

Summary
• Whole genome duplications (WGDs) are widespread and prevalent in vascular plants and frequently coincide with major episodes of global and climatic upheaval, including the mass extinction at the Cretaceous–Tertiary (KT) boundary (c. 65 million yr ago (Ma)) and during more recent periods of global aridification in the Miocene (c. 10–5 Ma). Here, we explore WGDs in the diverse flowering plant clade Malpighiales.
• Using transcriptomes and complete genomes from 42 species we applied a multi-pronged phylogenomic pipeline to identify, locate, and determine the age of WGDs in Malpighiales using three means of inference: distributions of synonymous substitutions per synonymous site (Ks) among paralogs, phylogenomic (gene tree) reconciliation, and a likelihood based gene count method.
• We conservatively identify 22 ancient WGDs, widely distributed across Malpighiales subclades. Importantly, these events are clustered around the Eocene–Paleocene Transition (c. 54 Ma), during which time the planet was warmer and wetter than any period in the Cenozoic.
• These results establish that the Eocene Climate Optimum likely represents a previously unrecognized period of prolific WGDs in plants, and lends further support to the hypothesis that polyploidization promotes adaptation and enhances plant survival during episodes of global change, especially for tropical organisms like Malpighiales, which have tight thermal tolerances.
Keywords: climatic upheaval, flowering plants, genome evolution, global change, phylogenomics, speciation
Introduction
Whole genome duplication (WGD), or polyploidy, is an important evolutionary force that has shaped plant evolution. It has long been appreciated that the formation of recent polyploids in vascular plants is common (Stebbins, 1947; Barker et al., 2016), and mounting evidence suggests that ancient polyploids are more frequent than once thought. WGD has been identified in ferns (Wood et al., 2009) and seed plants, including in gymnosperms (Li et al., 2015) and more prevalently, in angiosperms. Well-cited examples of ancient WGDs have been associated with the origin of numerous hyperdiverse clades, including in the common ancestor of seed plants, flowering plants, monocots, orchids, core eudicots, mustards (Brassicaceae), legumes (Fabaceae), and sunflowers (Asteraceae; Blanc & Wolfe, 2004; Barker et al., 2008, 2009; Bertioli et al., 2009; Tang et al., 2010; Jiao et al., 2011; Jiao et al., 2012; Jiao et al., 2014; Cannon et al., 2015; Huang et al., 2016; Zhang et al., 2017). Among these ancient WGDs, several have been dated to the Cretaceous–Tertiary (KT) boundary (c. 65 million yr ago (Ma)), potentially linking these polyploidization events to plants’ abilities to survive abrupt global environmental change (Fawcett et al., 2009; Vanneste et al., 2014). Similarly, a large number of WGDs have also been reported in grasses during the late Miocene when arid, grass dominated landscapes expanded dramatically (Estep et al., 2014). In these cases, the potential adaptive value of WGDs is thought to arise from the origin of genetic novelties (Ohno, 1970; Lynch & Conery, 2000; Taylor & Raes, 2004; Lynch, 2007) and by masking the effects of deleterious mutations (Gu et al., 2003). Together, this may facilitate plant survival across periods of global disruption. Although debate exists as to the influence of WGD on species diversification rates (Wood et al., 2009; Mayrose et al., 2011, 2015; Soltis et al., 2014; Tank et al., 2015; Kellogg, 2016), it is generally accepted that chromosomal rearrangements from WGDs can significantly accelerate isolating barriers, thus promoting cladogenesis (Werth & Windham, 1991; Lynch & Force, 2000; Husband et al., 2016). In summary, it is established that WGDs are a prominent feature of vascular plant evolution, yet remain underexplored in many clades.
Here, we investigate WGDs in the large and diverse angiosperm order Malpighiales, which contains more than 16,000 mostly tropical species with tremendous morphological and ecological diversity. Members of this clade also include numerous economically important crops such as rubber, cassava, and flax. The Malpighiales have long been recognized as one of the most difficult clades to resolve in the flowering plant tree of life (Davis et al., 2005; Wurdack & Davis, 2009), which has been attributed in part to its rapid radiation in the mid-Cretaceous (Davis et al., 2005; _Xi et al., 2012b). However, recent efforts utilizing phylogenomic approaches have greatly increased our understanding of deep level relationships in the order (Xi et al., 2012b). Additionally, the clade includes numerous species that have previously been targeted for genomic investigation of WGDs. Eight genomes are currently available for interrogation, including Hevea brasiliensis (rubber), Manihot esculenta (cassava), Linum usitatissimum (flax), Ricinus communis (castor bean), Jatropha curcas (Barbados nut), Populus trichocarpa (black cottonwood), Salix suchowensis (shrub willow), and Salix purpurea (purple willow). Notably, three WGDs have been identified in Malpighiales using these data: in the common ancestor of Populus and Salix (35–65 Ma; Tuskan et al., 2006; Dai et al., 2014); in the common ancestor of Manihot and Hevea (35–47 Ma; Bredeson et al., 2016); and more recently in Linum usitatissimum (5–9 Ma; Z. Wang et al., 2012). In addition, studies using transcriptomes have identified an older WGD shared by all blue-flowered Linum species, including Linum usitatissimum, at 20–40 Ma (Sveinsson et al., 2014). More complicated polyploidy histories involving multiple rounds of WGDs and hybridization have also been reported using chromosome count data in the genera Passiflora (Mayrose et al., 2009) and Viola (Marcussen et al., 2012, 2014). In short, given the apparent propensity of WGDs in Malpighiales and the existing complete sequence data available for the order, this clade is an ideal study system for investigating the frequency and timing of WGDs.
Materials and Methods
Taxon sampling and transcriptome sequencing
We collected genomic and transcriptomic data for 36 species representing 21 families of Malpighiales, spanning all major clades sensu Wurdack and Davis (Wurdack & Davis, 2009; Chase et al., 2016) (Supporting Information Tables S1–S3). In addition, three closely related outgroups from Celastraceae (Celastrales), Elaeocarpaceae (Oxalidales), and Oxalidaceae (Oxalidales), plus four more distantly related outgroups (Cucumis sativus (Eurosid I), Theobroma cacao (Eurosid II), and Vitis vinifera (basal Rosid)) were used for rooting (Chase et al., 2016). We sequenced transcriptomes of 15 Malpighiales species following the protocol described by Xi et al. (2012a). Total RNA from leaf tissue was extracted using the RNAqueous and Plant RNA Isolation Aid kits (Ambion, Inc.), and treated with the TURBO DNA-free kit (Ambion, Inc.) at 37°C for 4 h to remove residual DNA. The cDNA library was synthesized from total RNA following the protocols of Novaes et al. (2008). Illumina paired-end libraries were prepared for cDNA following the protocols of Bentley et al. (2008). Each library was sequenced in a single lane of the Genome Analyzer II (Illumina, Inc.) with paired-end 150 base pairs (bp) read lengths. We additionally included 13 annotated transcriptomes from the OneKP project (Table S2) to complete our taxon sampling for Malpighiales (Matasci et al., 2014). These sequences were obtained following the protocol outlined by Wickett et al. (2014). Finally, we also obtained whole genome sequence data from eight published genomes of Malpighiales plus three outgroup species: Hevea brasiliensis (Willd. ex A.Juss.) Müll.Arg., Jatropha curcas L., Linum usitatissimum L., Manihot esculenta Crantz, Populus trichocarpa Torr. & A.Gray ex Hook., Ricinus communis L., Salix purpurea L., Salix suchowensis W.C.Cheng ex G.H.Zhu, Cucumis sativus L., Theobroma cacao L., Vitis vinifera L. (Table S3).
Transcriptome assembly
Raw sequencing reads were first corrected for errors using Rcorrector (Song & Florea, 2015). Reads marked as ‘unfixable’, generally constituting regions of low-complexity, were discarded. Sequencing and PCR adapters were identified and trimmed using TrimGlore v0.4.2 (Krueger, 2015). We examined the quality of trimmed reads using FastQC v0.11.5 (Andrews, 2010) and then assembled the reads using Trinity v2.1.1 (Grabherr et al., 2011). We used the longest isoform from each Trinity assembly and further reduced the redundancy generated from sequencing error or alternative splicing by performing a similarity-based clustering (-c 0.99 -n 10, threshold following Yang et al., 2015) via CD-HIT EST v4.6.4 (Li & Godzik, 2006). The completeness of our assemblies was assessed by comparison against the single-copy orthologs plant database, BUSCO (Simão et al., 2015; Fig. S1). Coding regions (CDS) of each putative transcript were predicted following the transdecoder workflow (Haas & Papanicolaou, 2012). Finally, to control for transcriptome quality in our subsequent assessments of WGD, we reanalyzed our combined transcriptome and complete genome data following the methods described below with only high-quality transcriptomes. Here, transcritomes with more than 40% (382/956) missing BUSCOs were removed, including Bhesa paniculata, Flacourtia jangomas, Galearia maingayi, Ixonanthes reticulate, Podostemum ceratophyllum, Rinorea anguifera, and Tristellateia australasiae.
