

Abstract
The synthesis of 4-methoxycarbonyl-3-methylisoquinolinequinone (1) and a variety of its substitution products with amino-, alkylamino and halogen groups on the quinone nucleus is reported. The series of 6-, 7- and 6,7-subtituted isoquinolinequinones were evaluated in vitro for their cytotoxic activity using the MTT colorimetric method. All the newly synthesized compounds showed moderate to high potency against MRC-5 healthy lung fibroblasts and four human tumor cell lines: AGS gastric adenocarcinoma, SK-MES-1 lung, J82 bladder carcinoma, and HL-60 leukemia cells. Among the series, compounds 4b, 12 and 13 exhibited interesting antitumor activity against human gastric adenocarcinoma, human lung and human bladder carcinoma cancer cells. 7-Amino-6-bromoisoquinoline-5,8-quinone (13) was found to be the most promising active compound against the tested cancer cell lines, with IC50 values in the 0.21−0.49 μM range, lower than the anti-cancer agent etoposide used as reference.
Keywords: aminoisoquinoline-5,8-quinones; regioselectivity; anticancer; SAR analysis
1. Introduction
The search for biologically active natural products from marine sources continues to be an important scientific field that offers promising opportunities for the development of new compounds endowed with pharmacological properties [1,2,3,4,5,6,7,8]. Among the natural products with a quinoide system isoquinolinequinones form an important class characterized by their varied biological properties. For instance, marine isoquinolinequinones such as caulibugulones A–D and mansouramycins A–C (Figure 1) display a significant antitumor profile in many tumor cell lines [9,10]. Caulibugulones A–D, evaluated for antitumor activity against the murine IC-2WT cell line [9] exhibit high potency. In these assays, the A–C members displayed IC50 values ranging from 0.22 to 0.34 μg/mL, whereas caulibugulone D showed less cytotoxic activity (IC50 = 1.67 μg/mL). According to these IC50 values against the tested cell line it can be deduced that insertion of bromine or chlorine atoms at the 6-position in caulibugulone A (0.34 μg/mL), as in caulibugulone B (0.22 μg/mL) and C (0.28 μg/mL), is not an important determinant of cytotoxicity. Furthermore, the replacement of the methylamino group in caulibugulone A by the 2-hydroxyethylamino group, as in caulibugulone D, produces a decrease of the cytotoxic activity. Evidences reported by Wipf and Lazo [11,12] indicate that caulibugulones are selective in vitro inhibitors of the Cdc25 family of cell cycle-controlling protein phosphatases.
Figure 1.

Structure of caulibugulones A–D and mansouramycins A–C.
The access to caulibugulones A–D via regioselective nucleophilic amination substitution at the 7-position in the reaction of isoquinoline-5,8-quinone with methylamine or 2-aminoethanol in ethanol as solvent in the presence of CeCl3·7H2O has been simultaneously reported by Wipf [11] and Tamagnan [13]. These amination reactions yield caulibugulones A and D accompanied by low amounts of their corresponding 6-amino-substituted regioisomers. Further reaction of caulibugulone A with N-chlorosuccinimide and N-bromosuccinimide provides access to caulibugulones B and C, respectively.
Regarding mansouramycins A–C, screening on a panel of up to 36 human tumor cell lines displayed significant cytotoxicity, with pronounced selectivity for non-small cell lung cancer, breast cancer, melanoma, and prostate cancer cells [10]. Mansouramycin C (4) proved to be the most active compound, with an overall potency of 0.089 μM. It was followed by mansouramycin B (mean IC50 = 2.7 μM) and then mansouramycin A, with a mean IC50 value of 13.44 μM. These data indicate that insertion of a methoxycarbonyl group at C-1 or a chlorine atom at C-6 into the 7-methylaminoisoquinolinequinone pharmacophore induces an increase of antitumor activity.
As part of our research program aimed at the synthesis and structure-activity relationship (SAR) of novel aminoquinones as potential antitumor agents [14,15,16,17,18] we report herein the synthesis and the in vitro antitumor evaluation of a variety of amino-, alkylamino and alkylamino-haloisoquinolinequinone derivatives, structurally related with the above mentioned marine isoquinolinequinones, as well the SAR of this series.
2. Results and Discussion
Isoquinolinequinone 1, unsubstituted at the 1-position and containing a methoxycarbonyl group on the heterocyclic ring, as in mansouramycin C, was selected as a suitable precursor of the designed aminoisoquinolinequinones. The synthesis of the target compound 1 was accomplished in 86% yield from commercially available 2,5-dihydroxybenzaldehyde and methyl aminocrotonate, through a one-pot procedure previously developed in our laboratory [19] (Scheme 1).
Scheme 1.

Synthesis and reaction of isoquinolinequinone 1 with methylamine.
The amination reaction of quinone 1 was firstly examined with methylamine hydrochloride-sodium acetate in ethanol at room temperature. The reaction went to completion in 3 h to give an 80:20 mixture of regioisomers 2a and 2b (as evaluated by 1H-NMR) in 74% yield. Further separation by column chromatography on silica gel afforded pure compound 2a in 52% yield (Scheme 1). Our efforts to isolate a pure sample of regioisomer 2b by chromatography were unsuccessful. Then we examined the amination reaction of quinone 1 with 2-aminoethanol under the same conditions used with methylamine. Analysis of the progress of the reaction by TLC reveals that the amination reaction of 1 proceeds slowly (>7 h) and extensive decomposition of quinone 1 was detected. According to precedents on the use of a Lewis acid to facilitate the reaction of quinones with amines [20,21,22,23,24], compound 1 was allowed to react with 2-aminoethanol in ethanol in the presence of 5 mmol% of CeCl3·7H2O. Under these conditions, the reaction went to completion in 3 h to give an 80:20 mixture of regioisomers 3a and 3b (as evaluated by 1H-NMR) in 65% yield. Further separation by column chromatography on silica gel afforded the major regioisomer 3a pure in 47% yield (Scheme 2). Attempts to isolate a pure sample of the minor regioisomer 3b by chromatography were unsuccessful.
