

Abstract
The increasing application of positron emission tomography (PET) in nuclear medicine has stimulated the extensive development of a multitude of novel and versatile bioorthogonal conjugation techniques especially for the radiolabeling of biologically active high molecular weight compounds like peptides, proteins or antibodies. Taking into consideration that the introduction of fluorine-18 (t1/2 = 109.8 min) proceeds under harsh conditions, radiolabeling of these biologically active molecules represents an outstanding challenge and is of enormous interest. Special attention has to be paid to the method of 18F-introduction. It should proceed in a regioselective manner under mild physiological conditions, in an acceptable time span, with high yields and high specific activities. For these reasons and due to the high number of functional groups found in these compounds, a specific labeling procedure has to be developed for every bioactive macromolecule. Bioorthogonal strategies including the Cu-assisted Huisgen cycloaddition and its copper-free click variant, both Staudinger Ligations or the tetrazine-click reaction have been successfully applied and represent valuable alternatives for the selective introduction of fluorine-18 to overcome the afore mentioned obstacles. This comprehensive review deals with the progress and illustrates the latest developments in the field of bioorthogonal labeling with the focus on the preparation of radiofluorinated building blocks and tracers for molecular imaging.
Keywords: Staudinger ligation, Huisgen click reaction, bioorthogonal, radiolabeling, tetrazine
1. Introduction
Positron emission tomography (PET) is an outstanding instrument for the quantification and localization of physiological as well as pathophysiological activities and processes in vivo which were analyzed by tracing the appropriate biochemical fundamentals [1]. The basics of PET rely on the coincidental detection of annihilation photons in nearly 180° originating from positrons of the parent radionuclide inside the tracer [2]. Measurements and quantifications of the tracer distribution were accelerated noninvasively in living organisms. For that purpose, fluorine-18 produced by a cyclotron was chosen as the ideal radionuclide due to its favorable half-life. Furthermore, the low positron energy and short ranges in tissues lead to high image resolution [3]. However, tracers for PET imaging purposes are restricted by the kind of fluorinated molecules that researchers can prepare.
In principle, radiolabeling of biologically active compounds and especially high molecular weight compounds like peptides, proteins and antibodies with fluorine-18 still represents a considerable challenge and has therefore generated immense interest. As a result of harsh reaction conditions for the direct introduction of fluorine-18 at high specific activity levels, novel radiolabeling strategies comprising fluorine-18-containing prosthetic groups, also referred as 18F-labeling building blocks, are necessary. Due to this fact and due to the multitude of functional groups found in bioactive macromolecules, a labeling strategy has to be developed for almost every one of these compounds. To overcome the above mentioned obstacles, bioorthogonal click-labeling reactions were developed and successfully applied to allow the selective introduction of fluorine-18 under mild conditions, with high radiochemical yields (RCY) and high specific activity (AS) [4,5,6,7,8].
The concept of bioorthogonal syntheses refers to all reactions which can proceed inside of living systems without any interference with native biochemical processes. Several requirements have to be fulfilled for a reaction to be considered as bioorthogonal [9]. First and most prominently, selective reactions should be applied to avoid side-reactions with other functional groups found in the biological starting compounds. Both functional groups of the participating reaction partners must be inert to the residual biological moieties and should only react with each other. Secondly, the resulting covalent bond should be strong and chemically inert to biological reactions, and, in addition, it should not affect the (native) biological behavior of the desired molecule. Thirdly, the reaction should benefit from fast reaction kinetics in the case that covalent ligations are achieved prior to probe metabolism and clearance and the reaction must be run on the time scale of cellular processes to prevent biological side reactions. This point is especially important both for the work with short-living positron emitters and further for an application of in vivo pre-targeting strategies. At least, the non-toxicity of the reaction partner as well as catalysts are of high importance for in vivo applications, and the reaction should proceed under biological conditions taking into account pH, aqueous environments, and temperature.
Giving consideration to the criteria of bioorthogonal reactions, Kolb, Finn and Sharpless [10,11] defined the click chemistry as a “set of powerful, highly reliable, and selective reactions for the rapid synthesis of useful new compounds and combinatorial libraries” and pointed out the following requirements for click reactions: modularity, wideness in scope, very high yielding, stereospecificity (but not necessarily enantioselectivity) and simple product isolation (separation from harmless by-products by non-chromatographic methods). In addition to these criteria, these reactions should proceed using simple reaction conditions (solvent free or solvents like water) including simply accessible starting materials, and the final product has to be stable under physiological conditions [12]. Selected examples of click chemistry reactions are pointed out in Scheme 1.
Scheme 1.

Selected examples of chemical conjugations associated with click chemistry. All reactions highlighted in grey fulfill the bioorthogonality criteria.
In recent years, the terms “bioorthogonal reactions” and “click chemistry” have entered into the field of radiochemistry and radiopharmacy. Bioorthogonal reactions in general, and click chemistry in particular, are generic terms for a set of labeling reactions, which make use of several selective and modular building blocks and enable chemoselective ligations to radiolabel biologically relevant compounds. In this context, the Cu(I)-mediated triazole formation from azides and terminal alkynes according to the 1,3-dipolar Huisgen cycloaddition is a particularly powerful ligation reaction, due to its high degree of specificity and the biocompatibility of both starting materials. In the same way, the strain promoted Huisgen click-reaction as well as both variants of the Staudinger Ligation were applied. The absence of catalysts makes these reactions together with the inverse Diels-Alder tetrazine-click reaction tremendously attractive for radiolabeling purposes. Neither azides nor phosphanes, alkenes or tetrazines react with other functional groups commonly present in biopolymers like peptides or proteins. Hence, there is no need for protective group chemistry. Moreover, the desired linking moieties (triazoles, amides or cyclooctapyridazines) are metabolically stable under physiological conditions. As a consequence, click chemistry is a very attractive approach for the design and synthesis of novel potent radiotracers for molecular imaging purposes.
2. Fluorine-18 in General
Fluorine-18, with a medium half-life of 109.8 min, is one of 17 known radioisotopes [13] and it was introduced as the most prominent radionuclide for PET imaging [14,15,16,17,18,19,20,21]. The low positron energy (Eβ+ = 0.64 MeV) combined with a short range of the positron in tissue (max. 2.4 mm) provides high resolution images. It can be prepared via cyclotron using the 18O(p,n)18F nuclear reaction in a maximum specific activity (AS) of 63.3 TBq/µmol no carrier added (n.c.a.) as [18F]fluoride or by the 20Ne(d,α)18F nuclear reaction carrier added (c.a.) as [18/19]F2 [22]. These two species constitute the basis for nucleophilic ([18F]fluoride) as well as electrophilic ([18/19F]F2) labeling reactions. In general, the respective radiotracers were achieved with a lower AS using electrophilic labeling reactions due to the addition of F2-gas into the Ne-target [23]. For a successful application of [18F]fluoride for radiolabeling purposes, its solubility, nucleophilicity and lipophilicity has to be increased. Standard conditions for the nucleophilic introduction of fluorine-18 generally require: (i) a base such as carbonate, bicarbonate, hydroxide or oxalate, (ii) a phase transfer catalyst such as Kryptofix® K 2.2.2. or tetraalkylammonium salts and finally (iii) a dipolar aprotic solvent such as acetonitrile, DMSO, DMF [24] or alternatively, tert-butanol [25].
In most of the cases, radiolabeling with fluorine-18 is achieved by the bioisosterical exchange of a single hydrogen or OH groups. This is due to the fact that fluorine is absent in most biologically active compounds. Only weak steric disturbances occur because fluorine is only slightly larger (1.47 Å) than protons (1.20 Å) and somewhat smaller than oxygen (1.52 Å). Covalently bound fluorine occupies a smaller volume than methyl, amino, or hydroxyl groups, making it favorable for incorporation [26,27]. Next, the resulting C-F bond (453 kJ/mol) is stronger in contrast to the corresponding C-H bond (432 kJ/mol) or C-O bond (378 kJ/mol) [28]. Further, fluorine can also be involved to form hydrogen bonds, but with the difference that fluorine is only a hydrogen bond acceptor compared to a OH group. Notably, a change of the physiological and biological behavior of the fluorine-containing molecule to the desired biological target is often observed in combination of its metabolic stability and lipophilicity.
3. Cu-Catalyzed 1,3-Dipolar Huisgen Cycloaddition
1893 was the year of birth of the 1,3-dipolar Huisgen cycloaddition as Arthur Michael described for the first time the preparation of 1,2,3-triazoles from phenylazide and dimethyl but-2-ynedioate [29]. The reaction of hydrogen azide with acetylene to give the corresponding 1,2,3-triazole was first described by Dimroth and Fester 17 years later [30]. Based on this work, Huisgen investigated and specified the character of this reaction as a 1,3-dipolar addition in 1963 [31]. Arbitrary alkynes are used as first reaction partners and serve as dipolarophiles in this reaction. Azides as second reaction partners are ambivalent compounds only describable as zwitterions in an all-octet formula. The driving force for the formation of triazoles in particular is the disappearance of the charges in the azide [32].
Normally, organic azides react with alkynes at high temperatures to give 1,2,3-triazoles in the Huisgen cycloaddition with formation of both 1,4- and 1,5-disubstituted regioisomers. In 2002, the groups of Sharpless et al. [33] and Meldal et al. [34] independently introduced a revolutionary innovation into the field of 1,3-dipolar Huisgen cycloaddition due to the application of Cu(I)-species as catalyst. The so called copper-catalyzed azide alkyne cycloaddition (CuAAC) proceeds faster, delivers the respective 1,4-isomer exclusively and can be performed at ambient temperatures. Both groups assumed a mechanism in which Cu(I) coordinates first to the terminal alkyne forming a copper(I)acetylide. Computer calculations confirmed these presumptions and predicted a non-concerted mechanism through a six-membered copper(III) metallacycle [35]. Newer findings using continuous-flow analysis cannot exclude a primary addition of the Cu(I) to the azide moiety [36]. Nowadays, Worrel et al. have suggested a new mechanism discovered by utilizing 63Cu-enriched catalysts. Thus, two copper atoms are incorporated in the catalytic cycle [37] as seen in Scheme 2.
Scheme 2.

Proposed mechanism of the copper-catalyzed 1,3-dipolar Huisgen cycloaddition (CuAAC) catalyzed by two copper atoms.
As a result of its favorable properties and reaction conditions, the CuAAC has gained enormous popularity and has been applied in diverse fields of chemistry [38]. Since 2006, the CuAAC is introduced for labeling purposes of biologically and pharmacologically relevant molecules with fluorine-18 (Scheme 3). Up until now, over 60 works have been published using this 1,3-dipolar Huisgen cycloaddition for 18F-labeling of small organic compounds, peptides, oligonucleotides, proteins and nanoparticles which were partially used for PET studies [5,6,39,40]. The first application of click chemistry for the design and synthesis of radiometal-based radiotracers to form novel multidentate triazole ligand scaffolds for an efficient chelation of the 99mTc(CO)3 core was reported by the group of Schibli in 2006 [41]. In 2008, the first application of the CuAAC for the introduction of carbon-11 using [11C]methyl azide was published by Schirrmacher and co-workers [42].
Scheme 3.

A selection of various conventional and bioorthogonal labeling building blocks for the incorporation of fluorine-18.
The bioorthogonality of both functional groups used in the Huisgen cycloaddition makes this reaction to connect 18F-containing building blocks to sensitive bioactive molecules like peptides or proteins highly attractive. Problematically, copper is known to be cytotoxic towards prokaryotic and eukaryotic cells. It is reported that viruses and oligonucleotides are damaged or destroyed in its presence [43]. Other side effects that it is associated with include hepatitis, Alzheimer’s disease or neurological disorders. Therefore, it is of enormous importance for in vivo applications to remove the copper completely from the desired radiotracer or to develop copper-free, bioorthogonal alternatives [44].
The first application of the CuAAC in the field of fluorine-18-radiochemistry and -radiopharmacy was published by Marik and Sutcliffe in 2006. They described the synthesis of three 18F-containing alkynes [18F]2a–c with different chain lengths for the radiolabeling of azide-functionalized peptides [45]. For this purpose, three tosylated alkynes 1a–c were treated with [18F]fluoride/K2CO3/K 222 in acetonitrile at 100 °C (Scheme 4). Due to their fugacity, [18F]2a–c were purified by co-distillation with acetonitrile into a second vial which was cooled down to −78 °C. It was figured out that the formation of 2b resulted in the highest radiochemical yield (RCY) of 81% and radiochemical purity (RCP) of 98%. Subsequently, an azide-functionalized model peptide with amino acid sequence YGGFL was radiolabeled with [18F]2a–c under CuAAC conditions within 10 min. [18F]3b was obtained with the highest yield (97%) after optimizations. In addition, two other peptides [18F]4 (97%) and [18F]5 (99%) were prepared; the results are shown in Table 1. All peptides were obtained with AS > 35GBq/μmol after isolation using a C18 Sep-Pak extraction. Unreacted building blocks [18F]2a–c were evaporated together with the eluent to yield all 18F-labeled peptides in excellent radiochemical purity. It was also mentioned that the use of bipyridine or bathophenanthroline based ligands for the copper species further improve the yields.
Scheme 4.

Radiolabeling and distillation procedure followed by the CuAAC according to conditions published by Meldal et al. [34].
Table 1.
Results of the radiolabeling with [18F]2a–c.
| Building block | Resulting [18F]fluoropeptide | Yield [%] | Purity [%] |
|---|---|---|---|
| [18F]2a | (18F-alkyne, n = 1)-YGGFL [18F]3a | 54 | 95 |
| [18F]2b | (18F-alkyne, n = 2)-YGGFL [18F]3b | 97 | 98 |
| [18F]2c | (18F-alkyne, n = 3)-YGGFL [18F]3c | 62 | 99 |
| [18F]2b | AGDLHVLR-Ebes-Lys-(18F-alkyne, n = 2) [18F]4 | 97 | 81 |
| [18F]2b | (18F-alkyne, n = 2)-AGDLHVLR [18F]5 | 99 | 87 |
Next, Ross et al. applied the CuAAC for the synthesis of 18F-labeled folic acid derivative [18F]6 using 6-[18F]fluorohexyne ([18F]2c) [46]. After an overall synthesis time of approx. 90 min (Scheme 5), the isolated product [18F]6 was obtained in a RCY of 25–35%, with a RCP > 99% and a AS of 160 ± 70 GBq/μmol. Afterwards, [18F]6 was applied to mice bearing a folate receptor(FR)-expressing KB tumor. Activity accumulation in KB tumors at 45 min p.i. amounted to 3.13% ± 0.83 ID/g. Strong hepatobiliary excretion of the lipophilic tracer led to elevated accumulation (16.53% ID/g) of the activity in the abdominal region.
Scheme 5.

18F-labeled folic acid derivative [18F]6.
In 2008, Hausner et al. used 18F-pentyne [18F]2b for the radiolabeling of a αvβ6 specific peptide A20FMDV2 [47] and compared it with solid-phase radiolabeling using 4-[18F]fluorobenzoic acid [18F]FBA as well as 2-[18F]fluoropropionic acid [18F]FPA. In addition to that case, effects of these labeling moieties on PET imaging and pharmacokinetics were evaluated. In conclusion, the [18F]FBA-labeled peptide was obtained in 22 ± 4% RCY and the [18F]FPA-incorporated peptide was prepared in 13 ± 3% RCY. In contrast, labeling using [18F]2b resulted in only 10% RCY for the respective labeled peptide. Therefore, conventional labeling on solid support should be preferred for longer peptides. Additionally, male athymic nude mice bearing αvβ6-positive and αvβ6-negative (control) cell xenografts were employed for biodistribution studies. The click-labeled peptide shows similar tumor uptake but a different metabolism than ulterior 18F-labeled peptides.
In the same year, Kim et al. compared a two-step click labeling approach (method 1) with a direct radiofluorination (method 2) for the labeling of β-azido-d-glucose (7) as alternative for a radiolabeling with [18F]FDG (Scheme 6) [48]. For method 1 (click approach), 4-[18F]fluoro-1-butyne ([18F]2a) was prepared from the appropriate tosylate precursor and was co-distilled into a second vial containing glucopyranosyl azide 7, CuI, sodium ascorbate and 2,6-lutidine as base. Stirring at 90 °C for 10 min, followed by HPLC purification ensued glucose derivative [18F]9 with a RCY of 30% (d.c.).
Scheme 6.