Gene family clustering and orthology inference
To assign sequences into orthologous gene families, we used an integrated method that takes into account sequence similarity and species phylogeny. We first constructed whole genome/transcriptome homology scans using Proteinortho v5.13 (Lechner et al., 2011) with default parameter settings. This program extends the reciprocal best BLAST hit method and is computationally efficient. Clusters were searched to identify gene families containing at least 22 (>60%) ingroup species. This resulted in 8465 candidate homolog clusters. This similarity-based homology search can sometimes be erroneous due to deep paralogs, misassembly, or frame shifts (Yang & Smith, 2014). To reduce such errors in orthology inference, we further applied a tree-based method to sort genes into orthology groups (Yang & Smith, 2014). This method does not rely on a known species tree, but rather iteratively searches for the subtree with the highest number of ingroup taxa to assign as orthology groups (Fig. S2). Here, we first aligned the protein sequences of each homolog cluster with MAFFT v7.299 (Katoh & Standley, 2013) using the local alignment algorithm (--localpair --maxiterate 1000). The resulting protein alignments were converted into the corresponding codon alignments using pal2nal v14 (Suyama et al., 2006). A gene family tree of each codon alignment was then reconstructed using RAxML v8.1.5 (Stamatakis, 2014b) with ten random starting points. To sort homologs into ortholog groups, we first pruned exceptionally long and short branches within each gene family tree because we suspected such branches to be incorrect homologs, sequencing errors, or transcript isoforms. In addition, short branches may cause problems of over fitting in the subsequent penalized likelihood dating method (Sanderson, 2004). Along these lines, branches that were ten times longer than the ‘5% trimmed mean branch length’, or shorter than an absolute value of 1e-15, were pruned. The ‘5% trimmed mean branch length’ was defined as the mean branch length after discarding the lowest and highest 5% of the branch length distribution in each gene family. Orthology was then inferred based on this pruned gene family tree using the ‘RT’ method (‘prune_paralogs_RT.py’) following Yang & Smith (2014). The resulting 5113 orthology clusters were realigned as amino acids using the method described above. Finally, the back-translated nucleotide alignments were used for subsequent phylogenetic analysis.
Phylogeny reconstruction and molecular dating
To infer the phylogeny of each orthology group, we first removed sites containing >80% gaps using trimAL v1.4.15 (Capella-Gutiérrez et al., 2009). We then applied RAxML v8.1.5 to reconstruct maximum likelihood (ML) trees under the GTR+Γ model with 20 random starting points. We chose the GTR+ Γ model because it accommodates rate heterogeneity among sites, while the other available GTR model in RAxML, the GTRCAT model, is less appropriate due to our small taxon sampling size (Stamatakis, 2014a). We then filtered each gene tree to eliminate exceptionally long and short branches using the method outlined above. The remaining gene accessions were realigned and a final round of ML tree inference was conducted. Statistical confidence of each gene tree was assessed by performing 100 bootstrap (BS) replicates with branch length (-N 100 -k).
In addition to determining the phylogeny of orthology groups, we also estimated the divergence time of each orthologous tree for the purpose of dating WGDs. We applied the penalized likelihood method for its high efficiency when dealing with large data sets including thousands of genes. By contrast, Bayesian divergence time estimation is computationally intensive and difficult to implement with a data set of this size. We estimated molecular divergence times for each ML gene tree as well as the bootstrap trees with penalized likelihood as implemented in r8s v1.7 (Sanderson, 2003). The following four calibration points were applied to each tree: (1) the root age was fixed at 109 Ma, representing the approximate age of the crown group divergence in Malpighiales (Xi et al., 2012b); (2) the minimum age of crown group clusioids (including Calophyllum macrocarpum, Clusia rosea, Podostemum ceratophyllum, Garcinia oblongifolia, Hypericum perforatum, and Mammea americana) was set to be 89 Ma, representing the oldest known fossil in Malpighiales, Palaeoclusia chevalieri (Ruhfel et al., 2013); (3) two additional secondary minimum age constraints from Xi et al. (2012b) were used to constrain stem group Euphorbiaceae (97 Ma) and Salicaceae (69 Ma). For each clade, the age constraint was placed on the most recent common ancestor of all gene accessions forming a monophyletic group for that clade. The optimal smoothing parameter for each gene tree was determined within the range of parameter space (1e-4.5, 1e4.5) by cross-validation (Sanderson, 2003). Trees were subsequently dated under the assumption of a relaxed molecular clock by applying a semi-parametric penalized likelihood approach using a truncated Newton optimization algorithm in r8s.
Species tree estimation
We inferred a single reference species tree for our analysis of WGD applying a summary coalescent method as implemented in ASTRAL v4.10.5 (Mirarab & Warnow, 2015). As input gene trees for our species tree analysis we utilized all 5113 gene trees derived from each orthology cluster described above. Before species tree inference we applied an additional branch trimming process to remove duplicated taxa from individual gene trees following Yang & Smith (2014). At each node where two decedent clades contain overlapping taxa, the branch with the smaller number of taxa was pruned. These pruned gene trees were subsequently used in ASTRAL for species tree estimation. We additionally processed the one hundred bootstrap trees for each gene using the same pruning method described above. These bootstrap trees are used in ASTRAL analyses to conduct bootstrap replicates for species tree estimation. Molecular divergence time estimates were subsequently inferred for the species tree using the penalized likelihood method described above using a concatenated sequence matrix derived from all 40 genes containing at least 34 ingroup taxa.
Overview of the methods applied to infer WGD
We utilized three lines of inference described in the following three subsections to identify, locate, and determine the age of WGDs in Malpighiales. Each of these methods has been commonly applied in angiosperms and elsewhere, and includes: distribution of Ks among paralogs (Cui et al., 2006; Barker et al., 2008), phylogenomic (gene tree) reconciliation (Jiao et al., 2011; Li et al., 2015), and a likelihood based gene count method (Rabier et al., 2014; Tiley et al., 2016).
Ks-based method for WGD identification
This method identifies a proliferation of duplicated genes from WGDs under the assumption that synonymous substitutions between duplicate genes accrue at a relatively constant rate. Here, each species was subjected to a reciprocal BLAST search to identify putative paralogous gene pairs in their protein coding sequences. Paralogous pairs were identified as sequences that demonstrated 40% sequence similarity over at least 300 bp from a discontiguous MegaBlast search (Zhang et al., 2000; Barker et al., 2008). Each paralogous protein sequence pair was aligned using MAFFT (Katoh & Standley, 2013) and then back translated to their coding sequences using pal2nal (Suyama et al., 2006). All sites containing gaps were removed from the alignment. Ks values for each duplicate pair were calculated using the maximum likelihood method implemented in codeml of the PAML package (Yang, 1997) under the F3×4 model (Goldman & Yang, 1994). To infer WGDs from the Ks distribution, we employed the one sample K-S goodness of fit test followed by 100 bootstrap resampling to assess statistical confidence (Cui et al., 2006). A significant p value (<0.05) rejected the null hypothesis of a birth-death process of gene duplication, thus supporting evidence of WGD. Because peaks produced by paleopolyploidy are expected to be approximately Gaussian (Blanc & Wolfe, 2004; Schlueter et al., 2004), we applied the EM algorithm to fit mixtures of Gaussian distributions to our data using the normalmixEM() function in the R package mixtools (Benaglia et al., 2009). Estimated mean peak values for each taxon are reported in Figs S3, S4. Alternative splicing can confound the signal of WGD using the Ks method (Barker et al., 2008). To alleviate this concern, sites containing gaps were removed from paralog alignments, thus transcript isoforms generated from alternative splicing will receive a Ks value of zero. All pairs with a Ks value of less than 0.001, which would include these transcript isoforms as well as recent tandem duplications, were discarded and not considered in the Ks distribution. Finally, to further explore the sensitivity of misassemblies and tandem gene duplications on WGD identification, we conducted the same Ks analysis above but applied a more stringent 0.95 threshold in the CD-HIT EST analysis (-c 0.95 -r 10).
Placing and dating WGDs using phylogenetic reconciliation and molecular divergence time estimation
We applied a phylogenetic approach to identify more precise placements of WGDs and to determine the approximate age of these events. We first reconciled each orthology tree to the species tree under the duplication-transfer-loss model (DTL) in Notung v2.9 (Chen et al., 2000). Here, total numbers of gene duplications inferred from well-supported gene tree nodes (>70 BS) are summarized onto the species tree. For each branch in the species tree, we calculated the percentage of genes duplicated along that branch (total number of inferred gene duplications along the branch / total number of genes containing at least one descended copy on that branch (i.e., from both single and duplicated gene copies)). The range of duplicated genes along branches where WGD was inferred was 10.0% to 84.3% (Table S4). Our threshold percentage for identifying a WGD was 10.0%, which is the lowest percentage for a terminal WGD as determined by our Ks analysis (in species Bhesa). Terminal WGDs are likely to exhibit a higher percentage of retained duplicated genes, and thus represent a more conservative filter for WGD assessment. This threshold is also well above the percentage identified from fully sequenced genomes and transcriptomes that do not exhibit recent WGDs. These fully sequenced genomes and transcriptomes instead show maximally only 1.2 to 2.2% of duplicated genes where tandem duplications, not WGDs, have been inferred (calculated from Chrysobalanus and Jatropha, Table S4). Together, this represents a reasonable approach for estimating WGDs in Malpighiales.