Scheme 2.

Reaction of isoquinolinequinone 1 with amines induced by acid-catalysis.
The amination reactions of quinone 1 with cyclohexylamine and morpholine under the above acid-induced conditions were also explored. The experiments provided access to a mixture of the corresponding alkylaminoisoquinolinequinones 4a,b and 5a,b (Scheme 2). In these cases, pure samples of the regioisomers of each pair were isolated by column chromatography.
According to the amination reaction of quinone 1 with the studied alkyamines we can conclude that the addition of nitrogen nucleophiles across the quinone double bond occurs with high regioselective preference to give the 7-substituted regioisomer as the main product (Scheme 2).
The structure of the new compounds was established on the basis of their nuclear magnetic resonance (1H-NMR, 13C-NMR, 2D-NMR) and high resolution mass spectra (HRMS). The position of the nitrogen substituent in these aminoquinones was determined by means of HMBC experiments. For example, the location of the nitrogen group at C-7 in compound 4a was deduced by the 3JC,H couplings between the carbon at C-8 (δ 180.83) with the protons at C-1 (δ 9.17), at C-6 (δ 5.77) and that of the NH group (δ 5.96). In the case of aminoquinone 4b, the location of the nitrogen substituent at C-6 was established by 3JC,H coupling between the carbon at C-5 (δ 181.71) with the proton at C-7 (δ 5.79) and the proton of the NH group (δ 5.75). Also, the 3JC,H coupling between the carbon at C-8 (δ 181.49) with the proton at C-1 (δ 9.29) can be observed (Figure 2).
Figure 2.

HMBC correlations of regioisomers 4a and 4b.
Aminoisoquinolinequinones 2a, 3a, 4a and 5a were submitted to halogenations with N-halosuccinimide to yield the corresponding alkylamino-haloisoquinolinequinones (Table 1).
Table 1.
Preparation of aminohaloisoquinolinequinones 6–11.
| Substrate | Product | N° | Yield (%) * | Substrate | Product | N° | Yield (%) * |
|---|---|---|---|---|---|---|---|
| 2a | 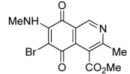 |
6 | 80 | 3a | 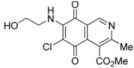 |
9 | 78 |
| 2a | 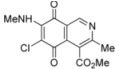 |
7 | 85 | 4a | 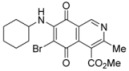 |
10 | 84 |
| 3a | 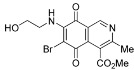 |
8 | 75 | 5a | 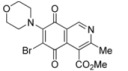 |
11 | 70 |
* Isolated yields.
We also explored the preparation of amino- and aminobromoisoquinolinequinones in order to include them in the biological assays. Treatment of quinone 1 with sodium azide in acetic acid, according to a previously reported procedure [15], afforded aminoisoquinolinequinone 12 as the sole regioisomer in 51% yield (Scheme 3). The structure of compound 12 was established by means of HMBC experiments. Further bromination of 12 with N-bromosuccinimide, provides aminobromoisoquinolinequinone 13 in 86% yield.
Scheme 3.

Preparation of amino- and aminobromoisoquinolinequinones 12 and 13.
Isoquinolinequinones 1, 2a, 3a, 4a, 4b, 5a, 6–13 were evaluated for their in vitro cytotoxic activity against MRC-5 (healthy lung fibroblasts) and four human cancer cell lines: AGS (gastric), SK-MES-1 (lung), J82 (bladder), and HL-60 (leukemia), using the conventional microculture tetrazolium reduction assay [25,26,27]. The broad variety of the synthesized compounds was designed in order to gain insight into the influence of nitrogen and halogen groups on the quinone nucleus of the isoquinolinequinone pharmacophore on the biological activity. Table 2 summarizes the data from these evaluations. According to the IC50 values collected in Table 2, it is evident that insertion of the nitrogen substituents in quinone 1, as in 2a,3a,4a,4b,5a and 5b, increases the cytotoxic activity in all the evaluated cell lines, compared to those of precursor 1. The initial structure-activity relationship (SAR) was focused on the nature and location of alkylamino at the quinone nucleus of the isoquinolinequinone pharmacophore. The data of Table 2 for compounds 2a, 3a, 4a and 5a reveal that the shape and polarity of the alkylamino group at the 7-position in the isoquinolinequinone moiety does not significantly influence the biological activity. Concerning the effect of the position of the alkylamino groups, the IC50 values for the regioisomers 4a/4b and 5a/5b indicate that substitution of the nitrogen group at the 6-position of the quinone pharmacophore induces a greater effect on the antitumor activity than at the 7-position. It can be seen that the effect of such substitution on the antitumor potency on gastric adenocarcinoma and lung cancer cells reaches values nearly seven times higher compared to regioisomer 4a.
Table 2.
