

Abstract
Halogenated quinazolinones and quinazolines are versatile synthetic intermediates for the metal-catalyzed carbon–carbon bond formation reactions such as the Kumada, Stille, Negishi, Sonogashira, Suzuki-Miyaura and Heck cross-coupling reactions or carbon-heteroatom bond formation via the Buchwald-Hartwig cross-coupling to yield novel polysubstituted derivatives. This review presents an overview of the application of these methods on halogenated quinazolin-4-ones and their quinazolines to generate novel polysubstituted derivatives.
Keywords: halogenoquinazolin-4-ones, halogenoquinazolines, cross-coupling reactions
1. Introduction and Scope
In recent years, the development of strategies to efficiently functionalize presynthesized halogenated quinazolinones and quinazolines or their tosylate derivatives via metal-catalyzed cross-coupling reactions to afford novel polycarbo- or polyheteroatom-substituted derivatives with potential application in pharmaceuticals and materials has attracted considerable interest. The 3-substituted 2,6-diarylquinazolin-4(3H)-ones 1 and 2 (Figure 1), for example, were previously prepared via Suzuki-Miyaura cross-coupling of the corresponding 6-halogenated 4(3H)-oxo precursors and were found to be ghrelin receptor and vasopressin V1b receptor antagonists, respectively [1,2].
Figure 1.

Examples of biologically-relevant polycarbo-substituted quinazolin-4(3H)-ones.
Lapatinib 3, a 6-heteroaryl substituted 4-anilinoquinazoline derivative is an oral dual tyrosine kinase inhibitor (TKI) that targets both EGFR and HER2 to inhibit the proliferation of breast cancer cells [3]. The 6-alkynylated 4-aminoquinazolines 4 and 5 (Figure 2), on the other hand, serve as selective inhibitors of Aurora A [4]. Likewise, the 4-anilinoquinazoline derivative 6 (CP-724,714) is a selective ErbB2 angiogenesis inhibitor under investigation for the treatment of breast, ovarian and other types of cancer [5].
Figure 2.

Carbo-substituted 4-(aryl/alkylamino)quinazolines of biological importance.
During the course of the exploration of non-anilinoquinazoline scaffold, trisubstituted quinazoline derivatives such as 4-(3-bromophenyl)-8-(trifluoromethyl)-2-phenylquinazoline 7a and 4-(3-bromophenyl)-2-(thiophen-2-yl)-8-(trifluoromethyl)quinazoline 7b (Figure 3) were prepared as part of a series of liver X-receptor modulators [6]. Likewise, the 4-alkynylquinazolines 8 were found to be potent epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors [7].
Figure 3.

