

Abstract
A series of thirteen C-3 functionalized isobenzofuran-1(3H)-ones (phtalides) was synthesized via condensation, aromatization, and acetylation reactions. NMR (one and two dimensional experiments), IR, and mass spectrometry analysis allowed confirmation of the identity of the synthesized compounds. The substances were submitted to in vitro bioassays against U937 (lymphoma) and K562 (myeloid leukemia) cancer cell lines using the MTT cytotoxicity assay. Some derivatives inhibited 90% of cell viability at 100 µM. Also, two phtalides presented biological activity superior than that of etoposide (VP16), a commercial drug used as a positive control in the assays. In silico drug properties of the evaluated compounds were calculated and the results are discussed.
Keywords: isobenzofuran-1(3H)-one, phtalides, antiproliferative activity
1. Introduction
Several compounds possessing a γ-lactone moiety fused to an aromatic ring, known as isobenzofuran-1(3H)-ones (phtalides), exhibit many biological activities: antioxidant, presented by compounds 1 and 2 [1]; antifungal, displayed by isopestacin (3) [2]; anti-platelet observed with the brominated derivative 4 [3]; and anticonvulsant activity related to compound 5 [4] (Figure 1). The phytotoxic compound 6 [5], the photosynthetic inhibitor 7 [6] and the cytotoxic substances 8 and 9 are other examples of bioactive phtalides. As can be seen in Figure 1, various phtalides are functionalized at C-3 position (see structure 1 for numbering).
Figure 1.

Structures of isobenzofuran-1(3H)-ones 1–9.
The isobenzofuranones 8 and 9 were prepared from anarchadic acids, the major natural phenolic lipid in cashew (Anacardium occidentale) nut-shells [7]. Cytotoxicity screening of compounds 8 and 9 against different human cancer cell lines (HL-60 leukemia, SF295 glioblastoma and MDA-MB435 melanoma) by the MTT assay showed that they are active. Compound 8 exhibited low activity against HL-60 (IC50 21.00 µg/mL) and SF295 (IC50 > 25 µg/mL) cells, and moderate activity against MDA-MB435 cells (IC50 12.17 µg/mL), while compound 9 exhibited a significant cytotoxic effect against HL-60 cells (IC50 3.24 µg/mL), and moderate activity on SF295 (IC50 10.09 µg/mL) and MDA-MB435 (IC50 8.70 µg/mL). The two compounds exhibited weak to moderate activities against PBMC cells (8, IC50 > 25 µg/mL; 9, IC50 > 14.51 µg/mL).
A natural extension of this investigation would be the synthesis and cytotoxic screening of isobenzofuranones structurally similar to compounds 8 and 9 but containing alicyclic and aromatic functionalities attached to C-3 position. Considering the aforementioned cytotoxic effects of 8 and 9 and our interest in the development of bioactive compounds [8,9,10,11], we detail in this paper the synthesis and evaluation of the antiproliferative activity of a series of alicyclic and aromatic C-3 functionalized isobenzofuranones. In silico drug properties of the evaluated compounds were determined and the results are also discussed.
2. Results and Discussion
2.1. Preparation of Isobenzofuran-1(3H)-ones
The synthetic route used to prepare compounds 11–22, functionalized at C-3 position of the isobenzofuranone core, is outlined in Scheme 1.
Scheme 1.
Synthetic route involved in the preparation of compounds 11–22.

Reagents and Conditions: (i) DBU, CHCl3, reflux; (ii) DBU, CHCl3, r.t.; (iii) DBU, CH3CN, reflux; (iv) Hg(OAc)2, NaOAc, CH3COOH, reflux; (v) DMAP, Ac2O, CHCl3, r.t.
DBU-promoted condensation of commercially available phthalaldehydic acid (10) and different 1,3-dicarbonyl compounds afforded derivatives 11–13 and 20–22 in yields varying from 53 to 95% [12,13]. Phenolic compounds 14–16 were prepared via Hg(OAc)2 mediated aromatization [14]. Treatment of substances 14–16 with Ac2O/DMAP resulted in the formation of acetylated derivatives 17–19. The compounds were fully characterized by IR, NMR, and MS analysis. High resolution mass spectrometry confirmed the molecular formula of the compounds. A combination of two dimensional NMR analyses (HSQC and HMBC) allowed complete hydrogen and carbon assignments. Taking compound 14 into consideration, some of the major long-range correlations (J2 and J3) observed in the HMBC contour plot are depicted in Scheme 2.
Scheme 2.
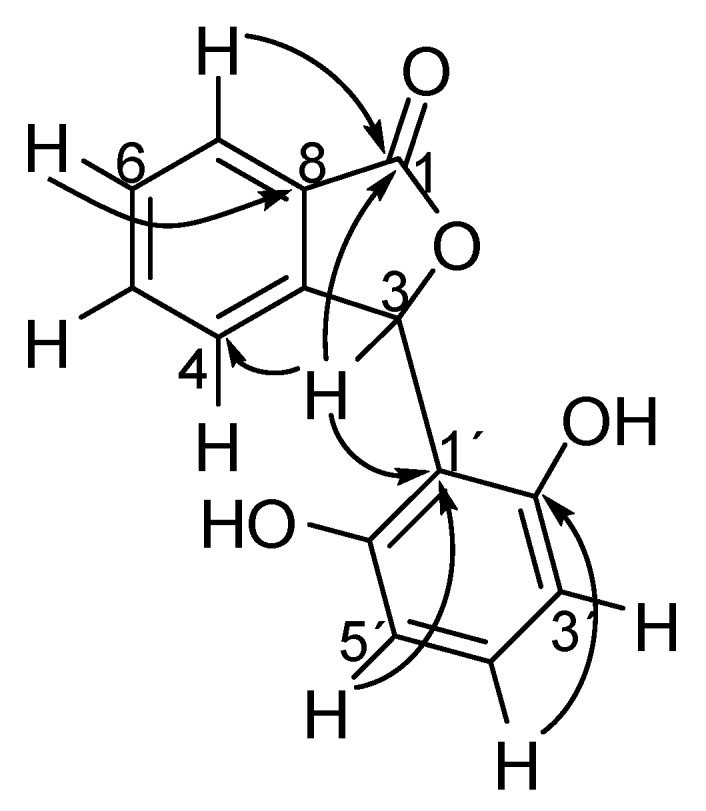
Some long range correlations found in the HMBC contour plot of compound 14.
2.2. Evaluation of Antiproliferative Activity
Isobenzofuran-1(3H)-ones 10–22 were biologically evaluated against U937 (lymphoma) and K562 (myeloid leukemia) cell lines in comparison to DMSO (1%) and etoposide (VP16), used as negative and positive controls, respectively. After 48 h of treatment, compounds 16–18 were the most effective in decreasing the viability of both the cell lines tested (Table 1 and Table 2). Compound 19 also inhibited proliferation, though only against K562 cells. Although the IC50 values obtained for compounds 16–18 suggest a moderate effect on U937 cells, a strong inhibitory activity was observed for compounds 16 and 18 against K562 cells (IC50 2.79 and 1.71 µM, respectively; Table 3 and Figure 2). More importantly, these inhibitory activities were even higher than those observed for etoposide (VP16) (IC50 7.06 µM for K562), an anticancer drug used in several chemotherapy regimes, including against leukemia.
