

Abstract
Fullerene epoxides, C60On, having epoxide groups directly attached to the fullerene cage, constitute an interesting class of fullerene derivatives. In particular, the chemical transformations of fullerene epoxides are expected to play an important role in the development of functionalized fullerenes. This is because such transformations can readily afford a variety of mono- or polyfunctionalized fullerene derivatives while conserving the epoxy ring arrangement on the fullerene surface, as seen in representative regioisomeric fullerene polyepoxides. The first part of this review addresses the synthesis and structural characterization of fullerene epoxides. The formation of fullerene epoxides through different oxidation reactions is then explored. Adequate characterization of the isolated fullerene epoxides was achieved by concerted use of NMR and LC-MS techniques. The second part of this review addresses the substitution of fullerene epoxides in the presence of a Lewis acid catalyst. Most major substitution products have been isolated as pure compounds and their structures established through spectroscopic methods. The correlation between the structure of the substitution product and the oxygenation pattern of the starting materials allows elucidation of the mechanistic features of this transformation. This approach promises to lead to rigorous regioselective production of various fullerene derivatives for a wide range of applications.
Keywords: fullerene, epoxidation, regioselectivity, nucleophilic substitution, Lewis acid
1. Introduction
Since the first detection of fullerene epoxides via mass spectrometry in a fullerene mixture generated by the arc discharge of graphite in 1991, many studies on fullerene oxides have been performed for the purpose of developing new materials. The epoxidation of fullerene can proceed readily in the presence of oxidants such as ozone [1], organic peroxide [2], dimethyldioxirane [3], methyltrioxorhenium-hydrogen peroxide [4], and cytochrome P450 [5]. Under several circumstances, in different oxidations fullerene C60 has been shown to give higher oxides (C60On, n ≥ 2) that possess only a few regioisomers. For instance, although there are indeed eight possible regioisomers of C60O2 and 43 isomers of C60O3 given the multiple reaction sites available on the C60 cage [6], the actual products of most oxidations are only two isomers of C60O2 and three isomers of C60O3. However, few C60On isomers have been isolated and identified thus far, and experimental data on the regioselective epoxidation of C60 are also scarce.
Meanwhile, the structures of C60On isomers were first studied theoretically on the basis of the thermodynamic stability of ground state molecules and on the dynamic behavior of the molecules via the transition states. Those results regarding the structures of C60O and the predominant isomers of C60O2 can explain these experimental observations. Manoharan showed computationally that multiple epoxidations of C60 preferentially proceeds at the adjacent rather than distant double bonds of the existing epoxide group, and predicted that multiple epoxidations should occur on one benzenoid ring of C60 to form the C60O3 isomer with C3v symmetry. Feng et al. studied three isomers of C60O3 by using the semiempirical quantum mechanical INDO method, and predicted that the three types of C60O3 isomers with C3v, Cs and C2 symmetries, respectively, should be very stable, and reported their calculated electronic spectra [7]. Curry et al. predicted that the three lowest-energy isomers (C3v, Cs and C2) of C60O3 should exist in equilibrium at room temperature by using a modified and extended Hűckel method [8].
Previously, we found three types of C60O3 isomers in a reaction solution of C60 with m-CPBA by means of a chromatographic technique involving the use of two different columns [9]. Electronic spectroscopy and mass-spectroscopy examinations of these isomers were mostly consistent with the calculated results by Feng et al. However, the precise structures of these isomers could not be confirmed by 13C-NMR and X-ray studies, which are unavailable due to the low solubility and poor crystallinity. We carried out measurements of FT-IR and 13C-NMR spectra precisely after the isolation and further purification of two types of C60O2 isomers and three types of C60O3 isomers. Simultaneously, we demonstrated experimentally the regioselectivity of the epoxidation of C60 by means of the identification of products from each oxidation of the isolated isomers.
Fullerene epoxides exhibit interesting properties applicable to new materials development. However, the chemical transformations of fullerene epoxides have been studied sparingly, despite the general recognition that they could serve as convenient starting materials for the synthesis of functionalized fullerene derivatives [10]. We first succeeded in converting fullerene epoxide stoichiometrically into a 1,3-dioxolane derivative [11]. Reaction of C60O with benzaldehyde in the presence of a Lewis acid led to the formation of a 1,3-dioxolane derivative of C60 in high yield. This implies the possibility of other nucleophilic substitutions of the epoxy rings on a fullerene cage. The chemical transformation of fullerene epoxide is expected to play an important role in the development of functionalized fullerenes, because such transformations can readily afford a variety of mono- or polyfunctionalized fullerene derivatives, such as regioisomeric fullerene polyepoxides, while conserving the epoxy ring arrangement on the fullerene surface. The recent development of large-scale production techniques for fullerene epoxide [12] thus prompted us to develop a new methodology to synthesize polyfunctionalized fullerene derivatives by means of efficient chemical transformation of regioisomerically pure fullerene polyepoxides. Then, we also achieved the efficient formation of fullerene derivatives from C60O with aromatic nucleophilic compounds by Lewis acid-assisted nucleophilic substitution of the epoxy ring. The direct substitution of epoxide oxygen atoms on the fullerene epoxide—a versatile and advantageous synthetic methodology we report here—provides highly regioselective access to a variety of fullerene adducts.
2. Epoxidation of Fullerene C60 and Their Regioisomeric Structure
The epoxidation of C60 generally affords the higher fullerene epoxides (C60On, n ≥ 2), which have only a few types of regioisomers. For instance, the epoxidation of C60 with m-chloroperoxybenzoic acid (m-CPBA) forms a few C60O2 regioisomers (see Figure 1 for positional notation of C60O) that can be separated into two fractions by high-performance liquid chromatography (HPLC) (peaks A or B in Figure 2), although from the standpoint of the multiple reacting sites available on a C60 cage there are eight possible regioisomers of C60O2 [6]. A major C60O2 fraction is known to be composed of only one isomer with both oxygen atoms positioned over 6:6 ring junctions on a common six-membered ring of the carbon cage, namely the cis-1 adduct [5]. Meanwhile, a minor fraction of C60O2 has been considered to contain more than one isomer [13]. These isomers could not be separated by any HPLC column and their precise structures have hardly been ascertained by 13C-NMR or X-ray studies because of the low yield and the non-crystalline nature of the products.
Figure 1.

Positional notation of the fullerene epoxide, C60O.
Figure 2.

HPLC reversible-phase chromatograms (Develosil C30 RPFULLERENE, 34:66 acetonitrile/toluene, 335 nm detection) for reaction mixtures resulting from the m-CPBA of C60 (in toluene, 80 °C, 30 min).
The splitting pattern of each fraction in HPLC varies with both the position of the epoxy group on the C60 cage and its structural symmetry. A detailed comparison of these patterns is informative in relation to the structure of existing C60O2 isomers. In order to explain the isomeric structure of the preferentially formed C60O2, we also performed calculation of the static reaction index of oxygen addition sites on the C60 cage, and compared the experimental observations with the calculated results [14].
