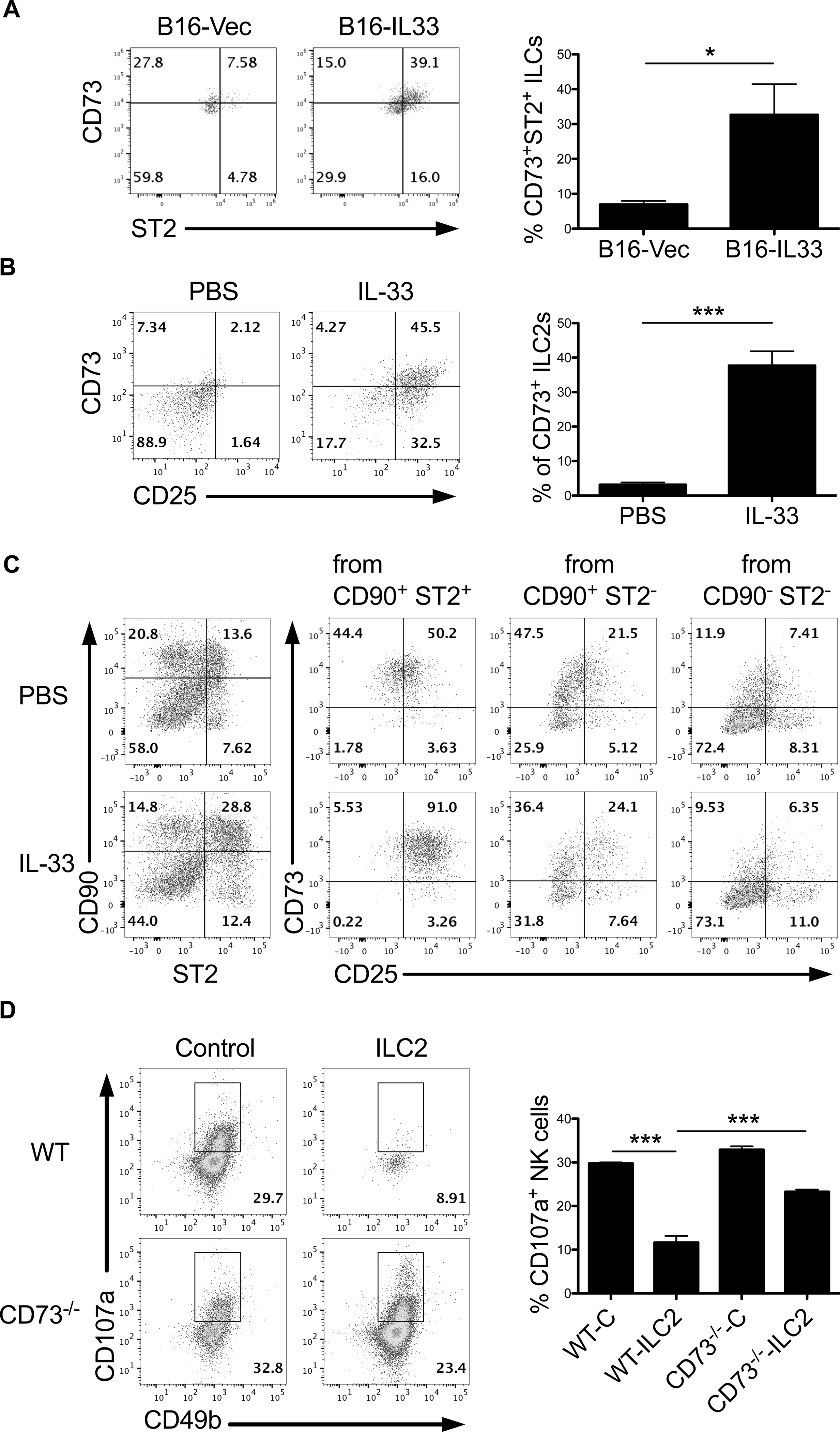
(A) Evaluation of ST2 and CD73 expression by intratumoral ILC2s from B16-Vec or B16-IL33 tumor-bearing Rag1−/− mice (n=4 mice per group). (B) Flow cytometric analysis of CD73 expression on activated splenic CD25+ ILC2 from PBS and IL-33-treated tumor-bearing Rag1−/− mice (n=4 mice per group). (C) Enriched splenic CD90+ ILCs from Rag1−/− mice were stimulated by PBS or recombinant IL-33 for 24h in vitro, and expression of CD73 and ST2 on CD90+ ILCs was determined by flow cytometry (left). Expression of CD73 and CD25 was further measured among ST2+CD90+, ST2-CD90+ and ST2-CD90- subsets (right). (D) CD107a expression among NK cells in coculture with WT or CD73−/− ILC2s. Control samples included BM cells cultured without IL-33 to serve as “control” ILC2s. ILC2s and NK cells were cocultured at a 1:1 ratio in 50μM AMP and 10ng/ml IL-33. Data are shown as mean ± SEM. * P< 0.05, *** P<0.001 as determined using a Student’s t-test.