Comparison of conventional nucleophilic labeling with click approach.
Method 2: Tosylate precursor 8 was radiolabeled under conventional conditions with [18F]fluoride in a mixture of acetonitrile and tert-butanol (v:v = 1:6). [18F]9 was obtained in a RCY of 21% (d.c.) after subsequent removal of acetyl groups with sodium methanolate in methanol and HPLC purification. Surprisingly, the achieved AS for the click approach was higher (59.9 GBq/µmol) compared to the conventional approach (23.5 GBq/µmol). Further, in vitro evaluation demonstrated that a triazole moiety at the C1 position of [18F]9 is tolerated neither by the hexokinase nor by the Glut-1 transporter. A slight uptake of [18F]9 in tumor cells (0.2%) could not be blocked.
Based on previous studies of Marik and Sutcliffe [45], Kim et al. further evaluated 4-[18F]fluoro-1-butyne ([18F]2a) as synthon for 18F-click radiolabeling with various azido compounds [49]. They pointed out, that the conditions of the nucleophilic substitution of butyne precursor 1a with K[18F]F delivered a by-product (Scheme 7) in a remarkable percentage which was evidenced as vinyl acetylene (9) among the desired 18F-butyne [18F]2a (approx. ratio of 9 and [18F]2a 79:21 in t-BuOH and 87:13 in acetonitrile) while the same reaction using pentyne 1b exclusively resulted 18F-pentyne [18F]2b. Thus, they underscored the thesis that alkyne-precursors with chain lengths longer than four carbon atoms are better synthons for the preparation of 18F-alkyne building blocks due to the suppression of elimination as a side-reaction.
Scheme 7.

Formation of vinyl acetylene (9).
Later, Zhang et al. presented an automated synthesis route for 18F-pentyne [18F]2b which was then applied for 18F-labeling of an apoptosis marker (Scheme 8) [50]. [18F]2b was obtained with a RCY of 51% and was automatically distilled into a second vial containing the peptide precursor 10. The conversion of 18F-pentyne [18F]2b into 18F-peptide [18F]11 amounts nearly 99% after a total synthesis time of 70 min (RCY: 21.0 ± 4.5%, RCP > 99%, AS = 870 GBq/μmol). A549 lung cells were utilized for an in vitro evaluation. After treatment with carboplatin, the A549 cell uptake of 18F-peptide [18F]11 was found to be 0.86 ± 0.04% together with a cell apoptosis rate of 10.23 ± 2.43%. In contrast, the cell uptake of untreated control cells was found to be 0.06 ± 0.01% (cells apoptosis rate = 1.02 ± 0.31%). From this point of view, 18F-peptide [18F]11 could be a promising candidate for future apoptosis imaging.
Scheme 8.

Automated synthesis of 18F-labeled peptide [18F]11 for apoptosis imaging.
In 2011, Balentova et al. presented an interesting work concerning the 18F-labeling of alkyne functionalized model peptide 14 under CuAAC-conditions utilizing a novel and 18F-Si-based azide [18F]13 [51]. In general, the high affinity of fluorine for silicon allows a facile introduction of fluorine-18 under mild conditions as well. For this purpose, precursor 12 was prepared and labeled with n-dodecyltrimethylammonium [18F]fluoride in DMSO at 90 °C for 20 min (RCY 64% for [18F]13, d.c.). Subsequently, labeling of peptide 14 was accomplished at ambient temperature for 15 min using CuI, sodium ascorbate and DIEA as base. Peptide [18F]15 was obtained in 75% RCY (d.c.) and with a RCP > 97% (Scheme 9).
Scheme 9.

18F-labeling of alkyne-functionalized model peptide 14 utilizing 18F-Si-based azide [18F]13.
Problematic, the thermodynamically stable F-Si bond is known to be hydrolyzed in water under physiological conditions unless there are no bulky alkyl groups attached to the silicon. Unfortunately, 18F-Si-labeled peptide [18F]15 as well as the building block [18F]13 had a low hydrolytic half-life (<13 min) in buffers at different pH values. The silicon fluorine bond is not enough stabilized by the presence of the two isopropyl groups.
3.1. 18Fluoro-PEGX-Derivatives
This part of the review deals with PEGylated 18F-labeled building blocks. In general, PEGylation describes the process of covalent connection of polyethylene glycol (PEG) moieties to drugs or therapeutic compounds. These compounds are “masked” from the host’s immune system and the water solubility of hydrophobic derivatives is enhanced. Due to the increase of the size its circulatory time is often prolonged by reducing renal clearance.
The main advantage of PEGylated 18F-labeled building blocks is their reduced volatility compared to the smaller counterparts. Hence, these compounds are easier to handle and the corresponding bioconjugates show a higher blood clearance which is important for in vivo studies. In 2007, Sirion et al. presented a set of four various 18F-labeled building blocks [18F]16 to [18F]19 for the labeling of molecules using the CuAAC (Scheme 10) [52], [18F]19 contains a PEG-like moiety. In the first step, all mesylate precursors were reacted with [18F]fluoride in t-BuOH at 100 °C for 20 min in the presence of TBAHCO3 to obtain the corresponding [18F]acetylenes [18F]16 to [18F]19 with RCY ≥ 90%. Further, these building blocks are thermally stable, nearly nonvolatile, and are therefore suitable for one-pot, two-step syntheses.
Scheme 10.

Building blocks evaluated by Sirion and co-workers.
In the next step, different functionalized biomolecule-like compounds were successfully labeled within 10 min with an excellent 18F-conversion of 97–100%. Small non-polar organic compounds needed longer reaction times (30 min) and resulted in limited 18F-conversion up to 71%. In most of the cases, the two-step reaction was completed within 40 min from the end of bombardment. The sample labeling reaction of azide functionalized deoxythymidine 20 utilizing alkyne [18F]19 is pointed out in Scheme 11.
Scheme 11.

18F-labeling of azide functionalized thymidine derivative utilizing [18F]19.
Li et al. described the preparation of [18F]FPTA-RGD2 [18F]25 which was labeled with 3-(2-(2-(2-[18F]-fluoroethoxy)-ethoxy)ethoxy)prop-1-yne (18F-PEG3) ([18F]23) [53]. This labeling building block [18F]23 was obtained in a non decay-corrected yield of 65.0 ± 1.9% from 22. To increase the affinity the RGD moiety was twice connected to a glutamate which was further functionalized with azide. The major advantage of the click approach was the shortening of the labeling time compared to the conventional labeling with [18F]SFB from 180 min to 110 min combined with an increase of the RCY to 53.8% (d.c.) for [18F]FPTA-RGD2 [18F]25 (Scheme 12). In vivo evaluations of [18F]25 using athymic nude mice bearing a U87MG tumor demonstrated a lower αvβ3-integrin affinity in contrast to other 18F-labeled RGD peptides due to the relatively long PEG3 moiety.
Scheme 12.

18F-labeling of an azide functionalized RGD-dimer utilizing a 18F-PEG3-alkyne.
In 2009, the group of Devaraj et al. used the same 18F-building block 18F-PEG3 [18F]23 for the preparation of 18F-CLIO – a fluorine-18-containing trimodal nanoparticle [54]. The basis of this particle consists of an aminated cross-linked dextran iron oxide (CLIO) decorated with diethylene triamine pentaacetic acid. The advantage of 18F-CLIO was the ability of a triple detection by PET, fluorescence molecular tomography as well as magnetic resonance imaging. The 18F-nanoparticle was prepared as briefly follows: 18F-PEG3 [18F]23 was added to the azido-CLIO in a PBS buffer. This mixture was then treated with CuSO4/bathophenanthrolinedisulfonic acid (BPDS, as disodium salt) and sodium ascorbate in distilled water. The reaction mixture was incubated for 40 min at 40 °C and filtrated to remove the resulting 18F-CLIO from unreacted 18F-PEG3 with a RCY of 58% (d.c.).
First evaluations of 18F-CLIO suspended in the agar phantom pointed out a detection threshold of 0.025 µg Fe/mL for PET-CT imaging. This is approximately 200 times lower than by T2*-weighted MR imaging. The lowest observed concentration of the nanoparticles by MRI was 5 µg Fe/mL (15), demonstrating a PET detection threshold 200 times more sensitive than MRI at current specific activities. A high in vivo stability for 18F-CLIO was demonstrated due to the low renal and bladder uptake, no excretion via the kidneys was observed.
In 2011, Gill and Marik used optimized conditions and switched the bioorthogonal functionalities for radiolabeling of N-alkynylated peptide 28 (4-pentynoyl-Val-Asp-Asn-Lys-Phe-Asn-Lys-Glu-Nle-Gln-Asn-Ala-Tyr-Ala-Ile-Glu-Ile-Ala-Leu-Leu-Pro-Asn-NH2) using 18F-PEG3-azide [18F]27 (Scheme 13) [55]. After purification by HPLC, 18F-labeled peptide [18F]29 could be obtained with RCY of 31% and a AS = 1.44 GBq/µmol. The removal of BPDS was difficult using solid phase extraction (SPE) which lowered the chemical purity. Therefore, optimized reaction conditions without BPDS provided 96% conversion after 60 °C for 10 min when using 1.6 µmol Cu(CH3CN)4PF6 and 10 nmol of 28 at high concentrations (125 µL). Noteworthy, a maximum AS and RCP were only obtainable when 28 was quantitatively converted to 18F-labeled peptide [18F]29 due to their similar molar mass and retention times in HPLC and SPE. Furthermore, the copper source should be considered very crucial, since PF6- can lower the AS due to an isotopic exchange.
Scheme 13.

18F-labeling of an alkyne functionalized peptide 28 utilizing 18F-PEG3-azide [18F]27.
In 2012, Lee et al. used the same 18F-PEG3-azide [18F]27 for the radiolabeling of ZnO nanoparticles (20 nm) in order to observe their behavior and accumulation in organic tissues after oral administration using PET [56]. 18F-labeling of ZnO particles resulted in AS = 0.73 MBq/mg and RCP > 95%. PET images indicated that 18F and 18F-ethoxyazide showed radioactivity in the bone and bladder 3 h after oral administration, whereas radioactivity for 18F-labeled ZnO nanoparticles was seen only in the gastrointestinal (GI) tract. At 5 h post-administration, biodistribution studies demonstrate that 18F-labeled ZnO nanoparticles showed radioactivity in the lung, liver and kidney including the GI tract.
3.2. [18F]Fluoroethylazide
[18F]Fluoroethylazide–18F-FEA ([18F]33) is certainly the most popular building block for radiolabeling purposes using the CuAAC. The first approach was evaluated and applied by Glaser and Årstad in 2007 [57]. 2-Azidoethyl-4-toluenesulfonate (32) was used as starting material and the subsequent nucleophilic fluorination with anhydrous n.c.a. K 222, K[18F]F in acetonitrile at 80 °C for 15 min yielded the desired azide [18F]33 (RCY = 55%, d.c.). The isolation was done by distillation. The reaction sequence for 18F-FEA [18F]33 is pointed out in Scheme 14.
Scheme 14.

Reaction sequence for the preparation of (18F-FEA) [18F]33.
In most of the cases, the isolation of [18F]33 was accomplished by co-distillation with acetonitrile. Notably, tosylate 32 is also co-distilled when using higher temperatures (>90 °C), and 32 reacts with the alkyne precursor as well in the subsequent labeling step. At temperatures below 90 °C the transfer of tosylate 32 to the cooled receiver vial was negligible, but the distillation time had to be extended [58]. Next, the somewhat difficult handling due to the volatility of the building block during the radiosynthesis brought different reaction set-ups in order to make the handling of 18F-FEA [18F]33 easier. Up to now, the [18F]FEA-synthesis and the following labeling step with the respective alkyne were transferred into a remotely controlled module using a cooling trap [58].
The convenient and fast access of 18F-FEA [18F]33 led to multitude of radiotracers, e.g., for the imaging of apoptosis with [18F]MitoPhos_01 [18F]34 [59] or isatine derivatives like [18F]ICMT-11 [18F]35 [60,61] connected with a GMP-applicable automated synthesis [62]. Furthermore, tracers for proliferation like [18F]deoxyuridine [18F]36 [63] or [18F]FOT and [18F]FTT [18F]37a,b [64] were developed. 18F-labeled quinazoline derivatives [18F]38 and [18F]39 for the EGFR activity imaging were prepared [58,65]. [18F]FET-G-TOCA [18F]40 for the somatostatin receptor [66], [18F]haloethylsulfoxides like [18F]41 [67] and six 18F-labeled nitroaromates based on [18F]MISO [68] to visualize hypoxia were evaluated. Further, it is possible to address the αvβ3 receptor with 18F-containing RGD peptides e.g., [18F]42a,b [69,70,71]. [18F]AFETP [18F]43 was introduced as a tracer for the imaging of brain tumors [72] and mouse DBT glioma [73]. Galante et al. used 18F-FEA [18F]33 for the radiofluorination of a series of 6-halopurines in a one-pot automated synthesis approach [74]. They used the copper chelator bathophenanthrolinedisulfonate to accelerate the cycloaddition reaction. An overview of all these tracers is given in Scheme 15.
Scheme 15.

Overview of radiotracers prepared by CuAAC with 18F-FEA [18F]33.
The last approach using [18F]fluoroethylazide ([18F]33) was published by Jia et al. in 2013 [75]. This group demonstrated the simultaneous labeling of various alkynes with 18F-FEA [18F]33 in a one-pot procedure. In general, two or more different alkynes were mixed in a DMF-solution together with CuSO4 and sodium ascorbate. Afterwards, a solution of [18F]33 in acetonitrile was added, the resulting mixture was shaken at 60 °C for 15 min and then analyzed/purified via HPLC after filtration. This procedure is pointed out in Scheme 16. Importantly, an appropriate HPLC separation system has to be developed for the separation of the resulting tracers prior the radiolabeling.
Scheme 16.

The technical route of the multiple labeling procedure.
In 2012 Glaser et al. reported, that 18F-FEA [18F]33 is able to be reduced to give 18F-fluoroethylamine by the use of copper wire under acidic conditions, which could be a reason for the sometimes low RCY in click reactions [76]. Another approach published by Zhou et al. in 2012 used 18F-FEA [18F]33 for the synthesis of a [18F]SFB analog [77]. They pointed out that base labile NHS-esters tolerate the “click” conditions well. Moreover, the resulting compound can be prepared in a shorter synthesis time (~ 35 min) in contrast to [18F]SFB (70 min) and in a higher RCY.
Two novel and universally applicable building blocks BFP [18F]46 and AFP [18F]47 were prepared by Pretze et al. in 2013 (Scheme 17) [78]. In addition to the Huisgen-click reaction, it is also possible to apply the Staudinger Ligation as well for radiolabeling purposes when using [18F]AFP [18F]47. Furthermore, an automated module synthesis was evaluated for both building blocks. The introduction of spiro salts 44 and 45 as precursors led to a simplified separation and purification from the resulting building blocks via elution using RP18 or Si cartridges in a short timespan.
Scheme 17.

Preparation of BFP [18F]46 and AFP [18F]47.
Both building blocks [18F]46 and [18F]47 were used for the radiolabeling of azide as well as alkyne functionalized peptides with SNEW sequence for an imaging of the EphB2 receptor [79]. Problematically, Glaser coupling was observed as a side reaction using [18F]BFP [18F]46 for the labeling of peptides (Scheme 18). To overcome this fact, [18F]AFP [18F]47 was used instead and the labeling procedure was accomplished on resin (Scheme 19). A second important fact is the removal of the copper. It was found that the copper forms strong complexes with this kind of peptides. For this reason, Cu-containing peptides do not behave like “normal” peptides. To fix this obstacle, bispidine [80], a strong chelating agent, was used to wash the resin after the click reaction, while other chelating agents failed.
Scheme 18.

Glaser coupling with [18F]BFP [18F]46 as side reaction.
Scheme 19.