In the past, the structure of C60On isomers was studied on the basis of the thermodynamic stability of ground state molecules and the dynamic behavior of the molecules via the transition states. The results of these studies can explain the experimental observations regarding the structures of C60O and the predominant isomer of C60O2. Manoharan demonstrated computationally that the multiple epoxidations of C60 preferentially proceeds at the adjacent rather than distant double bonds of the existing epoxide group [15]. By using a modified extended Hűckel method, Curry et al. also correctly predicted for C60O2 that the cis-1 adduct has exceptionally low energy compared to the next stable isomers [8]. Feng et al. studied the possible structure of C60O2 isomers by using the semiempirical quantum mechanical INDO method, and showed that three types of C60O2 with the regioisomeric structures of cis-1, cis-2, and trans-4 adduct, respectively, should be very stable [7]. A more detailed theoretical elucidation of the regioselective epoxidation on fullerene, however, is required to provide information concerning the possible structures of other experimentally obtained C60O2 isomers.
The reactivity for regioselective epoxidation on fullerene is assumed to be predicted by calculating the static reaction index for addition sites. On the basis of theoretical studies on the epoxidation of alkenes with peroxide [16,17], we hypothesized that the reaction index should fall between fullerene and m-CPBA. The electrophilicity of peroxide was attributed to its relatively weak O-O bond, which can provide an empty σ* orbital that can mix with the nucleophilic π-bond of fullerene. The electrophilic oxygen tends to attack the bonds with a high electron density. In the same manner, the epoxidation of fullerene by the electrophilic oxygen of m-CPBA is likely to occur on electron rich double bonds. Thus the electron density of a bond is expected to give a good index of a probable reaction site. A similar calculation has been reported for the first oxygen addition sites on fullerenes [18].
Since the epoxidation of fullerene is a step-wise reaction, the yield for C60On (n ≥ 2) can be calculated by considering the yield of the parent isomer C60On−1. The electron density of a bond has been determined by semiempirical molecular orbital calculations (PM3). The yield of the j-th isomer of C60On, Y(n,j), can then be determined by the summation of the product of the yield of i-C60On−1, Y(n−1,i), and the branching probability from i-C60On-1 to j-C60On, p(i,j) as follows:
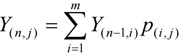
where m denotes the number of the parent isomer. If the branching probability increases linearly with the difference in electron density, p(i,j) is defined by utilizing the difference between the electron density of the bond of the addition site and the minimum electron density of the bond that undergoes epoxidation as follows:
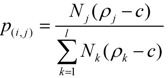
where ρj and Nj denotes the electron density of the bond of the addition site j, and the number of the symmetrically equivalent bond of j, respectively. The constant c is the minimum electron density that undergoes epoxidation. The variables l denotes the number of equivalent bonds of the possible addition sites.
Table 1 shows the calculated yield for the C60O2 isomers, and the candidates for the dominant isomers based on the calculated yield. The cis-1 adduct overwhelmingly dominated the other isomers. The next dominant isomer of C60O2 was predicted to be the equatorial adduct. The structure of the predominant isomer, the cis-1 adduct (1b in Figure 3), agreed with the structure actually confirmed by X-ray analysis [2], whereas the next dominant isomer is differed from a previous suggestion [1] that the C60O2 isomers in fraction B are probably the cis-2 or trans-4 adducts. From both our experiment and calculations, at a minimum, we expect that the main product in fraction B is not the trans-1 but rather the equatorial adduct of C60O2 (1c in Figure 3).
Table 1.
Calculated percentage yield and point group of constitutional isomers of diepoxidized fullerene C60O2.
| Structure | Symmetry | Yield / % |
|---|---|---|
| cis-1 | C s | 88 |
| equatorial | C s | 12 |
| trans-4 | C s | <0 |
| trans-2 | C 2 | <0 |
| trans-1 | D 2h | <0 |
Figure 3.

Fullerene mono-epoxide and regioisomeric di-epoxides isolated by means of preparative HPLC from a mixture of C60(O)n.
3. Lewis Acid-Assisted Reaction of Fullerene Epoxides with Nucleophiles
Epoxides are well-known to undergo heterolytic C–O bond cleavage in the presence of an acid catalyst to generate active carbocationic species for further reactions with nucleophiles. These active species react with various types of nucleophile such as carbonyl compounds, alcohols, and amines to afford 1,3-dioxolanes, 1,2-hydroxyethers, and 1,2-aminoalcohols, respectively. We have studied the reaction of fullerene epoxide 1a with various nucleophiles in the presence of a Lewis acid. Reaction with a carbonyl compound led to the formation of a 1,3-dioxolane derivative in high yield; interestingly, reaction with a phenol or aniline derivative afforded an O- or N-heterocycle-fused fullerene derivative. These reactions are considered to proceed via a nucleophilic reaction on the carbocationic active species generated on the carbon atom of fullerene epoxide 1a (Scheme 1). The formation of the heterocycle-fused fullerene derivative can be accounted for by the ring closure reaction induced by intramolecular dehydration of the initially formed 1,2-substituted product. In contrast, a Lewis acid-assisted nucleophilic reaction of C60O with an aromatic nucleophile such as toluene gave a 1,4-bisadduct. Application of these reactions to a regioisomeric fullerene polyepoxide would provide a convenient synthetic route to a variety of polyfunctionalized fullerene derivatives with a regioisomerically pure structure. These chemical transformations of fullerene epoxides are expected to play an important role in the development of functionalized fullerene cages. In this section, we review our recent progress in the work on the acid-assisted reaction of fullerene epoxides with various types of nucleophile.
Scheme 1.

Generation of active carbocationic species from 1a in the presence of an acid catalyst.
3.1. Acetalization of Fullerene Epoxides
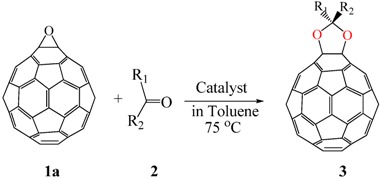
Several methods have been reported for the preparation of fullerene-fused 1,3-dioxolane derivatives, such as the reactions of C60 fullerene with dioxiranes [3], acyl hypohalogenites [19], and diacyl peroxides [20], or the reaction of C60 fullerene with a sodium alkoxide in the presence of air [21]. However, all of these reactions afforded fullerene-fused 1,3-dioxolane derivatives in only low to moderate yields, (i.e., <50%). As described in the previous section, we recently developed a preparative HPLC isolation method for some regioisomeric fullerene epoxides (Figure 3) [7]. Isolation of these epoxides prompted us to employ them as a starting material for efficient synthesis of fullerene-fused 1,3-dioxolane derivatives. We have found that the acetalization reactions of fullerene epoxide 1a with various carbonyl compounds occur in the presence of a Lewis acid catalyst, an ion-exchange resin such as Amberlite, and a clay mineral such as montmorillonite to afford the corresponding 1,3-dioxolane derivatives in very high yields (Table 2) [9,22].
Table 2.
Acetalization of fullerene epoxide 1a with carbonyl compound in the presence of an acid catalyst.
 | ||||
|---|---|---|---|---|
| Entry | 2 | Catalyst (equiv./amount) | Reaction Time | Yield a of 3 / % |
| 1 |
|
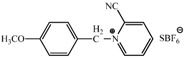 4a (0.29) 4a (0.29) |
60 min | 92 |
| 2 | 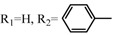 |
 4b (0.28) 4b (0.28) |
90 min | 91 |
| 3 | 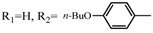 |
4a (0.29) | 60 min | 95 |
| 4 | 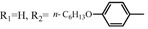 |
4b (0.28) | 4.5 h | 88 |
| 5 | 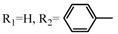 |
BF3·Et2O (one drop) | 60 min | 89 |
| 6 | 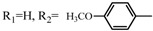 |
Amberlyst 15® (268 mg/0.02 mmol) | 3 h | 96 |
| 7 | 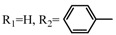 |
Montmorillonite (250 mg/0.02 mmol) | 4 h reflux | 60 |
| 8 |  |
4a (0.27) | 30 min | 45 |
| 9 | R1=CH3, R2=C2H5 | 4a (0.27) | 30 min | 44 |
| 10 | Cyclohexanone | Amberlyst 15® (250 mg/0.02 mmol) | 65 °C, 5 h in benzene | 60 |
| 11 | γ-Butyrolactone | BF3·Et2O (one drop) | 3 h in benzene | 75 |
a Isolated yield.