Following our phylogenetic localization of WGDs we applied a customized R script to extract the divergence times from our r8s analyses to summarize the age of gene duplications along each branch of the species tree. Only ages of nodes supported with >70 BS were used. The inferred distribution of divergence times was fitted to a mixture of Gaussian models using the R package mixtools as described above to estimate mean age of each WGD. In several cases, the best model inferred two WGDs along the same branch. These cases were independently supported by our phylogenetic analysis but with smaller percentages of duplications, presumably due to gene loss or missing data (Table S4). Estimated mean peak values for each taxon are reported in Table S4 and Fig. S5. Confidence intervals for mean age of each WGD were estimated using the 100 bootstrapped trees for each gene as outlined above (Table S4). To further explore the impact of root age on divergence time estimation using this penalized likelihood method, we fixed the root age to be 125 Ma, which is the maximum age of crown group eudicots based on the fossil record (Hughes & McDougall, 1990; Doyle & Hotton, 1991; Magallón et al., 2015). Moreover, this age is over 20 Ma older than the optimal age estimates for crown group Malpighiales (103.6 Ma) as inferred by Magallón et al. (2015). We applied the same set of minimum age constraints described above to conduct the divergence time estimation (Table S4).
Gene-count based method for confirmation of WGDs
To further assess statistical confidence for the 24 putative WGDs inferred from the Ks method and using the phylogenetic approach above, we applied a maximum likelihood method to test the number and placement of WGDs using gene count data (Rabier et al., 2014). This method estimates the likelihood of pre-specified putative WGDs on a phylogeny using a gene count matrix summarized across all orthologous genes. It is advantageous because it suffers less from false positive rates due to tandem duplication and assembly error (Rabier et al., 2014), which could create an artificial signal of polyploidization. We first tested the utility of this method by examining three independent WGDs previously identified from fully sequenced genomes using synteny analyses. These three WGDs were identified in (1.) Populus and Salix (Hanley et al., 2006; Tuskan et al., 2006); (2) Hevea and Manihot (Bredeson et al., 2016; Tang et al., 2016); (3) and in Linum (Z. Wang et al., 2012). Filtering of the gene count matrix to avoid missing data is critical for this method, which may otherwise lead to biased estimates (Rabier et al., 2014). To accomplish this, our dated species trees was first pruned to contain only species with fully sampled genomes, including these five species, plus Jatropha and Ricinus (where WGDs have not been previously detected) and Vitis (as outgroup). Next, a gene count matrix was summarized for all species across all ortholog trees (Fig. S6). We further conditioned the data matrix for all gene families to contain one or more gene copies descended from the branch along which the WGD was tested. Along these lines, gene families that were missing in the taxa affected by WGD were removed in each test. The conditional likelihoods were subsequently estimated for models with and without the WGD of interest using a prior geometric mean of 1.5 (Tiley et al., 2016). After convergence of the likelihood scores for all runs, we performed a series of likelihood ratio tests to determine the significance of individual WGDs. All three previously identified WGDs were successfully identified with confidence (LRT statistic >> 9.55, probability of type I error << 0.001). In addition, Ricinus and Jatropha showed no evidence of WGD (Table S4) suggesting a low false positive rate of this method.
We tested the remaining WGDs by sequentially adding species associated with each WGD to the seven-species phylogeny. In each case, we added all of the species from which a single WGD to be tested were descended and tested for a WGD along the added branch of interest. We generated the conditioned gene count matrix containing varying numbers of gene families as described above (Table S4).
Clustering of WGDs in time
To assess whether WGDs were clustered in time, we tested for the optimum number of clusters in age distribution using the finite Gaussian mixture modeling in R package mclust (Fraley & Raftery, 2002). The EM algorithm was used for mixture estimation and the Bayesian Information Criterion (BIC) was used for comprehensive clustering. In order to assess the confidence of the clustering, we conducted the same analysis on WGD age distributions derived from 100 bootstrap replicates. We conducted this Gaussian mixture modeling for age estimations derived from r8s analyses with fixed root age at 109 Ma as well as age estimations with fixed root age at 125 Ma. To further explore the clustering time of the more ancient WGDs, we excluded WGDs younger than 20 Ma and conducted the same Gaussian mixture modeling analysis.
Synteny based assessment of WGD in Linum
Synteny analysis serves as the gold standard for inferring WGDs, but is only amenable to the mostly completely assembled genomes. Our phylogenetic and Ks approach identified two WGDs in Linum. Here, we subsampled gene families containing three or four gene copies of Linum consistent with two duplications. The most closely related paralogous gene pairs of Linum were expected to arise from the most recent WGD, while their relationship with the remaining copy(ies) arose from the more ancient WGD (Fig. S7). We then mapped these paralogs onto the genome using MCScanX_h (Y. Wang et al., 2012) and visualized the result using RCircos (Zhang et al., 2013).
Results
Our final data set included 36 ingroup taxa derived from eight genomes and 28 transcriptomes (15 newly acquired for this study) plus six outgroup species (Table S1–3). Summary statistics of the transcriptome assemblies were reported in Table S5. The taxon sampling includes 21 traditionally recognized families in the order, thus representing the broad outline of phylodiversity within Malpighiales (sensu APG IV; Wurdack & Davis, 2009; Chase et al., 2016).
Our analyses identified a total of 22–24 WGDs broadly distributed across the Malpighiales phylogeny (Fig. 1). In nearly all cases these events are corroborated by all three methods. Our Ks analysis identified WGD in 22 species regardless of the sequence similarity threshold (Figs 1, S3, S4 for the 0.99 CD-HIT EST threshold; Fig S8 for the 0.95 CD-HIT EST threshold); our phylogenomic reconciliation analysis identified 24 WGDs (Table S4); and 22 WGDs were verified with the likelihood-based gene count method (LRT statistic > 9.55; Table S4). Moreover, these results are robust to data quality and phylogenetic uncertainty (by applying our bootstrap resampling procedure described earlier).
Fig. 1.
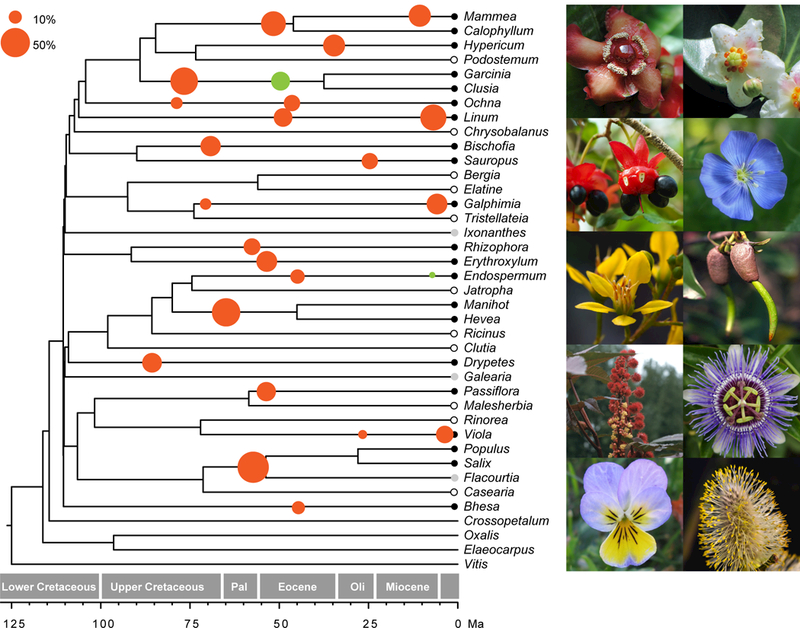
Phylogenetic distribution of whole genome duplications (WGDs) in Malpighiales. Species tree of Malpighiales inferred from 5113 gene trees using a summary coalescent method. WGDs identified from Ks analysis are illustrated with solid black dots on the terminal branches of corresponding species; species indicated with grey dots have transcriptomes that are potentially insufficient for adequate assessment of WGD using the Ks method. Two Salix species are collapsed into one terminal branch for simplicity. Circles illustrated along branches are dated WGDs (divergence time of parental genomes) from our phylogenomic reconciliation analysis. The radius of each circle is proportional to the percentage of orthologus genes supporting the WGD as determined from phylogenomic reconciliation (scale, top left). Solid red circles are significant WGDs as confirmed by our gene-count analysis; solid green circles indicate WGDs that do not receive significant support using the gene count method. Photos on the right are representatives of major Malpighiales clades sampled for investigating whole genome duplications, including from top to bottom (beginning with the left column): Garcinia, Ochna, Tristellateia, Manihot, Viola, Clusia, Linum, Rhizophora, Passiflora, and Salix. Ma, million years ago.