Discussion
To date, studies exploring IL-33/ST2 signaling in tumor growth have yielded mixed results (35, 36). Here, we have clearly identified a robust antitumor effect of IL-33 in established preclinical tumor models consistent with previous studies(5–7). Further, as demonstrated through the utilization of Rag1−/− mice, IL-33 is capable of exerting its antitumor effect solely through the innate immune system.
IL-33 is released as an alarmin by epithelial cells, endothelial cells, and fibroblasts in responses to cellular damage and acts as an early inducer of inflammation(1–3). IL-33 coordinates a wide range of immune responses through direct activation of many different cellular targets including T helper and regulatory cells, mast cells, eosinophils, and natural killer cells (1, 4). Part of the confusion regarding IL-33’s role in tumor development lies in the far-reaching nature and context-specificity of IL-33’s effects. One study demonstrated a protumor role of IL-33 in breast cancer that involved the recruitment and activation of immunosuppressive cells such as myeloid-derived suppressor cells and T regulatory cells(36). Conversely, IL-33 has also been shown to impede melanoma development through the activation of tumor-killing CD8+ T cells and NK cells(5). Depending on the context, the interplay between IL-33-mediated protumor and antitumor elements may yield different outcomes for tumor development.
Our results, however, consistently demonstrate a potent antitumor role for IL-33. This antitumor effect is observable in both systemic administrations of recombinant IL-33 and local ectopic expression of IL-33 within tumors. NK cells are crucial to IL-33 mediated tumor inhibition since antibody removal of NK cells significantly accelerated melanoma growth in IL-33-treated Rag−/− mice. NK cells have long been described as effective mediators of innate antitumor immunity(8, 9). Our results support the previous reports of NK cells contributing to antitumor immunity in tumors with ectopic expression of IL-33(5). We further demonstrate an antitumor role of NK cells in treatment with exogenous recombinant IL-33.
This study has established the role of ILC2s by dissecting their contribution in IL-33-mediated tumor suppression particularly in the absence of adaptive immune system. Numerous studies have observed IL-33-driven activation and expansion of type 2 innate lymphoid cells in in allergic inflammation (37, 38). However, the pathologic role of ILC2 in type 2 inflammatory diseases might be detrimental in the context of tumors. Indeed, a recent study reported that IL-33 treatment facilitated the expansion of IL-13-producing Lin−Sca-1+ ILCs in spleens and mammary tumors (36). Similarly, we show that IL-33 induced the expansion of IL-5/IL-13 producing Lin−CD25+ ILC2s in spleens and melanomas likely through ST2. As ILC2 produce type 2 cytokines, mainly IL-13 and IL-5, which are correlated with an environment promoting tumor formation and progression. A recent study supports this notion, reporting a link between the ILC2/IL-13 axis and human bladder cancer recurrence (39). ILC2 were also reported to be implicated in an experimental model of cholangiocarcinoma with an exogenous administration of IL-33 (40). Though these studies speculate ILC2s may facilitate tumor growth, no evidence to date has shown a direct tumor-promoting role for ILC2s (41, 42). To our knowledge, through the depletion of ILC2s, we provide the first evidence that ILC2s can act in a protumor growth manner. Beyond offering further insight into the diverse effects of IL-33 in tumor development, our findings help illuminate a novel role for a newly characterized immune cell subset. Further investigations may evaluate whether IL-33 amplifies a preestablished immunosuppressive tumor contexture through the interaction between ILC2 and other inhibitory cell populations such as myeloid-derived suppressor cells (MDSCs), as was recently suggested (39).
A recent study asserts IL-33 actually elicits an anti-tumor roll from ILC2s, mediated by CXCR2(25). However, with the ongoing characterization of innate lymphoid cells and their numerous subtypes, careful consideration needs to be given to how ILC2s—and innate lymphoid cells in general—are defined(43). Differing methods of ILC2 characterization and selection could potentially yield different subsets of innate lymphoid cells. Here, we identified ILC2s using characteristics consistent with current literature (20). The indication of the existence of different ILC2 subsets (44) adds to our understanding of the complexity of ILC2 biology and makes necessary an analysis of the relationship between the fate and functionality of distinct ILC2 subsets during tumor progression.