The reaction of a toluene solution of fullerene epoxide 1a with excess amounts (200 equiv.) of a benzaldehyde derivative at 75 °C in the presence of a Lewis acid catalyst led to the formation of 1,3-dioxolane 3 in very high yield. Table 2 also shows that a similar acetalization of 1a with other ketone compounds such as acetophenone, methyl ethyl ketone or cyclohexanone took place to give the corresponding 1,3-dioxolanes in moderate yields. Furthermore, a similar reaction of 1a with γ-butyrolactone in benzene afforded an ortho-ester type 1,3-dioxolane derivative in high yield, although in toluene, it resulted in the formation of a 1,4-addition product of toluene to 1a. This fact suggests that the nucleophilicity of a carbonyl group of an ester compound is not as strong as that of an aldehyde or a ketone, such that toluene instead acts preferentially as a nucleophile to the carbocationic species to yield a 1,4-addition product (1,4-bis(p-tolyl)-1,4-dihydro[60]fullerene). The Lewis acid catalyzed 1,4-addition reaction of aromatic nucleophiles to 1a will be discussed in Section 3.2. Recently Gan and Zhang et al. reported that a Lewis acid such as BF3·Et2O catalyzes the acetalization reaction of the fullerene epoxide bearing t-butylperoxo groups to yield the corresponding 1,3-dioxolane derivatives in moderate yields [23].
An epoxy ring is known to undergo a Lewis acid catalyzed acetalization to afford 1,3-dioxolane [24,25,26]. Generally the acetalization is assumed to follow a concerted SN2-like mechanism involving backside attack of a carbonyl oxygen on an epoxide carbon followed by rotation of the C–C bond of the epoxide and then the second C–O bond formation to produce a 1,3-dioxolane [26]. However, such a backside attack on a fullerene epoxide is not expected to occur owing to occupation of the entire side opposite the epoxy ring by the fullerene cage. According to this view, an acetalization of a fullerene epoxide is not expected to occur unless an alternative mechanism is involved in a front side attack of a carbonyl compound on the epoxy ring of the fullerene epoxide. The acetalization reaction of fullerene epoxide 1a with benzaldehyde at 75 °C for 1 d did not proceed at all in the absence of a Lewis acid and resulted in the complete recovery of 1a. This result suggests that the Lewis acid catalyzes the acetalization reaction to proceed by a stepwise SN1-like mechanism (Scheme 2). A Lewis acid induces a catalytic C–O bond cleavage of the epoxy ring to generate a carbocation on the carbon atom of the fullerene moiety. This is followed by nucleophilic attack of a carbonyl compound on the carbocation. An energetically unfavorable SN1-like attack of a carbonyl compound on the carbocation from the fullerene surface would be forced to occur with the requirement of relatively high activation energy for the C–O bond cleavage of the epoxy ring.
Scheme 2.

Stepwise SN1-like acetalization of fullerene epoxide 1a with benzaldehyde derivative.
The activation energy for the reaction of 1a with benzaldehyde was determined to be 112.7 kJ·mol−1 from the Arrhenius plots of ln k vs. 1/T, where k is the pseudo-first order rate constant and T is the reaction temperature. Based on the activation energy determined, the rate constant at room temperature (293 K) can be calculated to be 9.5 × 10−7 s−1, which is more than 103 times smaller than that at 75 °C (k = 1.43 × 10−3 s−1). Therefore, that the Lewis acid catalyzed acetalization of 1a with benzaldehyde did not occur at room temperature is unsurprising, although a similar acetalization of oxiranes such as but-2-ene epoxide or styrene oxide readily proceeds at room temperature and even at 0 °C to yield 1,3-dioxolanes according to an energetically favorable SN2-like mechanism [24,25,26].
The application of the above acetalization reaction to the regioisomerically pure fullerene di-epoxide 1b (cis-isomer) or 1c (equatorial isomer) would lead to chemical transformation of the two epoxy rings while conserving their arrangement on the fullerene cage. Under reaction conditions similar to those for mono-epoxide 1a, the fullerene di-epoxides 1b and 1c were also subjected to acetalization with benzaldehyde derivatives to give bis-1,3-dioxolanes 5a and 5b, respectivelyinhigh yields (Table 3) [9,21]. The visible absorption spectra of 5a and 5b show the bands characteristic to those of the di-epoxides 1b and 1c, respectively, that is, 5a showed a sharp band at 423 nm, which is the characteristic band for the cis-isomer 1b [11,15]. This fact suggests that during the acetalization reaction, no migration of the carbocation on the fullerene moiety occurs and that the acetalization reaction of the fullerene di-epoxide proceeds while the rearrangement of the two epoxy rings on the fullerene cage is conserved.
Table 3.
Acetalization of fullerene epoxide 1b or 1c with benzaldehyde derivatives 2 in the presence of pyridinium salt 4a or 4b shown in Table 2.
 | |||
|---|---|---|---|
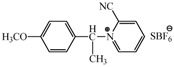
| Epoxide | 2 | Catalyst (equiv.) | Yield of 5 / % |
| 1b | R3 = H |
 4b (0.28) 4b (0.28) |
5a (R3 = H), 92 |
| 1b | R3 = n-BuO |
 4a (0.29) 4a (0.29) |
5a (R3 = n-BuO), 91 |
| 1c | R3 = H | 4b (0.28 equiv.) | 5b (R3 = H), 76 |
| 1c | R3 = n-BuO | 4a (0.27 equiv.) | 5b (R3 = n-BuO), 73 |
| 1c | R3 = n-C3H7 | 4a (0.27 equiv.) | 5b (R3 = n-C3H7), 82 |
The 1H-NMR spectrum of 5a (R3 = n-BuO) showed the two unequivalent acetal methine protons of equal intensity. In addition, it showed two sets of four phenyl protons and nine n-butoxy protons, also of equal intensity. Thus, the 1H-NMR spectrum of 5a clearly demonstrates that the two phenyl groups are unsymmetrically disposed with respect to the fullerene moiety. The 13C-NMR spectrum of 5a showed 51 signals for the fullerene sp2 carbons, indicating a lack of symmetry of the fullerene cage. Based on the above NMR analysis, bis-1,3-dioxolane 5a is confirmed to have the structure of the stereoisomer depicted in Table 3. However, the configuration of the two phenyl groups in 5b remains undetermined because no definitive information suggesting their configuration could be obtained from the 1H-NMR spectra of 5b. We assume that 5b is likely to be a mixture of two stereoisomers.
The fullerene di-epoxide 1b (cis-isomer) or 1c (equatorial isomer) also reacts with a ketone compound such as cyclohexanone in the presence of a Lewis acid catalyst or Amberlyst 15® to yield the corresponding bis-1,3-dioxolane derivative in moderate yields (50–70%) [27]. These acetalization reactions are presumed to proceed with conservation of their arrangement on the fullerene cage.