Cytotoxic activity of 1 and their aminoisoquinoline-5,8-quinone derivatives.
| IC50 ± SEM a (μM) | ||||||
|---|---|---|---|---|---|---|
| N° | Structure | MRC-5 b | AGS c | SK-MES1 d | J82 e | HL-60 f |
| 1 | 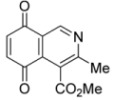 |
4.5 ± 0.3 | 17.34 ± 1.64 | 25.9 ± 1.6 | 14.81 ± 0.74 | 14.81 ± 0.74 |
| 2a | 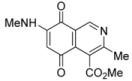 |
8.1 ± 0.3 | 3.5 ± 0.2 | 5.22 ± 0.38 | 5.1 ± 0.4 | 9.74 ± 0.76 |
| 3a | 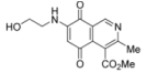 |
7.9 ± 0.3 | 2.3 ± 0.1 | 5.99 ± 0.36 | 7.2 ± 0.4 | 6.16 ± 0.36 |
| 4a | 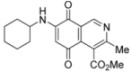 |
10.1 ± 0.6 | 4.0 ± 0.2 | 5.33 ± 0.26 | 12.0 ± 1.0 | 10.67 ± 0.64 |
| 4b | 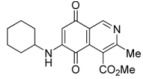 |
2.2 ± 0.1 | 0.59 ± 0.03 | 1.69 ± 0.09 | 3.0 ± 0.2 | 1.57 ± 0.13 |
| 5a | 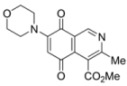 |
6.7 ± 0.4 | 1.75 ± 0.09 | 4.00 ± 0.22 | 8.56 ± 0.27 | 2.99 ± 0.15 |
| 5b | 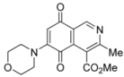 |
3.77 ± 0.21 | 1.20 ± 0.06 | 1.76 ± 0.11 | 2.99 ± 0.12 | 2.11 ± 0.13 |
| 6 | 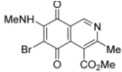 |
2.6 ± 0.2 | 1.8 ± 0.1 | 1.66 ± 0.08 | 2.0 ± 0.1 | 2.64 ± 0.21 |
| 7 | 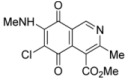 |
1.9 ± 0.2 | 1.9 ± 0.0 | 1.15 ± 0.05 | 1.2 ± 0.1 | 2.63 ± 0.13 |
| 8 | 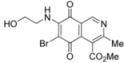 |
2.7 ± 0.2 | 1.1 ± 0.0 | 2.13 ± 0.13 | 2.2 ± 0.2 | 3.57 ± 0.16 |
| 9 | 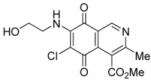 |
7.1 ± 0.5 | 2.6 ± 0.2 | 1.29 ± 0.08 | 1.2 ± 0.1 | 2.34 ± 0.14 |
| 10 | 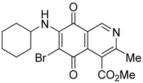 |
2.2 ± 0.2 | 0.56 ± 0.03 | 2.14 ± 0.13 | 2.3 ± 0.1 | 2.15 ± 0.12 |
| 11 | 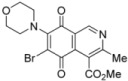 |
9.53 ± 0.38 | 8.15 ± 0.49 | 1.46 ± 0.06 | 6.97 ± 0.35 | 2.61 ± 0.18 |
| 12 | 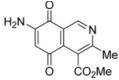 |
1.8 ± 0.1 | 1.2 ± 0.1 | 0.93 ± 0.03 | 1.1 ± 0.1 | 2.16 ± 0.09 |
| 13 | 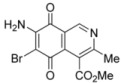 |
0.28 ± 0.01 | 0.26 ± 0.01 | 0.21 ± 0.01 | 0.29 ± 0.01 | 0.49 ± 0.02 |
| Etoposide | 0.33 ± 0.02 | 0.58 ± 0.02 | 1.83 ± 0.09 | 3.49 ± 0.16 | 2.23 ± 0.09 | |
a Data represent mean average values for six independent determinations; b Normal human lung fibroblasts cells; cHuman gastric adenocarcinoma cell line; d Human lung cancer cell line; e Human bladder carcinoma cell line; f Leukemia cell line.
Next, the SAR analysis was focused on the effects of insertion of chlorine and bromine atoms at the 6-position of the 7-substituted alkylaminoisoquinolinequinones 2a, 3a, 4a and 5a. Comparison of the IC50 values of halo derivatives 6–11 with those of their corresponding precursors indicates that the substitution of halogen atom at the 6-position enhances the antitumor potency in all the evaluated cell lines, except for compound 11 on lung fibroblasts and gastric cancer cells.
According to the data of Table 2, aminoisoquinolinequinone 12 showed more cytotoxic activity (0.93–2.16 µM) on the cancer cell lines than the N-alkyl 7-substituted analogs 2a, 3a, 4a and 5a. It is noteworthy that compound 12 exhibits higher antitumor potency on lung and bladder cell lines than the anticancer drug etoposide.
Comparison of the cytotoxic potency of compound 12 (0.93−2.16 µM) with that of its bromoderivative 13 (IC50: 0.21−0.49 µM) reveals that insertion of the bromine atom at the 6-position of 12 induces a high increase of the cytotoxic activity in all the evaluated cell lines. It is noteworthy that compounds 12 and 13 exhibit higher antitumor potency and selectivity index (SI = IC50 fibroblasts/IC50 cancer cells. 12: 0.8–1.9; 13: 0.5–1.3) in almost all cell lines than the anticancer drug etoposide (SI: 0.1–0.6). Among the tested analogs of the series, compounds 4b, 12 and 13 are the most significant antitumor members. Compound 13 was selected as a promising lead compound due to its high potency on the tested tumor cell lines and selectivity index.
3. Experimental
3.1. General
All reagents were commercially available reagent grade and were used without further purification. Melting points were determined on a Stuart Scientific SMP3 apparatus and are uncorrected. 1H-NMR spectra were recorded on Bruker AM-200 and Avance-400 instruments in deuterochloroform (CDCl3). 13C-NMR spectra were obtained in CDCl3 at 50 and 100 MHz. Bidimensional NMR techniques and DEPT were used for signal assignment. Chemical shifts are expressed in ppm downfield relative to tetramethylsilane and the coupling constants (J) are reported in Hertz. The HRMS spectra were obtained on a Thermo Finnigan spectrometer, model MAT 95XP. Silica gel Merck 60 (70–230 mesh) was used for preparative column chromatography and TLC aluminum foil 60F254 for analytical TLC.