Radiolabeling of a SNEW peptide using [18F]AFP [18F]47.
3.3. 18Fluoro-Aryl Building Blocks
The 18F-labeling of aryl precursors is a common task for radiochemists. In general, fluoroaryl compounds possess a higher metabolic stability in vivo than fluoroalkyl moieties due to the fluorine bound to the sp2-carbon atom. In most cases, the radiolabeled products show a higher lipophilicity due to the aryl moiety and therefore play a key role in the design of lipophilic radiotracers. A multitude of different aryl click building blocks were evaluated in the past. An overview is shown in Scheme 20.
Scheme 20.

Overview of [18F]fluoroaryl building blocks.
The first approach for the radiofluorination of biomolecules utilizing the sulfonamide-based alkyne 4-[18F]fluoro-N-(prop-2-ynyl)benzamide–[18F]F-SA ([18F]51) as 18F-labeling building block was presented by Ramenda et al. in 2007 [81]. [18F]F-SA [18F]51 was synthesized within 80 min including HPLC purification with RCY of 32 ± 5% (RCP > 95%, AS = 120–570 GBq/µmol) in a remotely controlled module. Next, [18F]51 was applied for the radiolabeling of an azide functionalized neurotensin(8-13); the labeled peptide was obtained with a RCY = 66% as determined by radio-HPLC. The in vitro binding affinity (IC50) of 18F-neurotensin(8-13) derivative was determined to be 66 nM.
Furthermore, the same building block [18F]F-SA [18F]51 was applied for 18F-labeling of azide-functionalized human serum albumin (HSA) 52 [82]. 18F-Labeled HSA [18F]53 was purified via size-exclusion chromatography (SEC) and was obtained with a RCY of 55% in a total synthesis time of 120 min starting from [18F]fluoride (Scheme 21). Finally, metabolite analyses of 18F-HSA [18F]53 were accomplished using nude mice. After 120 min, most of the activity was found in the gall bladder, intestines and in the bladder. The half-life of blood activity of [18F]53 was calculated to be 31 min. Further, the introduction of the azide residue into HSA and subsequent the radiolabeling has significantly altered the structural and functional integrity of HSA.
Scheme 21.

Preparation of [18F]51 and subsequent radiolabeling of HSA 52.
In 2013, the same group extended their repertoire and presented the radiolabeling of an azide-modified phosphopeptide (H-Met-Gln-Ser-pThr-Pro-Leu-OH) and an l-oligonucleotide (17-mer l-RNA: 5’-CCGCACCGCACAGCCGC-3’) with [18F]F-SA [18F]51 [83]. They pointed out that the lipophilicity (logP 1.7) of [18F]F-SA [18F]51 allows radiolabeling in aqueous solutions. After the click reaction, the 18F-phosphopeptide was obtained in 73 to 77% RCY at low precursor amounts (100–400 µg). Additionally, the 18F-labeled oligonucleotide was isolated in 44% RCY as determined by radio-HPLC after 20 min click-reaction time.
The first approach for radiolabeling with pyridine-based 18F-heteroaryl building blocks was presented by Inkster et al. in 2008 [84]. For this purpose, 2-[18F]fluoro-3-(hex-5-ynyloxy)pyridine–[18F]FPy5yne ([18F]55) was prepared via nucleophilic (hetero-)aromatic substitution from either the corresponding 2-nitro or 2-trimethylammonium pyridine 54a,b. Both precursors delivered [18F]55 in a high RCY of > 90% after 15 min synthesis time and 110 °C in DMSO or acetonitrile. Further, model peptide N3-(CH2)4-CO-Tyr-Lys-Arg-Ile-OH 56 was treated with [18F]55 under click-conditions to obtain [18F]57 in a RCY of 18.7% (d.c.) within a total preparation time of 160 min (start of synthesis). Tris(benzyltriazolylmethyl)amine (TBTA) was used as stabilizing agent for Cu(I). Results are summarized in Scheme 22.
Scheme 22.

Radiosynthesis of [18F]FPy5yne [18F]55 and CuAAC with model peptide 56.
Furthermore, [18F]FPy5yne [18F]55 was applied for the first radiofluorination of an azide-functionalized DNA analog [85]. For that reason, a 5’-azide-modified DNA sequence antisense to mdr1 mRNA was prepared and treated with [18F]55 in the presence of TBTA and 2,6-lutidine instead of DIPEA. 18F-Labeled DNA sequence was isolated with 24.6% ± 0.5% RCY (d.c., after EOB) in a shortest preparation time of 276 min from start of synthesis. The copper source was CuBr (99.9999% purity). The DNA sequence antisense to mRNA transcribed by the mdr1 gene could be used for imaging of breast cancer.
Moreover, the group of Valdivia et al. presented an approach for the synthesis of 2-[18F]fluoro-3-pent-4-yn-1-yloxypyridine–[18F]FPyKYNE which is one CH2 group shorter than the aforementioned [18F]FPy5yne [18F]55. An automated synthesis procedure was evaluated for [18F]FPyKYNE and the subsequent radiolabeling of an RGD peptide. The labeled peptide was obtained in a total RCY of 12–18% with a RCP > 99% after 125 min in a two-step, two-reactor process [86].
Finally, 6-[18F]fluoro-2-ethynylpyridine [18F]59 was prepared as the third heteroaromatic example from the corresponding bromo precursor 58 within 10 min at 130°C with a RCY of 27.5 ± 6.6% (d.c.) with high RCP ≥ 98% by Daumar et al. in 2012 [87]. A d-amino acid analogue of WT-pHLIP and an l-amino acid peptide K-pHLIP were used, both functionalized at the N-terminus with 6-azidohexanoic acid. The subsequent click-labeling was performed at 70 °C in a mixture of water and ethanol using Cu-acetate and sodium ascorbate (Scheme 23). [18F]-d-WT-pHLIP [18F]61a and [18F]-l-K-pHLIP [18F]61b were obtained with RCYs between 5−20% after HPLC purification in a total reaction time of 85 min including the formulation for the biological evaluation. In vitro stability tests in human and mouse plasma revealed high stability. After 120 min, 65% of [18F]61a and 85% of [18F]61b remained intact. PET imaging and biodistribution studies in LNCaP and PC-3 xenografted mice revealed pH-dependent tumor retention.
Scheme 23.

18F-labeling procedure for two pHLIP peptide analogs [18F]61a and [18F]61b.
Next, two approaches for the preparation of (azidomethyl)-[18F]fluorobenzenes like [18F]67 were presented by Thonon et al. [88] and Chun et al. [89] (Scheme 24). The first group used a four-step synthesis path starting from 4-trimethylammonium benzaldehyde (62). Fluorine-18 was introduced in the first step followed by reduction of the aldehyde [18F]63, conversion into the bromo compound [18F]65 and final functionalization with azide. 34% RCY (d.c.) of [18F]67 could be obtained within 75 min starting from [18F]fluoride. The second group used a single step strategy. Thus, diaryliodonium salts like 66 were introduced as precursor. Electron-rich aryl moieties, such as a 4-methoxyphenyl or 2-thienyl, are generally known to direct the [18F]fluoride into the other relatively electron-deficient ring of this diaryliodonium salt [90]. Each precursor was subjected to labeling with [18F]fluoride (n.c.a.) in the microfluidic device. The respective building blocks were obtained after 94−188 s in high RCY’s (35−45% for [18F]67) (d.c.) whereas the by-product was only formed in <3%.
Scheme 24.

Comparison between labeling of trimethylammonium precursors and diaryliodonium salts.
The group of Thonon et al. [88] used [18F]67 for subsequent click labeling of a 4-ethynyl-l-phenylalanine-containing peptide. For this purpose, the click-conditions were optimized and the ligation reaction was finished in less than 15 min. Later in 2011, the same group presented a general method for the labeling of siRNA [91]. Two complementary building blocks 1-(azidomethyl)-4-[18F]fluorobenzene [18F]67 and 1-azido-4-(3-[18F]fluoropropoxy)benzene [18F]68 have been produced with 41% and 35% RCY (d.c.), respectively. The whole labeling procedure takes 120 min with a RCY of 15 ± 5% (d.c.) for both pure [18F]siRNA derivatives [18F]69 and [18F]70 (Scheme 25).
Scheme 25.

Labeling procedure of siRNA [18F]69 and [18F]70.
In 2012, Flagothier et al. investigated the influence of the linkers connected to the oligonucleotides and presented a new alkyne-functionalized linker for siRNA [92]. For subsequent labeling purposes, [18F]67 was prepared in 45 min with 84% RCY using a remotely controlled device (GE Fastlab®). The siRNA concentration was used in a low concentration of about 10-5 M and represents an important advantage of the method considering the high cost of this precursor. [18F]siRNA was prepared under the same conditions as pointed out by Mercier et al. [91] in a comparable RCY of 12%.
Next, two further labeling building blocks based on the 4-[18F]fluorobenzoate skeleton were developed. First, propargyl 4-[18F]fluorobenzoate–[18F]PFB was prepared by the group of Vaidyanathan et al. from the respective trimethylammonium precursor in a RCY of 58 ± 31% (d.c.) within 15 min. Afterwards, several model compounds and a transglutaminase-reactive peptide (RCY = 37 ± 31%) were labeled using [18F]PFB [93]. The second approach consists of the preparation of 4-[18F]fluoro-3-nitro-N-2-propyn-1-yl-benzamide – [18F]FNPB published by Li et al. in 2012 [94]. [18F]FNPB was synthesized with a RCY of 58% (AS > 350 GBq/µmol, RCP > 98%) within 40 min. In vitro stability tests using mouse plasma revealed no radiodefluorination over 2 h. Afterwards, three different azide-modified peptides were labeled with RCYs between 87 and 93%.
Further, an unconventional and promising approach was introduced by Li et al. in 2013 consisting of a radiolabeling procedure using alkyne functionalized aryltrifluoroborate [18F]72 and either azide-modified bombesin 73 or RGD peptide 74 [95,96]. Advantageously, arylboronates like 71 capture [18F]fluoride directly and rapidly under aqueous conditions (20 min, 20–40°C, pH 2–3, then quench to pH 7.5) under formation of a water-soluble, non-coordinating aryltrifluoroborate anion [18F]72 that is highly polar (logP < −4). The alkyne-[18F]ArBF3− [18F]72 was subsequently conjugated to both peptides within 25 min without need for prior work-up in this two-step one-pot procedure (Scheme 26).
Scheme 26.

Arylboronate [18F]72 used for radiofluorination purposes.
Later, the specific activity for the building block as well as for the labeled peptides was improved. This was possible due to the stoichiometry of the introduction of n.c.a. [18F]fluoride into the precursor. Investigations to determine the AS were done using a rhodamine-N3 conjugate. After this improvement, it was possible to prepare alkyne-[18F]ArBF3− [18F]72 with a AS = 148 GBq/μmol and the resulting RGD peptide [18F]76 with a AS = 444 GBq/μmol. The tumor-uptake and corresponding good PET imaging of both peptides was accompanied by a three times higher AS which was statistically verified [97].
3.4. 18Fluoro-Gluco-Derivatives
The glycosylation of peptides represents an important tool for the enhancement of their in vivo behavior like blood clearance and stability. For this reason, labeling building blocks based on carbohydrates for the glycosylation of potential radiotracers using the CuAAC were evaluated by Maschauer and Prante in 2009 [98]. In this context, different α- and β-anomeric azides and alkynes were designed for radiolabeling purposes. They demonstrated that the radiolabeling of triflated precursors with fluorine-18 proceed better when using β-anomeric derivatives like 77 (RCY of [18F]78 was 71 ± 10%). Afterwards, a proof-of-principle reaction of the 18F-building block [18F]78 was accomplished by labeling Fmoc-l-propargylglycine with a RCY of 60% for unprotected building block and a RCY of 76% for the peracetylated building block.
Finally in 2010, they applied [18F]78 as the building block with the highest RCY to label different peptide moieties like RGD or neurotensin derivatized with l-propargylglycine (Scheme 27). RCYs ranged from 17 to 20% and AS = 55–210 GBq/µmol were obtained for the labeled conjugates in a total synthesis time of 75 min [99]. Furthermore, in vivo investigations with the [18F]FGlc-RGD peptide were carried out using U87MG-bearing mice. Because of the glycosylation they had a better blood clearance and stability in αvβ3-integrin expressing tumors. The specific tumor uptake of [18F]FGlc-RGD was 0.49% ID/g (60 min p.i.).
Scheme 27.

Preparation of mannopyranosyl azide 77 and subsequent 18F-glycosylation with [18F]78.
In 2012, Fischer et al. adapted this method to label a novel folic acid conjugate in order to image the folate receptor, which is overexpressed in various tumor entities [100]. After 3 h of synthesis, [18F]fluorodeoxyglucosyl folate [18F]79 was obtained in RCY = 5−25% (d.c.), with a AS = 90 ± 38 GBq/μmol and a RCP > 95% (Scheme 28). The biodistribution and PET imaging studies in KB tumor-bearing mice showed a high and specific uptake of the radiotracer in FR-positive tumors (10.03 ± 1.12% ID/g, 60 min p.i.) and kidneys (42.94 ± 2.04% ID/g, 60 min p.i.).
Scheme 28.

18F-glycosylation of a folate derivative.
4. Strain-Promoted Huisgen Cycloaddition
Strain-promoted and copper-free variants of the Huisgen cycloaddition of cyclic alkynes with azides are of great interest as powerful tools in reagent/catalyst-free bioconjugations. This type of reaction was applied especially for labeling purposes on living cells or in vivo studies due to the mild reaction conditions and the bioorthogonal character expressed by a high specificity and selectivity. Advantageously, the use of cytotoxic Cu(I) was not mandatory in contrast to the CuAAC. Cyclooctyne is the smallest stable cyclic alkyne and has a high reaction potential to react with azides as demonstrated by Blomquist and Liu in 1953 [101] and by Wittig and Krebs in 1961 [102]. The first connection of this type of click-reaction with cyclooctyne connected with a biological application was recognized by Agard et al. in 2004 [103].
With the entry of copper-free 1,3-dipolar cycloaddition reactions in radiopharmacy, several strain-promoted systems, such as cyclooctynes and dibenzocyclooctynes, have been developed for the incorporation of fluorine-18. Additionally, a few examples of radiometal labeling were reported [104].
In general, two strategies were pointed out. The first comprises the preparation of fluorine-18-containing cyclooctyne derivatives which were applied for radiolabeling of azide-functionalized molecules of interest. Alongside, 18F-azide building blocks were synthesized and used for the labeling of cyclooctyne-bearing bioactive compounds. In general, two regioisomers are obtained in both strategies of this reaction which could indicate a different biological behavior in certain circumstances depending on the remaining molecule residue.
The first example of a biological application in combination with 18F-radiolabeling was published in 2011 by Bouvet et al. [105]. Thus, a fluorine-18-labeled aza-dibenzocyclooctyne (DBCO) building block [18F]82 was synthesized through acylation of commercially available N-(3-amino-propionyl)-5,6-dihydro-11,12-didehydrodibenzo-[b,f]azocine (80) with N-succinimidyl 4-[18F]fluoro-benzoate–[18F]SFB ([18F]81) (Scheme 29). For this purpose, amino compound 80 and [18F]SFB [18F]81 were dissolved in acetonitrile and reacted for 30 min at 40 °C monitored by radio-HPLC. After separation using semipreparative radio-HPLC, cyclooctyne [18F]82 was obtained with a RCY of 85% and a RCP > 95%. To determine the in vivo stability of [18F]82, metabolite analyses was performed using Balb/C mice. After 60 min p.i., 60% of compound [18F]82 was still intact. Further, dynamic small animal PET studies showed rapid clearance of [18F]FB-DBCO [18F]82 from the blood (blood clearance half-life: 53 s) and from most other tissues and organs. Most activity was found in the bladder, gall bladder and intestines.
Scheme 29.