Table 1.
Inhibitory effects (%) a of compounds 10–22 on K562 cell viability (mean ± SD; n = 4).
| Comp. | 1 µM | 50 µM | 100 µM |
|---|---|---|---|
| 10 | 44.16 ± 3.59 | 41.71 ± 1.01 | 26.71 ± 3.91 |
| 11 | 49.69 ± 0.68 | 45.85 ± 3.07 | 36.18 ± 1.00 |
| 12 | 31.42 ± 1.18 | 38.38 ± 3.04 | 38.54 ± 4.24 |
| 13 | 32.87 ± 3.20 | 23.93 ± 3.04 | 31.64 ± 2.93 |
| 14 | 37.71 ± 2.20 | 49.33 ± 4.70 | 56.82 ± 1.36 |
| 15 | 40.59 ± 2.47 | 44.32 ± 1.08 | 43.64 ± 3.85 |
| 16 | 20.33 ± 1.13 | 65.61 ± 2.42 | 77.73 ± 2.01 |
| 17 | 26.77 ± 0.99 | 56.10 ± 3.58 | 63.70 ± 0.21 |
| 18 | 37.88 ± 3.86 | 69.63 ± 0.92 | 80.05 ± 0.28 |
| 19 | 12.48 ± 3.57 | 54.56 ± 2.97 | 71.76 ± 3.73 |
| 20 | 39.87 ± 4.43 | 43.86 ± 1.92 | 49.38 ± 2.50 |
| 21 | 20.64 ± 1.91 | 18.60 ± 2.65 | 21.84 ± 3.39 |
| 22 | 46.11 ± 4.1 | 45.9 ± 3.39 | 37.61 ± 0.38 |
a inhibition (%) after 48 h of continuous treatment (n = 4), normalized against K562 cells treated with 1% DMSO (n = 4).
Table 2.
Inhibitory effects (%) a of compounds 10–22 on U937 cell viability (mean ± SD; n = 4).
| Comp. | 1 µM | 50 µM | 100 µM |
|---|---|---|---|
| 10 | 28.00 ± 2.75 | 30.73 ± 2.12 | 28.81 ± 2.05 |
| 11 | 32.34 ± 1.93 | 28.58 ± 1.62 | 29.43 ± 2.76 |
| 12 | 20.38 ± 3.04 | 27.01 ± 3.68 | 24.72 ± 1.48 |
| 13 | 26.75 ± 2.12 | 45.31 ± 1.91 | 39.10 ± 0.92 |
| 14 | 25.57 ± 2.50 | 31.10 ± 3.53 | 29.86 ± 1.70 |
| 15 | 35.03 ± 2.05 | 27.03 ± 0.61 | 22.37 ± 1.13 |
| 16 | 11.64 ± 0.28 | 58.86 ± 2.62 | 90.82 ± 0.43 |
| 17 | 33.87 ± 0.92 | 61.33 ± 1.98 | 83.91 ± 0.92 |
| 18 | 36.63 ± 1.98 | 67.68 ± 1.77 | 90.78 ± 0.21 |
| 19 | 23.14 ± 3.19 | 25.46 ± 3.13 | 23.12 ± 3.43 |
| 20 | 24.33 ± 0.47 | 30.98 ± 1.04 | 34.14 ± 0.49 |
| 21 | 5.72 ± 1.27 | 21.17 ± 1.48 | 29.29 ± 2.02 |
| 22 | 27.63 ± 0.92 | 31.85 ± 0.99 | 33.58 ± 2.71 |
a inhibition (%) after 48 h of continuous treatment (n = 4), normalized against U937 cells treated with 1% DMSO (n = 4).
Table 3.
IC50 values (µM) a obtained when K562 and U937 cells were treated with compounds 16–18 and positive control etoposide (VP16).
| Cell line | Comp. 16 | Compound 17 | Compound 18 | Etoposide (VP16) |
|---|---|---|---|---|
| K562 | 2.79 | 66.81 | 1.71 | 7.06 |
| U937 | 62.97 | 71.39 | 46.63 | 0.35 |
a IC50: compound concentration required to inhibit 50% of cell viability, determined after 48 h of continuous treatment (n = 4).
Figure 2.


Dose-response profiles of compounds 16–18 and positive control VP16 on viability of K562 (A) and U937 (B) cells. K562 and U937 cell lines were treated for 48 h with increasing amounts of the indicated substances. All the assays were performed in quadruplicates and the results were normalized considering the cultures treated with the vehicle alone (1% DMSO).
2.3. Structure-Activity Relationship and in Silico Calculations of Drug-Like Properties
Considering the therapeutic potential of the isobenzofuranone derivatives described so far, a computational study was carried out to predict the physical chemical properties of compounds 10–22. These parameters influence pharmacokinetic properties, such as absorption, distribution, metabolism, and excretion (ADME). A screening was conducted to evaluate whether the substances can fulfill the characteristics of candidate drugs, based on Lipinski’s Rule of Five [15] and other related criteria added later by Veber and co-workers [16]. These characteristics theoretically determine if a compound presents absorptivity on and permeability across membranes, properties that would likely make it an orally active drug in humans.
Molecular attributes analyzed were n-octanol/water partition coefficient (LogP ≤ 5), the amount of hydrogen bond donors (HBD ≤ 5), the amount of hydrogen bond acceptor (HBA ≤ 10), the molecular weight of the drugs (MW ≤ 500), number of rotatable bonds (nRotb < 10) and polar surface area (PSA < 140 Å2). Values in parentheses represent the ideal values according to Lipinski [15] and Veber [16]. Physicochemical parameters were calculated by using Osiris Property Explorer [17] and Molinspiration [18], good free computational tools which help to predict pharmacokinetic properties of candidate drugs used by several research groups [19,20,21,22].
The Osiris Property Explorer allows evaluation of chemical and physicochemical data that influence the pharmacokinetic properties of a drug. The Osiris calculations for toxicity and drug-likeness follow a fragment-based approach. The software can calculate lipophilicity, inferred from LogP value, solubility in water, expressed as LogS, molecular weight, drug-likeness indices and drug scores. Low hydrophilicity, and therefore high LogP values, may cause poor absorption or permeation. Also, Osiris software calculates various attributes of the drugs, such as toxicity risks (mutagenic, tumorigenic, irritant and reproductive effects), drug likeness, and drug score [23,24]. Calculation of LogP and molecular weight, as well as total polar surface area (TPSA), number of rotatable bonds (nRotB), number of hydrogen bond donor (HBD), number of hydrogen bond acceptor (HBA) and bioactivity scores [25] can also be determined in the Molinspiration package. Table 4 and Table 5 show the results of the calculations based on these computational packages.
Table 4.