3.2. Chemistry
4-Methoxycarbonyl-3-methylisoquinoline-5,8-quinone (1). A suspension of 2,5-dihydroxybenzaldehyde (1 mmol), methyl 3-aminocrotonate (1 mmol), Ag2O (2 mmol) and MgSO4 (0.5 g) in CH2Cl2 (25 mL) was stirred at rt for 3 h. The mixture was filtered, the solids were washed with CH2Cl2 and the solvent removed under reduced pressure. The residue was column cromatographed over silica gel (90:10 CH2Cl2/AcOEt) to yield pure quinone 1 (86%) as yellow solid, mp 109–111.5 °C; IR νmax 1731 (C=O ester), 1668 (C=O quinone); 1H-NMR (400 MHz, CDCl3): δ 2.65 (s, 3H, Me), 4.03 (s, 3H, CO2Me), 7.04 (s, 2H, 6- and 7-H), 9.22 (s, 1H, 1-H); 13C-NMR (100 MHz): δ 22.69, 53.11, 122.18, 125.15, 133.39, 138.38, 138.58, 148.57, 161.83, 167.68, 183.38, 183.60; HRMS (M+): m/z calcd for C12H9NO4: 231.05315; found: 231.05229.
7-Methylamino-4-methoxycarbonyl-3-methylisoquinoline-5,8-quinone (2a). A suspension of quinone 1 (1 mmol), methylamine hydrochloride (2 mmol) and AcONa (2 mmol) in ethanol (20 mL) was stirred at rt for 1.5 h. The solvent was removed under reduced pressure to give an orange solid containing regioisomers 2a and 2b (74%) in 80:20 proportion. The major regioisomer 2a was isolated by column chromatography over silica gel (90:10 CH2Cl2/AcOEt): red solid (52 %), mp 209.5–211 °C; IR νmax 3279 (N-H), 1740 (C=O ester), 1681 (C=O quinone); 1H-NMR: (400 MHz, DMSO): δ 2.66 (s, 3H, Me), 2.95 (d , 3H, J = 5.4 Hz, NHMe), 4.04 (s, 3H, CO2Me), 5.75 (s, 1H, 6-H), 6.09 (br s, 1H, NH), 9.18 (s, 1H, 1-H); 13C-NMR (100 MHz) δ: 23.02, 29.36, 53.21, 101.16, 122.08, 126.38, 136.38, 148.10, 148.82, 163.02, 168.94, 180.24, 180.55; HRMS (M+): m/z calcd for C13H12N2O4: 260.0797; found: 260.0731.
7-(2-Hydroxyethylamino)-4-methoxycarbonyl-3-methylisoquinoline-5,8-quinone (3a). A suspension of quinone 1 (1 mmol), 2-aminoethanol (2 mmol), CeCl3.7H2O (0.05 mmol) and ethanol (25 mL) was left with stirring at rt for 3 h. The solvent was removed under reduced pressure to give an orange solid containing regioisomers 3a and 3b (65%) in 80:20 proportion. The major regioisomer 3a was isolated by column chromatography over silica gel (90:10 CH2Cl2/AcOEt); red solid (47%), mp 195.5–197 °C; IR νmax 3399 (O-H), 3186(N-H), 1700 (C=O ester), 1600 (C=O quinone); 1H-NMR (400 MHz, DMSO): δ 2.52 (s, 3H, Me), 3.27 (dt, 2H, J = 5.8, 11.7 Hz, NHCH2), 3.59 (dt, 2H, J = 5.6, 11.2 Hz, CH2OH), 3.88 (s, 3H, CO2Me), 4.89 (t, 1H, J = 5.7 Hz, OH), 5.79 (s, 1H, 6-H), 7.75 (t, 1H, J = 5.9 Hz, NH), 9.06 (s, 1H, 1-H); 13C-NMR (100 MHz): δ 22.35, 44.77, 52.63, 58.47, 99.87, 122.43, 125.18, 135.92, 147.36, 149.04, 161.13, 168.25, 178.91, 180.24; HRMS (M+): m/z calcd for C14H14N2O5: 290.0903; found: 290.0891.
6- and 7-(Cyclohexylamino)-4-methoxycarbonyl-3-methylisoquinoline-5,8-quinone (4a,4b). A suspension of quinone 1 (1 mmol), cyclohexylamine (2 mmol), CeCl3·7H2O (0.05 mmol) and ethanol (25 mL) was left with stirring at rt for 2 h. The solvent was removed under reduced pressure to give an orange solid containing regioisomers 4a and 4b in 86:14 ratio. These compounds were isolated by column chromatography over silica gel (90:10 CH2Cl2/AcOEt).
Compound 4a (less polar, 80%): orange solid, mp 161.5–163.5 °C; IR νmax 3,246 (N-H), 2,927 and 2,856 (CH), 1741 (C=O ester), 1685 (C=O quinone); 1H-NMR (400 MHz, CDCl3): δ 1.35 (m, 5H, CH2), 1.68 (m, 1H, CH2), 1.81 (m, 2H, CH2), 2.03 (m, 2H, CH2), 2.65 (s, 3H, Me), 3.30 (m,1H, CH), 4.03 (s, 3H, CO2Me), 5.77 (s, 1H, 6-H), 5.96 (d, 1H, J = 7.5 Hz, NH), 9.17 (s, 1H, 1-H); 13C-NMR (100 MHz): δ 22.99, 24.54 (2C), 25.42, 31.83 (2C), 51.44, 53.16, 101.25, 122.11, 126.23, 136.34, 146.52, 148.03, 162.89, 168.93, 180.13, 180.83; HRMS (M+): m/z calcd for C18H20N2O4: 328.14227; found: 328.14083.