The present work has clearly shown that the epoxy moiety of fullerene epoxides can be readily converted to 1,3-dioxolane derivatives in high yield by treatment with a carbonyl compound in the presence of a Lewis acid, ion-exchange resin or clay mineral. The acetalization reaction proceeds by a stepwise SN1-like mechanism which involves the nucleophilic attack of a carbonyl moiety at the carbocationic active species generated on the carbon atom of fullerene epoxides. Furthermore, the efficient bis-acetalization of a regioisomerically pure fullerene di-epoxide occurs under conservation of the arrangement of the two epoxy moieties to yield bis-1,3-dioxolane derivatives in high yield. These results could lead to development of a new methodology for facile synthesis of a variety of regioisomeric polyfunctionalized fullerene derivatives by means of efficient chemical transformation of a specific regioisomeric fullerene polyepoxide. In order to elucidate the reactivity of the carbocationic active species generated from fullerene epoxides, the next part of this section is oriented toward studies on the reaction of fullerene epoxides with nucleophiles other than carbonyl compounds.
3.2. Nucleophilic Substitution of Fullerene Epoxide with Aromatic Compounds
The reaction of a toluene solution of fullerene epoxide 1a in the presence of 5 equiv. of boron trifluoride etherate (BF3·OEt2) at r.t. for 60 min led to the formation of 1,4-bis(p-tolyl)-1,4-dihydro [60]fullerene (7a) in 80.8% yield together with a very small amount of pristine fullerene C60 based on HPLC analysis [28]. Compound 7a was purified by flash column chromatography on a silica gel with n-hexane as an eluent. A detailed spectral characterization of 7a revealed that two p-tolyl moieties were introduced to the C60 core in a 1,4-addition pattern. The FT-IR spectrum was in good agreement with that in an earlier report [29,30]. The characteristic vibrations for the 1,4-adduct were found at 1,430, 1,187, 573, and 527 cm−1, and the C-H vibrations for toluene showed strong vibrations at 2,963 and 810 cm−1. The atmospheric pressure photochemical ionization (APPI) mass spectrum showed a molecular ion peak at m/z 902 (i.e., 166 units more than 1a), corresponding to a 1:2 adduct of C60 and toluene-H. In the 1H-NMR spectrum, in addition to two doublets at 7.94 (2H), 7.93 (2H), 7.28 (2H), and 7.27 (2H) ppm for eight phenyl protons, one singlet methyl proton was observed at 2.46 (6H) ppm. The 13C-NMR showed the characteristic absorption of aromatic carbons in the p-tolyl group at 138.25 (2C), 136.91 (2C), 129.52 (4C), and 126.94 (4C) ppm, and two quaternary carbons of the C60 moiety were observed at 60.86(2C) ppm. The UV-VIS spectrum showed a characteristic absorption band around 450 nm. The 1,4-bisadduct structure for 7a was evidently confirmed by comparing these spectra with FT-IR, 1H-NMR and 13C-NMR spectra for similar compounds [29,30].
Reactions of 1 with other aromatic compounds were carried out under the same conditions as described above, the results of which are listed in Table 4. In anisole, o-xylene and m-xylene the reaction produced the corresponding 1,4-bisadducts (7b, 7c and 7d) in 45–95% yields, whereas neither the 1,2-(6e) nor 1,4-bisadduct (7e) was produced in p-xylene.
Table 4.
Substitution of 1 with aromatic compounds.
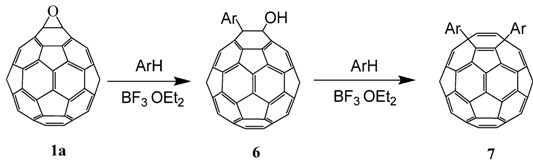 | |||
|---|---|---|---|
| Aromatic Compound a | Isolated Yield of 6 / % b | Isolated Yield of 7 / % c | |
| a | toluene | 0 | 80.8 |
| b | anisole | 0 | 95.8 |
| c | o-xylene | 0 | 76.5 |
| d | m-xylene | Trace | 45.3 d |
| e | p-xylene | 0 | 0 |
| f | 1,3,5-trimethylbenzene | 35.3 | Trace |
| g | chlorobenzene | 0 | 0 |
| h | benzene | 0 | 0 |
a All reactions were performed according to the procedure described in the text; b,c All reactions were continued until complete disappearance of the starting material 1; d Overall yield of the structural isomeric mixture of 1,4-adducts.
Although the purification of 1,4-bisadducts 7a, 7b, and 7c was easily carried out with the HPLC apparatus, 3d was not well separated owing to the formation of structural isomers. In the case of 1,3,5-trimethylbenzene, the corresponding 1-(substituted phenyl)-2-hydroxyl-1,2-dihydro[60]fullerene (6f) was dominantly formed instead of the 1,4-bisadduct, as identified by UV-VIS, APPI mass, 1H- and 13C-NMR spectra. Usually the sp3 carbons of the C60 cage were observed to shift downfield ca. 10 ppm for 1,2-adducts relative to the 1,4-adducts. The two peaks at 86.9 and 72.0 ppm for the two sp3 carbons of the fullerene core in 6f indicate a typical structure of 1,2-adduct. This suggests that the formation of 1,4-bisadduct from 1a proceeds stepwise via 1,2-adduct 6 as an intermediate. In fact, the formation of 6d can be observed as a transient intermediate in the course of the reaction in m-xylene. With respect to the UV-VIS spectrum beyond wavelengths of 300 nm, 6d is identical to 6f. The substitution reaction scarcely proceeded in benzene and chlorobenzene, even above 75 °C, and pristine C60 was observed instead of 1,4-adducts. The formation of pristine C60 without epoxy group was observed. Thus, it appears that the progress of the reaction depends considerably on the nucleophilicity of the aromatic compound. In the absence of BF3℘OEt2, no reaction was observed in any solvent. From this general behavior, we may conclude that the first substitution of 1a to obtain 6 occurs via a carbocationic intermediate generated with the assistance of the Lewis acid, followed by a nucleophilic attack of an aromatic compound on the cation. In the transformation of 6 into 7, a reasonable assumption is that the substitution of the hydroxyl group proceeded by an SN1- or SN2-type mechanism with allylic rearrangement (Scheme 3).
Scheme 3.

Proposed mechanism for the substitution of C60O with aromatic compounds. Part of the fullerene surface is shown.
Generally, the Lewis acid-assisted substitution of the epoxy group is assumed to follow a concerted SN2-like mechanism involving a backside attack of a nucleophilic reagent on an epoxide carbon atom [26]. However, such an attack is rather unrealistic, because the fullerene cage occupies the entire side opposite the epoxy ring. The first substitution of the epoxy ring on a fullerene, therefore, would occur with an energetically unfavorable SN1-like attack of a nucleophilic reagent on the carbocation generated by the C−O bond cleavage of the epoxy ring. On the other hand, the second substitution for 6 might occur by an SN2'-type mechanism, in which the nucleophilic attack on the residual hydroxyl group occurs first, followed by the elimination of -OH on the side with an allylic rearrangement [31]. The fact that no reaction proceeded in the absence of the Lewis acid catalyst suggests that the second substitution occurs by an SN1- or SN2-type mechanism with allylic rearrangement, where 7 is formed via a carbocationic intermediate with an allylic resonance [32,33,34,35,36], generated with the assistance of BF3·OEt2. In no case was the formation of 1,2-bis(substituted phenyl)adduct observed, presumably because of the steric hindrance between the two substituents.