Predicted drug-likeness properties and toxicity risks of compounds 10–22 and epotoside calculated by Osiris package.
| Comp. | Bioavailability and Drug Score | Lipinski’s violations | Toxicity risks a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cLogP | LogS | MW | HBD | HBA | Drug-likeness | Drug-Score | M | T | I | R | ||
| 10 | 0.64 | −1.68 | 150.0 | 1 | 3 | −4.65 | 0.49 | 0 | - | - | - | - |
| 11 | 1.78 | −2.71 | 244.0 | 1 | 4 | −6.63 | 0.46 | 0 | - | - | - | - |
| 12 | 1.98 | −2.87 | 258.0 | 1 | 4 | −3.61 | 0.46 | 0 | - | - | - | - |
| 13 | 2.78 | −3.30 | 286.0 | 1 | 4 | −4.20 | 0.43 | 0 | - | - | - | - |
| 14 | 2.55 | −2.66 | 242.0 | 2 | 4 | −4.26 | 0.46 | 0 | - | - | - | - |
| 15 | 2.87 | −3.01 | 256.0 | 2 | 4 | −5.25 | 0.44 | 0 | - | - | - | - |
| 16 | 3.65 | −3.53 | 284.0 | 2 | 4 | −6.33 | 0.39 | 0 | - | - | - | - |
| 17 | 3.10 | −3.54 | 326.0 | 2 | 6 | −6.34 | 0.40 | 0 | - | - | - | - |
| 18 | 3.41 | −3.88 | 340.0 | 2 | 6 | −7.52 | 0.38 | 0 | - | - | - | - |
| 19 | 4.19 | −4.41 | 368.0 | 2 | 6 | −8.59 | 0.32 | 0 | - | - | - | - |
| 20 | 2.25 | −3.05 | 272.0 | 1 | 4 | −3.58 | 0.45 | 0 | - | - | - | - |
| 21 | 1.29 | −2.42 | 230.0 | 0 | 4 | −3.95 | 0.48 | 0 | - | - | - | - |
| 22 | 2.32 | −4.04 | 278.0 | 0 | 4 | −4.12 | 0.41 | 0 | - | - | - | - |
| Etoposide | 0.53 | −3.95 | 588.0 | 3 | 13 | −0.28 | 0.39 | 2 | - | - | - | - |
cLogP, calculated lipophilicity; LogS, logarithm of aqueous solubility measured in mol.L−1; MW, molecular weight; HBD, number of hydrogen bond donor; HBA, number of hydrogen bond acceptor; M, mutagenic effect; T, tumorigenic effect; I, irritating effect; R, reproductive effect. a Ranked according to: (−), no bad effect; (±), medium bad effect; (+), bad effect.
Table 5.
Drug-likeness calculations of compounds 10–22 by using Molinspiration package.
| Comp. | Molinspiration calculations | Calculations of Bioactivity Scores a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| cLogP | TPSA b | NONI c | Volume | nRotB | GPCRL | ICM | KI | NRL | PI | EI | |
| 10 | 0.895 | 46.533 | 1 | 126.015 | 0 | −0.59 | −0.04 | −1.12 | −0.62 | −0.95 | 0.14 |
| 11 | 1.741 | 63.604 | 1 | 211.979 | 1 | −0.60 | −0.30 | −1.40 | −0.42 | −0.68 | −0.11 |
| 12 | 1.982 | 63.604 | 1 | 228.566 | 1 | −0.58 | −0.41 | −1.35 | −0.32 | −0.60 | −0.14 |
| 13 | 3.483 | 63.604 | 1 | 261.955 | 2 | −0.39 | −0.29 | −1.15 | −0.14 | −0.35 | −0.08 |
| 14 | 3.016 | 66.761 | 2 | 205.442 | 1 | −0.29 | −0.42 | −0.31 | −0.20 | −0.56 | −0.17 |
| 15 | 3.416 | 66.761 | 2 | 222.003 | 1 | −0.29 | −0.49 | −0.30 | −0.17 | −0.56 | −0.21 |
| 16 | 4.48 | 66.761 | 2 | 255.392 | 2 | −0.13 | −0.37 | −0.20 | 0.03 | −0.37 | −0.11 |
| 17 | 2.183 | 100.903 | 2 | 243.409 | 3 | −0.01 | −0.34 | −0.18 | −0.01 | −0.25 | −0.08 |
| 18 | 2.584 | 100.903 | 2 | 259.970 | 3 | −0.04 | −0.41 | −0.21 | −0.01 | −0.27 | −0.14 |
| 19 | 3.647 | 100.903 | 2 | 293.359 | 4 | 0.03 | −0.32 | −0.19 | 0.08 | −0.17 | −0.07 |
| 20 | 2.394 | 63.604 | 1 | 244.803 | 1 | −0.50 | −0.39 | −1.31 | −0.27 | −0.66 | −0.20 |
| 21 | 0.652 | 60.447 | 0 | 195.554 | 1 | −0.26 | −0.20 | −0.85 | −0.27 | −0.49 | 0.04 |
| 22 | 2.394 | 60.447 | 0 | 233.259 | 1 | −0.12 | −0.35 | −0.54 | −0.05 | −0.45 | 0.09 |
| d Etop. | 0.698 | 160.861 | 3 | 493.508 | 5 | 0.18 | −0.48 | −0.38 | −0.33 | 0.12 | 0.30 |
a GPCRL: GPCR ligand; ICM: Ion channel modulator; KI: Kinase inhibitor; NRL: Nuclear receptor ligand; PI: Protease inhibitor; EI: Enzyme inhibitor; b TPSA: Total polar surface area; c NONI: number OH-NH interaction; d Etop: Etoposide.
A parameter to determine good absorption of compounds is the value of cLogP. On this basis, for a reasonable probability of good absorption, LogP value must not be greater than 5.0. Notably, the cLogP values of phtalides 10–22 are greater than the etoposide value, suggesting that the hydrophobicity of isobenzofuranone derivatives is greater than that of the compound used as positive control in the biological assays. Normally, the drugs interacting with enzyme inside the human body have LogP values between 2 and 5 [26]. In this respect, most of the studied compounds present values within this range (Table 4 and Table 5).
Toxicity risk alerts are an indication that the structure of a compound may be harmful. Analysis of theoretical toxicity risks for these series using Osiris software revealed that all the compounds examined were non-mutagenic, non-tumorigenic, non-irritating and without effects on mammalian reproduction (Table 4). In addition, these compounds were evaluated as potential drugs through the drug-likeness calculations, showing negative values (between −8.59 and −3.58, Table 4), indicating that compounds 10–22 do not contain fragments that are frequently present in commercial drugs [23,24].
Aqueous solubility of a compound significantly affects its absorption and distribution characteristics. In general, it is desirable to obtain compounds which are soluble in a biological environment. A parameter to evaluate the solubility is LogS. More than 80% of the drugs in the market have estimated LogS values greater than −4 [19]. In the case of compounds 19 and 22, LogS values (−4.41 and −4.04, respectively) are lower, compared to others in the series (Table 4).
Drug score values can be used to judge the compound’s overall potential to qualify it for a drug. The values are the combination of drug likeness, toxicity risk, and some physicochemical parameters, such as cLogP, solubility, and molecular weight [23,24]. These values would be used to evaluate the potential of a candidate drug. As can be seen in Table 4, drug score values of compounds 10–22 are comparable or higher in relation to the commercial drug etoposide.
Hydrogen-bonding capacity (the number of HBA) was estimated by considering the number of nitrogen and oxygen atoms in the chemical structure. The number of HBD atoms was the sum of hydrogen atoms bound to oxygen and to nitrogen atoms [23]. The values of these parameters for compounds 10–22 are within the limit set by the Lipinski rules. This rule also suggests that two or more violations in a compound show probability of bio-availability problems [27]. All the compounds in Table 4 present zero violations of the rules, unlike epotoside, that has two rule violations.