Compound 4b (9.5%): red solid, mp 158.5–160 °C; IR νmax 3246 (N-H); 2927 and 2856 (CH); 1741 (C=O ester); 1685 (C=O quinone); 1H-NMR (400 MHz, CDCl3): δ 1.35 (m, 5H, CH2), 1.68 (m, 1H, CH2), 1.81 (m, 2H, CH2), 2.03 (m, 2H, CH2), 2.65 (s, 3H, Me), 3.30 (m, 1H, CH), 4.05 (s, 3H, CO2Me), 5.75 (d, 1H, J = 7.8 Hz, NH), 5.79 (s, 1H, 6-H), 9.28 (s, 1H, 1-H), 13C-NMR (100 MHz): δ 22.64, 24.54 (2C), 25.47, 31.86 (2C), 51.39, 53.26, 101.23, 123.05, 124.92, 132.44, 146.31, 148.73, 159.91, 168.51, 181.49, 181.71;HRMS (M+): m/z calcd for C18H20N2O4: 328.14227; found: 328.14070.
6- and 7-Morpholino-4-methoxycarbonyl-3-methylisoquinoline-5,8-quinone (5a,5b). A suspension of quinone 1 (1 mmol), morpholine (2 mmol), CeCl3·7H2O (0.05 mmol) and ethanol (25 mL) was left with stirring at rt for 2 h. The solvent was removed under reduced pressure to give an orange solid containing regioisomers 5a and 5b in 87:13 proportion. These compounds were isolated by column chromatography over silica gel (90:10 CH2Cl2/AcOEt).
Compound 5a (less polar, 72%): dark orange solid, mp 169.5–171.5 °C; IR νmax 1722 (C=O ester), 1,696 and 1,638 (C=O quinone); 1H-NMR (400 MHz, CDCl3): δ 2.65 (s, 3H, Me), 3.59 (t, 4H, J = 4.7 Hz, N-CH2), 3.87 (t, 4H, J = 4.7 Hz, O-CH2), 4.03 (s, 3H, CO2Me), 6.01 (s, 1H, 6-H), 9.14 (s, 1H, 1-H); 13C-NMR (100 MHz): δ 22.90, 49.21 (2C), 53.28, 66.49 (2C), 110.92, 123.98, 125.36, 134.86, 148.72, 152.78, 161.98, 168.72, 181.16, 181.75; HRMS (M+): m/z calcd for C16H16N2O5: 316.10593; found: 316.10521.
Compound 5b (17%): dark red solid, mp 159.5–162 °C; IR νmax 1724 (C=O ester), 1,678 and 1,634 (C=O quinone); 1H-NMR (400 MHz, CDCl3): δ 2.66 (s, 3H, Me), 3.47 (t, 4H, J = 4.8, N-CH2), 3.85 (t, 4H, J = 4.8 Hz, O-CH2), 4.03 (s, 3H, CO2Me), 6.01 (s, 1H, 6-H), 9.23 (s, 1H, 1-H); 13C-NMR (100 MHz): δ 22.89, 49.10 (2C), 53.25, 66.44 (2C), 111.30, 122.43, 125.41, 135.43, 148.35, 153.21, 160.76, 168.54, 182.10, 182.30; HRMS (M+): m/z calcd for C16H16N2O5: 316.10593; found: 316.10445.
3.3. General Procedure for the Synthesis of 6-Bromo (or 6-chloro)-7-aminoisoquinolinequinone Derivatives
A solution of the 7-aminoisoquinolinequinone derivative (1 mmol), the corresponding N-bromosuccinimide (NBS) or N-chlorosuccinimide (NCS) (1 mmol) and methanol (15 mL) was left with stirring at rt after completion of the reaction as indicated by TLC. The solvent was removed under reduced pressure and the residue was column cromatographed over silica gel (90:10 CH2Cl2/AcOEt) to yield the corresponding 7-amino-6-haloisoquinolinequinone derivative.
6-Bromo-4-methoxycarbonyl-7-methylamino-3-methylisoquinoline-5,8-quinone (6). Prepared from 2a and NBS (30 min, 80%): dark red solid, mp 170–172 °C; IR νmax 3331 (N-H); 1730 (C=O ester), 1687 (C=O quinone); 1H-NMR (400 MHz, CDCl3): δ 2.65 (s, 3H, Me), 3.48 (d , 3H, J = 5.8 Hz, NHMe), 4.05 (s, 3H, CO2Me), 6.39 (br s, 1H, NH), 9.16 (s, 1H, 1-H); 13C-NMR (100 MHz): δ 23.09, 33.34, 53.39, 121.29, 126.56, 134.72, 146.86, 148.62, 163.23, 168.42, 174.40, 178.84 (the signal of the C-6 was not observed in this experiment); HRMS (M+): m/z calcd for C13H11N2O4Br: 337.99023; found: 337.98951.
6-Chloro-4-methoxycarbonyl-7-methylamino-3-methylisoquinoline-5,8-quinone (7). Prepared from 2a and NCS (3 h, 85%): dark red solid, mp 158–160 °C; IR νmax 3331 (N-H); 1773 (C=O ester), 1693 (C=O quinone); 1H-NMR (400 MHz, CDCl3): δ 2.65 (s, 3H, Me), 3.48 (d , 3H, J = 6.0 Hz, NHMe), 4.05 (s, 3H, CO2Me), 6.30 (br s, 1H, NH), 9.15 (s, 1H, 1-H); 13C-NMR (100 MHz): δ 23.09, 32.82, 53.37, 121.25, 126.52, 135.11, 144.83, 148.50, 163.41, 168.36, 174.83, 179.27 (the signal of the C-6 was not observed in this experiment); HRMS (M+): m/z calcd for C13H11N2O4Cl: 294.0407; found: 294.04002
6-Bromo-2-(hydroxyethylamino)-4-methoxycarbonyl-3-methylisoquinoline-5,8-quinone (8). Prepared from 3a and NBS (30 min, 75%): dark red solid, mp 134–136 °C; IR νmax 3463 (O-H), 3320 (N-H), 1720 (C=O ester), 1685 (C=O quinone); 1H-NMR (400 MHz, DMSO): δ 2.63 (s, 3H, Me), 3.92 (dt, 2H, J = 5.1, 11.2 Hz, CH2OH), 4.06 (s, 3H, CO2Me), 4.11 (dt, 2H, J = 5.3, 10.8 Hz, NHCH2), 6.65 (br s, 1H, NH), 9.13 (s, 1H, 1-H); 13C-NMR (100 MHz): δ 22.98, 47.25, 53.40, 61.36, 121.49, 126.41, 134.60, 146.84, 148.61, 163.02, 168.52, 174.19, 178.66, (the signal of the C-6 was not observed in this experiment); HRMS (M+): m/z calcd for C14H13N2O5Br: 368.00079; found: 368.00005.