The role of NK cells in anti-tumor immunity has been established. However, such a role for other ILC populations in interacting with NK cells remains largely unknown. Our results indicate ILC2s support tumor growth at least in-part by inhibiting NK cell activity. This ILC2-mediated suppression of NK cells is significantly influenced by CD73 expressed on ILC2s, likely in a similar manner as CD73 expressed on tumor cells or other immune cell populations. In this regard, CD73, in combination with CD39, catabolizes the breakdown of ATP to adenosine to counteract anti-tumor immunity (32–34).
The detection of IL-10 by tumoral ILC2s is a particularly interesting observation. IL-10 expression by ILC2s has recently been observed and has been described to downregulate some proinflammatory genes (45). The impact of IL-10 on tumor development has long-been the focus of intense study, and an immunosuppressive, pro-tumor role has been well-described (46, 47). The role of ILC2-derived IL-10 on tumor progression, however, is currently unknown and worthy of further investigation.
Collectively, our data present a new understanding of IL-33’s complex involvement in tumor development. Whether by local expression or systemic administration, IL-33 consistently played an overall inhibitory role in tumor growth, even in the absence of an adaptive immune system. However, IL-33 regulation of ILC2s can antagonize the function of NK cells during tumor growth via CD73, indicating a role of IL-33 in balancing anti- and pro-tumor immune components within the tumor. In conclusion, our data indicate a close interaction of ILC2s and NK cells in IL-33-mediated antitumor effects and emphasize the biological activities of IL-33 are cell context-dependent for tumor regression or progression.
Supplementary Material
Acknowledgments
This research was in part supported by National Institutes of Health grant CA149669, CA208354 and CA222963, Melanoma Research Alliance Pilot Award, Northwestern University RHLCCC Flow Cytometry Facility, a Cancer Center Support Grant (NCI CA060553) and the Walter S. and Lucienne Driskill Immunotherapy Research fund.
Abbreviations used in this article:
- IL-33
interleukin 33
- ILC2
Type 2 innate lymphoid cells
- MDSCs
myeloid-derived suppressor cells
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NK
natural killer
- NKG2D
natural-killer group 2, member D
- PBS
phosphate-buffered saline
- SEM
standard error of the mean
- WT
wild type
References
- 1.Liew FY, Pitman NI, and McInnes IB 2010. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nature reviews. Immunology 10: 103–110. [DOI] [PubMed] [Google Scholar]
- 2.Haraldsen G, Balogh J, Pollheimer J, Sponheim J, and Kuchler AM 2009. Interleukin-33 - cytokine of dual function or novel alarmin? Trends in immunology 30: 227–233. [DOI] [PubMed] [Google Scholar]
- 3.Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G, and Girard JP 2007. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proceedings of the National Academy of Sciences of the United States of America 104: 282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palmer G, and Gabay C 2011. Interleukin-33 biology with potential insights into human diseases. Nature reviews. Rheumatology 7: 321–329. [DOI] [PubMed] [Google Scholar]
- 5.Gao K, Li X, Zhang L, Bai L, Dong W, Gao K, Shi G, Xia X, Wu L, and Zhang L 2013. Transgenic expression of IL-33 activates CD8(+) T cells and NK cells and inhibits tumor growth and metastasis in mice. Cancer Lett 335: 463–471. [DOI] [PubMed] [Google Scholar]
- 6.Gao X, Wang X, Yang Q, Zhao X, Wen W, Li G, Lu J, Qin W, Qi Y, Xie F, Jiang J, Wu C, Zhang X, Chen X, Turnquist H, Zhu Y, and Lu B 2015. Tumoral expression of IL-33 inhibits tumor growth and modifies the tumor microenvironment through CD8+ T and NK cells. J Immunol 194: 438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Villarreal DO, Wise MC, Walters JN, Reuschel EL, Choi MJ, Obeng-Adjei N, Yan J, Morrow MP, and Weiner DB 2014. Alarmin IL-33 acts as an immunoadjuvant to enhance antigen-specific tumor immunity. Cancer Res 74: 1789–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Childs RW, and Carlsten M 2015. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakens. Nat Rev Drug Discov 14: 487–498. [DOI] [PubMed] [Google Scholar]