3.3. Formation of Indolino[2',3':1,2][60]fullerene Derivatives from C60O and Aniline Derivatives
As shown in section 3.1, the reactions of C60Ox with nucleophiles in the presence of acidic compounds have uncovered a means to achieve the regioselective functionalization of various fullerene derivatives. In extending this procedure, we found that a reaction of C60O with aniline derivatives in the presence of acidic compounds afforded the indoline-fused fullerene derivatives: indolino[2',3':1,2]-1,2-dihydro[60]fullerene (Figure 4), in moderate to excellent isolated yields (ca. 60–80%) [37]. To the best of our knowledge, only a few cases have thus far been reported on the preparation of indolino[60]fullerene analogues [38,39,40,41,42]. This method enabled access to a wide variety of indolino[60]fullerene derivatives because numerous anilines are commercially available.
Figure 4.
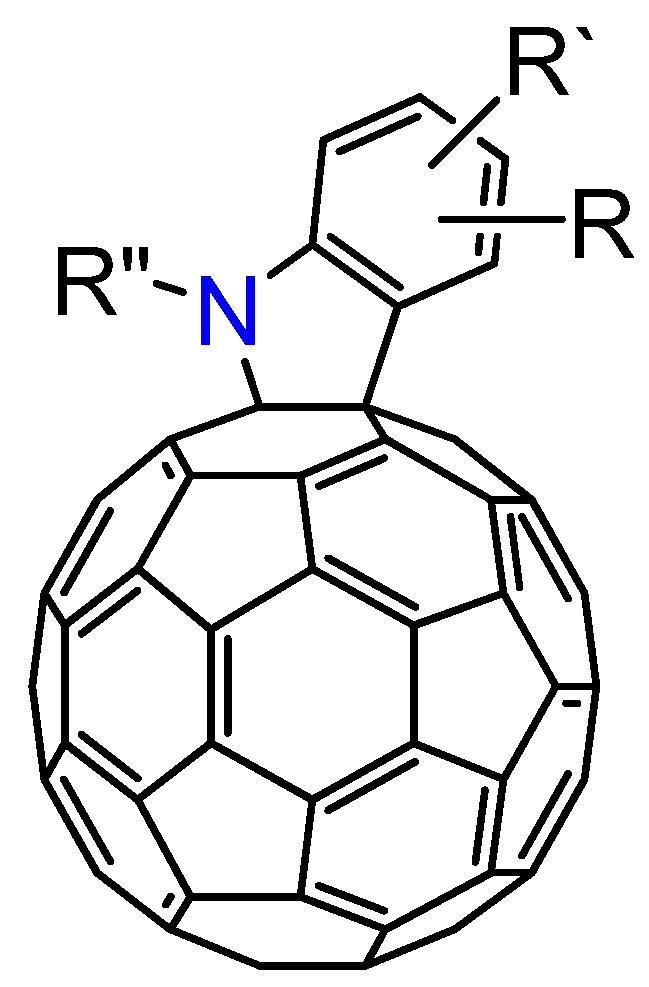
Molecular structure of indolino[2',3':1,2]-1,2-dihydro[60]fullerene derivatives 8.
First, we examined the catalytic activities of different types of acidic compound for this reaction by using 4-dodecylaniline as a model compound. The examined acidic compounds were BiCl3, BF3·OEt2, Montmorillonite K10, Sepiolite, TsOH, and Amberlyst 15®. The Brønsted acids TsOH and Amberlyst 15® did not exhibit catalytic activities for the reactions, and all of the C60O was converted to pristine C60. Although the Lewis acid compound; BiCl3 gave products with moderate production yields (−50%), BF3·OEt2 did not give products and only the pristine C60 was obtained. The clay compounds, Montmorillonite K10 and Sepiolite exhibited excellent catalytic activities toward the cyclization reactions (Table 5).
Table 5.
Catalytic activities of acidic compounds on formation of 5-dodecylindolino[60]fullerene.
| Catalyst | Type | Time | Yield of 8a / % a |
|---|---|---|---|
| Montmorillonite K10 | clay mineral | 6 h | 56.7 |
| Sepiolite | clay mineral | 6 h | 63.8 |
| BiCl3 | Lewis acid | 2 h | 50.3 |
| BF3·OEt2 | Lewis acid | 2 h | 0 (C60 was given) |
| p-TsOH | Brønsted acid | over 5 days | 0 (C60 was given) |
| Amberlyst 15® | Brønsted acid | over 5 days | trace (C60 was given) |
a Isolated yields, estimated from HPLC areas. Reaction condition: C60O 100 mg, 0.136 mmol; 4-dodecylaniline 355 mg, 1.357 mmol; Catalyst ×2,000 wt%, chlorobenzene 100 mL, 100 °C.
The lack of catalytic activity of the Brønsted acid may be attributed to a neutralization process between the Brønsted acid and anilines. For BiCl3, the Lewis acidity may be slightly strong; thus, the epoxy oxygen may have been removed from the C60 core more quickly than with the nucleophilic addition of anilines. In contrast, BF3·OEt2 forms a Lewis acid-base complex with aniline. The capped aniline may have lost its nucleophilicity, and thus completely prevented the reaction from proceeding. Cray minerals presumably have moderate Lewis acidity relative to BF3, and may thus be appropriate for the reaction.
Subsequently, in order to examine the versatility of this reaction, we carried out the reactions with a wide variety of anilines. The examined anilines and results are summarized in Table 6. Each aniline examined gave the corresponding indolino[60]fullerene derivative in good yield except for the reaction with N-Me-p-toluidine (Entry 8f). For 8f, the steric hindrance of the methyl group on the nitrogen atom might inhibit nucleophilic addition to the carbon atom of the fullerene core.
Table 6.
Isolated yields of indolino[60]fullerene derivatives.
| Entry | Aromatic Amine | 5- | N- | Yield / % a |
|---|---|---|---|---|
| 8a | 4-Dodecylaniline | n-C12H25 | H | 77.7 |
| 8b | 4- n-Butylaniline | n-C4H9 | H | 64.1 |
| 8c | p-Toluidine | Me | H | 84.0 |
| 8d | 4-Fluoroaniline | F | H | 75.7 |
| 8e | Aniline | H | H | 78.9 |
| 8f | N-Me-p-toluidine | Me | Me | 29.3 |
a Isolated yield, estimated from HPLC areas. Reaction conditions: C60O 100 mg, 0.136 mmol; aromatic amines 1.357 mmol; Sepiolite 2.0 g; chlorobenzene 100 mL, 100 °C.
Next, we attempted to reveal the reaction mechanism of the formation of indolino[60]fullerene from C60O and anilines. By tracing the reaction with an HPLC-MS system, we revealed that the cyclization reaction proceeded via 1,2-anilinoalcohol fullerene derivative. The time course of the HPLC chart and the change in the relative peak area for each compound are shown in Figure 5.
Figure 5.

Time course of the HPLC chart for the reaction of C60O with 4-dodecylaniline.