In addition to LogP, total polar surface area (TPSA) is an important descriptor for prediction of drug transport properties, including intestinal absorption, Caco-2 monolayer permeability, and blood-brain barrier penetration [28,29]. This parameter was calculated using Molinspiration software based on the sum of the surface belonging to polar atoms (usually oxygens, nitrogens, and attached hydrogens) [25]. Compounds with TPSA ≥ 140 A2 are thought to have poor oral bioavailability and TPSA ≤ 61 A2 are thought to have good bioavailability [28]. Considering the TPSA values, compounds 10–22 (Table 5) are expected to exhibit good bioavailability, based on the acceptable range. However, it must be mentioned that TPSA ≤ 140 A2 is a necessary but not sufficient criterion for good bioavailability.
The number of rotatable bonds is a simple topological parameter of molecular flexibility. It has been shown to be a very good descriptor of oral bioavailability of drugs [17]. Compounds 17, 18 and 19 exhibit greater flexibility among the compounds tested with nRotB = 3, 3, and 4, respectively, while the commercial drug etoposide presented a value equal to 5 (Table 5).
Bioactivity of all the 13 compounds and etoposide were analyzed under different regular human body receptors, with six criteria of known successful drug activity being observed in the areas of GPCR ligand activity, ion channel modulation, kinase inhibition activity, nuclear receptor ligand activity, protease inhibition activity, and enzyme inhibition activity. The higher the value of the score, the better the probability of a molecule being active. Results of these parameters are presented (Table 5) by numerical assignments. Like etoposide, all the compounds studied have consistent negative numerical values for ion channel modulator and kinase inhibitor. In the other categories, the values for some isobenzofuranones were positive.
Although no plane correlation could be found between the physical chemical properties of isobenzofuranone derivatives and antiproliferative activity, some generalizations can be made. For instance, the presence of the acetyl group on the benzene ring of compounds 14–19 seems to increase biological activity. There is an apparent correlation between the values of cLogP with the antiproliferative activity of these compounds, since the most active compounds have cLogP values in the range of 3.1–4.19. The other compounds show lower lipophilicity values.
3. Experimental
3.1. General
Analytical grade phthalaldehydic acid, 8-diazabicyclo [5.4.0]undec-7-ene (DBU), cycloexane-1,3-dione, 5-methylcyclohexane-1,3-dione, 5,5-dimethyl cicloexane-1,3-dione (dimedone), 5-isopropylcyclohexane-1,3-dione and dymethylaminopyridine (DMAP) were purchased from Aldrich (St. Louis, MO, USA). Mercury (II) acetate, sodium acetate, acetic acid, acetonitrile and chloroform were purchased from Vetec (Rio de Janeiro, Brazil). 1H and 13C-NMR spectra were recorded on a Bruker AVANCE DRX 400 spectrometer at 400 and 100 MHz, respectively, using MeOH-d4, CDCl3 and DMSO-d6 as solvents. Infrared spectra were recorded on a Varian 660-IR, equipped with GladiATR scanning from 4000 to 500 cm−1. HRMS data were recorded under ESI conditions on a micrOTOF-QII Brucker spectrometer. Melting points are uncorrected and were obtained with a MQAPF-301 melting point apparatus (Microquimica, Campinas, Brazil). Analytical thin layer chromatography was carried out on TLC plates recovered with 60GF254 silica gel. Column chromatography was performed over silica gel (60–230 mesh).
3.2. Synthesis of Compounds 11–13, Exemplified by the Synthesis of 3-(2-Hydroxy-6-oxocyclohex-1-enyl)isobenzofuran-1(3H)-one (11)
Cyclohexane-1,3-dione (0.891 g, 7.95 mmol), acetonitrile (10 mL) and DBU (1.06 mL, 7.1 mmol) were added to a round-bottomed flask (50 mL). The mixture was magnetically stirred at room temperature for 5 min. Phthaldehydic acid (10, 1.070 g; 7.1 mmol) was added and the mixture was refluxed for 7 h. The reaction was quenched by the addition of aqueous 10% HCl (10 mL), followed by ethyl acetate (100 mL). The layers were separated and the organic layer was washed with water (2 × 20 mL) and brine (10 mL), dried over anhydrous sodium sulphate and concentrated under reduced pressure. The resulting material was recrystallized from ethyl acetate. Compound 11 was obtained as a white solid in 64% yield (1.121 g; 4.6 mmol). The structure of the compound is supported by the following data: TLC Rf = 0.20 (ethyl acetate). mp 217.5–218.0 °C. IR (ATR, cm−1): 2960, 2917, 2887, 2555 (broad band), 1758, 1565, 1380, 1281, 1056, 1025, 944. 1H-NMR (400 MHz, DMSO-d6): δ 1.83–1.91 (m, 2H, H-4′), 2.26–2.45 (m, 4H, H-3′/H-5′), 6.59 (s, 1H, H-3), 7.30 (d, 1H, J = 7.6 Hz, H-4), 7.50 (dd, 1H, J = 7.6, 7.2 Hz, H-5), 7.65 (dd, 1H, J = 7.6, 7.2 Hz, H-6), 7.78 (d, 1H, J = 7.6 Hz, H-7). 13C-NMR (100 MHz, DMSO-d6): δ 20.1 (C-3′/C-5′), 32.6 (C-4′), 74.3 (C-3), 109.1 (C-1′), 121.4 (C-4), 124.2 (C-7), 126.5 (C-8), 128.0 (C-6), 133.6 (C-5), 150.7 (C-9), 170.7 (C-1), 188.0 (C-2′). HREIMS m/z (M+H+): Calculated for C14H13O4, 245.0814; found: 245.0840.
Compounds 12 and 13 were prepared using a procedure similar to that described for compound 11.
3-(2-Hydroxy-4-methyl-6-oxocyclohex-1-enyl)isobenzofuran-1(3H)-one (12): The compound was obtained as a white solid in 68% yield. The structure of the compound is supported by the following data: TLC Rf = 0.50 (ethyl acetate). mp. 214.6–218.3 °C. IR (ATR, cm−1): 2960, 2917, 2886, 2565 (broad band), 1757, 1564, 1380, 1281, 1060, 944, 787, 740, 690, 538. 1H-NMR (400 MHz, MeOH-d4): δ 1.08 (d, 3H, J = 4.8, -CH3), 2.13–2.55 (m, 5H, H-4′, H-3′/H-5′), 6.69 (s, 2H, H-3), 7.31 (d, 1H, J = 7.6 Hz, H-4), 7.49 (dd, 1H, J = 7.6, 7.2 Hz, H-5), 7.64 (dd, 1H, J = 7.6, 7.2 Hz, H-6), 7.81 (d, 1H, J = 7.6 Hz, H-7); 13C-NMR (100 MHz, MeOH-d4): δ 21.0 (-CH3), 29.5 (C-3′/C-5′), 42.2 (C-4′), 76.6 (C-3), 110.8 ( C-1′), 122.7 (C-4), 125.7 (C-7), 128.2 (C-8), 129.5 (C-6), 135.2 (C-5), 152.4 (C-9), 174.2 (C-1), 189.7 (C-2′). HREIMS m/z (M+H+): Calculated for C15H15O4, 259.0970; found: 259.0998.