We additionally analyzed a reduced data set containing only completely sequenced genomes and high quality transcriptomes to further verify these results. Even with this more conservative data set, we still identified 22 WGDs using all three methods (Table S6). Three of the WGDs we identify validate those from previous studies in the common ancestor of Populus (Tuskan et al., 2006) and Salix (Sterck et al., 2005; Hanley et al., 2006), in the common ancestor of Manihot (Bredeson et al., 2016) and Hevea (Tang et al., 2016), and in Linum (Z. Wang et al., 2012). In addition, recent analyses have inferred two independent WGDs in Linum using Ks distributions (Z. Wang et al., 2012; Sveinsson et al., 2014). These events, however, have not previously been corroborated simultaneously due to the limited power of this method. Our reconciliation and gene-count method, by contrast, identifies these two duplications in the lineage leading to Linum. Furthermore, we verified these two independent WGDs using a phylogeny-guided synteny analysis (Jiao et al., 2014) based on 879 gene families containing three or four gene copies in Linum (Fig. S7). Here, we identified 15 syntenic regions across large scaffolds reflecting the four-parted paralogous relationship created by two independent WGDs in the Linum lineage.
Our divergence time estimates indicated that the timing of WGDs range broadly from 3.2–85.0 Ma (Fig. 2; Table S4). Surprisingly, however, these events are not randomly distributed in time. Instead, our Gaussian mixture model (Fraley & Raftery, 2002) indicates that the inferred ages of WGDs display a bimodal distribution (Bayesian Information Criterion, BIC −225.05, compared to BIC −227.30 for the univariate normal distribution) with peaks at the Eocene-Paleocene (mean age 53.89 Ma) and late Miocene (mean age 7.39 Ma), respectively. Our assessment using bootstrap replicates provide confident statistical support for this interpretation: 98% of the replicates support a bimodal distribution of WGDs with the mean age of each cluster ranging from 6.06–13.78 Ma and 52.26–57.79 Ma. In our case, an overwhelming number of WGDs (n = 19) occur during the older, early Eocene time period, versus only five during the more recent late Miocene period. When we excluded WGDs with mean age younger than 20 Ma, the 19 WGDs were estimated to cluster at 54.21 Ma according to the Gaussian mixture model analysis. The ages of these WGDs are also robust to alternative age constraints. When we fixed the root age of each gene tree at 125 Ma, age estimations of most WGDs were 0–9.9 Ma older than previous estimations except the recent WGD in Endospermum, which is 0.3 Ma younger (Table S4). However, regardless of these older ages our Gaussian mixture model still supported a clustering age of 56.8 Ma (54.81–60.42 Ma in the 100 bootstrap replicates), which remains closer to the Eocene–Paleocene transition than to the KT boundary.
Fig. 2.
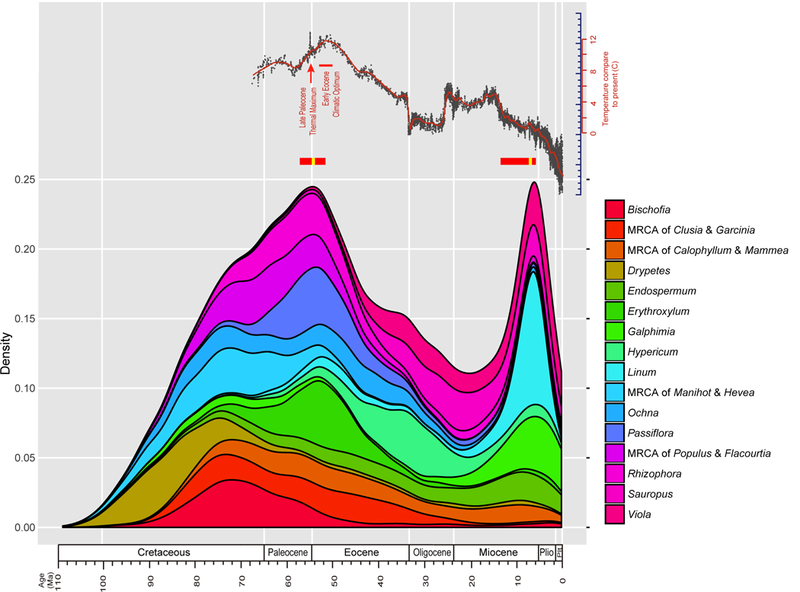
Age distributions of whole genome duplications (WGDs) among clades of Malpighiales. Density of divergence time of duplicated genes by taxa are plotted in millions of years (Ma). Varying colors refer to different clades exhibiting WGDs (Bhesa excluded for readability). Zachos et al. (2001) curve of global temperature fluctuations during the Cenozoic is redrawn at the top. Mean ages of Gaussian mixture model and variations estimated from bootstrap replicates are indicated by yellow and red bars below the Zachos curve, respectively.
Discussion
Massive WGDs in Malpighiales
The WGDs we identified are broadly distributed across 18 branches of the Malpighiales phylogeny. All of these WGDs are inferred in the crown groups of major clades where phylogenetic uncertainty is minimum. As a result, although the backbone of the phylogeny in Malpighiales is still challenging to resolve (species tree with bootstrap support Fig. S9), the phylogenetic placements of these WGDs are clear. Moreover, we identified the same set of WGDs in our full dataset as compared with the more stringent, filtered dataset (cf. Bhesa, which we excluded in the more conservative analysis). This result indicates that low-quality transcriptomes do not have a major impact on our assessment of WGD in Malpighiales.
Interestingly, these WGDs are commonly associated with the most diverse clades in the order, including in the clusioids, ochnoids, euphorbioids, phyllanthoids, violets, and passion flowers. Additionally, we note that some species-poor clades show no evidence of duplication (e.g., Malesherbia, Rinorea, and Elatinaceae, among others; Fig. 1), despite having species-rich sister clades. This lends tentative support to suggestions that WGDs may fuel species diversification (Tank et al., 2015), possibly via the establishment of reproductive barriers (Husband et al., 2016). However, other studies have concluded that although polyploidization is important to cladogenesis in plants, it likely does not enhance species diversification rates (Wood et al., 2009; Estep et al., 2014). We cannot adequately address this question with existing data (Kellogg, 2016), but regardless, our analyses set the stage for establishing the finer scale taxon sampling necessary to pinpoint these events to clarify the association between WGDs and the tempo of diversification in Malpighiales. Namely, do WGDs precede prolific diversification of Malpighiales subclades?
One WGD requires more detailed exploration. In our phylogenetic reconciliation and gene-count analysis, a WGD is inferred to predate the common ancestor of Populus, Salix, and Flacourtia (the Ks analysis is inconclusive for Flacourtia; Figs 1, S4). Chromosome count data, however, do not support such an early WGD. Instead, the chromosome number of Populus and Salix are approximately twice that of Flacourtia (2n = 38; versus 2n =20 or 22 in Flacourtia; IPCN Chromosome Reports, http://www.tropicos.org/Project/IPCN) suggesting that this WGD event likely occurred more recently, and is thus restricted to the common ancestor of Populus and Salix. A similar discrepancy in yeast involves the identification of an older WGD using phylogenetic reconciliation versus a more recent WGD inferred using a gold standard synteny-guided genome comparison (Scannell et al., 2007; Marcet-Houben & Gabaldón, 2015). Here, the authors provide reasonable evidence that the older WGD is spurious and confounded by an allopolyploidization event resulting from hybridization. The nature of this allopolyploidization resulted in a deeper, yet spurious, phylogenetic placement of the WGD. It is important to recognize here that gene-tree data are limited in some respects since they provide an estimate of the divergence times of the parental diploid genomes, but are less conclusive around exactly when the hybridization and polyploidization event occurred (Gaut & Doebley, 1997; Doyle & Egan, 2010); dates in Table S4 are thus likely older than the actual polyploidization events. In light of these results, a plausible hypothesis is that the WGD shared by Populus and Salix results from an allopolyploidization in which an ancestor of the Flacourtia lineage served as one parental lineage. Testing and evaluating this hypothesis remains a challenge (Goulet et al., 2017), and is an obvious avenue for future research. Regardless, we do not anticipate this phenomenon to be a pervasive problem for our analysis given that the origin of viable polyploids derived from widely disparate phylogenetic lineages appears to be rare, and thus not likely to greatly influence our placement and dating of the large number of WGDs identified here.
These results further point toward a propensity for pervasive and widespread WGDs in angiosperms. Recent and ongoing investigations incorporating vast nuclear genomic data and extended taxon sampling indicate that other similarly diverse clades, including Asteraceae (Barker et al., 2008; Huang et al., 2016), Poaceae (Estep et al., 2014), and Caryophyllales (Yang et al., 2015; Yang et al., 2017) also show histories characterized by prolific WGDs. Collectively, these results suggest that Malpighiales and other previously examined clades represent a more pervasive pattern of WGDs characteristic of possibly hundreds of major angiosperm clades (and extending to other vascular plant clades, including gymnosperms and ferns; Wood et al., 2009; Li et al., 2015). Further investigation is required to address this question more broadly, but this seems a plausible hypothesis in light of recent findings.