6-Chloro-2-(hydroxyethylamino)-4-methoxycarbonyl-3-methylisoquinoline-5,8-quinone (9). Prepared from 3a and NCS (4 h, 78%): dark red solid, mp 123–125 °C; IR νmax 3461 (O-H), 3325 (N-H), 1716 (C=O ester), 1687 (C=O quinone); 1H-NMR (400 MHz, CDCl3): 1H-NMR (400 MHz, CDCl3): δ 2.65 (s, 3H, Me), 3.92 (m, 2H, NHCH2), 4.05 (s, 3H, CO2Me), 4.07 (m, 2H, CH2OH), 6.58 (br s, 1H, NH), 9.16 (s, 1H, 1-H); 13C-NMR (100 MHz): δ 23.09, 4.82, 53.42, 61.71, 121.38, 126.48, 134.99, 144.53, 148.59, 163.35, 168.41, 174.67, 179.22, (the signal of the C-6 was not observed in this experiment); HRMS (M+): m/z calcd for C14H13N2O5Cl: 324.0513; found: 324.05073.
6-Bromo-7-(cyclohexylamino)-4-methoxycarbonyl-3-methylisoquinoline-5,8-quinone (10). Prepared from 4a and NBS (15 min, 84% yield): dark red solid, mp 132.5–134.5 °C; IR νmax 3312 (N-H), 3008 and 2858 (C-H), 1735 (C=O ester), 1687 (C=O quinone); 1H-NMR (400 MHz, CDCl3): δ 1.34 (m, 5H, CH2), 1.68 (m, 1H, CH2), 1.81 (m, 2H, CH2), 2.03 (m, 2H, CH2), 2.64 (s, 3H, Me), 4.05 (s, 3H, CO2Me), 4.58 (m,1H, CH), 6.24 (br s, 1H, NH), 9.15 (s, 1H, 1-H); 13C-NMR (100 MHz): δ 23.05, 24.52 (2C), 25.29, 34.50 (2C), 53.32, 61.58, 121.36, 126.59, 134.80, 148.65, 149.99, 163.16, 168.38, 174.32, 178.90, (the signal of the C-6 was not observed in this experiment); HRMS (M+): m/z calcd for C18H19N2O4Br: 406.05280; found: 406.05196.
6-Bromo-7-morpholino-4-methoxycarbonyl-3-methylisoquinoline-5,8-quinone (11). Prepared from 5a and NBS (30 min, 70%): violet solid, mp 127.5–129 °C; IR νmax 2955 and 2856 (C-H), 1722 (C=O ester), 1696 and 1638 (C=O quinone); 1H-NMR (400 MHz, CDCl3): δ 2.66 (s, 3H, Me), 3.70 (t, 4H, J = 4.5 Hz, N-CH2), 3.90 (t, 4H, J = 4.5 Hz, O-CH2), 4.05 (s, 3H, CO2Me), 9.19 (s, 1H, 1-H); 13C-NMR (100 MHz): δ 22.87, 52.59 (2C), 53.42, 67.51 (2C), 115.51, 122.43, 126.05, 133.51, 149.00, 152.08, 162.25, 168.21, 176.25, 180.24; HRMS (M+): m/z calcd for C16H15N2O5Br: 394.01643; found: 394.01457.
7-Amino-4-methoxycarbonyl-3-methylisoquinoline-5,8-quinone (12). A solution of sodium azide (1.2 mmol) in water (5 mL) was added, with stirring, to a previously heated solution (~40 °C) of quinone 1 (1 mmol) in glacial acetic acid (10 mL). The mixture was stirred at rt for 3.5 h, diluted with water and then extracted with AcOEt (3 × 15 mL). The organic phase was washed with saturated aqueous solution of NaHCO3, dried (MgSO4) and the solvent was removed under reduced pressure. The residue was column cromatographed over silica gel (80:20 CH2Cl2/AcOEt) to yield aminoquinone 12 as an orange solid (51%), mp 189–191 °C; IR νmax 3376 and 3140 (N-H), 1732 (C=O ester), 1691 (C=O quinone); 1H-NMR (400 MHz, DMSO): δ 2.66 (s, 3H, Me), 4.05 (s, 3H, CO2Me), 5.38 (br s, 2H, NH2), 6.03 (s, 1H, 6-H), 9.20 (s, 1H, 1-H); 13C-NMR (100 MHz): δ 23.37, 52.68, 102.44, 122.47, 125.17, 135.92, 147.19, 150.88, 161.00, 168.34, 179.30, 180.65; HRMS (M+): m/z calcd for C12H10N2O4: 246.06406; found: 246.06207.
7-Amino-6-bromo-4-methoxycarbonyl-3-methylisoquinoline-5,8-quinone (13). Prepared from 12 and NBS (20 min, 86%): mp 146–148 °C; IR νmax 3423 and 3326 (N-H), 1726 (C=O ester), 1683 (C=O quinone); 1H-NMR (400 MHz, CDCl3): δ 2.67 (s, 3H, Me), 4.06 (s, 3H, CO2Me), 5.45 (br s, 1H, NH), 6.26 (br s, 1H, NH), 9.22 (s, 1H, 1-H); 13C-NMR (100 MHz): δ 23.18, 53.48, 105.07, 121.08, 126.91, 134.81, 147.20, 148.50, 163.58, 168.26, 174.53, 177.24; HRMS (M+): m/z calcd for C12H9N2O4Br: 323.97455; found: 323.97419.