3-(2-Hydroxy-4-isopropyl-6-oxocyclohex-1-enyl)isobenzofuran-1(3H)-one (13): The compound was obtained as a white solid in 69% yield. The structure of the compound is supported by the following data: TLC Rf = 0.08 (hexane-ethyl acetate 1:3 v/v). mp 176.7–177.8 °C. IR (ATR, cm−1): 2964, 2872, 2534 (broad band), 2034, 1758, 1555, 1466, 1370, 1341, 1252, 1090, 1063, 950, 727. 1H-NMR (300 MHz, MeOH-d4): δ 0.94 (d, 6H, J = 6.9 Hz, -CH(CH3)2), 1.57–1.67 (m, 1H, -CH(CH3)2), 1.83–1.96 (m, 1H, H-4′), 2.22–2.51 (m, 4H, H-3′ and H-5′), 6.68 (s, 1H, H-3), 7.31 (dd, 1H, J = 7.5, 0.9, H-4), 7,50 (t, 1H, J = 7.5, H-5), 7.64 (td, 1H, J = 7.5, 1.2, H-6), 7.80 (d, 1H, J = 7.5, H-7). 13C-NMR (75 MHz, MeOH-d4): δ 18.7 (-CH(CH3)2), 31.7 (-CH(CH3)2), 36.8 (C-3′/C-5′), 39.5 (C-4′), 75.3 (C-3), 109.4 (C-1′), 121.4 (C-4), 124.4 (C-7), 126.8 (C-8), 128.2 (C-6), 139.9 (C-5), 151.0 (C-9), 172.9 (C-1), 189,4 (C-2′). HREIMS m/z (M+H+): Calculated for C17H19O4, 287.1283; found: 287.1260.
3.3. Synthesis of Compounds 14–16, Exemplified by the Synthesis of 3-(2,6-Dihydroxyphenyl)-isobenzofuran-1(3H) one (14)
A 50 mL round-bottomed flask was charged with compound 11 (477 mg, 1.95 mmol), acetic acid (20 mL), Hg(OAc)2 (1,897 mg; 5.93 mmol) and NaOAc (481 mg, 5.86 mmol). The resulting mixture was stirred under reflux for 5 h. The reaction mixture was cooled and 5 mL of HCl 1 mol L−1 was added and stirred for an additional 15 min. The mixture was filtered through a Celite pad and the filtrate diluted with ethyl acetate (60 mL). The organic layer was separated, washed with saturated sodium bicarbonate aqueous solution (2 × 10 mL), water (5 mL), brine (5 mL) and 0.200 mol L−1 sodium EDTA aqueous solution (2 × 15 mL). The organic layer was dried over sodium sulphate, filtered, concentrated under reduced pressure and the residue purified by silica gel column chromatography (hexane-ethyl acetate; 1:3 v/v). Compound 11 was obtained as a white solid in 68% yield (320 mg; 1.32 mmol). The structure of the compound is supported by the following data: TLC Rf = 0.70 (hexane-ethyl acetate 1:3 v/v). mp 238.6–239.8 °C. IR (ATR, cm−1): 3470 (broad band), 3296 (broad band), 1712, 1614, 1469, 1011, 945, 715, 587. 1H-NMR (400 MHz, CDCl3 + DMSO-d6): δ 6.29 (d, 2H, J = 8.0 Hz, H-3′/H-5′), 6.92 (t, 1H, J = 8.0 Hz, H-4), 7.02 (s, 1H, H-3), 7.31 (d, 1H, J = 7.2 Hz, H-4), 7.47 (t, 1H, J = 7.6 Hz, H-6), 7.59 (t, 1H, J = 7.2 Hz, H-5), 7.81 (d, 1H, J = 7.6 Hz, H-7). 13C-NMR (100 MHz, CDCl3 + DMSO-d6): δ 75.6 (C-3), 106.9 (C-3′/C-5′), 108.6 (C-1′), 121.8 (C-4), 124.2 (C-7), 127.2 (C-8), 127.9 (C-6), 130.2 (C-4′), 133.3 (C-5), 151.1 (C-9), 157.5 (C-2′/C-6′), 171.5 (C-1). HREIMS m/z (M+H+): Calculated for C14H11O4, 243.0657; found: 243.0658.
Compounds 15 and 16 were prepared using a procedure similar to that described for compound 14.
3-(2,6-Dihydroxy-4-methylphenyl)-isobenzofuran-1(3H)-one (15): The compound was obtained as a pale yellow solid via aromatization of compound 12 in 71% yield (purified by silica gel column chromatography, using hexane-ethyl acetate 1:3 v/v as the eluting solvent). The structure of the compound is supported by the following data: TLC Rf = 0.82 (hexane-ethyl acetate 1:3 v/v). mp 228–231 °C. IR (ATR, cm−1): 3579, 3201 (broad band), 2924, 2853, 1721, 1618, 1595, 1305, 1046, 952, 821, 734, 684, 535. 1H RMN (200 MHz, CDCl3 + MeOH-d4): δ 1.94 (s, 3H, -CH3), 5.94 (s, 2H, H-3′/H-5′), 6.82 (s, 1H, H-3), 7.11 (d, 1H, J = 7.2 Hz, H-4); 7.16–7.44 (m, 2H, H-5 e H-6), 7.62 (d, 1H, J = 7.2 Hz, H-7). 13C-NMR (50 MHz, CDCl3 + MeOH-d4): δ 22.5 (-CH3), 72.2 (C-3), 106.9 (C-1′), 109.1 (C-3′/C-5′), 123.2 (C-4), 125.4 (C-6), 128.5 (C-8), 128.9 (C-7), 134.4 (C-5), 141.9 (C-4′), 152.6 (C-9), 158.3 (C-2′/C-6′), 173.2 (C-1). HREIMS m/z (M+H+): Calculated for C15H13O4, 257.0814; found: 257.0880.
3-(2,6-Dihydroxy-4-isopropylphenyl)-isobenzofuran-1(3H)-one (16): The compound was obtained as a viscous orange oil via aromatization of compound 13 in 94% yield (purified by silica gel column chromatography, using hexane-ethyl acetate 1:1 v/v as the eluting solvent). The structure of the compound is supported by the following data: TLC Rf = 0.44 (hexane-ethyl acetate 1:1 v/v). IR (ATR, cm−1): 3305 (broad band), 2959, 2924, 2870, 2159, 1726, 1621, 1596, 1433, 1034, 732. 1H-NMR (300 MHz, MeOH-d4): δ 1,17 (d, 6H, J = 6.9 Hz, -CH(CH3)2), 2.68 (hept, 1H, J = 6.9 Hz; -CH(CH3)2), 6.17 (s, 2H; H-3′/H-5′), 7.02 (s, 1H, H-3); 7.30 (dd, 1H, J = 7.5, J = 0.9 Hz, H-4), 7.49 (t, 1H, J = 7.2 Hz, H-5), 7.63 (td, 1H, J = 7.5, 1.2 Hz, H-6), 7.82 (d, 1H, J = 7.5 Hz, H-7). 13C-NMR (75 MHz, MeOH-d4) δ: 22.9 (-CH(CH3)2), 34.2 (-CH(CH3)2), 76.8 (C-3), 104.6 (C-3/C-5′), 105.8 (C-1′), 121.8 (C-4), 124.1 (C-6), 127.0 (C-8), 128.0 (C-7), 133.7 (C-5), 151.8 (C-9), 152.3 (C-4′), 157.6 (C-2′/C-6′), 173.4 (C-1). HREIMS m/z (M+H+): Calculated for C17H17O4, 285.1127; found: 285.1116.