[18F]Fluorobenzoate-functionalization of DBCO 80.
Subsequently, various azide-functionalized sample molecules and bioactive compounds of interest like carbohydrates and geldanamycine were conjugated and radiolabeled with [18F]82 (for an overview see Scheme 30). For the preparation of the non-radioactive references, 1.5 to 2.0 equiv. of respective azides were treated with 1 equiv. of FB-DBCO. All reactions resulted in the formation of two distinct regioisomers (1,4- and 1,5-triazole regioisomer). Products were isolated using HPLC-purification; but in some cases both regioisomers were not separable. Five different reaction conditions were tested for the radiolabeling procedure with [18F]82 (a: methanol, room temperature, 15 min; b: phosphate buffer, 40 °C, 30 min; c: 3.50% bovine serum albumin (BSA) solution in water, room temperature, 60 min; d: DMSO/water (1/1), 40 °C, 60 min.; e: water, 40 °C, 30 min) and all tracers were obtained in 69−98% RCY (determined by radio-TLC).
Scheme 30.

Scope of the labeling reaction with [18F]80.
Furthermore, the work of Carpenter et al. was focused on the synthesis of a new 18F-labeled cyclooctyne building block [106] based on ADIBO which was recently synthesized by Popik et al. [107]. They used a 6-aminohexamido spacer and [18F]SFB [18F]81 for the introduction of fluorine-18 into the building block. After purification via a C18-SPE cartridge, the 18F-labeled cyclooctyne [18F]88 was obtained in 64 ± 15% RCY and > 80% RCP. As proof-of-principle, the following click-reaction was tested with benzyl azide and an azido-functionalized PEGylated acid at 37 °C in DMF to form the two regioisomers (ratio 1:1) [18F]89 (74% RCY after 1 h) and [18F]90 (64% after 2 h) (Scheme 31). HPLC-purified cyclooctyne [18F]88 and triazoles [18F]89 and [18F]90 showed no decomposition or radiolysis over 6 h. Moreover, the stability of these products in PBS/saline buffer as well as in rat serum was tested. After 1 h, over 98% of intact radiotracer was found for both in rat serum. Additionally, formulations of both radiotracers were stable over 4 h in buffer.
Scheme 31.

Labeling scheme for the labeling with [18F]88.
In 2013, the same group evaluated an integrin αvβ6-specific peptide and labeled it with fluorine-18 using this click-chemistry approach on the basis of the previously shown results. For that purpose, peptide N3-PEG7-A20FMDV2 (91) was yielded after azide-modification and preparation via solid-phase peptide synthesis [108]. Peptide 91 selectively targets the integrin αvβ6-receptor which is located at the cell surface and has been identified as a prognostic indicator for several cancer entities [109,110].
The click-reaction for the preparation of radiolabeled peptide [18F]93 was performed in ethanol by treating peptide-precursor 91 with building block [18F]92 for 10 min at 35–45°C (Scheme 32). After HPLC-purification, [18F]93 was obtained as a regioisomeric mixture with 11.9% RCY (based on [18F]92) with > 99% RCP and with a AS of 68 ± 25 GBq/μmol. The nonradioactive standard [19F]93 was prepared from HPLC-purified [19F]FBA-C6-ADIBO [19F]92 and N3-PEG7-A20FMDV2 (91) in 69% yield. Thus, equimolar amounts of the azide and the alkyne were combined in DMF and stirred at rt. Next, the in vitro stability of [18F]93 was tested. After 60 min 94.6% of the tracer remained stable in rat serum, but after 2 h 80% of intact tracer was found. Cell binding studies of [18F]93 revealed a strong αvβ6-targeted binding (DX3puroβ6 cells, 15 min: 43.2% binding). In vivo studies using a mouse model demonstrated low binding to the DX3puroβ6-tumor (1 h: 0.47 ± 0.28% ID/g, 4 h: 0.14 ± 0.09% ID/g) and clearing from the bloodstream resulted via the renal and hepatobiliary routes. These results can be explained with the undesirable effects on pharmacokinetics which were expressed by the high lipophilicity and the large size of the labeling prosthetic group.
Scheme 32.

Labeling of N3-PEG7-A20FMDV2 (91) with [18F]FBA-C6-ADIBO [18F]92.
The next publication regards the preparation of [18F]ADIBO [18F]95 by direct radiolabeling with [18F]fluoride [111]. Thus, ADIBO was modified with a 6-tosyloxyhexanoyl residue. Labeling was carried out via nucleophilic introduction of [18F]fluoride in the presence of Et4NHCO3 in acetonitrile at 100 °C for 15 min. [18F]ADIBO [18F]95 was obtained in a RCY of 65%. Subsequent radiolabeling of an azide-functionalized Tyr3-octreotate (TATE) peptide was accomplished in ethanol. More than 95% of [18F]95 was converted into [18F]96 after 30 min (Scheme 33).
Scheme 33.

Preparation of [18F]ADIBO [18F]95 and labeling of Tyr3-octreotate.
All following works used a change of functionalities to 18F-labeled azides which underwent a click-reaction with different cyclooctynes connected to the bioactive molecule of interest. The advantage of labeled azides is their versatile applicability. They can be used for Cu-mediated click-reaction, the variants of the Staudinger Ligation and for copper-free click reaction with cyclooctyne. To demonstrate the versatility, the group of Campbell-Verduyn et al. [112] modified lys[3]-bombesin 97 with aza-dibenzocyclooctyne 98 which was previously functionalized with a succinimidyl ester. Resulting peptide 99 was then reacted with three different fluorine-18-containing azides at room temperature for 15 min in DMF with radiochemical yields of 19−37% (Scheme 34). The binding affinities of these labeled bombesin-derivatives to gastrin-releasing peptide receptors were determined using human PC3 prostate cancer cells which revealed a high affinity despite the modifications (IC50 values: 29 nM to 40 nM).
Scheme 34.

Preparation of Aza-DBCO-bombesin 99 and radiolabeling with three 18F-building blocks.
In 2012, Evans and co-workers demonstrated that CFC reactions can be carried out between [18F]fluoroethylazide ([18F]33) and five derivatives with different cyclooctyne scaffolds as pointed out in Scheme 35 [113]. The process of radiolabeling was optimized. Highest RCYs were obtained when using acetonitrile as solvent, a reaction time of 15 min, and a temperature of 90 °C. In addition, the biological behavior of [18F]33 was analyzed using BALB/c nude mice.
Scheme 35.

Cyclooctyne moieties for the radiolabeling with [18F]fluoroethylazide ([18F]33).
The group of Kim et al. presented a protocol for the radiolabeling of peptides with fluorine-18 under physiological reaction conditions [114]. Four fluorine-18-labeled biologically active peptides for tumor targeting such as cyclic Arg-Gly-Asp (cRGD) peptide, bombesin (BBN), c-Met binding peptide (cMBP) and apoptosis targeting peptide (ApoPep) were employed for this purpose. Advantageously, the resulting 18F-peptides were provided as direct injectable solutions without any HPLC purification and/or formulation processes due to the application of a novel azide-functionalized scavanger resin after the labeling procedure. The remaining cyclooctyne precursor was cached with the azide-functionalized resin for purification purposes; the overview is given in Scheme 36.
Scheme 36.

Overview of the general labeling and purification procedure.
In the first step, the desired peptides (cyclic Arg-Gly-Asp (cRGD) peptide, bombesin (BBN), c-Met binding peptide (cMBP) and apoptosis targeting peptide (ApoPep)) were modified with the cyclooctyne moiety. The next step involved the radiolabeling with [18F]103 in an ethanol/water mixture (v/v = 1/1) at ambient temperature for 15 min. Afterwards, the azide-functionalized scavenger resin was added and the mixture maintained for 20 min to allow the remaining ADIBO-peptide to conjugate with the resin. After filtration and washing of the resin with PBS solution, all peptides were produced within approx. 35 min total reaction time in 92% RCY (d.c. for [18F]104) with > 98% of RCP as a direct injectable solution for further studies without any HPLC purification. cRGD-ADIBOT-18F [18F]104 was shown as a representative example in Scheme 37.
Scheme 37.

cRGD-ADIBOT-18F [18F]104.
5. Staudinger Ligation
In 1919, Hermann Staudinger and Jules Meyer investigated the reaction of organic azides with phosphane derivatives [115]. Phosphane imines and elemental nitrogen were obtained from that reaction under absence of water. When water was added, the respective primary amines were yielded. This reaction is known as Staudinger reduction [116]. Conjugations with the respective phosphane imines are possible to execute using other electrophiles like carbonyl compounds instead of water. Based on these results, ligation reactions were developed which allow a bioorthogonal conjugation between azide functionalized molecules and phosphane species [117,118,119,120,121,122,123,124]. Two versions of this ligation type are known [125]. The non-traceless variant was introduced by Bertozzi et al. in the year 2000 [126]. Modified therephtalate compounds with a diphenylphosphanyl residue serve as fundament of this reaction. These modified phosphanes were reacted with organic azides and led to the desired ligation products after hydrolysis. Notably, the formed ligation product still contains the phosphane species which is oxidized during the reaction (Scheme 38: path A).
Scheme 38.

Variants of the Staudinger Ligation.
In the same year, the traceless variant of the Staudinger Ligation was independently and simultaneously developed by Raines and co-workers [127] and the group of Bertozzi [128]. In this case, organic azides were used as starting material as well. In contrast to the non-traceless ligation, phosphanes were introduced which are functionalized in the ortho-position relative to the phosphorus on one of the aromatic rings. The phosphane oxide residue which is formed during the reaction is excluded and both reaction partners are connected exclusively via a native amide (peptide) bond (Scheme 38: path B). Therefore, this reaction type is often adapted in peptide syntheses [129,130]. Both ligations proceed in a wide pH range using organic or aqueous solvents.
The non-traceless Staudinger Ligation is seldom used for radiolabeling purposes with fluorine-18. To date, only one application is known. In 2006, the preparation of a fluorine-18 containing phosphane and the ligation to an azide containing carbohydrate was described [131] as shown in Scheme 39. In this case, the labeled glucose derivative served as an alternative for [18F]FDG and the application for the pretarget PET imaging of cancer was designated.
Scheme 39.

Building blocks for the radiolabeling using the non-traceless Staudinger Ligation.
In contrast to the non-traceless variant, several examples for the traceless Staudinger Ligation were published in the past. In general, two strategies were pointed out depending on the incorporation of fluorine-18 into the starting material. In the first case, phosphanes were evaluated containing the fluorine-18. For this purpose, biologically active molecules were modified with azide (direct approach). In the second case, the phosphane residue was introduced into the biologically active molecule and the fluorine-18 was connected to an azide functionalized molecule (indirect approach). Both ways are introduced in Scheme 40.
Scheme 40.

Direct and indirect approach of the traceless Staudinger Ligation.
The direct approach was intensively investigated by Mamat and co-workers starting in 2009 [132]. For that purpose, phosphanes with various benzoate residues functionalized in the para-position were prepared [133]. Hence, two reaction paths were developed. First, the conventional esterification of phosphanol 107 with benzoyl chlorides 108a–c in the presence of a base or via Steglich esterification (for 111d) delivered the substituted phosphanes 109a–d (yield: 38−92%). However, application of these reaction conditions sometimes led to low yields of the desired phosphanes or the formation of by-products. Due to this fact, functionalized 4-iodophenyl derivatives 110a-h were applied as an alternative. Phosphanes 109a–h were obtained in 58−89% yield from the Pd-catalyzed cross coupling of 110a–h with diphenylphosphane (HPPh2); the findings are summarized in Scheme 41. This approach should serve as a possibility for the introduction of the 4-[18F]fluorobenzoate moiety (alternatively for [18F]SFB), but without success.
Scheme 41.

Preparation of various phosphane building blocks.
Later, a different labeling strategy was evaluated [134] based on 2-(diphenylphosphano)phenyl 4-hydroxybenzoate (112) which was reacted with 2-[18F]fluoroethyl tosylate ([18F]113) to yield the desired phosphane [18F]114. In the second step, the respective azide was added to yield the desired radiotracer. This reaction was carried out as a three-step/one-pot procedure starting from ethylene ditosylate as seen in Scheme 42. The phosphane precursor was added in a one-pot two-step procedure after the generation of [18F]113 to yield the labeled phosphane species. Subsequently, the respective azido compound was added for labeling purposes within the Staudinger Ligation to yield [18F]115-[18F]118 (17−21% RCY, d.c.).
Scheme 42.

Radiofluorinations with [18F]114 using the traceless Staudinger Ligation.
Notably, a two-step sequence was required for the preparation of the 18F-containing phosphane which generally lowers the RCY as well as the AS of the final tracers. For this purpose, a one-step approach for the preparation of fluorine-18 containing phosphane building blocks like [18F]120 (RCY = 65%, d.c.) was developed based on aliphatic phosphanyl esters [135,136]. A chain length of five carbon atoms was chosen for precursor 119. Different bases as well as fluorination agents and solvents were evaluated for optimization. The best results were obtained using [18F]fluoride in the presence of TBAOH which was used as base in a mixture of acetonitrile/t-BuOH (v/v = 1:4) [25] for 10 min at 100 °C using 21 mg of precursor 119. Several model compounds [18F]118-[18F]121 including a carbohydrate moiety and (+)-biotin were first azide functionalized and subsequently labeled using the traceless Staudinger Ligation (Scheme 43) with RCYs from 12 to 31% (d.c.).
Scheme 43.

One pot preparation of phosphane building block [18F]120 and subsequent radiolabeling of azide functionalized molecules using the traceless Staudinger Ligation.
In 2010, Gaeta et al. reported the preparation and radiolabeling of GABAA receptor binding 4-quinolones with Ki from 0.7 to 3.7 nM [137]. For this purpose, the bioactive site of molecule 125 was modified and (diphenylphosphino)methanethiol moiety 126 was introduced (indirect approach). For the radiolabeling, 2-[18F]fluoroethyl azide ([18F]33) was applied as the labeling building block. The following Staudinger Ligation was accomplished in a mixture of acetonitrile and DMF for 15 min at 130 °C and [18F]128 was obtained after purification using preparative HPLC with 75% RCY (30–50 MBq, not d.c.) and a AS = 0.9 GBq/μmol (radiochemical purity > 95%); see Scheme 44. Biological data showed that only a modest fraction of the compound crossed the blood–brain barrier, with peak uptake no greater than 0.12% of the injected dose (0.11% ID/g). Ex vivo autoradiographic analysis of prefrontal cortex, striatum and thalamus slices indicated significant differential binding of [18F]128 consistent with the distribution of GABAA receptors.
Scheme 44.