Timing of WGDs coincides with events of climate upheaval
WGDs in Malpighiales show a bimodal distribution through time, which is supported by 98% of our bootstrap replicates. The majority of the WGDs were identified to cluster around the Eocene–Paleocene Transition (mean age 53.89 Ma) during which time the planet was warmer and wetter than any period in the Cenozoic. The age of this older peak is estimated to range between 52.26–57.79 Ma based on our bootstrap replicates, which falls entirely within the prolonged warming trend from the Late Paleocene through the early Eocene (58–50 Ma, Zachos et al., 2008). Moreover, the late Paleocene-early Eocene age of this peak is also robust when we excluded WGDs younger than 20 Ma (mean age 54.21 Ma) or apply an older root age of 125 Ma consistent with the oldest eudicots (mean age 56.8 Ma). Previously, WGD has been hypothesized to buffer plants though episodes of major global and climatic upheavals (Fawcett et al., 2009). Studies have identified WGDs associated with the earlier KT boundary (c. 65 Ma) when a large meteor impacted off the Yucatán Peninsula disrupting the global climate, precipitating a major reorganization of the terrestrial biota (Fawcett et al., 2009; Vanneste et al., 2014). Similarly, the late Miocene-early Pliocene (c. 10–5 Ma) has been implicated as another period of climatic instability when WGDs were pervasive (Estep et al., 2014). The expansion of C4 grassland as a result of widespread global aridification (Cerling et al., 1997; Arakaki et al., 2011) in particular, has been inferred to be correlated with numerous polyploidizations in grasses, which are among the most important members of these arid and cooler habitats that bear their namesake, i.e., grassland and steppe biomes (Estep et al., 2014).
By contrast, relatively little is known about WGDs during the Eocene. This is surprising because the Eocene-Paleocene transition is associated with an extended and prolonged period of intense warming. Most notably, the Palaeocene-Eocene Thermal Maximum (PETM; 56 Ma) and the subsequent Eocene Climatic Optimum (49 Ma) constitute the warmest and most humid period during the Cenozoic. During this time, mean global temperatures increased by 5 to 10°C due to massive release of 13C-depleted carbon (Pagani et al., 2006; Zeebe et al., 2009). This dramatic climate upheaval is thought to have stimulated profound reshuffling of the terrestrial biome spurring plant migrations, extensive species turnover, and accelerated species diversification in numerous plant and animal clades (Clyde & Gingerich, 1998; Wilf, 2000; Bowen et al., 2002; Wing et al., 2005; McKenna & Farrell, 2006; Ramírez et al., 2007; Schuettpelz & Pryer, 2009; Jaramillo et al., 2010). Our results establish a record of at least 19 WGDs during this period, suggesting a role in adaptation during Palaeocene-Eocene warming. We hypothesize that this may be common for predominantly tropical groups, like Malpighiales (Davis et al., 2005), which are likely much more impacted by warming given the relatively tight thermal tolerances exhibited by many such groups (Janzen, 1967; Tewksbury et al., 2008; Wright et al., 2009).
What may have stimulated interactions that facilitated increased polyploid formation during the warmer, wetter period of the Eocene beyond the generally increased rates of angiosperm diversification during this window of time? One hypothesis, coined the ‘neutral’ process (Van de Peer et al., 2017), posits that these sorts of upheavals increase the formation of unreduced gametes and therefore result in an excess of polyploids. It is widely appreciated that external stimuli, temperature in particular, has a pronounced effect on unreduced gamete formation (De Storme & Geelen, 2014). In this case, it appears that both high and low temperatures can promote unreduced gametes in various taxa as demonstrated in Arabidopsis (De Storme et al., 2012) and some roses (Pécrix et al., 2011), respectively. In addition, a recent study in Arabidopsis thaliana demonstrated that supernumerary sperm fusion could generate viable polyploid offspring that exhibited vegetative vigor (Nakel et al., 2017). Thus, the extensive early Eocene warming and even the Miocene aridification might have significantly increased unreduced gametes, perhaps contributing to enhanced polyploid formation. This is supported by evidence of increased levels of unreduced gametes in gymnosperms and lycophytes during comparable upheavals, including during the Triassic–Jurassic (Kürschner et al., 2013) and Permian–Triassic (Visscher et al., 2004; Foster & Afonin, 2005) transitions.
Although such polyploidizations might have initially arisen more neutrally, it is also possible that polyploids were adapted for survival in these changing landscapes (the ‘adaptive’ processes; Van de Peer et al., 2017). Of course, both neutral and adaptive processes can contribute simultaneously to the formation and establishment of polyploids. Polyploids are often viewed as evolutionary dead ends owing to their small population sizes, relatively restricted distributions, high extinction risks, and seemingly sparser representation in the deep angiosperm phylogeny (Stebbins, 1970; Comai, 2005; Mayrose et al., 2011). However, these may not apply under less stable environments, such as during major climatic upheavals when polyploids may outperform their diploid progenitors (Van de Peer et al., 2017). It has been hypothesized that such genomic novelty and epigenetic repatterning may result in phenotypic variability, including variants that confer selective advantages in stressful conditions (Wendel, 2000; Comai, 2005; Madlung, 2013; Van de Peer et al., 2017). In particular, the advantages of especially allopolyploids include altered gene expression leading to hybrid vigor and increased genetic variation (Comai, 2005; Lynch, 2007). Along these lines, polyploids have been reported to be more frequent at higher latitudes and in xeric environments, which may mimic such upheavals (Löve & Löve, 1949; Löve, 1953; Hanelt, 1966; Brochmann et al., 2004). Polyploids also occur with greater frequency among invasive plants, which commonly become established on disturbed grounds (Prentis et al., 2008; te Beest et al., 2011). In support of this argument, Brochmann et al. (2004) reported an unexpected overabundance of recently formed polyploids in newly deglaciated areas in the Arctic. Additionally, megaflora fossils from Wyoming, United States indicate that the dynamics of plant community assembly after dramatic warming during the PETM is very similar to late and postglacial floras (Wing et al., 2005), suggesting that observations for the Arctic may represent similar responses to warmer and wetter periods during the Eocene transition. Regardless of these competing ideas, the striking propensity and clustered distribution of WGDs in time, strengthens the hypothesis that polyploidization may be an important means of lineage persistence during episodes of major global change.
Supplementary Material
BUSCO assessment of transcriptome and genomic completeness of 42 sampled species.
Bioinfomatic pipeline depicting transcriptome assembly, homology and orthology inferences, and three methods for WGD identification, placement, and dating.
Histograms of the synonymous substitutions per synonymous site (Ks) of duplicated gene pairs among 22 Malpighiales taxa showing WGDs.
Histograms of the synonymous substitutions per synonymous site (Ks) of duplicated gene pairs among 11 Malpighiales taxa where WGD is absent (or inconclusive in the case of three taxa).
Histograms depicting divergence time estimations inferred using penalized likelihood for 22 species that exhibited WGDs (summarized in Fig. S3).
Gene count matrix for 5113 ortholog groups of 36 Malpighiales species.
Phylogeny-guided synteny analyses demonstrate successive WGDs in Linum.
Histograms of the synonymous substitutions per synonymous site (Ks) of duplicated gene pairs among 22 Malpighiales taxa showing WGDs.
Cladogram of 50% majority-rule bootstrap tree of Malpighiales inferred from 5113 gene trees using ASTRAL.
Voucher and GenBank information for 15 species in Malpighiales used for de novo transcriptome assembly
Taxa sampled from the OneKP data set and the corresponding OneKP library ID
Taxa sampled with complete genomes and corresponding reference
Whole genome duplications (WGDs) in Malpighiales identified with complete taxon sampling, including their phylogenetic placement, percentage of gene families supporting gene duplication, log likelihood, and age estimations of each WGD
Summary statistics for the transcriptome assembly in Malpighiales
Whole genome duplications (WGDs) in Malpighiales identified with more conservative taxon sampling, including their phylogenetic placement, percentage of gene families supporting gene duplication, log likelihood, and age estimations of each WGD
Please note: Wiley Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
We thank S. Edwards, G. Giribet, E. Kellogg, A. Knoll, and members of the Davis laboratory for technical assistance and valuable discussions. D. Soltis provided access to the Malpighiales, Celastrales, and Oxalidales sampling from the 1KP project and read a final version of the draft in October 2017. Funding for this study came from US National Science Foundation (NSF) Assembling the Tree of Life grant DEB-0622764, DEB-1120243, and DEB-0544039 (to C.C.D.) and from Harvard University. J.S.R. acknowledges financial support from the US National Institutes of Health/National Institute of General Medical Sciences grant R01 GM108904. A.M.A. acknowledges financial support from Brazil National Council for Scientific and Technological Development (CNPq) PROTAX Malpighiales grant 440543/2015–0 and Research Productivity Fellowship grant 310717/2015–9.
Footnotes
Author contributions
L.C. and C.C.D. designed the research; L.C. analyzed the data; Z.X. collected the data; L.C. and C.C.D. wrote the initial paper draft with input from A.M.A., M.S., J.S.R. and L.L.