- 9.Rezvani K, and Rouce RH 2015. The Application of Natural Killer Cell Immunotherapy for the Treatment of Cancer. Frontiers in immunology 6: 578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lanier LL 1997. Natural killer cells: from no receptors to too many. Immunity 6: 371–378. [DOI] [PubMed] [Google Scholar]
- 11.Raulet DH, Vance RE, and McMahon CW 2001. Regulation of the natural killer cell receptor repertoire. Annual review of immunology 19: 291–330. [DOI] [PubMed] [Google Scholar]
- 12.Farag SS, Fehniger TA, Becknell B, Blaser BW, and Caligiuri MA 2003. New directions in natural killer cell-based immunotherapy of human cancer. Expert opinion on biological therapy 3: 237–250. [DOI] [PubMed] [Google Scholar]
- 13.Clement MV, Haddad P, Soulie A, Legros-Maida S, Guillet J, Cesar E, and Sasportes M 1990. Involvement of granzyme B and perforin gene expression in the lytic potential of human natural killer cells. Research in immunology 141: 477–489. [DOI] [PubMed] [Google Scholar]
- 14.Street SE, Cretney E, and Smyth MJ 2001. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood 97: 192–197. [DOI] [PubMed] [Google Scholar]
- 15.Trinchieri G 1995. Natural killer cells wear different hats: effector cells of innate resistance and regulatory cells of adaptive immunity and of hematopoiesis. Seminars in immunology 7: 83–88. [DOI] [PubMed] [Google Scholar]
- 16.Chester C, Fritsch K, and Kohrt HE 2015. Natural Killer Cell Immunomodulation: Targeting Activating, Inhibitory, and Co-stimulatory Receptor Signaling for Cancer Immunotherapy. Frontiers in immunology 6: 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Licona-Limon P, Kim LK, Palm NW, and Flavell RA 2013. TH2, allergy and group 2 innate lymphoid cells. Nat Immunol 14: 536–542. [DOI] [PubMed] [Google Scholar]
- 18.Walker JA, and McKenzie AN 2013. Development and function of group 2 innate lymphoid cells. Current opinion in immunology 25: 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McHedlidze T, Waldner M, Zopf S, Walker J, Rankin AL, Schuchmann M, Voehringer D, McKenzie AN, Neurath MF, Pflanz S, and Wirtz S 2013. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 39: 357–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roediger B, Kyle R, Yip KH, Sumaria N, Guy TV, Kim BS, Mitchell AJ, Tay SS, Jain R, Forbes-Blom E, Chen X, Tong PL, Bolton HA, Artis D, Paul WE, Fazekas de St Groth B, Grimbaldeston MA, Le Gros G, and Weninger W 2013. Cutaneous immunosurveillance and regulation of inflammation by group 2 innate lymphoid cells. Nature immunology 14: 564–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spooner CJ, Lesch J, Yan D, Khan AA, Abbas A, Ramirez-Carrozzi V, Zhou M, Soriano R, Eastham-Anderson J, Diehl L, Lee WP, Modrusan Z, Pappu R, Xu M, DeVoss J, and Singh H 2013. Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat Immunol 14: 1229–1236. [DOI] [PubMed] [Google Scholar]
- 22.Klein Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, and Hendriks RW 2012. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. European journal of immunology 42: 1106–1116. [DOI] [PubMed] [Google Scholar]
- 23.Dominguez D, Ye C, Geng Z, Chen S, Fan J, Qin L, Long A, Wang L, Zhang Z, Zhang Y, Fang D, Kuzel TM, and Zhang B 2017. Exogenous IL-33 Restores Dendritic Cell Activation and Maturation in Established Cancer. J Immunol 198: 1365–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen S, Wang L, Fan J, Ye C, Dominguez D, Zhang Y, Curiel TJ, Fang D, Kuzel TM, and Zhang B 2015. Host miR155 promotes tumor growth through a myeloid-derived suppressor cell-dependent mechanism. Cancer Res 75: 519–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim J, Kim W, Moon UJ, Kim HJ, Choi HJ, Sin JI, Park NH, Cho HR, and Kwon B 2016. Intratumorally Establishing Type 2 Innate Lymphoid Cells Blocks Tumor Growth. Journal of immunology 196: 2410–2423. [DOI] [PubMed] [Google Scholar]
- 26.Dominguez D CS, Fan J, Qin L, Long A, Zhang Y, Kuzel TM, Zhang B. Exogenous IL-33 restores dendritic cell activation and maturation in established cancer. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Artis D, and Spits H 2015. The biology of innate lymphoid cells. Nature 517: 293–301. [DOI] [PubMed] [Google Scholar]