Preparation and radiolabeling of quinolone-based precursor 127.
A second work was published in 2011 by the group of Gouverneur et al. [138] using a similar strategy with 2-[18F]fluoroethyl azide ([18F]33) as labeling agent and phosphane functionalized biologically active molecules 129–133 (indirect approach). In this case, various amino acid derivatives were labeled as shown in Table 2 [labeling conditions: (A) THF/H2O (4/1), 120 °C, 15 min; (B) DMF/H2O (6/1), 120 °C, 15 min].
Table 2.
Scope of the labeling reaction under Staudinger conditions.
| Starting material | Cond. | RCY‡ | Product |
|---|---|---|---|
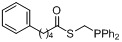 129
129
|
A | > 95% |
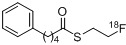 134
134
|
 130
130
|
A | > 95% |
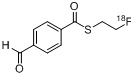 135
135
|
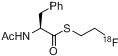
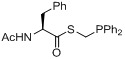 131
131
|
A B |
> 95% > 95% |
 136
136
|
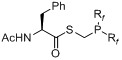 132
132
|
B | > 95% |
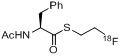 136
136
|
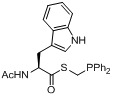 133
133
|
A | > 95% |
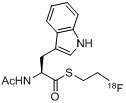 137
137
|
‡ = conversion from 2-[18F]fluoroethyl azide ([18F]33), Rf = C6H4CH2CH2C4F9.
The purification step of this labeling reaction was improved using fluorous solid phase extraction (FSPE). For this purpose, derivative 132 was prepared with a fluorous tag [139] (C4F9 chain on the aromatic ring of the phosphane) attached onto the phosphino fragment. Adversely, this compound was faster oxidized to the corresponding phosphine than the non fluorous counterpart. Thus, the purification step of 132 was done under absence of air. The following Staudinger Ligation with 2-fluoroethyl azide using standard conditions led to the formation of 136 in 27% yield after purification of the reaction mixture using FSPE [140]. The decrease in isolated yield could be attributed to the propensity of the phosphane precursor 132 towards oxidation. The radiofluorination was performed in acetonitrile using [18F]–/K+/K 222 followed by FSPE to separate 2-[18F]fluoroethyl azide ([18F]33) (AS~10 GBq/mmol) from the excess fluorous precursor 132, no distillation was necessary. Pleasingly, the labelling of both the non-fluorous and fluorous alanine precursor 131 and 132 with 2-[18F]fluoroethyl azide ([18F]33) in DMF–H2O (6 : 1) at 120°C for 15 min afforded [18F]136 in an excellent RCY of >95% (entries 3–4, Table 2). The purification using FSPE for the labeling of 132 led to an improvement, but HPLC was still necessary to obtain an analytically pure sample due to the breakthrough of fluorous material into the fluorophobic eluted fraction.
6. Tetrazine-Click
Investigated first by the group of Fox and co-workers, the tetrazine ligation is a fast bioconjugation method based on inverse-electron-demand Diels−Alder reactions [141], a proposed mechanism is pointed out in Scheme 45. The main advantage of this kind of reaction involves the unusually fast reaction rates (k2 = 2,000 M−1·s−1, solvent MeOH/water: 9/1) without need for catalysis. A further advantage is the non-reversibility due to the loss of elemental nitrogen, in contrast to most other Diels-Alder reactions. These reactions tolerate a broad range of functionality and proceed with high yield in organic solvents, water, buffer, cell media, or lysate. This fast reactivity enables protein modification at low concentration. The resulting cycloocta[d]pyridazines were obtained as conjugation products from the cycloaddition of s-tetrazine and trans-cyclooctene derivatives. Mechanistically, the tetrazine ligation proceeds in two steps. An inverse-demand Diels-Alder reaction occurs followed by a retro-Diels-Alder reaction to eliminate nitrogen gas.
Scheme 45.

Proposed reaction mechanism of the tetrazine-click reaction.
Based on computational work by Bach, E-cyclooctene has a highly twisted double bond resulting in a strain energy of 17.9 kcal/mol compared to Z-cyclooctenes or cyclooctane [142]. Based on these results, highly strained trans-derivatives were used as reactive dienophiles. Further, 3,6-diaryl-s-tetrazines serve as dienes which have been substituted in order to resist immediate reaction with water. The fact that the reaction is not only tolerant of water is also important especially for fast radiolabeling reactions. It has also been found that the rate increases in aqueous media. In addition, norbornenes were introduced as dienophiles at second order rates on the order of 1 M−1·s−1 in aqueous media. The labeling of live cells [143,144,145] or polymer coupling [146,147] were prominent examples of an application of this kind of conjugation reaction. Alongside, an elegant work concerning the radiolabeling of peptides using 111In-DOTA were published by Robillard [148] and a modular strategy for the preparation of 64Cu and 89Zr radiometalated antibodies was previously published [149].
One of the first studies to apply the tetrazine-click reaction for radiofluorinations was achieved in the year 2010 by the group of Fox and co-workers (Scheme 46) [150]. First, they tried to label tetrazine derivatives with fluorine-18 for a conjugation with the trans-cyclooctene containing bioactive molecule. The instability of the tetrazines (RCY~1%) prompted the group to change the functionalities. Thus, [18F]TCO [18F]139 as fluorine-18-containing cyclooctene derivative was prepared (RCY 71%) from the respective nosylate precursor 138 in a reaction time of 15 min. [18F]139 was then used for the conjugation with the tetrazine containing sample molecule 140 within 10 s of mixing at ambient temperature with a RCY > 98%.
Scheme 46.

First example of the tetrazine-click reaction.
Based on these results, a cyclic RGD peptide was developed as an αvβ3 targeted PET tracer [151]. Tetrazine-RGD conjugate 143 was prepared as the precursor by treatment of c(RGDyK) with a NHS-ester. Radiolabeling was done with [18F]TCO [18F]139 at ambient temperature for 5 min in a mixture of DMSO and EtOH (1/3). [18F]RGD-tracer [18F]144 was obtained in >95% RCY (based on [18F]139), see Scheme 47. microPET analyses using a U87MG xenograft model showed a tumor uptake of 4.4%ID/g after 1 h. Blocking experiments revealed a specific binding to integrin αvβ3.
Scheme 47.

Labeling of tetrazine-modified RGD peptide 143 using [18F]139.
Next, the tetrazine residue was functionalized with a maleimide moiety for the radiolabeling of other cyclic RGD peptides without lysine residue [152]. Two strategies were evaluated for that purpose. In the first, the tetrazine-maleimide linker was conjugated with a cysteine-modified RGD peptide and finally labeled with [18F]TCO [18F]139. The labeled peptide was obtained in > 95% RCP with a AS of approx. 111 to 222 GBq/µmol. To compare the efficiency of this ligation type, [18F]TCO [18F]139 was directly reacted with the maleimide functionalized tetrazine. The original RGD peptide was then treated with this conjugate; however, this reaction proceeded within 20 min whereas the first labeling way took only 5 min. In addition, a higher peptide concentration was mandatory for the second way. MicroPET studies of U87MG tumor-bearing mice showed an uptake of 18F-RGD [18F]144 of approx. 2% ID/g in the tumor which is comparable with the previous findings of this group.
Next, based on previous work [153], the syntheses and in vivo imaging of two 18F-labeled PARP1 inhibitors were presented by Reiner et al. in 2011 [154,155]. In the first publication, they compared the conventional direct nucleophilic introduction of [18F]fluoride with the click-labeling using [18F]TCO [18F]139. For this purpose, a fully automated synthesis (41 min) of [18F]TCO [18F]139 was developed which yielded the building block in 44.7 ± 7.8% RCY (d.c., > 93% RCP, conditions: [18F]fluoride, n-Bu4NHCO3, 90 °C). Subsequent radiofluorination with tetrazine 145 was accomplished in a DMSO/DCM solution for 3 min. [18F]146 was provided in 59.6 ± 5.0% RCY (d.c., n = 3, RCP > 96%) after HPLC purification. In contrast, the direct radiofluorination of 147 delivered [18F]148 in only 1% RCY. Next, the target affinity of both tracers was studied. Thus, a colorimetric assay was employed to measure PARP1 activity with the result that [18F]148 had an IC50 of 5.2 ± 1.1 nM whereas the clicked product [18F]146 had an IC50 of 17.9 ± 1.1 nM. This results show impressively the dependence of the introduced moiety on the affinity of a molecule (Scheme 48).
Scheme 48.

Comparison between conventional and tetrazine-click labeling.
An improvement was done regarding the purification of the resulting radiotracer [18F]146 from the precursor 145 [156]. A magnetic TCO resin was developed and used for a subsequent incubation after the radiolabeling step. The following separation was done using a magnet. An overview was pointed out in Scheme 49. This method was applied to successfully remove the excess of precursor 145 and to avoid lengthy HPLC purifications. After separation of the magnetic resin, the radiotracer [18F]146 was obtained in 92 ± 0.4% RCY (d.c.).
Scheme 49.

Overview of the general labeling and purification procedure.
In 2012, an efficient radiofluorination strategy regarding exendin-4 analogues of imaging of beta cells was described (Scheme 50) [157]. For this purpose, (E)-5-(2-[18F]fluoroethoxy)cyclooct-1-ene ([18F]139) was used on the one hand and aminobenzyl tetrazine 149 was functionalized with maleimide 150 for a conjugation with SH-groups of peptides like cysteine. Therefore, a natural occurring lysine at position C12 was exchanged for a cysteine in an exendin-4-affine peptide (E4C12). Next, E4C12 peptide was modified with the tetrazine containing cross linker 151 to give E4C12Tz 152. Similar to the above mentioned techniques, E4C12Tz 152 was incubated with [18F]TCO [18F]139 at rt for 20 min and the labeled peptide [18F]153 was obtained with > 94% RCP after separation using the aforepublished magnetic scavenger resin.
Scheme 50.

Radiolabeling of E4 peptide [18F]153 using tetrazine-click.
7. Miscellaneous
In addition to the classical CuAAC, the 1,3-dipolar cycloaddition of nitrones with alkenes which led to isoxazolidines or further to isoxazoles belongs to the [3+2]-dipolar cycloadditions and was evaluated for radiofluorination purposes by the group of Zlatopolsky et al. in 2012 [158]. Mild reaction conditions, regioselectivity, absence of protection groups, acceptable yields and metabolic stability of the formed isoxazoles are the main advantages. Both starting materials are stable under a wide range of conditions (pH, temperature, microwave irradiation, reagents) [159] and this reaction can be carried out in different solvents, including aqueous media [160,161]. Of high importance is the absence of metal catalysts like Cu(I) in contrast to the classical CuAAC. In the first sample reaction, the fluorine-18 containing nitrone was simply prepared from the appropriate aldehyde [18F]154 and N-substituted hydroxyl amines like 155. Based on this strategy, the resulting labeling building block [18F]154 was prepared in 74% RCY after 10 min reaction time at ambient temperature (Scheme 51).
Scheme 51.

Preparation of C-(4-[18F]fluorophenyl)-N-phenyl nitrone ([18F]156).
The next step includes the radiolabeling of three sample molecule. For this purpose, these molecules were modified with a maleimide residue. The subsequent labeling step was carried out in a one-pot synthesis starting from 4-[18F]benzaldehyde ([18F]154), phenyl hydroxylamine (155) and the respective malinide species in RCYs > 80% (Scheme 52). The respective 18F-labeled diastereomers were formed as endo- and exo-isomers (ratio approx. 2/1 = endo/exo) in all cases and were easy separable.
Scheme 52.

One-pot radiolabeling of model compound 157.
A further improvement of the labeling reaction was done in the same year [162]. In contrast to the first approach, hydroxylamine (as its HCl salt) was used instead of N-functionalized derivatives for the preparation of 4-[18F]benzonitrile oxide [18F]159 as 18F-containing building block. [18F]159 was obtained in 92% RCY with >90% RCP after 10 min in aqueous methanol at 40 °C from [18F]benzaldehyde [18F]154 and hydroxylamine. For radiofluorination purposes, [18F]159 was treated with phenyl iodine bis(trifluoroacetate) (PIFA) for 5–10 s. Afterwards, different dipolarophiles were chosen and even less reactive derivatives like styrene or 4-pentyn-1-ol showed high yields of the cycloaddition product (Scheme 53).
Scheme 53.

Overview of the modified reaction.
Using this reaction, three fluorine-18 labeled indomethacin derivatives [18F]162-[18F]164 were prepared to enable visualization of the COX-2 expression in inflammation of cancer. For this purpose, the indomethacine scaffold was functionalized with maleimide, propyne and cyclooctyne. The highest results were obtained with 81% RCY using the cyclooctyne derivative. Results are shown in Scheme 54.
Scheme 54.