References
- Andrews S 2010. FastQC: a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Arakaki M, Christin P- A, Nyffeler R, Lendel A, Eggli U, Ogburn RM, Spriggs E, Moore MJ, Edwards EJ. 2011. Contemporaneous and recent radiations of the world’s major succulent plant lineages. Proceedings of the National Academy of Sciences of the United States of America 108(20): 8379–8384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker MS, Arrigo N, Baniaga AE, Li Z, Levin DA. 2016. On the relative abundance of autopolyploids and allopolyploids. New Phytologist 210(2): 391–398. [DOI] [PubMed] [Google Scholar]
- Barker MS, Kane NC, Matvienko M, Kozik A, Michelmore RW, Knapp SJ, Rieseberg LH. 2008. Multiple paleopolyploidizations during the evolution of the Compositae reveal parallel patterns of duplicate gene retention after millions of years. Molecular Biology and Evolution 25(11): 2445–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker MS, Vogel H, Schranz ME. 2009. Paleopolyploidy in the Brassicales: analyses of the Cleome transcriptome elucidate the history of genome duplications in Arabidopsis and other Brassicales. Genome Biology and Evolution 1: 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benaglia T, Chauveau D, Hunter D, Young D. 2009. mixtools: an R package for analyzing finite mixture models. Journal of Statistical Software 32(6): 1–29. [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456(7218): 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertioli DJ, Moretzsohn MC, Madsen LH, Sandal N, Leal-Bertioli SC, Guimarães PM, Hougaard BK, Fredslund J, Schauser L, Nielsen AM. 2009. An analysis of synteny of Arachis with Lotus and Medicago sheds new light on the structure, stability and evolution of legume genomes. BMC Genomics 10(1): 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc G, Wolfe KH. 2004. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. The Plant Cell 16(7): 1667–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen GJ, Clyde WC, Koch PL, Ting S, Alroy J, Tsubamoto T, Wang Y, Wang Y. 2002. Mammalian dispersal at the Paleocene/Eocene boundary. Science 295(5562): 2062–2065. [DOI] [PubMed] [Google Scholar]
- Bredeson JV, Lyons JB, Prochnik SE, Wu GA, Ha CM, Edsinger-Gonzales E, Grimwood J, Schmutz J, Rabbi IY, Egesi C. 2016. Sequencing wild and cultivated cassava and related species reveals extensive interspecific hybridization and genetic diversity. Nature Biotechnology 34(5): 562–570. [DOI] [PubMed] [Google Scholar]
- Brochmann C, Brysting A, Alsos I, Borgen L, Grundt H, Scheen A- C, Elven R. 2004. Polyploidy in arctic plants. Botanical Journal of the Linnean Society 82(4): 521–536. [Google Scholar]
- Cannon SB, McKain MR, Harkess A, Nelson MN, Dash S, Deyholos MK, Peng Y, Joyce B, Stewart CN, Rolf M. 2015. Multiple polyploidy events in the early radiation of nodulating and nonnodulating legumes. Molecular Biology and Evolution 32(1): 193–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- <J/>Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics (Oxford, England) 25(15): 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerling TE, Harris JM, MacFadden BJ, Leakey MG, Quade J, Eisenmann V, Ehleringer JR. 1997. Global vegetation change through the Miocene/Pliocene boundary. Nature 389(6647): 153–158. [Google Scholar]
- Chase M, Christenhusz M, Fay M, Byng J, Judd W, Soltis D, Mabberley D, Sennikov A, Soltis P, Stevens P. 2016. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Botanical Journal of the Linnean Society 181(1): 1–20. [Google Scholar]
- Chen K, Durand D, Farach-Colton M. 2000. NOTUNG: a program for dating gene duplications and optimizing gene family trees. Journal of Computational Biology 7(3–4): 429–447. [DOI] [PubMed] [Google Scholar]
- Clyde WC, Gingerich PD. 1998. Mammalian community response to the latest Paleocene thermal maximum: an isotaphonomic study in the northern Bighorn Basin, Wyoming. Geology 26(11): 1011–1014. [Google Scholar]
- Comai L 2005. The advantages and disadvantages of being polyploid. Nature Reviews Genetics 6(11): 836–846. [DOI] [PubMed] [Google Scholar]
- Cui L, Wall PK, Leebens-Mack JH, Lindsay BG, Soltis DE, Doyle JJ, Soltis PS, Carlson JE, Arumuganathan K, Barakat A. 2006. Widespread genome duplications throughout the history of flowering plants. Genome Research 16(6): 738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Hu Q, Cai Q, Feng K, Ye N, Tuskan GA, Milne R, Chen Y, Wan Z, Wang Z. 2014. The willow genome and divergent evolution from poplar after the common genome duplication. Cell Research 24(10): 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CC, Webb CO, Wurdack KJ, Jaramillo CA, Donoghue MJ. 2005. Explosive radiation of Malpighiales supports a mid-Cretaceous origin of modern tropical rain forests. The American Naturalist 165(3): E36–E65. [DOI] [PubMed] [Google Scholar]
- De Storme N, Copenhaver GP, Geelen D. 2012. Production of diploid male gametes in Arabidopsis by cold-induced destabilization of postmeiotic radial microtubule arrays. Plant Physiology 160(4): 1808–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Storme N, Geelen D. 2014. The impact of environmental stress on male reproductive development in plants: biological processes and molecular mechanisms. Plant, Cell & Environment 37(1): 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JA, Hotton CL. 1991. Diversification of early angiosperm pollen in a cladistic context. Pollen and spores: patterns of diversification 169: 195. [Google Scholar]
- Doyle JJ, Egan AN. 2010. Dating the origins of polyploidy events. New Phytologist 186(1): 73–85. [DOI] [PubMed] [Google Scholar]
- Estep MC, McKain MR, Diaz DV, Zhong J, Hodge JG, Hodkinson TR, Layton DJ, Malcomber ST, Pasquet R, Kellogg EA. 2014. Allopolyploidy, diversification, and the Miocene grassland expansion. Proceedings of the National Academy of Sciences of the United States of America 111(42): 15149–15154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JA, Maere S, Van de Peer Y. 2009. Plants with double genomes might have had a better chance to survive the Cretaceous–Tertiary extinction event. Proceedings of the National Academy of Sciences of the United States of America 106(14): 5737–5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster C, Afonin S. 2005. Abnormal pollen grains: an outcome of deteriorating atmospheric conditions around the Permian–Triassic boundary. Journal of the Geological Society 162(4): 653–659. [Google Scholar]
- Fraley C, Raftery AE. 2002. Model-based clustering, discriminant analysis, and density estimation. Journal of the American Statistical Association 97(458): 611–631. [Google Scholar]
- Gaut BS, Doebley JF. 1997. DNA sequence evidence for the segmental allotetraploid origin of maize. Proceedings of the National Academy of Sciences of the United States of America 94(13): 6809–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman N, Yang Z. 1994. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Molecular Biology and Evolution 11(5): 725–736. [DOI] [PubMed] [Google Scholar]
- Goulet BE, Roda F, Hopkins R. 2017. Hybridization in plants: old ideas, new techniques. Plant Physiology 173(1): 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 29(7): 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Steinmetz LM, Gu X, Scharfe C, Davis RW, Li W- H. 2003. Role of duplicate genes in genetic robustness against null mutations. Nature 421(6918): 63–66. [DOI] [PubMed] [Google Scholar]
- Haas B, Papanicolaou A 2012. Transdecoder. https://transdecoder.github.io/.