3.4. Anticancer Assay
The cell lines used in this work were obtained from the American Type Culture Collection (ATCC. Manasas, VA, USA). They included MRC-5 normal human lung fibroblasts (CCL-171), AGS human gastric adenocarcinoma cells (CRL-1739), SK-MES-1 human lung cancer cells (HTB-58) and J82 human bladder carcinoma cells (HTB-1). After the arrival of the cells, they were proliferated in the corresponding culture medium as suggested by the ATCC. The cells were stored in medium containing 10% glycerol in liquid nitrogen. The viability of the cells after thawing was higher than 90%, as assessed by trypan blue exclusion test. Cells were sub-cultured once a week and the medium was changed every two days. Cells were grown in the following media: MRC-5, SKMES-1, and J82 in Eagle's minimal essential medium (EMEM) and AGS cells in Ham F-12. The EMEM medium contained 2 mM L-glutamine, 1 mM sodium pyruvate and 1.5 g/L sodium hydrogen carbonate. Ham F-12 was supplemented with 2 mM L-glutamine and 1.5 g/L sodium hydrogen carbonate. All media were supplemented with 10% heat-inactivated FBS, 100 IU/mL penicillin and 100 μg/mL streptomycin in a humidified incubator with 5% CO2 in air at 37 °C. For the experiments, cells were plated at a density of 50,000 cells/mL in 96-well plates. One day after seeding, the cells were treated with the medium containing the compounds at concentrations ranging from 0 up to 100 μM during 3 days and finally the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay was carried out. The final concentration of MTT was 1 mg/mL. The compounds were dissolved in DMSO (1% final concentration) and complete medium. Untreated cells (medium containing 1% DMSO) were used as controls. Each experiment was carried out in sextuplicate.
4. Conclusions
We have carried out the synthesis of 4-methoxycarbonyl-3-methylisoquinoline-5,8-quinone (1) from 2,5-dihydroxybenzaldehyde, methyl aminocrotonate and silver (I) oxide by employing a highly efficient one-pot procedure. A variety of new isoquinolinequinones substituted with amino, alkylamino and halogen groups were prepared from quinone 1. The members of this series expressed moderate to high in vitro cytotoxic activity against MRC-5 (healthy lung fibroblasts) and four human cancer cell lines: AGS (gastric), SK-MES-1 (lung), J82 (bladder), and HL-60 (leukemia) cell lines. From the current investigation, structure activity relationships of the aminoisoquinolinequinone series demonstrate that nitrogen and halogen substituents at the quinone nucleus of pharmacophore show increased antitumor activity compared with precursor 1. The effect of such substitutions is more significant in enhancing the antitumor activity for those members containing the cyclohexylamino group at C-6 (compound 4b), the amino group at C-7 (compound 12) and the amino and bromine substituents at the 6- and 7-positions (compound 13). Due to the high incidence of undesirable side-effects induced by the majority of current anticancer drugs and by considering the selectivity index values of aminoquinones 4b (SI: 0.7–3.7), 12 and 13 (Figure 3), they appear as promising and interesting leads endowed with potential anticancer activity. These results prompt us to design and synthesize more new members of the aminoisoquinoline-5,8-quinones series in order to discover more active and selective anticancer agents. Studies aimed to understand the mechanisms of bioactivity of aminoisoquinolinequinones at cellular level are currently in progress.
Figure 3.

Structures of the isoquinolinequinones selected as the most significant antitumor agents.
Acknowledgements
This research were supported by FONDECYT (Grants N°: 1060591 and 1100376) and the Vicerrectoria de Investigación (VRI) of the Pontificia Universidad Católica de Chile (V.D.; POSTDOC/N°1/2010).
Footnotes
Sample Availability: Samples of all the compounds are available from the authors.
References and Notes
- 1.Bhatnagar I., Kim S.-K. Immense essence of excellence: marine microbial bioactive compounds. Mar. Drugs. 2010;8:2673–2701. doi: 10.3390/md8102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cragg G.M., Grothaus P.G., Newman D.J. Impact of natural products on developing new anti-cancer agents. Chem. Rev. 2009;109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 3.Villa F.A. Gerwick., L. Marine natural product drug discovery: Leads for treatment of inflammation, cancer, infections, and neurological disorders. Immunopharm. Immunot. 2010;32:228–237. doi: 10.3109/08923970903296136. [DOI] [PubMed] [Google Scholar]
- 4.Gordaliza M. Cytotoxic terpene quinones from marine sponges. Mar. Drugs. 2010;8:2849–2870. doi: 10.3390/md8122849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Molinski T.T., Dalisay D.S., Lievens S.L., Saludes J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009;8:69–85. doi: 10.1038/nrd2487. [DOI] [PubMed] [Google Scholar]