3.4. Synthesis of Compounds 17–19, Exemplified by the Synthesis of 2-(3-Oxo-1,3-dihydroisobenzofuran-1-yl)-1,3-phenylene diacetate (17)
A round-bottomed flask (25 mL) was charged with compound 14 (0.109 g, 0.45 mmol), chloroform (5 mL), acetic anhydride (0.16 mL, 1.62 mmol), and a catalytic amount of DMAP (0.005 g, 0.045 mmol). The reaction mixture was stirred at room temperature for 12 h. The solvent was removed and compound 17 was purified by recrystallization from ethyl acetate affording a white solid in 44% yield (0.064 g, 0.20 mmol). The structure of the compound is supported by the following data: TLC Rf = 0.54 (hexane-ethyl acetate 1:1 v/v). mp. 175.9–177.8 °C. IR (ATR, cm−1): 1756, 1611, 1465, 1369, 1285, 1170, 1023, 975; 1H-NMR (200 MHz, DMSO-d6): δ 2.01 (brs, 6H, 2 × -COCH3), 6.85 (s, 1H, H-3), 7.08 (d, 2H, J = 8.2 Hz, H-3′/H-5′), 7.25 (d, 1H, J = 6.8 Hz, H-4), 7.53 (t, 1H, J = 8.2 Hz, H-4′), 7.58–7.75 (m, 2H, H-5 and H-6), 7.95 (d, 1H, J = 7.4 Hz, H-7). 13C-NMR (50 MHz, DMSO): δ 22.0 (-COCH3), 76.0 (C-3), 122.3 (C-3′/C-5′), 123.2 (C-1′), 124.4 (C-4), 126.6 (C-7), 127.3 (C-8), 131.2 (C-6), 132.5 (C-4′), 136.5 (C-5), 150.67 (C-9), 151.61 (C-2′/C-6′), 170.5 (C-1), 172.0 (-COCH3). HREIMS m/z (M+H+): Calculated for C18H15O6, 327.0869; found: 327.0848.
Compounds 18 and 19 were prepared using a procedure similar to that described for compound 17.
5-Methyl-2-(3-oxo-1,3-dihydroisobenzofuran-1-yl)-1,3-phenylene diacetate (18): The compound was obtained as a pale yellow solid from compound 15 in 93% yield (after purification by silica gel column chromatography, using hexane-ethyl acetate 1:2 v/v as the eluting solvent). The structure of the compound is supported by the following data: TLC Rf = 0.8 (hexane-ethyl acetate 1:2 v/v) mp. 171.5–172.3 °C. IR (ATR, cm−1): 2925, 2855, 1758, 1621, 1172, 1040, 974, 692, 738. 1H-NMR (200 MHz, DMSO-d6): δ 1.99 (brs, 6H, 2 × -COCH3), 2.31 (s, 3H, CH3), 6.78 (s, 1H, H-3), 6.94 (s, 2H, H-3′/H-5′), 7.23 (d, 1H, J = 7.4 Hz, H-4), 7.61 (dd, 1H, J = 7.4, 7.2 Hz, H-5), 7.73 (dd, 1H, J = 7.4, 7.2 Hz, H-6), 7.93 (d, 1H, J = 7.2 Hz, H-7). 13C-NMR (50 MHz, DMSO-d6): δ 22.0 (-COCH3), 22.3 (-CH3), 76.0 (C-3), 119.3 (C-3′/C-5′), 123.6 (C-1′), 124.4 (C-4), 126.5 (C-6), 127.3 (C-8), 131.0 (C-7), 136.4 (C-5), 142.9 (C-9′), 150.8 (C-4′), 151.4 (C-2′/C-6′), 170.5 (C-1), 171.9 (-COCH3). HREIMS m/z (M+H+): Calculated for C19H17O6, 341.1025; found: 341.1030.
5-Isopropyl-2-(3-oxo-1,3-dihydroisobenzofuran-1-yl)-1,3-phenylene diacetate (19): This compound was obtained as a solid in 78% yield (after purification by recrystallization from ethyl acetate) from compound 15. The structure of the compound is supported by the following data: TLC Rf = 0.81 (hexane-ethyl acetate 1:1 v/v). mp. 157.5–158.1 °C. IR (ATR, cm−1): 2963, 2930, 2870, 1756, 1620, 1366, 1288, 1179, 1030, 973, 739, 562. 1H-NMR (200 MHz, DMSO-d6): δ 1.16 (d, 1H, J = 7.0 Hz, -CH(CH3)2), 1.98 (brs, 6H, 2 × -COCH3), 2.89 (hept, 1H, J = 7.0 Hz, -CH(CH3)2, 6.78 (S, 1H, H-3), 7.00 (s, 2H, H-3′/H-5′), 7.26 (d, 1H, J = 7.2 Hz, H-4), 7.54–7.78 (m, 2H, H-5 and H-6), 7.93 (d, 1H, J = 7.2 Hz, H-7). 13C-NMR (50 MHz, DMSO-d6): δ 22.0 (-COCH3), 25.0 (-CH(CH3)2), 34.80 (-CH(CH3)2), 76.0 (C-3), 119.6 (C-3′/C-5′), 121.0 (C-1′), 124.5 (C-4), 126.5 (C-6), 127.3 (C-8), 131.1 (C-7), 136.4 (C-5), 150.7 (C-9), 151.5 (C-2′/C-6′), 153.9 (C-4′) 170.5 (C-1), 171.9 (-COCH3). HREIMS m/z (M+H+): Calculated for C21H21O6, 369.1338; found: 369.1418.
3.5. Synthesis of 3-(2-Hydroxy-4,4-dimethyl-6-oxo-cyclohexen-1-yl)isobenzofuran-1(3H)-one (20)
Dimedone (500 mg; 3.57 mmol), chloroform (5 mL), and DBU (0.53 mL; 3.57 mmol) were placed into a 25 mL round-bottomed flask. The resulting mixture was stirred at room temperature for 5 min followed by the addition of 5-hydroxyisobenzofuran-1(3H)-one (19) (495 mg; 3.30 mmol). The reaction mixture was stirred at room temperature for 5 h, quenched with 10% HCl (5 mL), and diluted with ethyl acetate (100 mL). The organic layer was separated, washed with water (2 × 20 mL) and brine (10 mL), dried over sodium sulphate, filtered and concentrated under reduced pressure. Compound 18 was obtained as a white solid in 95% yield (850 mg; 3.12 mmol) after recrystallization from ethyl acetate. The structure of the compound is supported by the following data: TLC Rf = 0.10 (hexano-ethyl acetate 1:1 v/v). mp 212.1–213.0 °C. IR (ATR, cm−1): 2962, 2934, 2884, 2535 (broad band), 1763, 1569, 1382, 1321, 1121, 1059, 942, 738, 694, 613, 574. 1H-NMR (400 MHz, MeOH-d4): δ 1.09 (s, 6H, 2 × CH3), 2.35 (s, 4H, H-3′/H-5′), 6.70 (s, 1H, H-3), 7.31 (d, 1H, J = 7.6 Hz, H-4), 7.50 (t, 1H, J = 7.6 Hz, H-5), 7.66 (t, 1H, J = 7.6 Hz, H-6), 7.81 (d, 1H, J = 7.6 Hz, H-7); 13C-NMR (100 MHz, MeOH-d4): δ 28.5 (-CH3), 33.1 (C-4′), 47.9 (C-3′/C-5′), 76.6 (C-3), 110.1 (C-1′), 122.6 (C-4), 125.8 (C-7), 128.2 (C-8), 129.5 (C-6), 135.2 (C-5), 152.4 (C-9), 174.2 (C-1), 188.9 (C-2′). HREIMS m/z (M+H+): Calculated for C16H17O4, 273.1127; found: 273.1124.