- Hanelt P 1966. Polyploidie-Frequenz und geographische Verbreitung bei hohereren Pflanzen. Biologische Rundschau 4: 183–196. [Google Scholar]
- Hanley S, Mallott M, Karp A. 2006. Alignment of a Salix linkage map to the Populus genomic sequence reveals macrosynteny between willow and poplar genomes. Tree Genetics & Genomes 3(1): 35–48. [Google Scholar]
- Huang C- H, Zhang C, Liu M, Hu Y, Gao T, Qi J, Ma H. 2016. Multiple polyploidization events across Asteraceae with two nested events in the early history revealed by nuclear phylogenomics. Molecular Biology and Evolution: msw 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes NF, McDougall AB. 1990. Barremian-Aptian angiospermid pollen records from southern England. Review of Palaeobotany and Palynology 65(1–4): 145–151. [Google Scholar]
- Husband BC, Baldwin SJ, Sabara HA. 2016. Direct vs. indirect effects of whole-genome duplication on prezygotic isolation in Chamerion angustifolium: Implications for rapid speciation. American Journal of Botany 103(7): 1259–1271. [DOI] [PubMed] [Google Scholar]
- Janzen DH. 1967. Why mountain passes are higher in the tropics. The American Naturalist 101(919): 233–249. [Google Scholar]
- Jaramillo C, Ochoa D, Contreras L, Pagani M, Carvajal-Ortiz H, Pratt LM, Krishnan S, Cardona A, Romero M, Quiroz L. 2010. Effects of rapid global warming at the Paleocene-Eocene boundary on neotropical vegetation. Science 330(6006): 957–961. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Leebens-Mack J, Ayyampalayam S, Bowers JE, McKain MR, McNeal J, Rolf M, Ruzicka DR, Wafula E, Wickett NJ. 2012. A genome triplication associated with early diversification of the core eudicots. Genome Biology 13(1): 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Li J, Tang H, Paterson AH. 2014. Integrated syntenic and phylogenomic analyses reveal an ancient genome duplication in monocots. The Plant Cell 26(7): 2792–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS. 2011. Ancestral polyploidy in seed plants and angiosperms. Nature 473(7345): 97–100. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30(4): 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg EA. 2016. Has the connection between polyploidy and diversification actually been tested? Current Opinion in Plant Biology 30: 25–32. [DOI] [PubMed] [Google Scholar]
- Krueger F 2015. Trim galore. A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files. https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
- Kürschner WM, Batenburg SJ, Mander L. 2013. Aberrant Classopollis pollen reveals evidence for unreduced (2n) pollen in the conifer family Cheirolepidiaceae during the Triassic–Jurassic transition. Proceedings of the Royal Society of London. Series B, Biological Sciences 280(1768): 20131708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner M, Findeiß S, Steiner L, Marz M, Stadler PF, Prohaska SJ. 2011. Proteinortho: detection of (co-) orthologs in large-scale analysis. BMC Bioinformatics 12(1): 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22(13): 1658–1659. [DOI] [PubMed] [Google Scholar]
- Li Z, Baniaga AE, Sessa EB, Scascitelli M, Graham SW, Rieseberg LH, Barker MS. 2015. Early genome duplications in conifers and other seed plants. Science Advances 1(10): e1501084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löve Á 1953. Subarctic polyploidy. Hereditas 39(1–2): 113–124. [Google Scholar]
- Löve Á, Löve D. 1949. The geobotanical significance of polyploidy. I. Polyploidy and latitude. Portugaliae Acta Biologica Ser. A RB Goldschmidt: 273–352. [Google Scholar]
- Lynch M 2007. The origins of genome architecture: Sinauer Associates Sunderland. [Google Scholar]
- Lynch M, Conery JS. 2000. The evolutionary fate and consequences of duplicate genes. Science 290(5494): 1151–1155. [DOI] [PubMed] [Google Scholar]
- Lynch M, Force AG. 2000. The origin of interspecific genomic incompatibility via gene duplication. The American Naturalist 156(6): 590–605. [DOI] [PubMed] [Google Scholar]
- Madlung A 2013. Polyploidy and its effect on evolutionary success: old questions revisited with new tools. Heredity 110(2): 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magallón S, Gómez-Acevedo S, Sánchez-Reyes LL, Hernández-Hernández T. 2015. A metacalibrated time-tree documents the early rise of flowering plant phylogenetic diversity. New Phytologist 207(2): 437–453. [DOI] [PubMed] [Google Scholar]
- Marcet-Houben M, Gabaldón T. 2015. Beyond the whole-genome duplication: phylogenetic evidence for an ancient interspecies hybridization in the baker’s yeast lineage. PLOS Biology 13(8): e1002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcussen T, Heier L, Brysting AK, Oxelman B, Jakobsen KS. 2014. From gene trees to a dated allopolyploid network: insights from the angiosperm genus Viola (Violaceae). Systematic Biology 64 (1): 84–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcussen T, Jakobsen KS, Danihelka J, Ballard HE, Blaxland K, Brysting AK, Oxelman B. 2012. Inferring species networks from gene trees in high-polyploid North American and Hawaiian violets (Viola, Violaceae). Systematic Biology 61(1): 107–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matasci N, Hung L- H, Yan Z, Carpenter EJ, Wickett NJ, Mirarab S, Nguyen N, Warnow T, Ayyampalayam S, Barker M. 2014. Data access for the 1,000 Plants (1KP) project. GigaScience 3(1): 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayrose I, Barker MS, Otto SP. 2009. Probabilistic models of chromosome number evolution and the inference of polyploidy. Systematic Biology 59(2): 132–144. [DOI] [PubMed] [Google Scholar]
- Mayrose I, Zhan SH, Rothfels CJ, Arrigo N, Barker MS, Rieseberg LH, Otto SP. 2015. Methods for studying polyploid diversification and the dead end hypothesis: a reply to Soltis et al. (2014). New Phytologist 206(1): 27–35. [DOI] [PubMed] [Google Scholar]
- Mayrose I, Zhan SH, Rothfels CJ, Magnuson-Ford K, Barker MS, Rieseberg LH, Otto SP. 2011. Recently formed polyploid plants diversify at lower rates. Science 333(6047): 1257–1257. [DOI] [PubMed] [Google Scholar]
- McKenna DD, Farrell BD. 2006. Tropical forests are both evolutionary cradles and museums of leaf beetle diversity. Proceedings of the National Academy of Sciences of the United States of America 103(29): 10947–10951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirarab S, Warnow T. 2015. ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics 31(12): i44–i52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakel T, Tekleyohans DG, Mao Y, Fuchert G, Vo D, Groß-Hardt R. 2017. Triparental plants provide direct evidence for polyspermy induced polyploidy. Nature Communications 8(1): 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novaes E, Drost DR, Farmerie WG, Pappas GJ, Grattapaglia D, Sederoff RR, Kirst M. 2008. High-throughput gene and SNP discovery in Eucalyptus grandis, an uncharacterized genome. BMC Genomics 9(1): 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S 1970. Evolution by gene duplication: New York, NY, USA: Springer Science & Business Media. [Google Scholar]
- Pagani M, Caldeira K, Archer D, Zachos JC. 2006. An ancient carbon mystery. Science 314(5805): 1556–1557. [DOI] [PubMed] [Google Scholar]
- Pécrix Y, Rallo G, Folzer H, Cigna M, Gudin S, Le Bris M. 2011. Polyploidization mechanisms: temperature environment can induce diploid gamete formation in Rosa sp. Journal of Experimental Botany 62(10): 3587–3597. [DOI] [PubMed] [Google Scholar]
- Prentis PJ, Wilson JR, Dormontt EE, Richardson DM, Lowe AJ. 2008. Adaptive evolution in invasive species. Trends in Plant Science 13(6): 288–294. [DOI] [PubMed] [Google Scholar]
- Rabier C- E, Ta T, Ané C. 2014. Detecting and locating whole genome duplications on a phylogeny: a probabilistic approach. Molecular Biology and Evolution 31(3): 750–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez SR, Gravendeel B, Singer RB, Marshall CR, Pierce NE. 2007. Dating the origin of the Orchidaceae from a fossil orchid with its pollinator. Nature 448(7157): 1042–1045. [DOI] [PubMed] [Google Scholar]
- Ruhfel BR, Stevens PF, Davis CC. 2013. Combined morphological and molecular phylogeny of the clusioid clade (Malpighiales) and the placement of the ancient rosid macrofossil Paleoclusia. International Journal of Plant Sciences 174(6): 910–936. [Google Scholar]
- Sanderson MJ. 2003. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19(2): 301–302. [DOI] [PubMed] [Google Scholar]
- Sanderson MJ. 2004. r8s, version 1.70 user’s manual. http://loco.biosci.arizona.edu/r8s/r8s1.
- Scannell DR, Frank AC, Conant GC, Byrne KP, Woolfit M, Wolfe KH. 2007. Independent sorting-out of thousands of duplicated gene pairs in two yeast species descended from a whole-genome duplication. Proceedings of the National Academy of Sciences of the United States of America 104(20): 8397–8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlueter JA, Dixon P, Granger C, Grant D, Clark L, Doyle JJ, Shoemaker RC. 2004. Mining EST databases to resolve evolutionary events in major crop species. Genome 47(5): 868–876. [DOI] [PubMed] [Google Scholar]
- Schuettpelz E, Pryer KM. 2009. Evidence for a Cenozoic radiation of ferns in an angiosperm-dominated canopy. Proceedings of the National Academy of Sciences of the United States of America 106(27): 11200–11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19): 3210–3212. [DOI] [PubMed] [Google Scholar]
- Soltis DE, Segovia-Salcedo MC, Jordon-Thaden I, Majure L, Miles NM, Mavrodiev EV, Mei W, Cortez MB, Soltis PS, Gitzendanner MA. 2014. Are polyploids really evolutionary dead-ends (again)? A critical reappraisal of Mayrose et al. (2011). New Phytologist 202(4): 1105–1117. [DOI] [PubMed] [Google Scholar]
- Song L, Florea L. 2015. Rcorrector: efficient and accurate error correction for Illumina RNA-seq reads. GigaScience 4(1): 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A 2014a. The RAxML v8. 2. X Manual. https://sco.h-its.org/exelixis/resource/download/NewManual.pdf.