Radiolabeled indomethazin derivatives.
The last experiment contained the labeling of a dipeptide (β-Ala-Phe-OMe) either bound to the cyclooctyne residue or bound to the norbornene moiety. Both labeled peptides [18F]165 and [18F]166 are pointed out in Scheme 54.
8. Conclusions
This review has summarized the applications and the scope of bioorthogonal click reactions for the introduction of fluorine-18 in the field of radiopharmacy. This kind of reaction has become a very important tool, especially for the radiofluorination of high molecular weight pharmacologically relevant molecules like peptides or proteins but also for the radiofluorination of small compounds of interest. For labeling purposes, fast and high yielding reactions are required and strategies for a convenient purification in an acceptable time span.
After considering all results which were obtained in the last years a guideline for the choice of building blocks could be generated: first, a building block should be azide-functionalized for universal applicability and to prevent side-reactions like the Glaser coupling. This is important especially for radiolabeling purposes with alkyne functionalized building blocks using the CuAAC. In addition, when utilizing the CuAAC the use of Cu(I) stabilizing agents like TBTA, BTTAA and bathophenanthroline is crucial and should be at least 1.1 eq to the Cu-source. Second, the building blocks should contain 18F-propyl, 18F-ethyl or 18F-aryl groups for a higher metabolic stability. Third, these blocks should be directly radiolabeled by fluorine-18 for shorter preparation times and higher RCY and AS. Further, glycosylations or PEGylations of these building blocks enhance the in vivo availability of the corresponding radiolabeled bioactive molecule. An overview of building blocks is given in Table 5.
Table 5.
Reaction conditions and yields of selected 18F-containing building blocks.
| Building block | RCY
[%] |
RCP
[%] |
t
[min] |
AS
[GBq/µmol] |
Synthesis procedure | Literature |
|---|---|---|---|---|---|---|
| [18F]SFB | 25–38 | >95 | 70 | n.d. | automatically | [163] |
| [18F]FBAM | 29 | 94–98 | 40 | 13–17 | automatically | [164] |
| [18F]AFP [18F]47 | 25–50 | >97 | 40 | 3–6 | automatically | [78] |
| [18F]BFP [18F]46 | 25–50 | >98 | 40 | 5–9 | automatically | [78] |
| [18F]Fluorobutyne [18F]2a | 36 | >98 | 40 | n.d. | manually | [48] |
| [18F]Fluoropentyne [18F]2b | 51 | >99 | 40 | n.d. | automatically | [50] |
| [18F]FSi-based azide [18F]13 | 54 | >95 | 20 | n.d. | manually | [51] |
| [18F]PEG3-alkyne [18F]23 | 57 | >99 | 40 | n.d. | automatic | [54] |
| [18F]PEG3-azide [18F]27 | 62 | >99 | 40 | n.d. | manually | [55,56] |
| [18F]FEA [18F]33 | 55 | >99 | 40 | n.d. | automatically | e.g., [57,58] |
| [18F]FDG-β-Az [18F]78 | 71 | n.d. | 30 | n.d. | manually | [100] |
| [18F]F-SA [18F]51 | 32 | >95 | 80 | 120–570 | automatically | [81,82,83] |
| [18F]ArBF3- [18F]72 | 50 | 70 | 555 | manually | [95,96,97] | |
| [18F]Fluoroethynylpyridine [18F]59 | 28 | >98 | 35 | n.d. | manually | [87] |
| [18F]FB-DBCO [18F]82 | 85 (from SFB) | 95 | 60 (from SFB) | n.d. | manually | [105] |
| [18F]ADIBO [18F]47 | 73–95 | 95 | 55 | 3.8 | manually | [111] |
| [18F]FHP [18F]120 | 65 | n.d. | 10 | n.d. | manually | [135] |
| [18F]TCO [18F]139 | 46 | >94 | 102 | n.d. | automatically | [157] |
Moreover, it is necessary to separate and deplete catalysts like the copper species completely when using the Huisgen-click. This is important especially for in vivo applications due to the formation of complexes of Cu with the respective high molecular weight bioactive molecule during the labeling procedure. In this case and due to the toxic properties of copper species in vivo, copper-free variants should be applied. Thus, the CuAAC should be more preferred rather for small compounds. Last but not least, the non-radioactive reference building blocks as well as the precursors should be conveniently prepared from inexpensive chemicals.
Tetrazine-click as well as the Cu-free variant of the Huisgen cycloaddition show the best results in terms of the labeling time. This is important, especially for the labeling of high molecular weight compounds. Moreover, the removal of Cu or other catalysts is not needed. The size of the building block and the formation of regioisomers is a drawback, but the higher the molar mass of the molecule to be labeled the smaller is the influence of the building block in most of the cases.
The use of cartridges or scavenger resins for a fast separation and purification is a fundamental topic. This allows labeling reaction with high yields, high radiochemical purity and high specific activity in a very short time. A high hydrophilicity of the bioorthogonal building blocks should be considered for the radiolabeling of proteins and antibodies.
Acknowledgments
The authors thank the Fonds der Chemischen Industrie (FCI) for financial support. Moreover, the authors would like to thank Fang Xie for the translation of the Chinese papers and Alexandra Born for the extensive proofreading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Phelps M.E. PET: Molecular Imaging and Its Biological Applications. Springer; New York, NY, USA: 2004. [Google Scholar]
- 2.Vallabhajosula S. Molecular Imaging: Radiopharmaceuticals for PET and SPECT. Springer; Dordrecht, The Netherlands: 2009. [Google Scholar]
- 3.Schober O., Heindel W.L. PET–CT Hybrid Imaging. Thieme; Stuttgart, Germany: 2010. [Google Scholar]
- 4.Schirrmacher R., Wängler C., Schirrmacher E. Recent developments and trends in 18F-radiochemistry: Syntheses and applications. Mini-Rev. Org. Chem. 2007;4:317–329. doi: 10.2174/157019307782411699. [DOI] [Google Scholar]
- 5.Mamat C., Ramenda T., Wuest F.R. Recent applications of click chemistry for the synthesis of radiotracers for molecular imaging. Mini-Rev. Org. Chem. 2009;6:21–34. doi: 10.2174/157019309787316148. [DOI] [Google Scholar]
- 6.Glaser M., Robins E.G. ‘Click labelling’ in PET radiochemistry. J. Labelled Compd. Radiopharm. 2009;52:407–414. doi: 10.1002/jlcr.1656. [DOI] [Google Scholar]
- 7.Olberg D.E., Hjelstuen O.K. Labeling strategies of peptides with 18F for positron emission tomography. Curr. Topics Med. Chem. 2010;10:1669–1679. doi: 10.2174/156802610793176747. [DOI] [PubMed] [Google Scholar]
- 8.Lei M., Tian M., Zhang H. Click labeled peptides with fluorine-18 for PET imaging. Curr. Med. Imaging Rev. 2010;6:33–41. doi: 10.2174/157340510790693884. [DOI] [Google Scholar]
- 9.Bertozzi C.R. A decade of bioorthogonal chemistry. Acc. Chem. Res. 2011;44:651–653. doi: 10.1021/ar200193f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolb H.C., Finn M.G., Sharpless K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 11.Kolb H.C., Sharpless K.B. The Growing Impact of click chemistry on drug discovery. Drug Discov. Today. 2003;8:1128–1137. doi: 10.1016/S1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 12.Lahann J. Click Chemistry for Biotechnology and Material Science. John Wiley & Son; Chichester, UK: 2009. [Google Scholar]
- 13.Pfennig G., Klewe-Nebenius H., Seelmann-Eggebert W. Karlsruher Nuklidkarte. Haberbeck; Lage, Germany: 2009. [Google Scholar]
- 14.Elsinga P.H. Radiopharmaceutical chemistry for positron emission tomography. Methods. 2002;27:208–217. doi: 10.1016/S1046-2023(02)00076-2. [DOI] [PubMed] [Google Scholar]
- 15.Schubiger P.A., Lehmann L., Friebe M., editors. PET Chemistry—The Driving Force in Molecular Imaging. Springer; Berlin, Heidelberg, Germany: 2007. pp. 1–158. [Google Scholar]
- 16.Miller P.W., Long N.J., Vilar R., Gee A.D. Synthese von 11C-, 18F-, 15O- und 13N-radiotracern für die positronenemissionstomographie. Angew. Chem. 2008;120:9136–9172. doi: 10.1002/ange.200800222. [DOI] [Google Scholar]
- 17.Ametamey S.M., Honer M., Schubiger P.A. Molecular imaging with PET. Chem. Rev. 2008;108:1501–1516. doi: 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- 18.Serdons K., Verbruggen A., Bormans G.M. Developing new molecular imaging probes for PET. Methods. 2009;48:104–111. doi: 10.1016/j.ymeth.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 19.Li Z., Conti P.S. Radiopharmaceutical chemistry for positron emission tomography. Adv. Drug Deliv. Rev. 2010;62:1031–1051. doi: 10.1016/j.addr.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 20.Hjelstuen O.K., Svadberg A., Olberg D.E., Rosser M. Standardization of fluorine-18 manufacturing processes: New scientific challenges for PET. Eur. J. Pharm. Biopharm. 2011;78:307–313. doi: 10.1016/j.ejpb.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 21.Littich R., Scott P.J.H. Neue Strategien für die 18F-Radiochemie. Angew. Chem. 2012;124:1132–1135. doi: 10.1002/ange.201106785. [DOI] [Google Scholar]
- 22.Vértes A., Nagy S., Klencsár Z., Lovas R.G., Rösch F. Handbook of Nuclear Chemistry. 2nd ed. Volume 4. Springer; Dordrecht, Netherlands: 2011. pp. 2026–2031. [Google Scholar]
- 23.Vértes A., Nagy S., Klencsár Z., Lovas R.G., Rösch F. Handbook of Nuclear Chemistry. 2nd ed. Volume 4. Springer; Dordrecht, Netherlands: 2011. pp. 2032–2041. [Google Scholar]
- 24.Schirrmacher R., Wängler C., Schirrmacher E. Fluorine-18 Radiochemistry: Theroy and Practice. In: Wester H.J., editor. Pharmaceutical Radiochemistry (I) Volume 1. Scintomics Print Media and Publishing; Fürstenfeldbruck, Germany: 2010. pp. 5–73. [Google Scholar]
- 25.Kim D.W., Jeong H.-J., Lim S.T., Sohn M.H., Katzenellenbogen J.A., Chi D.Y.J. Facile nucleophilic fluorination reactions using tert-alcohols as a reaction medium: Significantly enhanced reactivity of alkali metal fluorides and improved selectivity. Org. Chem. 2008;73:957–962. doi: 10.1021/jo7021229. [DOI] [PubMed] [Google Scholar]
- 26.Böhm H.-J., Banner D., Bendels S., Kansy M., Kuhn B., Müller K., Obst-Sander U., Stahl M. Fluorine in medicinal chemistry. ChemBioChem. 2004;5:637–643. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 27.Bégué J.-P., Bonnet-Delpon D. Biological Impacts of Fluorination: Pharmaceuticals Based on Natural Products. In: Tressaud A., Haufe G., editors. Fluorine and Health. 1st ed. Elsevier; Amsterdam, Netherlands: 2008. pp. 553–576. [Google Scholar]
- 28.Yamazaki T., Taguchi T., Ojima I. Introduction: Basic Aspects of Fluorine in Chemistry and Biology. In: Ojima I., editor. Fluorine in Medicinal Chemistry and Chemical Biology. Wiley-Blackwell; Chichester, UK: 2009. pp. 3–46. [Google Scholar]
- 29.Michael A. Ueber die Einwirkung von Diazobenzolimid auf Acetylendicarbonsäuremethylester. J. Prakt. Chem. 1893;48:94–95. doi: 10.1002/prac.18930480114. [DOI] [Google Scholar]
- 30.Dimroth O., Fester G. Triazol und Tetrazol aus Stickstoffwasserstoffsäure. Ber. Dtsch. Chem. Ges. 1910;43:2219–2223. [Google Scholar]
- 31.Huisgen R. 1.3-dipolare Cycloadditionen. Angew. Chem. 1963;75:604–637. doi: 10.1002/ange.19630751304. [DOI] [Google Scholar]
- 32.Bräse S., Banert K. Organic Azides: Syntheses and Applications. Wiley; Chichester, UK: 2010. [Google Scholar]
- 33.Rostovtsev V.V., Green L.G., Fokin V.V., Sharpless K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. 2002;114:2708–2711. doi: 10.1002/1521-3757(20020715)114:14<2708::AID-ANGE2708>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 34.Tornøe C.W., Christensen C., Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 35.Himo F., Lovell T., Hilgraf R., Rostovtsev V.V., Noodleman L., Sharpless K.B., Fokin V.V. Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 2004;127:210–216. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- 36.Fuchs M., Goessler W., Pilger C., Kappe C.O. Mechanistic insights into copper(I)-catalyzed azide-alkyne cycloadditions using continuous flow conditions. Adv. Synth. Catal. 2010;352:323–328. doi: 10.1002/adsc.200900726. [DOI] [Google Scholar]
- 37.Worrell B.T., Malik J.A., Fokin V.V. Direct evidence of a dinuclear copper intermediate in Cu(I)-catalyzed azide-alkyne cycloadditions. Science. 2013;340:457–460. doi: 10.1126/science.1229506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gil M.V., Arévalo M.J., López Ó. Click chemistry–What’s in a name? Triazole synthesis and beyond. Synthesis. 2007:1589–1620. [Google Scholar]
- 39.Nwe K., Brechbiel M.W. Growing applications of “click chemistry” for bioconjugation in contemporary biomedical research. Cancer Biother. Radiopharm. 2009;24:289–302. doi: 10.1089/cbr.2008.0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wängler C., Schirrmacher R., Bartenstein P., Wängler B. Click-chemistry reactions in radiopharmaceutical chemistry: Fast & easy introduction of radiolabels into biomolecules for in vivo imaging. Curr. Med. Chem. 2010;17:1092–1116. doi: 10.2174/092986710790820615. [DOI] [PubMed] [Google Scholar]
- 41.Mindt T.L., Struthers H., Brans L., Anguelov T., Schweinsberg C., Maes V., Tourwe D., Schibli R. “Click to chelate”: Synthesis and installation of metal chelates into biomolecules in a single step. J. Am. Chem. Soc. 2006;128:15096–15097. doi: 10.1021/ja066779f. [DOI] [PubMed] [Google Scholar]
- 42.Schirrmacher R., Lakhrissi Y., Jolly D., Goodstein J., Lucas P., Schirrmacher E. Rapid in situ synthesis of [11C]methyl azide and its application in 11C click-chemistry. Tetrahedron Lett. 2008;49:4824–4827. doi: 10.1016/j.tetlet.2008.06.020. [DOI] [Google Scholar]
- 43.Wang T., Guo Z.J. Copper in medicine: Homeostasis, chelation therapy and antitumor drug design. Curr. Med. Chem. 2006;13:525–537. doi: 10.2174/092986706776055742. [DOI] [PubMed] [Google Scholar]
- 44.Ning K., Guo J., Wolfert M.A., Boons G.-J. Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angew. Chem. 2008;120:2285–2287. doi: 10.1002/ange.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marik J., Sutcliffe J.L. Click for PET: Rapid preparation of [18F]fluoropeptides using CuI catalyzed 1,3-dipolar cycloaddition. Tetrahedron Lett. 2006;47:6681–6684. doi: 10.1016/j.tetlet.2006.06.176. [DOI] [Google Scholar]
- 46.Ross T.L., Honer M., Lam P.Y.H., Mindt T.L., Groehn V., Schibli R., Schubiger P.A., Ametamey S.M. Fluorine-18 click radiosynthesis and preclinical evaluation of a new 18F-labeled folic acid derivative. Bioconjugate Chem. 2008;19:2462–2470. doi: 10.1021/bc800356r. [DOI] [PubMed] [Google Scholar]
- 47.Hausner S.H., Marik J., Karen M., Gagnon J., Sutcliffe J.L. In vivo positron emission tomography (PET) imaging with an αvβ6 specific peptide radiolabeled using 18F-“click” chemistry: Evaluation and comparison with the corresponding 4-[18F]fluorobenzoyl- and 2-[18F]fluoropropionyl-peptides. J. Med. Chem. 2008;51:5901–5904. doi: 10.1021/jm800608s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim D.H., Choe Y.S., Jung K.-H., Lee K.-H., Choi J.Y., Choi Y., Kim B.-T. A 18F-labeled glucose analog: Synthesis using a click labeling method and in vitro evaluation. Arch. Pharm. Res. 2008;31:587–593. doi: 10.1007/s12272-001-1197-2. [DOI] [PubMed] [Google Scholar]
- 49.Kim D.H., Choe Y.S., Kim B.-T. Evaluation of 4-[18F]fluoro-1-butyne as a radiolabeled synthon for click chemistry with azido compounds. Appl. Radiat. Isot. 2010;68:329–333. doi: 10.1016/j.apradiso.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 50.Zhang J., Zhang X., Zhang B., Liu J., Wang H., Sun Z., Tian J. Automated “click chemistry” synthesis of (18)F-Pen-peptide as an apoptosis imaging agent. Nucl. Techn. 2011;34:845–850. [Google Scholar]