At the start time, peaks corresponding to C60O at 9.3 min and slightly impure C60 at 11.8 min appeared. At first, a new peak (9a) appeared at 5.4 min in the LC chart, and then, a peak corresponding to 8a at 6.8 min increased as the peak area of 9a decreased. Finally, the peaks of the starting C60O and 9a were completely consumed and the peak 8a and a small peak corresponding to pristine C60 as a decomposed compound at 11.8 min were observed. Therefore, we surmised that 9a was an intermediate of the indolino[60]fullerene derivative. To confirm our assumption, isolated 9a with the catalyst under similar reaction conditions, whereupon the reaction gave 8a.
In order to assign each peak, we isolated and characterized compound 9a by means of IR, APPI-MS, and NMR spectra. In the IR spectrum, an absorption corresponding to O–H stretching was observed at 3,793 cm–1. The MS spectrum of 9a showed a peak at m/z = 998. The MS of the peak corresponds to the summation of C60O (mw = 736) and dodecylaniline (mw = 262). As a result of 1H-NMR, we observed in the range of 0.87–2.60 ppm, two doublet peaks of aryl protons corresponding to four aniline protons at 7.19 and 7.61 ppm, and one singlet peak of NH at 5.93 ppm. Taking these results into consideration, we assigned 9a to a 1-anilino-2-hydroxy[60]fullerene derivative (Scheme 4).
Scheme 4.

Plausible reaction mechanism of indolino[60]fullerene derivative formation from C60O and anilines.
Based on the experimental results, we proposed a plausible mechanism for the formation of indolino[60]fullerene (8) via 1,2-anilinohydroxy fullerene (9) (Scheme 4). At first, the epoxy ring is opened by adding a Lewis acid to the oxygen of the epoxide. Then, nucleophilic addition of anilines to the carbon atom underlying the epoxy oxygen forms a 1,2-anilinohydroxy fullerene derivatives. Consequently, the Lewis acid again adds to the oxygen atom of the hydroxyl group, and a C–C bond forms between the carbon of C60 and the ortho-position of the anilino group to give the indoline-fused fullerene derivative.
3.4. Formation of Benzo[b]furano[2',3':1,2][60]fullerene Derivatives from C60O and Phenols
A similar nucleophilic addition to the fullerene core and the consequent cascade cyclization reaction with phenols via a 1-hydroxy-2-phenolic fullerene derivative as an intermediate was also observed. This reaction gave benzo[b]furano[2',3':1,2][60]fullerene derivatives in good yields as well (Figure 6) [43].
Figure 6.
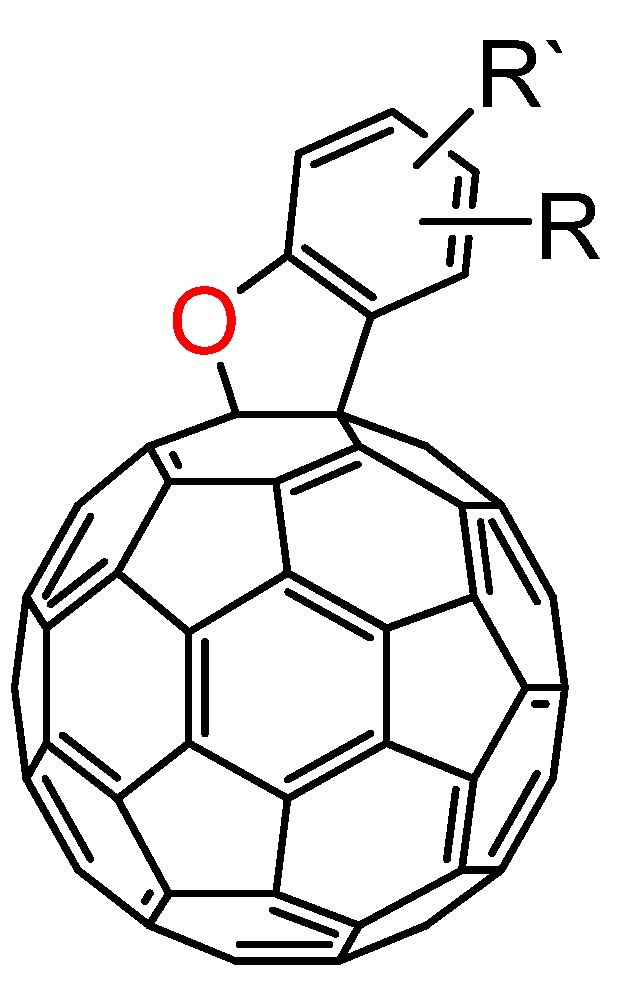
Molecular structure of benzo[b]furano[2',3':1,2][60]fullerene derivatives.
Unlike in the case of anilines, BF3·OEt2 exhibited superior catalytic activity for the reaction of C60O with phenols. The reaction of C60O with phenol derivatives in the presence of BF3·OEt2 was complete within 30 min for nearly all examined phenols. The different catalytic activities of the Lewis acids may be attributed to the Lewis acidity of a catalyst, the Lewis basicity of a nucleophile, or both. For the reaction with anilines, a complexation between BF3 and the amine occurs preferentially over the addition of BF3 to the epoxy oxygen. Therefore, either BF3 or the aniline is completely consumed, and the reaction cannot proceed at all. On the other hand, for benzo[b]furano[60]fullerenes, the Lewis basicity of nearly all phenols is weaker than that of anilines. BF3 thus effectively catalyzes the reaction of epoxy ring opening, nucleophilic addition, and cyclization.
The production yields of the benzo[b]furano[60]fullerene derivatives were subjected to the electron-donating and withdrawing properties of the substituent of the phenols. The isolated yields are summarized in Table 7.
Table 7.
Isolated yields of benzo[b]furano[60]fullerene derivatives.
| Entry | Phenols | Yield / % a |
|---|---|---|
| 10b | 4 -n-octylphenol | 91.5 |
| 10c | 4- n-butylphenol | 88.8 |
| 10d | 4-methylphenol | 91.5 |
| 10e | 4-methoxyphenol | 72.9 |
| 10g | 4-fluorophenol | 30.8 |
| 10h | phenol | 41.1 |
| 10i | 3,5-di-methoxyphenol | 82.2 |
| 10l | 4-nitrophenol | 0 |
a Isolated yields, estimated from HPLC areas. Reaction conditions: C60O 100 mg, 0.136 mmol; phenols 1.357 mmol; BF3·OEt2 37 μL 0.136 mmol; chlorobenzene 100 mL, 70 °C.
Unlike the case of anilines, the yields of benzo[b]furano[60]fullerene derivatives were strongly influenced by the type of substituent of the phenols. Figure 7 shows a plot of the isolated yields for benzo[b]furano[60]fullerene derivatives as a function of the Hammett constant for the 2-position of the precursor 4-substituted phenols [44]. Negative Hammett constants correspond to electron-withdrawing (EW) property, and positive ones correspond to the electron-donating (ED) property. The stronger ED property of the substituent increases the nucleophilicity of the oxygen atom of phenols and the carbon atoms of ortho-position of the phenols. In contrast, EW groups reduce the nucleophilicity of the phenolic oxygen and the carbon atoms of ortho-position. Therefore, ED functional groups improve the production yields of nucleophilic addition and cascade cyclization reactions. Conversely, 4-nitorophenol could not produce the benzo[b]furano[60]fullerene owing to the strong electron-withdrawing property of the nitro group. The low yield with pristine phenol (entry 10h) is presumably due to the competitive formation of the 1,4-bisadduct (Section 3.2), because the para-position of the phenol is not substituted.