- Stamatakis A 2014b. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9): 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins GL. 1947. Types of polyploids: their classification and significance. Advances in Genetics 1: 403–429. [DOI] [PubMed] [Google Scholar]
- Stebbins GL. 1970. Variation and evolution in plants: Progress during the past twenty years Essays in evolution and genetics in honor of Theodosius Dobzhansky: A supplement to evolutionary biology. Boston, MA, USA: Springer Science & Business Media, 173–208. [Google Scholar]
- Sterck L, Rombauts S, Jansson S, Sterky F, Rouzé P, Van de Peer Y. 2005. EST data suggest that poplar is an ancient polyploid. New Phytologist 167(1): 165–170. [DOI] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P. 2006. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Research 34(suppl_2): W609–W612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sveinsson S, McDill J, Wong GK, Li J, Li X, Deyholos MK, Cronk QC. 2014. Phylogenetic pinpointing of a paleopolyploidy event within the flax genus (Linum) using transcriptomics. Annals of Botany 113(5): 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Yang M, Fang Y, Luo Y, Gao S, Xiao X, An Z, Zhou B, Zhang B, Tan X. 2016. The rubber tree genome reveals new insights into rubber production and species adaptation. Nature Plants 2: 16073. [DOI] [PubMed] [Google Scholar]
- Tang H, Bowers JE, Wang X, Paterson AH. 2010. Angiosperm genome comparisons reveal early polyploidy in the monocot lineage. Proceedings of the National Academy of Sciences of the United States of America 107(1): 472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank DC, Eastman JM, Pennell MW, Soltis PS, Soltis DE, Hinchliff CE, Brown JW, Sessa EB, Harmon LJ. 2015. Nested radiations and the pulse of angiosperm diversification: increased diversification rates often follow whole genome duplications. New Phytologist 207(2): 454–467. [DOI] [PubMed] [Google Scholar]
- Taylor JS, Raes J. 2004. Duplication and divergence: the evolution of new genes and old ideas. Annual Review of Genetics 38: 615–643. [DOI] [PubMed] [Google Scholar]
- te Beest M, Le Roux JJ, Richardson DM, Brysting AK, Suda J, Kubešová M, Pyšek P. 2011. The more the better? The role of polyploidy in facilitating plant invasions. Annals of Botany 109(1): 19–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewksbury JJ, Huey RB, Deutsch CA. 2008. Putting the heat on tropical animals. Science 320(5881): 1296–1297. [DOI] [PubMed] [Google Scholar]
- Tiley GP, Ané C, Burleigh JG. 2016. Evaluating and characterizing ancient whole-genome duplications in plants with gene count data. Genome Biology and Evolution 8(4): 1023–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A. 2006. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313(5793): 1596–1604. [DOI] [PubMed] [Google Scholar]
- Van de Peer Y, Mizrachi E, Marchal K. 2017. The evolutionary significance of polyploidy. Nature Reviews Genetics 18(7): 411–424. [DOI] [PubMed] [Google Scholar]
- Vanneste K, Baele G, Maere S, Van de Peer Y. 2014. Analysis of 41 plant genomes supports a wave of successful genome duplications in association with the Cretaceous–Paleogene boundary. Genome Research 24(8): 1334–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher H, Looy CV, Collinson ME, Brinkhuis H, Van Konijnenburg-Van Cittert JH, Kürschner WM, Sephton MA. 2004. Environmental mutagenesis during the end-Permian ecological crisis. Proceedings of the National Academy of Sciences of the United States of America 101(35): 12952–12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tang H, DeBarry JD, Tan X, Li J, Wang X, Lee T-h, Jin H, Marler B, Guo H 2012. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Research 40(7): e49–e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Hobson N, Galindo L, Zhu S, Shi D, McDill J, Yang L, Hawkins S, Neutelings G, Datla R. 2012. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. The Plant Journal 72(3): 461–473. [DOI] [PubMed] [Google Scholar]
- Wendel JF 2000. Genome evolution in polyploids Plant Molecular Evolution. Dordrecht, the Netherlands: Springer Science & Business Media, 225–249. [PubMed] [Google Scholar]
- Werth CR, Windham MD. 1991. A model for divergent, allopatric speciation of polyploid pteridophytes resulting from silencing of duplicate-gene expression. The American Naturalist 137(4): 515–526. [Google Scholar]
- Wickett NJ, Mirarab S, Nguyen N, Warnow T, Carpenter E, Matasci N, Ayyampalayam S, Barker MS, Burleigh JG, Gitzendanner MA. 2014. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proceedings of the National Academy of Sciences of the United States of America 111(45): E4859–E4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilf P 2000. Late Paleocene–early Eocene climate changes in southwestern Wyoming: Paleobotanical analysis. Geological Society of America Bulletin 112(2): 292–307. [Google Scholar]
- Wing SL, Harrington GJ, Smith FA, Bloch JI, Boyer DM, Freeman KH. 2005. Transient floral change and rapid global warming at the Paleocene-Eocene boundary. Science 310(5750): 993–996. [DOI] [PubMed] [Google Scholar]
- Wood TE, Takebayashi N, Barker MS, Mayrose I, Greenspoon PB, Rieseberg LH. 2009. The frequency of polyploid speciation in vascular plants. Proceedings of the National Academy of Sciences of the United States of America 106(33): 13875–13879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SJ, Muller‐Landau HC, Schipper J. 2009. The future of tropical species on a warmer planet. Conservation Biology 23(6): 1418–1426. [DOI] [PubMed] [Google Scholar]
- Wurdack KJ, Davis CC. 2009. Malpighiales phylogenetics: gaining ground on one of the most recalcitrant clades in the angiosperm tree of life. American Journal of Botany 96(8): 1551–1570. [DOI] [PubMed] [Google Scholar]
- Xi Z, Bradley RK, Wurdack KJ, Wong K, Sugumaran M, Bomblies K, Rest JS, Davis CC. 2012a. Horizontal transfer of expressed genes in a parasitic flowering plant. BMC Genomics 13(1): 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z, Ruhfel BR, Schaefer H, Amorim AM, Sugumaran M, Wurdack KJ, Endress PK, Matthews ML, Stevens PF, Mathews S. 2012b. Phylogenomics and a posteriori data partitioning resolve the Cretaceous angiosperm radiation Malpighiales. Proceedings of the National Academy of Sciences of the United States of America 109(43): 17519–17524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Moore M, Brockington S, Mikenas J, Olivieri J, Walker J, Smith S. 2017. Improved transcriptome sampling pinpoints 26 paleopolyploidy events in Caryophyllales, including two Paleo-allopolyploidy events. New Phytologist. 217(2):855–870. [DOI] [PubMed] [Google Scholar]
- Yang Y, Moore MJ, Brockington SF, Soltis DE, Wong GK- S, Carpenter EJ, Zhang Y, Chen L, Yan Z, Xie Y. 2015. Dissecting molecular evolution in the highly diverse plant clade Caryophyllales using transcriptome sequencing. Molecular Biology and Evolution 32(8): 2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Smith SA. 2014. Orthology inference in nonmodel organisms using transcriptomes and low-coverage genomes: improving accuracy and matrix occupancy for phylogenomics. Molecular Biology and Evolution 31(11): 3081–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Bioinformatics 13(5): 555–556. [DOI] [PubMed] [Google Scholar]
- Zachos J, Pagani M, Sloan L, Thomas E, Billups K. 2001. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 292(5517): 686–693. [DOI] [PubMed] [Google Scholar]
- Zachos JC, Dickens GR, Zeebe RE. 2008. An early Cenozoic perspective on greenhouse warming and carbon-cycle dynamics. Nature 451(7176): 279. [DOI] [PubMed] [Google Scholar]
- Zeebe RE, Zachos JC, Dickens GR. 2009. Carbon dioxide forcing alone insufficient to explain Palaeocene–Eocene Thermal Maximum warming. Nature Geoscience 2(8): 576–580. [Google Scholar]
- Zhang G- Q, Liu K- W, Li Z, Lohaus R, Hsiao Y- Y, Niu S- C, Wang J- Y, Lin Y- C, Xu Q, Chen L- J. 2017. The Apostasia genome and the evolution of orchids. Nature 549(7672): 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Meltzer P, Davis S. 2013. RCircos: an R package for Circos 2D track plots. BMC Bioinformatics 14(1): 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Schwartz S, Wagner L, Miller W. 2000. A greedy algorithm for aligning DNA sequences. Journal of Computational Biology 7(1–2): 203–214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
BUSCO assessment of transcriptome and genomic completeness of 42 sampled species.
Bioinfomatic pipeline depicting transcriptome assembly, homology and orthology inferences, and three methods for WGD identification, placement, and dating.
Histograms of the synonymous substitutions per synonymous site (Ks) of duplicated gene pairs among 22 Malpighiales taxa showing WGDs.
Histograms of the synonymous substitutions per synonymous site (Ks) of duplicated gene pairs among 11 Malpighiales taxa where WGD is absent (or inconclusive in the case of three taxa).
Histograms depicting divergence time estimations inferred using penalized likelihood for 22 species that exhibited WGDs (summarized in Fig. S3).
Gene count matrix for 5113 ortholog groups of 36 Malpighiales species.
Phylogeny-guided synteny analyses demonstrate successive WGDs in Linum.
Histograms of the synonymous substitutions per synonymous site (Ks) of duplicated gene pairs among 22 Malpighiales taxa showing WGDs.
Cladogram of 50% majority-rule bootstrap tree of Malpighiales inferred from 5113 gene trees using ASTRAL.
Voucher and GenBank information for 15 species in Malpighiales used for de novo transcriptome assembly
Taxa sampled from the OneKP data set and the corresponding OneKP library ID
Taxa sampled with complete genomes and corresponding reference
Whole genome duplications (WGDs) in Malpighiales identified with complete taxon sampling, including their phylogenetic placement, percentage of gene families supporting gene duplication, log likelihood, and age estimations of each WGD
Summary statistics for the transcriptome assembly in Malpighiales
Whole genome duplications (WGDs) in Malpighiales identified with more conservative taxon sampling, including their phylogenetic placement, percentage of gene families supporting gene duplication, log likelihood, and age estimations of each WGD
Please note: Wiley Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.