- 6.Nagle D.G., Zhou Y.-D. Marine natural products as inhibitors of hypoxic signaling in tumors. Phytochem. Rev. 2009;8:415–429. doi: 10.1007/s11101-009-9120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordaliza M. Natural products as leads to anticancer drugs. Clin. Transl. Oncol. 2007;9:767–776. doi: 10.1007/s12094-007-0138-9. [DOI] [PubMed] [Google Scholar]
- 8.Simmons T.L., Andrianasolo E., McPhail K., Flatt P., Grewick W.H. Marine natural products as anticancer drugs. Mol. Cancer Ther. 2005;4:333–342. [PubMed] [Google Scholar]
- 9.Milanowski D.J., Gustafson K.R., Kelley J.A., McMahon J.B. Caulibugulones A-F, novel cytotoxic isoquinoline quinones and iminoquinones from the marine bryozoan caulibugula intermis. J. Nat. Prod. 2004;67:70–73. doi: 10.1021/np030378l. [DOI] [PubMed] [Google Scholar]
- 10.Hawas U.W., Shaaban M., Shaaban K.A., Speitling M., Maier A., Kelter G., Fiebig H.H., Meiners M., Helmke E., Laatsch H. Mansouramycins A-D, cytotoxic isoquinolinequinones from a marine streptomycete. J. Nat. Prod. 2009;72:2120–2124. doi: 10.1021/np900160g. [DOI] [PubMed] [Google Scholar]
- 11.Wipf P., Joo B., Nguyenb T., Lazo J.S. Synthesis and biological evaluation of caulibugulones A–E. Org. Biomol. Chem. 2004;2:2173–2174. doi: 10.1039/b408184f. [DOI] [PubMed] [Google Scholar]
- 12.Brisson M., Foster C., Wipf P., Joo B., Tomk R.J., Jr., Nguyen T., Lazo J.S. Independent Mechanistic Inhibition of Cdc25 Phosphatases by a Natural Product Caulibugulone. Mol. Pharmacol. 2007;71:184–192. doi: 10.1124/mol.106.028589. [DOI] [PubMed] [Google Scholar]
- 13.Alagille D.R., Baldwina M., Tamagnan G.D. Total synthesis of the marine cytotoxic caulibugulones A–D. Tetrahedron Lett. 2004;45:6179–6181. doi: 10.1016/j.tetlet.2004.06.007. [DOI] [Google Scholar]
- 14.Valderrama J.A., Colonelli P., Vásquez D., González M.F., Rodriguez J.A., Theoduloz C. Studies on quinones. Part 44: novel angucyclinone N-heterocyclic analogues endowed with antitumoral activity. Bioorg. Med. Chem. 2008;16:10172–10181. doi: 10.1016/j.bmc.2008.10.064. [DOI] [PubMed] [Google Scholar]
- 15.Valderrama J.A., Ibacache J.A., Arancibia V., Rodriguez J., Theoduloz C. Studies on quinones. Part 45: novel 7-aminoisoquinoline-5,8-quinone derivatives with antitumor properties on cancer cell lines. Bioorg. Med. Chem. 2009;17:2894–2901. doi: 10.1016/j.bmc.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 16.Vásquez D., Rodríguez J.A., Theoduloz C., Verrax J., Buc Calderon P., Valderrama J.A. Synthesis and antitumor evaluation of 8-phenylaminopyrimido[4,5-c]isoquinolinequinones. Bioorg. Med. Chem. Lett. 2009;19:5060–5062. doi: 10.1016/j.bmcl.2009.07.041. [DOI] [PubMed] [Google Scholar]
- 17.Benites J., Valderrama J.A., Bettega K., Curi Pedrosa R., Buc Calderon P., Verrax J. Biological evaluation of donor-acceptor aminonaphthoquinones as antitumor agents. Eur. J. Med. Chem. 2010;45:6052–6057. doi: 10.1016/j.ejmech.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 18.Valderrama J.A., Ibacache A., Rodríguez J.A., Theoduloz C., Benites J. Studies on quinones. Part 47. Synthesis of novel phenylaminophenanthridinequinones as potential antitumor agents. Eur. J. Med. Chem. 2011;46:3398–3409. doi: 10.1016/j.ejmech.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Valderrama J.A., González M.F., Colonelli P., Vásquez D. Design and synthesis of angucyclinone 5-aza analogues. Synlett. 2006:2777–2780. [Google Scholar]
- 20.Pratt Y.T. Quinolinequinones. VI. Reactions with aromatic amines. J. Org. Chem. 1962;27:3905–3910. doi: 10.1021/jo01058a036. [DOI] [Google Scholar]
- 21.Yoon E.Y., Choi H.Y., Shin K.J., Yoo K.H., Chi D.Y., Kim D.J. The regioselectivity in the reaction of 6,7-dihaloquinoline-5,8-diones with amine nucleophiles in various solvents. Tetrahedron Lett. 2000;41:7475–7480. doi: 10.1016/S0040-4039(00)01278-8. [DOI] [Google Scholar]
- 22.Defant A., Guella G., Mancini I. Regioselectivity in the multi-component synthesis of indolizinoquinoline-5,12-dione derivatives. Eur. J. Org. Chem. 2006;41:4201–4210. [Google Scholar]
- 23.Yoshida K., Ishiguro M., Honda H., Yamamoto M., Kubo Y. Regioselective 6-amination and 6-arylation of 5,8-quinolinedione promoted by metal ions. Bull. Chem. Soc. Jpn. 1988;61:4335–4340. doi: 10.1246/bcsj.61.4335. [DOI] [Google Scholar]
- 24.Valderrama J.A., Ibacache J.A. Regiochemical control in the amination reaction of phenanthridine-7,10-quinones. Tetrahedron Lett. 2009;54:4361–4363. doi: 10.1016/j.tetlet.2009.05.042. [DOI] [Google Scholar]
- 25.Alley M.C., Scudiero D.A., Monks A., Hursey M.L., Czerwinski M.J., Fine D.L., Abbott B.J., Mayo J.G., Shoemaker R.H., Boyd M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988;48:589–601. [PubMed] [Google Scholar]
- 26.Van de Loosdrecht A.A., Beelen R.H., Ossenkoppele G.J., Broekhoven M.G., Langenhuijsen M.M. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J. Immunol. Methods. 1994;174:311–320. doi: 10.1016/0022-1759(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 27.Scudiero D.A., Shoemaker R.H., Paull K.D., Monks A., Tierney S., Nofziger T.H., Currens M.J., Seniff D., Boyd M.R. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988;48:4827–4833. [PubMed] [Google Scholar]