3.6. Synthesis of Compounds 21 and 22, Exemplified by the Synthesis 3-(2-Hydroxy-5-oxocyclopent-1-enyl)isobenzofuran-1(3H)-one (21)
Cyclopentane-1,3-dione (0.357 g; 3.64 mmol), chloroform (5 mL), and DBU (0.56 mL, 3.70 mmol) were added to a 25 mL round-bottomed flask. The mixture was stirred at room temperature for 5 min. Phthalaldehydic acid (10) (0.533 g, 3.55 mmol) was added and the resulting mixture was refluxed for 5 h, quenched with 10% HCl (5 mL), and diluted with ethyl acetate (100 mL). The organic layer was separated, washed with water (2 × 20 mL) and brine (10mL), dried over sodium sulphate, filtered and concentrated under reduced pressure. Compound 21 was obtained as a white solid in 53% yield (0.448 g, 1.94 mmol) after recrystallization from ethyl acetate. The structure of the compound is supported by the following data: TLC Rf = 0.10 (hexane-ethyl acetate 1:1 v/v). mp. 203.6–205.2 °C. IR (ATR, cm−1): 2987, 2930, 2370 (broad band), 2366, 2339, 1763, 1648, 1557, 1368, 1281, 1048, 963. 1H-NMR (200 MHz, CDCl3 + DMSO-d6) δ 2.27 (brs, 4H, 2 × -CH2), 6.07 (s, 1H, H-3), 7.11 (d, 1H, J = 7.2 Hz, H-4), 7.23–7.45 (m, 2H, H-5 and H-6), 7.62 (d, 1H, J = 7.2 Hz, H-7). 13C-NMR (50 MHz, CDCl3 + DMSO-d6): δ 31.8 (-CH2), 74.5 (C-3), 112.6 (C-1′), 123.3 (C-4), 126.0 (C-7), 127.5 (C-8), 129.7 (C-6), 134.9 (C-5), 150.1 (C-9), 172.2 (C-1), 197.3 (C-2′). HREIMS m/z (M+H+): Calculated for C13H11O4, 231.0657; found: 231.0672.
Compound 22 was obtained as a brown solid in 69% yield, using a procedure similar to that described for compound 21. The structure of this compound is supported by the following data: TLC Rf = 0.10 (hexane-ethyl acetate 1:1 v/v). mp. 214.0–215.3 °C. IR (ATR, cm−1): 3068, 2922, 1770, 1741, 1702, 1266, 1221, 1054. 1H-NMR (400 MHz, CDCl3 + DMSO-d6) δ 4.12 (d, 1H, J = 2.4 Hz, H-1′), 6.20 (d, 1H, J = 2.4 Hz, H-3), 7.84 (m, 8H, H-4, H-5, H-6, H-7, H-3′, H-4′, H-5′, and H-6′).13C-NMR. 55,0 (C-1′), 77.3 (C-3), 124,2 (C-4), 123.1 (C-3′), 123.6 (C-6′), 125.3 (C-7), 125.8 (C-6), 129.5 (C-8), 134.4 (C-4′/C-5′), 136.1 (C-5), 142.1 (C-8′), 142.5 (C-9′), 147.4 (C-9), 169.4 (C-1), 194.9 (C-2’), 196.3 (C-7′). HREIMS m/z (M+H+): Calculated for C17H11O4, 279.0657; found: 279.0665.
3.7. Antiproliferative Activity Assays
The human cell lines U937 (lymphoma) and K562 (myeloid leukemia) were purchased from ATCC (Rockville, MD, USA). They were cultured in a RPMI 1640 medium (Sigma Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (LGC, Campinas, Brazil), 100 μg/mL streptomycin and 100 U/mL penicillin at 37 °C under 5.0% CO2 atmosphere. To evaluate the antiproliferative activity of compounds 10–22, U937 and K562 cells were grown in 96-well plates (TPP, Trasadingen, Switzerland) at a density of 2.0 × 104 cells per well at different compound concentrations (0–100 µM) and etoposide (VP-16) (0–100 µM). No precipitation was observed for any of the compounds at the assayed concentrations. Cell viability was determined by the modified colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma Aldrich) assay. After 24 and/or 48 h, MTT (0.5 mg/mL) was added to the wells (2 h, 37 °C), followed by the removal of MTT solution and addition of 100 µL/well of dimethyl suphoxide (DMSO, Sigma Aldrich) to solubilize the formazan crystals. Absorbances were measured at 540 nm. Each analysis was performed in quadruplicate and the results were normalized considering the cultures treated with the vehicle alone (1% DMSO). The data were analyzed as the mean ± SD and IC50 was determined using GraphPad Prism 5.0 software.
4. Conclusions
We have demonstrated that several isobenzofuranone possessing alicyclic and aromatic groups attached to C-3 position exhibit significant antiproliferative active against two cancer cell lines. In some cases, the biological effect was higher than that displayed by the commercial drug etoposide (VP-16). The in silico favorable drug-like parameters predicted and shown above suggest that the isobenzofuranones prepared in this work possess good physicochemical properties that qualify them as promising candidates for future studies. Further investigations to validate their potential as anti-tumor compounds, including evaluation of their toxicity against human PMBC cells and in vivo assays using murine animal models, are under way and will be published elsewhere.
Acknowledgments
The authors are grateful to the Brazilian agencies: Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for research grant (to G.C.B); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) research fellowhships (to W.L.P.); Fundação de Amparo a Pesquisa de Minas Gerais (FAPEMIG) for research grant (to R.R.T) and research fellowship (to F.M.O.).
Footnotes
Sample Availability: Samples of the compounds 11–22 are available from the authors.