Figure 7.

A plot of Hammett constant vs. production yields forbenzo[b]furano[60]fullerene derivatives.
4. Conclusions
In summary, this review presents an overview of our works on the nature and chemical transformation of fullerene epoxides. The structures of the formed fullerene epoxides, C60On, were characterized on the basis of their LC-MS and 13C-NMR spectral analyses. The number of carbon peaks and structural symmetry correlations enable the different oxygen addition sites on the fullerene framework to be distinguished. The obtained results show highly regioselective exohedral oxygenation of fullerene with oxidizing agents such as m-CPBA and ozone.
A variety of fullerene derivatives are easily prepared from fullerene epoxides, in which the epoxy groups on the fullerene framework are efficiently activated to undergo chemical transformations in the presence of a Lewis acid. We have found several Lewis acid-assisted reactions of fullerene epoxide with various types of nucleophiles. In all of the examined nucleophilic substitution reactions on fullerene epoxides, carbon-oxygen bond cleavage of the epoxy ring occurs with a front side attack of a nucleophile. Moreover, the substitution reaction of the fullerene di-epoxide proceeds while the rearrangement of the two epoxy rings on the fullerene cage is conserved. Fullerene epoxides are 0.39"materials in many industrial fields.
Acknowledgements
The author is deeply grateful to Kazuo Takeuchi (Senior Research Priority Planning Member of RIKEN’s Research Priority Committee) for his kind and continuous help, as well as to Takumi Hara, Takeshi Honma, Shizuka Seo, Yumiko Yamada, Jun-ichi Kawashima and Mikio Hoshino of RIKEN Nano-integration Materials Research Unit for their experimental contributions and helpful discussions. This work was financially supported by NEDO (Grant for Practical Application of University R&D Results under the Matching Fund Method).
References and Notes
- 1.Beck R.D., Stoermer C., Schultz C., Michel R., Weis P., Brauchle G., Kappes M.M. Enhanced coalescence upon laser desorption of fullerene oxides. J. Chem. Phys. 1994;101:3243–3250. [Google Scholar]
- 2.Balch A.L., Costa D.A., Lee J.W., Noll B.C., Olmstead M.M. Directing effects in a fullerene epoxide addition. formation and structural characterization of (eta.2-C60O)Ir(CO)Cl(P(C6H5)3)2. Inorg.Chem. 1994;33:2071–2072. doi: 10.1021/ic00088a002. [DOI] [Google Scholar]
- 3.Elemes Y., Silverman S.K., Sheu C., Kao M., Foote C.S., Alvare M.M., Whetten R.L. Reaction of C60 with dimethyldioxirane formation of an epoxide and a 1,3-dioxolane derivative. Angew. Chem. Int. Ed. Engl. 1992;31:351–353. doi: 10.1002/anie.199203511. [DOI] [Google Scholar]
- 4.Ogrin D., Barron A.R. Highly oxygenated fullerenes by catalytic epoxidation of c60 and single walled carbon nanotubes with methyltrioxorhenium-hydrogen peroxide. J. Mole. Catal. A Chem. 2006;244:267–270. doi: 10.1016/j.molcata.2005.09.017. [DOI] [Google Scholar]
- 5.Hamano T., Mashino T., Hirobe M. Oxidation of [60]fullerene by cytochrome p450 chemical models. J. Chem. Soc. Chem. Commun. 1995:1537–1538. [Google Scholar]
- 6.Balch A.L., Costa D.A., Bruce C., Noll B.C., Olmstead M.M. Oxidation of buckminsterfullerene with m-chloroperoxybenzoic acid. Characterization of a cs isomer of the diepoxide C60O2. J. Am. Chem. Soc. 1995;117:8926–8932. doi: 10.1021/ja00140a005. [DOI] [Google Scholar]
- 7.Feng J., Ren A., Tian W., Ge M., Li Z., Sun C., Zheng X., Zerner M.C. Theoretical studies on the structure and electronic spectra of some isomeric fullerene derivatives C60On (n = 2, 3) Int. J. Quantum Chem. 2000;76:23–43. doi: 10.1002/(SICI)1097-461X(2000)76:1<23::AID-QUA3>3.0.CO;2-P. [DOI] [Google Scholar]
- 8.Curry N.P., Doust B., Jelski D.A. Computational study of the combinatorial addition of oxygen to buckminsterfullerene. J. Clust. Sci. 2000;12:385–390. [Google Scholar]
- 9.Tajima Y., Takeuchi K. Discovery of C60O3 isomer having C3v symmetry. J. Org. Chem. 2002;67:1696–1698. doi: 10.1021/jo010908n. [DOI] [PubMed] [Google Scholar]
- 10.Barrow M.P., Tower N.J., Taylor R., Drewello T. Matrix-assisted laser-induced gas-phase aggregation of C60 oxides, Chem. Phys. Lett. 1998;293:302–308. doi: 10.1016/S0009-2614(98)00772-6. [DOI] [Google Scholar]
- 11.Shigemitsu Y., Kaneko M., Tajima Y., Takeuchi K. Efficient acetalization of epoxy rings on a fullerene cage. Chem. Lett. 2004;33:1604. doi: 10.1246/cl.2004.1604. [DOI] [Google Scholar]
- 12.The large-scale supply of fullerene epoxides was materialized by Frontier Carbon Corporation in Japan since 2007.