- 51.Balentova E., Collet C., Lamandé-Langle S., Chrétien F., Thonon D., Aerts J., Lemaire C., Luxen A., Chapleur Y. Synthesis and hydrolytic stability of novel 3-[18F]fluoroethoxybis (1−methylethyl)silyl]propanamine-based prosthetic groups. J. Fluorine Chem. 2011;132:250–257. doi: 10.1016/j.jfluchem.2011.01.011. [DOI] [Google Scholar]
- 52.Sirion U., Kim H.J., Lee J.H., Seo J.W., Lee B.S., Lee S.J., Oh S.J., Chi D.Y. An efficient F−18 labeling method for PET study: Huisgen 1,3-dipolar cycloaddition of bioactive substances and F−18-labeled compounds. Tetrahedron Lett. 2007;48:3953–3957. [Google Scholar]
- 53.Li Z.-B., Wu Z., Chen K., Chin F.T., Chen X. Click chemistry for 18F-labeling of RGD peptides and microPET imaging of tumor integrin αvβ3 expression. Bioconjugate Chem. 2007;18:1987–1994. doi: 10.1021/bc700226v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Devaraj N.K., Keliher E.J., Thurber G.M., Nahrendorf M., Weissleder R. 18F Labeled Nanoparticles for in vivo PET-CT imaging. Bioconjugate Chem. 2009;20:397–401. doi: 10.1021/bc8004649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gill H.S., Marik J. Preparation of 18F-labeled peptides using the copper(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition. Nat. Protoc. 2011;6:1718–1725. doi: 10.1038/nprot.2011.390. [DOI] [PubMed] [Google Scholar]
- 56.Lee C.-M., Jeong H.-J., Kim D.W., Sohn M.-H., Lim S.T. The effect of fluorination of zinc oxide nanoparticles on evaluation of their biodistribution after oral administration. Nanotechnology. 2012;23 doi: 10.1088/0957-4484/23/20/205102. [DOI] [PubMed] [Google Scholar]
- 57.Glaser M., Årstad E. “Click labeling” with 2-[18F]fluoroethylazide for positron emission tomography. Bioconjugate Chem. 2007;18:989–993. doi: 10.1021/bc060301j. [DOI] [PubMed] [Google Scholar]
- 58.Kobus D., Giesen Y., Ullrich R., Backes H., Neumaier B. A fully automated two-step synthesis of an 18F-labelled tyrosine kinase inhibitor for EGFR kinase activity imaging in tumors. Appl. Radiat. Isot. 2009;67:1977–1984. doi: 10.1016/j.apradiso.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 59.Haslop A., Gee A., Plisson C., Long L. Fully automated radiosynthesis of [1-(2-[18F]fluoroethyl)-1H[1,2,3]triazole 4-ethylene]triphenylphosphonium bromide as a potential positron emission tomography tracer for imaging apoptosis. J. Labelled Compd. Radiopharm. 2013;56:313–316. doi: 10.1002/jlcr.3024. [DOI] [PubMed] [Google Scholar]
- 60.Smith G., Glaser M., Perumal M., Nguyen Q.-D., Shan B., Årstad E., Aboagye E.O. Design, synthesis, and biological characterization of a caspase 3/7 selective isatin labeled with 2-[18F]fluoroethylazide. J. Med. Chem. 2008;51:8057–8067. doi: 10.1021/jm801107u. [DOI] [PubMed] [Google Scholar]
- 61.Glaser M., Goggi J., Smith G., Morrison M., Luthra S.K., Robins E., Aboagye E.O. Improved radiosynthesis of the apoptosis marker 18F-ICMT11 including biological evaluation. Bioorg. Med. Chem. Lett. 2011;21:6945–6949. doi: 10.1016/j.bmcl.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 62.Fortt R., Smith G., Awais R.O., Luthra S.K., Aboagye E.O. Automated GMP synthesis of [18F]ICMT-11 for in vivo imaging of caspase-3 activity. Nucl. Med. Biol. 2012;39:1000–1005. doi: 10.1016/j.nucmedbio.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 63.Ackermann U., O’Keefe G., Lee S.-T., Rigopoulos A., Cartwright G., Sachinidis J.I., Scott A.M., Tochon-Danguy H.J. Synthesis of a [18F]fluoroethyltriazolylthymidine radiotracer from [18F]2-fluoroethyl azide and 5-ethynyl-20-deoxyuridine. J. Labelled Compd. Radiopharm. 2011;54:260–266. doi: 10.1002/jlcr.1863. [DOI] [Google Scholar]
- 64.Smith G., Sala R., Carroll L., Behan K., Glaser M., Robins E., Nguyen Q.-D., Aboagye E.O. Synthesis and evaluation of nucleoside radiotracers for imaging proliferation. Nucl. Med. Biol. 2012;39:652–665. doi: 10.1016/j.nucmedbio.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 65.Pisaneschi F., Nguyen Q.-D., Shamsaei E., Glaser M., Robins E., Kaliszczak M., Smith G., Spivey A.C., Aboagye E.O. Development of a new epidermal growth factor receptor positron emission tomography imaging agent based on the 3-cyanoquinoline core: Synthesis and biological evaluation. Bioorg. Med. Chem. 2010;18:6634–6645. doi: 10.1016/j.bmc.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 66.Iddon L., Leyton J., Indrevoll B., Glaser M., Robins E.G., George A.J.T., Cuthbertson A., Luthra S.K., Aboagye E.O. Synthesis and in vitro evaluation of [18F]fluoroethyl triazole labelled [Tyr3]octreotate analogues using click chemistry. Bioorg. Med. Chem. Lett. 2011;21:3122–3127. doi: 10.1016/j.bmcl.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 67.Laurens E., Yeoh S.D., Rigopoulos A., Cao D., Cartwright G.A., O’Keefe G.J., Tochon-Danguy H.J., White J.M., Scott A.M., Ackermann U. Radiolabelling and evaluation of novel haloethylsulfoxides as PET imaging agents for tumor hypoxia. Nucl. Med. Biol. 2012;39:871–882. doi: 10.1016/j.nucmedbio.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 68.Bejot R., Carroll L., Bhakoo K., Declerck J., Gouverneur V. A fluorous and click approach for screening potential PET probes: Evaluation of potential hypoxia biomarkers. Bioorg. Med. Chem. 2012;20:324–329. doi: 10.1016/j.bmc.2011.10.084. [DOI] [PubMed] [Google Scholar]
- 69.Glaser M., Solbakken M., Turton D.R., Pettitt R., Barnett J., Arukwe J., Karlsen H., Cuthbertson A., Luthra S.K., Årstad E. Methods for 18F-labeling of RGD peptides: Comparison of aminooxy [18F]fluorobenzaldehyde condensation with ‘click labeling’ using 2-[18F]fluoroethylazide, and S-alkylation with [18F]fluoropropanethiol. Amino Acids. 2009;37:717–724. doi: 10.1007/s00726-008-0200-0. [DOI] [PubMed] [Google Scholar]
- 70.Li J., Shi L., Jia L., Jiang D., Zhou W., Hu W., Qi Y., Zhang L. Radiolabeling of RGD peptide and preliminary biological evaluation in mice bearing U87MG tumors. Bioorg. Med. Chem. 2012;20:3850–3855. doi: 10.1016/j.bmc.2012.04.037. [DOI] [PubMed] [Google Scholar]
- 71.Bejot R., Goggi J., Moonshi S.S., Robins E.G. A practical synthesis of [18F]FtRGD: An angiogenesis biomarker for PET. J. Labelled Compd. Radiopharm. 2013;56:42–49. doi: 10.1002/jlcr.3019. [DOI] [PubMed] [Google Scholar]
- 72.McConathy J., Zhou D., Shockley S.E., Jones L.A., Griffin E.A., Lee H., Adams S.J., Mach R.H. Click synthesis and biologic evaluation of (R)- and (S)-2-amino-3-[1-(2-[18F]fluoroethyl)-1H-[1,2,3]Triazol-4-yl]propanoic acid for brain tumor imaging with positron emission tomography. Mol. Imaging. 2010;9:329–342. [PubMed] [Google Scholar]
- 73.Sai K.K.S., Huang C., Yuan L., Zhou D., Piwnica-Worms D., Garbow J.R., Engelbach J.A., Mach R.H., Rich K.M., McConathy J. 18F-AFETP, 18F-FET, and 18F-FDG imaging of mouse DBT gliomas. J. Nucl. Med. 2013;54:1120–1126. doi: 10.2967/jnumed.112.113217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Galante E., Schoultz B.W., Koepp M., Årstad E. Chelator-accelerated one-pot ‘click’ labeling of small molecule tracers with 2-[18F]fluoroethyl azide. Molecules. 2013;18:5335–5347. doi: 10.3390/molecules18055335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jia L., Cheng Z., Shi L., Li J., Wang C., Jiang D., Zhou W., Meng H., Qi Y., Cheng D., et al. Fluorine-18 labeling by click chemistry: Multiple probes in one pot. Appl. Radiat. Isot. 2013;75:64–70. doi: 10.1016/j.apradiso.2013.01.033. [DOI] [PubMed] [Google Scholar]
- 76.Glaser M., Årstad E., Gaeta A., Nairne J., Trigg W., Robins E.G. Copper-mediated reduction of 2-[18F]fluoroethyl azide to 2-[18F]fluoroethylamine. J. Labelled Compd. Radiopharm. 2012;55:326–331. doi: 10.1002/jlcr.2945. [DOI] [Google Scholar]
- 77.Zhou D., Chu W., Dence C.S., Mach R.H., Welch M.J. Highly efficient click labeling using 2-[18F]fluoroethyl azide and synthesis of an 18F-N-hydroxysuccinimide ester as conjugation agent. Nucl. Med. Biol. 2012;39:1175–1181. doi: 10.1016/j.nucmedbio.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pretze M., Mamat C. Automated preparation of [18F]AFP and [18F]BFP: Two novel bifunctional 18F-labeling building blocks for Huisgen—Click. J. Fluorine Chem. 2013;150:25–35. doi: 10.1016/j.jfluchem.2013.02.028. [DOI] [Google Scholar]
- 79.Pretze M., Kuchar M., Bergmann R., Steinbach J., Pietzsch J., Mamat C. A bioorthogonal strategy using CuAAC-click for radiofluorinations of SNEW peptides and the role of copper-depletion. ChemMedChem. 2013;8:935–945. doi: 10.1002/cmdc.201300053. [DOI] [PubMed] [Google Scholar]
- 80.Juran S., Walther M., Stephan H., Bergmann R., Steinbach J., Kraus W., Emmerling F., Comba P. Hexadentate bispidine derivatives as versatile bifunctional chelate agents for copper(II) radioisotopes. Bioconjugate Chem. 2009;20:347–359. doi: 10.1021/bc800461e. [DOI] [PubMed] [Google Scholar]
- 81.Ramenda T., Bergmann R., Wuest F. Synthesis of 18F-labeled neurotensin(8-13) via copper-mediated 1,3-dipolar [3+2]cycloaddition reaction. Lett. Drug Des. Discov. 2007;4:279–285. doi: 10.2174/157018007784619998. [DOI] [Google Scholar]
- 82.Ramenda T., Kniess T., Bergmann R., Steinbach J., Wuest F. Radiolabelling of proteins with fluorine-18 via click chemistry. Chem. Commun. 2009:7521–7523. doi: 10.1039/b916075b. [DOI] [PubMed] [Google Scholar]
- 83.Ramenda T., Steinbach J., Wuest F. 4-[18F]Fluoro-N-methyl-N-(propyl-2-yn-1-yl)benzene-sulfonamide ([18F]F-SA): A versatile building block for labeling of peptides, proteins and oligonucleotides with fluorine-18 via Cu(I)-mediated click chemistry. Amino Acids. 2013;44:1167–1180. doi: 10.1007/s00726-012-1450-4. [DOI] [PubMed] [Google Scholar]
- 84.Inkster J.A.H., Guérin B., Ruth T.J., Adam M.J. Radiosynthesis and bioconjugation of [18F]FPy5yne, a prosthetic group for the 18F labeling of bioactive peptides. J. Labelled Compd. Radiopharm. 2008;51:444–452. doi: 10.1002/jlcr.1561. [DOI] [Google Scholar]
- 85.Inkster J.A.H., Adam M.J., Storr T., Ruth T.J. Labeling of an antisense oligonucleotide with [18F]FPy5yne. Nucleos. Nucleot. Nucl. 2009;28:1131–1143. doi: 10.1080/15257770903400691. [DOI] [PubMed] [Google Scholar]
- 86.Valdivia A.C., Estrada M., Hadizad T., Stewart D.J., Beanlands R.S., DaSilva J.N. A fast, simple, and reproducible automated synthesis of [18F]FPyKYNE-c(RGDyK) for αvβ3 receptor positron emission tomography imaging. J. Labelled Compd. Radiopharm. 2012;55:57–60. doi: 10.1002/jlcr.1948. [DOI] [Google Scholar]
- 87.Daumar P., Wanger-Baumann C.A., Pillarsetty N.V.K., Fabrizio L., Carlin S.D., Andreev O.A., Reshetnyak Y.K., Lewis J.S. Efficient 18F-labeling of large 37-amino-acid pHLIP peptide analogues and their biological evaluation. Bioconjugate Chem. 2012;23:1557–1566. doi: 10.1021/bc3000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thonon D., Kech C., Paris J., Lemaire C., Luxen A. New strategy for the preparation of clickable peptides and labeling with 1-(azidomethyl)-4-[18F]-fluorobenzene for PET. Bioconjugate Chem. 2009;20:817–823. doi: 10.1021/bc800544p. [DOI] [PubMed] [Google Scholar]
- 89.Chun J.-H., Pike V.W. Single-step radiosynthesis of “18F-labeled click synthons” from azide-functionalized diaryliodonium salts. Eur. J. Org. Chem. 2012:4541–4547. doi: 10.1002/ejoc.201200695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chun J.-H., Lu S.Y., Lee Y.-S., Pike V.W. Fast and high-yield microreactor syntheses of ortho-substituted [18F]fluoroarenes from reactions of [18F]fluoride ion with diaryliodonium salts. J. Org. Chem. 2010;75:3332–3338. doi: 10.1021/jo100361d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mercier F., Paris J., Kaisin G., Thonon D., Flagothier J., Teller N., Lemaire C., Luxen A. General method for labeling siRNA by click chemistry with fluorine-18 for the purpose of PET imaging. Bioconjugate Chem. 2011;22:108–114. doi: 10.1021/bc100263y. [DOI] [PubMed] [Google Scholar]
- 92.Flagothier J., Kaisin G., Mercier F., Thonon D., Teller N., Wouters J., Luxen A. Synthesis of two new alkyne-bearing linkers used for the preparation of siRNA for labeling by click chemistry with fluorine-18. Appl. Radiat. Isot. 2012;70:1549–1557. doi: 10.1016/j.apradiso.2012.04.022. [DOI] [PubMed] [Google Scholar]
- 93.Vaidyanathan G., White B.J., Zalutsky M.R. Propargyl 4-[18F]fluorobenzoate: A putatively more stable prosthetic group for the fluorine-18 labeling of biomolecules via click chemistry. Curr. Radiopharm. 2009;2:63–74. doi: 10.2174/1874471010902010063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li Y., Liu Y., Zhang L., Xu Y. One-step radiosynthesis of 4-[18F]flouro-3-nitro-N-2-propyn-1-yl-benzamide ([18F]FNPB): A new stable aromatic porosthetic group for efficient labeling of peptides with fluorine-18. J. Labelled Compd. Radiopharm. 2012;55:229–234. doi: 10.1002/jlcr.2931. [DOI] [Google Scholar]
- 95.Li Y., Guo J., Tang S., Lang L., Chen X., Perrin D.M. One-step and one-pot-two-step radiosynthesis of cyclo-RGD-18F-aryltrifluoroborate conjugates for functional imaging. Am. J. Nucl. Med. Mol. Imaging. 2013;3:44–56. [PMC free article] [PubMed] [Google Scholar]
- 96.Li Y., Liu Z., Harwig C.W., Pourghiasian M., Lau J., Lin K.-S., Schaffer P., Benard F., Perrin D.M. 18F-click labeling of a bombesin antagonist with an alkyne-18F-ArBF3−: In vivo PET imaging of tumors expressing the GRP-receptor. Am. J. Nucl. Med. Mol. Imaging. 2013;3:57–70. [PMC free article] [PubMed] [Google Scholar]
- 97.Liu Z., Li Y., Lozada J., Schaffer P., Adam M.J., Ruth T.J., Perrin D.M. Stoichiometric leverage: Rapid 18F-aryltrifluoroborate radiosynthesis at high specific activity for click conjugation. Angew. Chem. Int. Ed. 2013;53:2303–2307. doi: 10.1002/anie.201208551. [DOI] [PubMed] [Google Scholar]
- 98.Maschauer S., Prante O. A series of 2-O-trifluoromethylsulfonyl-d-mannopyranosides as precursors for concomitant 18F-labeling and glycosylation by click chemistry. Carbohydr. Res. 2009;344:753–761. doi: 10.1016/j.carres.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 99.Maschauer S., Einsiedel J., Haubner R., Hocke C., Ocker M., Hübner H., Kuwert T., Gmeiner P., Prante O. Labeling and glycosylation of peptides using click chemistry: A general approach to 18F-glycopeptides as effective imaging probes for positron emission tomography. Angew. Chem. Int. Ed. 2010;49:976–979. doi: 10.1002/anie.200904137. [DOI] [PubMed] [Google Scholar]
- 100.Fischer C.R., Müller C., Reber J., Müller A., Krämer S.D., Ametamey S.M., Schibli R. [18F]fluoro-deoxy-glucose folate: A novel PET radiotracer with improved in vivo properties for folate receptor targeting. Bioconjugate Chem. 2012;23:805–813. doi: 10.1021/bc200660z. [DOI] [PubMed] [Google Scholar]
- 101.Blomquist A.T., Liu L.H. Many-membered carbon rings.VII. Cycloöctyne. J. Am. Chem. Soc. 1953;75:2153–2154. doi: 10.1021/ja01105a039. [DOI] [Google Scholar]
- 102.Wittig G., Krebs A. Zur Existenz niedergliedriger Cycloalkine 1. Chem. Ber. 1961;94:3260–3275. doi: 10.1002/cber.19610941213. [DOI] [Google Scholar]