- 28.Raulet DH, and Guerra N 2009. Oncogenic stress sensed by the immune system: role of natural killer cell receptors. Nat Rev Immunol 9: 568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Regateiro FS, Cobbold SP, and Waldmann H 2013. CD73 and adenosine generation in the creation of regulatory microenvironments. Clinical and experimental immunology 171: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antonioli L, Pacher P, Vizi ES, and Hasko G 2013. CD39 and CD73 in immunity and inflammation. Trends in molecular medicine 19: 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eltzschig HK, Sitkovsky MV, and Robson SC 2012. Purinergic signaling during inflammation. N Engl J Med 367: 2322–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang B 2010. CD73: A Novel Target for Cancer Immunotherapy. Cancer Res 70: 6407–6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beavis PA, Stagg J, Darcy PK, and Smyth MJ 2012. CD73: a potent suppressor of antitumor immune responses. Trends Immunol 33: 231–237. [DOI] [PubMed] [Google Scholar]
- 34.Allard B, Longhi MS, Robson SC, and Stagg J 2017. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol Rev 276: 121–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Villarreal DO, and Weiner DB 2014. Interleukin 33: a switch-hitting cytokine. Curr Opin Immunol 28C: 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jovanovic IP, Pejnovic NN, Radosavljevic GD, Pantic JM, Milovanovic MZ, Arsenijevic NN, and Lukic ML 2014. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer 134: 1669–1682. [DOI] [PubMed] [Google Scholar]
- 37.Martinez-Gonzalez I, Steer CA, and Takei F 2015. Lung ILC2s link innate and adaptive responses in allergic inflammation. Trends Immunol 36: 189–195. [DOI] [PubMed] [Google Scholar]
- 38.Ealey KN, Moro K, and Koyasu S 2017. Are ILC2s Jekyll and Hyde in airway inflammation? Immunol Rev 278: 207–218. [DOI] [PubMed] [Google Scholar]
- 39.Chevalier MF, Trabanelli S, Racle J, Salome B, Cesson V, Gharbi D, Bohner P, Domingos-Pereira S, Dartiguenave F, Fritschi AS, Speiser DE, Rentsch CA, Gfeller D, Jichlinski P, Nardelli-Haefliger D, Jandus C, and Derre L 2017. ILC2-modulated T cell-to-MDSC balance is associated with bladder cancer recurrence. J Clin Invest 127: 2916–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Razumilava N, Gores GJ, Walters S, Mizuochi T, Mourya R, Bessho K, Wang YH, Glaser SS, Shivakumar P, and Bezerra JA 2014. Biliary repair and carcinogenesis are mediated by IL-33-dependent cholangiocyte proliferation. J Clin Invest 124: 3241–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tian Z, van Velkinburgh JC, Wu Y, and Ni B 2016. Innate lymphoid cells involve in tumorigenesis. International journal of cancer. Journal international du cancer 138: 22–29. [DOI] [PubMed] [Google Scholar]
- 42.Carrega P, Campana S, Bonaccorsi I, and Ferlazzo G 2016. The Yin and Yang of Innate Lymphoid Cells in Cancer. Immunology letters 179: 29–35. [DOI] [PubMed] [Google Scholar]
- 43.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, and Vivier E 2013. Innate lymphoid cells--a proposal for uniform nomenclature. Nature reviews. Immunology 13: 145–149. [DOI] [PubMed] [Google Scholar]
- 44.Huang Y, and Paul WE 2016. Inflammatory group 2 innate lymphoid cells. Int Immunol 28: 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seehus CR, Kadavallore A, Torre B, Yeckes AR, Wang Y, Tang J, and Kaye J 2017. Alternative activation generates IL-10 producing type 2 innate lymphoid cells. Nat Commun 8: 1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steinbrink K, Jonuleit H, Muller G, Schuler G, Knop J, and Enk AH 1999. Interleukin-10-treated human dendritic cells induce a melanoma-antigen-specific anergy in CD8(+) T cells resulting in a failure to lyse tumor cells. Blood 93: 1634–1642. [PubMed] [Google Scholar]
- 47.Sato T, Terai M, Tamura Y, Alexeev V, Mastrangelo MJ, and Selvan SR 2011. Interleukin 10 in the tumor microenvironment: a target for anticancer immunotherapy. Immunol Res 51: 170–182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.