- 13.Fusco C., Seraglia R., Curci R., Lucchini V. Oxyfunctionalization of non-natural targets by dioxiranes. 3. Efficient oxidation of buckminsterfullerene C60 with methyl-(trifluoromethyl)-dioxirane. J. Org. Chem. 1999;64:8363–8368. doi: 10.1021/jo9913309. [DOI] [PubMed] [Google Scholar]
- 14.Tajima Y., Osawa S., Takeuchi K. Polyoxidized fullerene. RIKEN Rev. 1998;17:53–54. [Google Scholar]
- 15.Manoharan M. Predicting efficient C60 epoxidation and viable multiple oxide formation by theoretical study. J. Org. Chem. 2000;65:1093–1098. doi: 10.1021/jo9915527. [DOI] [PubMed] [Google Scholar]
- 16.Bach R.D., Wolber G.J. Mechanism of oxygen transfer from oxaziridine to ethylene: The consequences of homo-homo interactions on frontier orbital narrowing. J. Am. Chem. Soc. 1984;106:1410–1415. doi: 10.1021/ja00317a036. [DOI] [Google Scholar]
- 17.Bach R.D., Owensby A.L., Gonzalez C., Schlegel B.W., McDouall J.J. Transition structure for the epoxidation of alkenes with peroxy acids. J. Am. Chem. Soc. 1991;113:2338–2339. [Google Scholar]
- 18.Taylor R. C60, C70, C76, C78 and C84: Numbering, π-bond order calculations and addition pattern considerations. J. Chem. Soc. Perkin Trans. 1993;2:813–824. [Google Scholar]
- 19.Troshin P.A., Peregudov A.S., Lyubovskaya R.N. Reaction of [60]fullerene with CF3COOHal affords an unusual 1,3-dioxolano-[60]fullerene. Tetrahedron Lett. 2006;47:2969–2972. doi: 10.1016/j.tetlet.2006.02.106. [DOI] [Google Scholar]
- 20.Yoshida M., Morinaga Y., Iyoda M., Kikuchi K., Ikemoto I., Achiba Y. Reaction of C60 with diacyl peroxides containing perfluoroalkyl groups. The first example of electron transfer reaction via C60+ in solution. Tetrahedron Lett. 1993;34:7629–7632. [Google Scholar]
- 21.Wang G.W., Shu L.H., Wu S.H., Wu H.M., Lao X.L. Reaction of sodium alkoxides with [60]fullerene: Formation of a 1,3-dixolane derivative and involvement of O2 in a nucleophilic addition reaction of C60. J. Chem. Soc. Chem. Commun. 1995:1071–1072. [Google Scholar]
- 22.Shigemitsu Y., Tajima Y. Method for Manufacturing Fullerene Derivatives. 7,671,219 B2. U.S. Patent. 2010 Mar 2;
- 23.Yang X.B., Huang S.H., Jia Z.S., Xiao Z., Jiang Z.P., Zhang Q.Y., Gan L.B., Zheng B., Yuan G., Zhang S.W. Reactivity of fullerene epoxide: Preparation of fullerene-fused thiirane, tetrahydrothiazolidin-2-one, and 1,3-dioxolane. J. Org. Chem. 2008;73:2513–2526. doi: 10.1021/jo7023587. [DOI] [PubMed] [Google Scholar]
- 24.Meskens F.A.J. Methods for the preparation of acetals from alcohols or oxiranes and carbonyl compounds. Synthesis. 1981;1981:501–522. doi: 10.1055/s-1981-29507. [DOI] [Google Scholar]
- 25.Lee S.B., Tanaka T., Endo T. N-Benzyl pyridinium salts as new useful catalysts for transformation of epoxides to cyclic acetals, orthoesters, and orthocarbonates. Chem. Lett. 1990;19:2019–2022. [Google Scholar]
- 26.Blackett B.N., Coxon J.M., Hartshorn M.P., Lewis A.J., Little G.R., Wright G.J. The mechanism of 1,3-dioxolane formation from the bf3-catalysed reaction of epoxides with carbonyl compounds. Tetrahedron. 1970;26:1311–1313. doi: 10.1016/S0040-4020(01)93001-7. [DOI] [Google Scholar]
- 27.Tajima, Y. RIKEN, Saitama, Japan, 2005, Unpublished data.
- 28.Tajima Y., Hara T., Honma T., Matsumoto S., Takeuchi K. Lewis acid-assisted nucleophilic substitution of fullerene epoxide. Org. Lett. 2006;8:3203–3206. doi: 10.1021/ol0610183. [DOI] [PubMed] [Google Scholar]
- 29.Chen Z.X., Wang G.W. One-pot sequential synthesis of acetoxylated [60]fullerene derivatives. J. Org. Chem. 2005;70:2380–2383. doi: 10.1021/jo047894g. [DOI] [PubMed] [Google Scholar]
- 30.Kadish K.M., Gao X., Caemelbecke E.V., Suenobu T., Fukuzumi S. Effect of addition pattern on the electrochemical and spectroscopic properties of neutral and reduced 1,2- and 1,4-(C6H5CH2)2C60 isomers. J. Phys. Chem. A. 2000;104:3878–3883. doi: 10.1021/jp993708c. [DOI] [Google Scholar]
- 31.Bordwell F.G. Are nucleophilic bimolecular concerted reactions involving four or more bonds a myth? Acc. Chem. Res. 1970;3:281–290. doi: 10.1021/ar50033a001. [DOI] [Google Scholar]
- 32.Reed C.A., Kim K.C., Bolskar R.D., Mueller L.J. Taming superacids: Stabilization of the fullerene cations HC60+ and C60+ Science. 2000;289:101–104. doi: 10.1126/science.289.5476.101. [DOI] [PubMed] [Google Scholar]
- 33.Mueller L.J., Elliott D.W., Kim K.C., Reed C.A., Boyd P.D.W. Establishing through-bond connectivity in solids with NMR: STRUCTURE and dynamics in HC60+ J. Am. Chem. Soc. 2002;124:9360–9361. doi: 10.1021/ja0266619. [DOI] [PubMed] [Google Scholar]
- 34.Kitagawa T., Sakamoto H., Takeuchi K. Electrophilic addition of polychloroalkanes to C60: Direct observation of alkylfullerenylcation intermediates. J. Am. Chem. Soc. 1999;121:4298–4299. doi: 10.1021/ja9900048. [DOI] [Google Scholar]
- 35.Murata Y., Cheng F., Kitagawa T., Komatsu K. Generation of fullerenylcation (EtO)2P+(OH)CH2−C60+ from RC60−H and from RC60−C60R (R=CH2P(O)(OEt)2) J. Am. Chem. Soc. 2004;126:8874–8875. doi: 10.1021/ja047483h. [DOI] [PubMed] [Google Scholar]
- 36.Kitagawa T., Lee Y., Hanamura M., Sakamoto H., Konno H., Takeuchi K., Komatsu K. Nucleophilic substitution of alkylchlorodihydro[60]fullerenes: Thermodynamic stabilities of alkylated C60 cation intermediates. Chem. Commun. 2002:3062–3063. doi: 10.1039/b210126b. [DOI] [PubMed] [Google Scholar]
- 37.Numata Y., Kawashima J., Hara T., Tajima Y. One-pot synthesis of indolino[2',3':1,2][60]-fullerenes from fullerene epoxide: Lewis acid-assisted nucleophilic addition followed by intramolecularcyclization. Chem. Lett. 2008;37:1018–1019. doi: 10.1246/cl.2008.1018. [DOI] [Google Scholar]
- 38.Chen C.P., Luo C., Ting C., Chuang S.C. Organic photovoltaics incorporating fulleroisoquinolinones as n-type materials. Chem. Commun. 2011;47:1845–1847. doi: 10.1039/c0cc04246c. [DOI] [PubMed] [Google Scholar]
- 39.Andersson C.-H., Berggren G., Ott S., Grennberg H. Synthesis and IR spectroelectrochemical studies of a [60]fulleropyrrolidine-(tricarbonyl)chromium complex: Probing C60 redox states by IR spectroscopy. Eur. J. Inorg. Chem. 2011:1744–1749. [Google Scholar]
- 40.Zhu B., Wang G.W. Synthesis of [60]fulleroindolines: Palladium-catalyzed heteroannulations of [60]fullerene with o-iodoanilines. J. Org. Chem. 2009;74:4426–4428. doi: 10.1021/jo900585u. [DOI] [PubMed] [Google Scholar]
- 41.Zhu B., Wang G.W. Palladium-catalyzed heteroannulation of fullerene with anilides via C–H bond activation. Org. Lett. 2009;11:4334–4337. doi: 10.1021/ol901675t. [DOI] [PubMed] [Google Scholar]
- 42.Nambo M., Segawa Y., Itami K. Aziridinofullerene: A versatile platform for functionalized fullerenes. J. Am. Chem. Soc. 2011;133:2402–2405. doi: 10.1021/ja111213k. [DOI] [PubMed] [Google Scholar]
- 43.Numata Y., Kawashima J., Tajima Y. Substituent effect on reduction potentials of heterocyclic-fused fullerene derivatives. ECS Trans. 2009;16:33–43. [Google Scholar]
- 44.Hammett L.P. The effect of structure upon the reactions of organic compounds. benzene derivatives. J. Am. Chem. Soc. 1937;59:96–103. doi: 10.1021/ja01280a022. [DOI] [Google Scholar]