References
- 1.Huang X.-Z., Zhu Y., Guan X.-L., Tian K., Guo J.-M., Wang H.-B., Fu G.-M. A novel antioxidant isobenzofuranone derivative from fungus Cephalosporium sp. AL031. Molecules. 2012;17:4219–4224. doi: 10.3390/molecules17044219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Strobel G., Ford E., Worapong J., Harper J.K., Arif A.M., Grant D.M., Fung P.C.W., Chau R.M.W. Isopestacin, an isobenzofuranone from Pestalotiopsis microspora possessing antifungal and antioxidant activities. Phytochemistry. 2002;60:170–183. doi: 10.1016/S0031-9422(02)00062-6. [DOI] [PubMed] [Google Scholar]
- 3.Ma F., Gao Y., Qiao H., Hu X., Chang J. Antiplatelet activity of 3-butyl-6-bromo-1(3H)-isobenzofuranone on rat platelet aggregation. J. Thromb. Thrombolysis. 2012;33:64–73. doi: 10.1007/s11239-011-0647-9. [DOI] [PubMed] [Google Scholar]
- 4.Cardozo J.A., Braz-Filho R., Rincón-Velandia J., Guerrero-Pabón M.F. 3-butil-isobenzofuranona: Un compuesto aislado de Apium graveolens com actividad anticonvulsivante. Rev. Colomb. Cienc. Quim. Farm. 2005;34:69–76. [Google Scholar]
- 5.Arnone A., Assante G., Nasini G., Strada S., Vercesi A. Cryphonectric acid and other minor metabolites from a hypovirulent strain of Cryphonectria parasítica. J. Nat. Prod. 2002;65:48–50. doi: 10.1021/np0103012. [DOI] [PubMed] [Google Scholar]
- 6.Demuner A.J., Barbosa L.C.A., Veiga T.A.M., Barreto R.W., King-Diaz B., Lotina-Hennsen B. Phytotoxic constituents from Nimbya alternantherae. Biochem. Syst. Ecol. 2006;34:790–795. doi: 10.1016/j.bse.2006.06.008. [DOI] [Google Scholar]
- 7.Logrado L.P.L., Santos C.O., Romeiro L.A.S., Costa A.M., Ferreira J.R.O., Cavalcanti B.C., de Moraes O.M., Costa-Lotufo L.V., Pessoa C. Synthesis and cytotoxicity screening of substituted isobenzofuranones designed from anarcadic acids. Eur. J. Med. Chem. 2010;45:3480–3489. doi: 10.1016/j.ejmech.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 8.Teixeira R.R., Barbosa L.C.A., Maltha C.R.A., Rocha M.E., Bezerra D.P., Costa-Lotufo L.V., Pessoa C., Moraes M.O. Synthesis and cytotoxic activity of some 3-benzyl-5-arylidenefuran-2(5H)-ones. Molecules. 2007;12:1101–1116. doi: 10.3390/12051101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbosa L.C.A., Pereira U.A., Maltha C.R.A., Teixeira R.R., Valente V.M.M., Ferreira J.R.O., Costa-Lotufo V., Moraes M.O., Pessoa C. Synthesis and Biological Evaluation of 2,5-bis(alkylamino)-1,4-benzoquinones. Molecules. 2010;15:5629–5643. doi: 10.3390/molecules15085629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barbosa L.C.A., Nogueira L.B., Maltha C.R.A., Teixeira R.R., Silva A.A. Synthesis and phytogrowth properties of oxabicyclic analogues related to helminsthosporin. Molecules. 2009;14:160–173. doi: 10.3390/molecules14010160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teixeira R.R., Barbosa L.C.A., Forlani G., Piló-Veloso D., de Mesquita Carneiro J.W. Synthesis of photosynthesis-inhibiting nostoclide analogues. J. Agric. Food Chem. 2008;56:2321–2329. doi: 10.1021/jf072964g. [DOI] [PubMed] [Google Scholar]
- 12.Pahari P., Senapati B., Mal D. Regiospecific synthesis of isopestacin, a naturally occurring isobenzofuranone antioxidante. Tetrahedron Lett. 2004;45:5109–5112. doi: 10.1016/j.tetlet.2004.04.175. [DOI] [Google Scholar]
- 13.Mal D., Pahari P., De S.R. Regiospecific synthesis of 3-(2,6-dihydroxyphenyl)phtalides: Application to the synthesis of isopestacin and cryphonectric acid. Tetrahedron. 2007;63:11781–11792. doi: 10.1016/j.tet.2007.08.048. [DOI] [Google Scholar]
- 14.Oliver J.E., Wilzer K.R., Waters R.M. Synthesis of 1-(2,6-Dihydroxyphenyl)-1-alkanones and Benzophenone by Aromatization of 2-Acyl-3-hydroxy-2-cyclohexene-1-ones with Mercuric Acetate. Synthesis. 1990;1990:1117–1119. doi: 10.1055/s-1990-27107. [DOI] [Google Scholar]
- 15.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997;23:3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 16.Veber D.F., Johnson S.R., Cheng H.-Y., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 17.Tetko I.V. Computing chemistry on the web. Drug Discov. Today. 2005;10:1497–1500. doi: 10.1016/S1359-6446(05)03584-1. [DOI] [PubMed] [Google Scholar]
- 18.Molinspiration Cheminformatics. [(accessed on September 2012)]; Available online: http://www.molinspiration.com/
- 19.Alafeefy A.M., Alqasoumi S.I., Ashour A.E., Masand V., Al-Jaber N.A., Hadda T.B., Mohameda M.A. Quinazoline-etyrphostin as a new class of antitumor agents, molecular properties prediction, synthesis and biological testing. Eur. J. Med. Chem. 2012;53:133–140. doi: 10.1016/j.ejmech.2012.03.044. [DOI] [PubMed] [Google Scholar]
- 20.Ali P., Meshram J., Sheikh J., Tiwari V., Dongre R., Hadda T.B. Predictions and correlations of structure activity relationship of some aminoantipyrine derivatives on the basis of theoretical and experimental ground. Med. Chem. Res. 2012;21:157–164. doi: 10.1007/s00044-010-9505-0. [DOI] [Google Scholar]
- 21.Mahajan D.T., Masand V.H., Patil K.N., Hadda T.B., Jawarkar R.D., Thakur S.D., Rastija V. CoMSIA and POM analyses of anti-malarial activity of synthetic prodiginines. Bioorg. Med. Chem. Lett. 2012;22:4827–4835. doi: 10.1016/j.bmcl.2012.05.115. [DOI] [PubMed] [Google Scholar]
- 22.Chohan Z.H., Sumrra S.H., Youssoufi M.H., Hadda T.B. Metal based biologically active compounds: Design, synthesis, and antibacterial/antifungal/cytotoxic properties of triazole-derived Schiff bases and their oxovanadium(IV) complexes. Eur. J. Med. Chem. 2010;45:2739–2747. doi: 10.1016/j.ejmech.2010.02.053. [DOI] [PubMed] [Google Scholar]
- 23.Lipinski C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 24.Proudfoot R. Drugs, leads, and drug-likeness: An analysis of some recently launched drugs. Bioorg. Med. Chem. Lett. 2012;12:1647–1650. doi: 10.1016/S0960-894X(02)00244-5. [DOI] [PubMed] [Google Scholar]
- 25.Jarrahpour A., Fathi J., Mimouni M., Hadda T.B., Sheikh J., Chohan Z., Parvez A. Petra, Osiris and molinspiration (POM) together as a successful support in drug design: Antibacterial activity and biopharmaceutical characterization of some azo schiff bases. Med. Chem. Res. 2012;21:1984–1990. doi: 10.1007/s00044-011-9723-0. [DOI] [Google Scholar]
- 26.Tambunan U.S.F., Bramantya N., Parikesit A.A. In silico modification of suberoylanilide hydroxamic acid (SAHA) as potential inhibitor for class II histone deacetylase (HDAC) BMC Bioinformatics. 2011;12:S13–S23. doi: 10.1186/1471-2105-12-S13-S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 28.Clark D.E. Rapid calculation of polar molecular surface area and its application to the prediction of transport phenomena. 1. Prediction of Intestinal Absorption. J. Pharm. Sci. 1999;88:807–814. doi: 10.1021/js9804011. [DOI] [PubMed] [Google Scholar]
- 29.Ertl P., Rohde B., Selzer P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000;43:3714–3717. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]