- 103.Agard N.J., Prescher J.A., Bertozzi C.R. A strain-promoted [3+2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 104.Chen K., Wang X., Lin W.Y., Shen C.K.F., Yap L.P., Hughes L.D., Conti P.S. Strain promoted catalyst free click chemistry for rapid construction of 64Cu labeled PET imaging probe. ACS Med. Chem. Lett. 2012;3:1019–1023. doi: 10.1021/ml300236m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bouvet V., Wuest M., Wuest F. Copper-free click chemistry with the short-lived positron emitter fluorine-18. Org. Biomol. Chem. 2011;9:7393–7399. doi: 10.1039/c1ob06034a. [DOI] [PubMed] [Google Scholar]
- 106.Carpenter R.D., Hausner S.H., Sutcliffe J.L. Copper-free click for PET: Rapid 1,3-dipolar cycloadditions with a fluorine-18 cyclooctyne. ACS Med. Chem. Lett. 2011;2:885–889. doi: 10.1021/ml200187j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kuzmin A., Poloukhtine A., Wolfert M.A., Popik V.V. Surface functionalization using catalyst-free azide-alkyne cycloaddition. Bioconjugate Chem. 2010;21:2076–2085. doi: 10.1021/bc100306u. [DOI] [PubMed] [Google Scholar]
- 108.Hausner S.H., Abbey C.K., Bold R.J., Gagnon M.K., Marik J., Marshall J.F., Stanecki C.E., Sutcliffe J. Targeted in vivo imaging of integrin αvβ6 with an improved radiotracer and its relevance in a pancreatic tumor model. Cancer Res. 2009;69:5843–5850. doi: 10.1158/0008-5472.CAN-08-4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang Z.-Y., Xu K.-S., Wang J.-S., Yang G.-Y., Wang W., Wang J.-Y., Niu W.-B., Liu E.-Y., Mi Y.-T., Niu J. Integrin αvβ6 acts as a prognostic indicator in gastric carcinoma. Clin. Oncol. 2008;20:61–66. doi: 10.1016/j.clon.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 110.Bandyopadhyay A., Raghavan S. Defining the role of integrin αvβ6 in cancer. Curr. Drug Targets. 2009;10:645–652. doi: 10.2174/138945009788680374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Arumugam S., Chin J., Schirrmacher R., Popik V.V., Kostikov A.P. [18F]Azadibenzo-cyclooctyne ([18F]ADIBO): A biocompatible radioactive labeling synthon for peptides using catalyst free [3+2] cycloaddition. Bioorg. Med. Chem. Lett. 2011;21:6987–6991. doi: 10.1016/j.bmcl.2011.09.126. [DOI] [PubMed] [Google Scholar]
- 112.Campbell-Verduyn L.S., Mirfeizi L., Schoonen A.K., Dierckx R.A., Elsinga P.H., Feringa B.L. Strain-promoted copper-free “click” chemistry for 18F-radiolabeling of bombesin. Angew. Chem. Int. Ed. 2011;50:11117–11120. doi: 10.1002/anie.201105547. [DOI] [PubMed] [Google Scholar]
- 113.Evans H.L., Slade R.L., Carroll L., Smith G., Nguyen Q.-D., Iddon L., Kamaly N., Stöckmann H., Leeper F.J., Aboagye E.O., Spivey A.C. Copper-free click – a promising tool for pre-targeted PET imaging. Chem. Commun. 2012;48:991–993. doi: 10.1039/c1cc16220a. [DOI] [PubMed] [Google Scholar]
- 114.Sachin K., Jadhav V.H., Kim E.M., Kim H.L., Lee S.B., Jeong H.J., Lim S.T., Sohn M.H., Kim D.W. F-18 labeling protocol of peptides based on chemically orthogonal strain-promoted cycloaddition under physiologically friendly reaction conditions. Bioconjugate Chem. 2012;23:1680–1686. doi: 10.1021/bc3002425. [DOI] [PubMed] [Google Scholar]
- 115.Staudinger H., Meyer J. Über neue organische phosphorverbindungen III. Phosphinmethylenderivate und phosphinimine. Helv. Chim. Acta. 1919;2:635–646. doi: 10.1002/hlca.19190020164. [DOI] [Google Scholar]
- 116.Staudinger H., Hauser E. Über neue organische phosphorverbindungen IV phosphinimine. Helv. Chim. Acta. 1921;4:861–886. doi: 10.1002/hlca.19210040192. [DOI] [Google Scholar]
- 117.Köhn M., Breinbauer R. The staudinger ligation–A gift to chemical biology. Angew. Chem. Int. Ed. 2004;43:3106–3116. doi: 10.1002/anie.200401744. [DOI] [PubMed] [Google Scholar]
- 118.Langenhan J.M., Thorson J.S. Recent carbohydrate-based chemoselective ligation applications. Curr. Org. Synth. 2005;2:59–81. doi: 10.2174/1570179052996937. [DOI] [Google Scholar]
- 119.Hackenberger C.P.R., Schwarzer D. Chemoselektive ligations- und modifikationsstrategien für peptide und proteine. Angew. Chem. 2008;120:10182–10228. doi: 10.1002/ange.200801313. [DOI] [Google Scholar]
- 120.Sletten E.M., Bertozzi C.R. Bioorthogonale chemie-Oder: In einem meer aus funktionalität nach selektivität fischen. Angew. Chem. 2009;121:7108–7133. doi: 10.1002/ange.200900942. [DOI] [Google Scholar]
- 121.Best M.D. Click chemistry and bioorthogonal reactions: Unprecedented selectivity in the labeling of biological molecules. Biochemistry. 2009;48:6571–6584. doi: 10.1021/bi9007726. [DOI] [PubMed] [Google Scholar]
- 122.Kalia J., Raines R.T. Advances in bioconjugation. Curr. Org. Chem. 2010;14:138–147. doi: 10.2174/138527210790069839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schilling C.I., Jung N., Biskup M., Schäpers U., Bräse S. Bioconjugation via azide–Staudinger ligation: An overview. Chem. Soc. Rev. 2011;40:4840–4871. doi: 10.1039/c0cs00123f. [DOI] [PubMed] [Google Scholar]
- 124.Van Berkel S.S., van Eldijk M.B., van Hest J.C.M. Staudinger ligation as a method for bioconjugation. Angew. Chem. Int. Ed. 2011;50:8806–8827. doi: 10.1002/anie.201008102. [DOI] [PubMed] [Google Scholar]
- 125.Sletten E.M., Bertozzi C.R. From mechanism to mouse: A tale of two bioorthogonal reactions. Acc. Chem. Res. 2011;44:666–676. doi: 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Saxon E., Bertozzi C.R. Cell surface engineering by a modified staudinger reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 127.Nilsson B.L., Kiessling L.L., Raines R.T. Staudinger ligation: A peptide from a thioester and azide. Org. Lett. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]
- 128.Saxon E., Armstrong J.I., Bertozzi C.R. A “traceless” staudinger ligation for the chemoselective synthesis of amide bonds. Org. Lett. 2000;2:2141–2143. doi: 10.1021/ol006054v. [DOI] [PubMed] [Google Scholar]
- 129.Nilsson B.L., Kiessling L.L., Raines R.T. High-yielding staudinger ligation of a phosphinothioester and azide to form a peptide. Org. Lett. 2001;3:9–12. doi: 10.1021/ol006739v. [DOI] [PubMed] [Google Scholar]
- 130.Liu L., Hong Z.-Y., Wong C.-H. Convergent glycopeptide synthesis by traceless staudinger ligation and enzymatic coupling. ChemBioChem. 2006;7:429–432. doi: 10.1002/cbic.200500437. [DOI] [PubMed] [Google Scholar]
- 131.Robillard M.S., Gruell H. Compounds, kits and methods for use in medical. WO2006/038184A2 Patent.
- 132.Mamat C., Pretze M., Steinbach J., Wüst F. Radiofluorination via staudinger ligation. J. Labelled Compd. Radiopharm. 2009;52:S142. [Google Scholar]
- 133.Mamat C., Flemming A., Köckerling M., Steinbach J., Wuest F.R. Synthesis of benzoate-functionalized phosphanes as novel building blocks for the traceless staudinger ligation. Synthesis. 2009:3311–3321. [Google Scholar]
- 134.Mamat C., Franke M., Peppel T., Köckerling M., Steinbach J. Synthesis, structure determination, and (radio-)fluorination of novel functionalized phosphanes suitable for the traceless Staudinger ligation. Tetrahedron. 2011;67:4521–4529. doi: 10.1016/j.tet.2011.04.091. [DOI] [Google Scholar]
- 135.Pretze M., Wuest F., Peppel T., Köckerling M., Mamat C. The traceless Staudinger ligation with fluorine-18: A novel and versatile labeling technique for the synthesis of PET-radiotracers. Tetrahedron Lett. 2010;51:6410–6414. doi: 10.1016/j.tetlet.2010.09.134. [DOI] [Google Scholar]
- 136.Pretze M., Flemming A., Köckerling M., Mamat C. Synthesis and radiofluorination of iodophenyl esters as tool for the traceless staudinger ligation. Z. Naturforsch. B. 2010;65b:1128–1136. [Google Scholar]
- 137.Gaeta A., Woodcraft J., Plant S., Goggi J., Jones P., Battle M., Trigg W., Luthra S.K, Glaser M. Use of 2-[18F]fluoroethylazide for the Staudinger ligation—Preparation and characterisation of GABAA receptor binding 4-quinolones. Bioorg. Med. Chem. Lett. 2010;20:4649–4652. doi: 10.1016/j.bmcl.2010.05.106. [DOI] [PubMed] [Google Scholar]
- 138.Carroll L., Boldon S., Bejot R., Moore J.E., Declerck J., Gouverneur V. The traceless Staudinger ligation for indirect 18F-radiolabelling. Org. Biomol. Chem. 2011;9:136–140. doi: 10.1039/c0ob00564a. [DOI] [PubMed] [Google Scholar]
- 139.Bejot R., Fowler T., Carroll L., Boldon S., Moore J.E., Declerck J., Gouverneur V. Fluorous synthesis of 18Fradiotracers with the [18F]fluoride ion: Nucleophilic fluorination as the detagging process. Angew. Chem. 2009;121:594–597. doi: 10.1002/ange.200803897. [DOI] [PubMed] [Google Scholar]
- 140.Boldon S., Moore J.E., Gouverneur V. Fluorous synthesis of allylic fluorides: C–F bond formation as the detagging process. Chem. Commun. 2008:3622–3624. doi: 10.1039/b804484h. [DOI] [PubMed] [Google Scholar]
- 141.Blackman M.L., Royzen M., Fox J.M. Tetrazine ligation: Fast bioconjugation based on inverse-electron-demand diels−alder reactivity. J. Am. Chem. Soc. 2008;130:13518–13519. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bach R.D. Ring strain energy in the cyclooctyl system. The effect of strain energy on [3+2] cycloaddition reactions with azides. J. Am. Chem. Soc. 2009;131:5233–5243. doi: 10.1021/ja8094137. [DOI] [PubMed] [Google Scholar]
- 143.Devaraj N.K., Weissleder R., Hilderbrand S.A. Tetrazine-based cycloadditions: Application to pretargeted live cell imaging. Bioconjugate Chem. 2008;19:2297–2299. doi: 10.1021/bc8004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Devaraj N.K., Upadhyay R., Haun J.B., Hilderbrand S.A., Weissleder R. Fast and sensitive pretargeted labeling of cancer cells through a tetrazine/trans-cyclooctene cycloaddition. Angew. Chem. Int. Ed. 2009;48:7013–7016. doi: 10.1002/anie.200903233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Devaraj N.K., Hilderbrand S., Upadhyay R., Mazitschek R., Weissleder R. Bioorthogonal turn-on probes for imaging small molecules inside living cells. Angew. Chem. Int. Ed. 2010;49:2869–2872. doi: 10.1002/anie.200906120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Barker I.A., Hall D.J., Hansell C.F., Du Prez F.E., O’Reilly R.K., Dove A.P. Tetrazine-norbornene click reactions to functionalize degradable polymers derived from lactide. Macromol. Rapid Commun. 2011;32:1362–1366. doi: 10.1002/marc.201100324. [DOI] [PubMed] [Google Scholar]
- 147.Hansell C.F., Espeel P., Stamenović M.M., Barker I.A., Dove A.P., Du Prez F.E., O’Reilly R.K. Additive-free clicking for polymer functionalization and coupling by tetrazine–norbornene chemistry. J. Am. Chem. Soc. 2011;133:13828–13831. doi: 10.1021/ja203957h. [DOI] [PubMed] [Google Scholar]
- 148.Rossin R., Verkerk P.R., van den Bosch S.M., Vulders R.C.M., Verel I., Lub J., Robillard M.S. In vivo chemistry for pretargeted tumor imaging in live mice. Angew. Chem. 2010;122:3447–3450. doi: 10.1002/anie.200906294. [DOI] [PubMed] [Google Scholar]
- 149.Zeglis B.M., Mohindra P., Weissmann G.I., Divilov V., Hilderbrand S.A., Weissleder R., Lewis J.S. Modular strategy for the construction of radiometalated antibodies for positron emission tomography based on inverse electron demand diels–alder click chemistry. Bioconjugate Chem. 2011;22:2048–2059. doi: 10.1021/bc200288d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li Z., Cai H., Hassink M., Blackman M.L., Brown R.C.D., Conti P.S., Fox J.M. Tetrazine–trans-cyclooctene ligation for the rapid construction of 18F labeled probes. Chem Commun. 2010;46:8043–8045. doi: 10.1039/c0cc03078c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Selvaraj R., Liu S., Hassink M., Huang C.-W., Yap L.-P., Park R., Fox J.M., Li Z., Conti P.S. Tetrazine-trans-cyclooctene ligation for the rapid construction of integrin αvβ3 targeted PET tracer based on a cyclic RGD peptide. Bioorg. Med. Chem. Lett. 2011;21:5011–5014. doi: 10.1016/j.bmcl.2011.04.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Liu S., Hassink M., Selvaraj R., Yap L.-P., Park R., Wang H., Chen X., Fox J.M., Li Z., Conti P.S. Efficient 18F labeling of cysteine-containing peptides and proteins using tetrazine–trans-cyclooctene ligation. Mol. Imaging. 2013;12:1536–0121. [PMC free article] [PubMed] [Google Scholar]
- 153.Reiner T., Earley S., Turetsky A., Weissleder R. Bioorthogonal small-molecule ligands for PARP1 imaging in living cells. ChemBioChem. 2010;11:2374–2377. doi: 10.1002/cbic.201000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Keliher E.J., Reiner T., Turetsky A., Hilderbrand S.A., Weissleder R. High-yielding, two-step 18F labeling strategy for 18F-PARP1 inhibitors. ChemMedChem. 2011;6:424–427. doi: 10.1002/cmdc.201000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Devaraj N.K., Thurber G.M., Keliher E.J., Marinelli B., Weissleder R. Reactive polymer enables efficient in vivo bioorthogonal chemistry. Proc. Natl. Acad. Sci. USA. 2012;109:4762–4767. doi: 10.1073/pnas.1113466109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Reiner T., Keliher E.J., Earley S., Marinelli B., Weissleder R. Synthesis and in vivo imaging of a 18F-labeled PARP1 inhibitor using a chemically orthogonal scavenger-assisted high-performance method. Angew. Chem. 2011;123:1963–1966. doi: 10.1002/ange.201006579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Keliher E.J., Reiner T., Thurber G.M., Upadhyay R., Weissleder R. Efficient 18F-labeling of synthetic exendin-4 analogues for imaging beta cells. Chem. Open. 2012;1:177–183. doi: 10.1002/open.201200014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zlatopolskiy B.D., Kandler R., Mottaghy F.M., Neumaier B. C-(4-[18F]fluorophenyl)-N-phenyl nitrone: A novel 18F-labeled building block for metal free [3+2]cycloaddition. Appl. Radiat. Isot. 2012;70:184–192. doi: 10.1016/j.apradiso.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 159.Jones R.C.F., Martin J.N. Nitrones. In: Padwa A., Pearson W.H., editors. The Chemistry of Heterocyclic Compounds: Synthetic Applications of 1.3-Dipolar Cycloaddition Chemistry toward Heterocycles and Natural Products. Volume 59. John Wiley and Sons; New York, NY, USA: 2002. pp. 1–81. [Google Scholar]
- 160.Moulay S., Touati A. Cycloaddition reactions in aqueous systems: A two-decade trend endeavour. Compt. Rend. Chim. 2010;13:1474–1511. doi: 10.1016/j.crci.2010.05.025. [DOI] [Google Scholar]
- 161.Butler R.N., Coyne A.G. Nature’s reaction enforcer-comparative effects for organic synthesis “in-water” and “on-water”. Chem. Rev. 2010;110:6302–6337. doi: 10.1021/cr100162c. [DOI] [PubMed] [Google Scholar]
- 162.Zlatopolskiy B.D., Kandler R., Kobus D., Mottaghy F.M., Neumaier B. Beyond azide–alkyne click reaction: Easy access to 18F-labelled compounds via nitrile oxide cycloadditions. Chem. Commun. 2012;48:7134–7136. doi: 10.1039/c2cc31335a. [DOI] [PubMed] [Google Scholar]
- 163.Kapty J., Kniess T., Wuest F., Mercer J.R. Radiolabeling of phosphatidylserine-binding peptides with prosthetic groups N-[6-(4-[18F]fluorobenzylidene)aminooxyhexyl]maleimide ([18F]FBAM) and N-succinimidyl-4-[18F]fluorobenzoate ([18F]SFB) Appl. Radiat. Isot. 2011;69:1218–1225. doi: 10.1016/j.apradiso.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 164.Kniess T., Kuchar M., Pietzsch J. Automated radiosynthesis of the thiol-reactive labeling agent N-[6-(4-[18F]fluorobenzylidene)aminooxyhexyl]maleimide ([18F]FBAM) Appl. Radiat. Isot. 2011;69:1226–1230. doi: 10.1016/j.apradiso.2011.03.043. [DOI] [PubMed] [Google Scholar]