Examples of biologically active 4-aryl- 7 and 4-alkynylquinazolines 8.
New findings on the biological and photophysical properties of polycarbo-substituted quinazolinones and quinazolines reveal a need to increase the diversity of substituents around these heterocyclic scaffolds. To this end, halogenated quinazolinones and quinazoline derivatives have established themselves as important intermediates for the synthesis of polycarbo- and polyheteroatom-substituted derivatives with potential application in material and medicinal chemistry. The versatility of the halogenated heterocycles as synthetic intermediates is a consequence of Csp2–X bonds, which enable further elaboration into polycarbo-substituted or polyheteroatom-substituted derivatives. Despite the importance of metal catalysed cross-coupling reactions in synthesis, there is no comprehensive review in the literature on the application of these methods on halogenated quinazolinones and quinazolines to afford polysubstituted derivatives. Only a few examples of transition metal mediated cross-coupling of halogenated quinazolinones or quinazolines are described in a monograph by de Vries [8] and the review paper by Connolly et al. [9]. Most of the review articles in the literature on quinazolinones and quinazolines, on the other hand, only focus on the conventional and microwave syntheses of heteroatom-substituted derivatives as well as their medicinal applications [10,11,12]. This prompted us to compile a comprehensive review on the application of the Kumada, Stille, Negishi, Sonogashira, Suzuki-Miyaura and Heck cross-coupling as well as the Buchwald-Hartwig cross-coupling and palladium-catalysed cyanation in the synthesis of polycarbo- or polyheteroatom-substituted quinazolinones and quinazolines derivatives from the corresponding halogenated precursors.
2. Reactivity of Halogenated Quinazolinones and Quinazoline Derivatives in Cross-Coupling Reactions
The use of halogenated quinazolinones and quinazolines as intermediates in transition metal-catalyzed cross-coupling reactions to form Csp2–Csp2, Csp2–Csp or Csp2–heteroatom bond(s) takes advantage of the ease of displacement of iodine, bromine, or chlorine atom(s) on the aryl or heteroaryl moiety by nucleophiles or metal catalysts. The order of reactivity of Csp2–halogen bonds in transitional metal-mediated cross-coupling for aryl/heteroaryl halides, C-I > C-Br >> C-Cl, generally allows selective coupling with iodides or bromides in the presence of chlorides [13,14]. Theoretical calculations at B3LYP level, on the other hand, reveal that the bond dissociation energy of the C(4)-Cl bond (84.8 kcal/mol) of 6-bromo-2,4-dichloroquinazoline is larger than that of the weaker Csp2-Br bond (83 kcal/mol at B3LYP) [15]. The Csp2–Cl bond of the 4-chloroquinazoline moiety is, however, highly activated relative to other chlorinated or brominated positions due to α-nitrogen effect and additional activation resulting from the coordination of palladium(0) with the N-3 lone pair electron density in the oxidative-addition step [15,16,17]. Among the cross-coupling reactions with organometallic reagents that involve halogenoquinazolinones and halogenoquinazolines as well as their tosylate derivatives, Sonogashira and Suzuki-Miyaura cross-coupling reactions and to some extent the Heck reaction are more prevalent. Only limited examples involving the application of the Kumada, Stille and Negishi cross-coupling reactions in the synthesis of polycarbo-substituted quinazolinones and quinazolines exist in the literature. This also applies for the miscellaneous methods such as Buchwald-Hartwig cross-coupling and palladium catalysed cyanation of halogenated quinazolinones and quinazoline derivatives.
3. Application of Cross-Coupling Reactions in the Synthesis of Substituted Quinazolinones and Quinazolines
The development of metal-catalyzed cross-coupling reactions over the past decade has revolutionized the way carbon-carbon and carbon-heteroatom bonds are formed. Kumada, Negishi, Heck, Suzuki-Miyaura, Stille and Sonogashira cross-coupling reactions represent powerful synthetic tools for the construction of carbosubstituted quinazolinones and/or quinazolines. The Buchwald-Hartwig cross-coupling and Pd-catalyzed cyanation, on the other hand, have also been employed for the synthesis of novel heteroatom- or cyano-substituted quinazolinones and quinazoline derivatives.
3.1. Application of Kumada Cross-Coupling Reaction in the Synthesis of Polysubstituted Quinazolines
Kumada cross-coupling involves the reaction of arylhalides or triflates with Grignard reagents in the presence of palladium catalyst. Despite its relatively early discovery, the application of the Kumada reaction is only limited to the transformation of halogenated quinazolines because of the incompatibility of Grignard reagents with the amide group of quinazolinones. The reducing ability of the Grignard reagents, on the other hand, has been found to cause the precipitation of palladium black and, in turn, arrest the catalytic turnover. A modification of this reaction involving the cross-coupling of 2,4-dichloroquinazoline (R = H) or its 2,4-dichloro-6,7-dimethoxyquinazoline derivative (R = OMe) 9 with tert-butylmagnesium chloride in the presence of copper(I) iodide as catalyst in tetrahydrofuran (THF) at room temperature afforded the 4-substituted quinazoline derivatives 10a (92%) and 10b (63%) in less than an hour (Scheme 1) [18]. The selectivity of Csp2–Csp2 bond formation in this case is attributed to the increased reactivity of the C-4 position of the quinazoline ring due to the α-nitrogen effect. Anhydrous manganese chloride (MnCl2) or manganese chloride tetrahydrate (MnCl2.4H2O) have also been employed to promote the cross-coupling of 4-chloro-2-phenylquinazoline with phenylmagnesium chloride in THF for 1.5 h to afford 2,4-diphenylquinazoline in 71% yield [19]. Despite its efficiency, this method makes use of an excess of the Grignard reagent (4 equiv.), which only tolerates limited functional groups on both of the coupling partners. The mild reaction conditions as well as the lower cost of copper(I) iodide and manganese chloride nevertheless, render these modified methods an attractive alternative for the synthesis of carbosubstituted quinazolines using palladium or nickel salts.
Scheme 1.
CuI-catalyzed cross-coupling of 9 with tert-butylmagnesium chloride.

Reagents and conditions: (i) tert-BuMgCl, CuI, THF, 0 °C to r.t., <1 h.
The high nucleophilicity of the readily accessible Grignard reagents generally limits the application of the Kumada cross-coupling towards the synthesis of novel polycarbo-substituted quinazolinones.
3.2. Application of Negishi Cross-Coupling in the Synthesis of Polysubstituted Quinazolines
The Negishi cross-coupling reaction of aryl or vinyl halides/triflates with organozinc reagents in the presence of palladium [Pd0, Pd2+] or nickel source [Ni0, Ni2+] as catalyst and a phosphine ligand is a versatile and efficient method for the synthesis of a variety of heterocyclic motifs [20]. Organozinc reagents can be prepared either from the corresponding organohalide (RX) by reductive metalation [21] or from other organometalic compounds, often RLi, by transmetalation [22]. The cross-coupling reaction normally occurs at or slightly above room temperature to avoid the degradation of the zinc compound at high temperature. This reaction has thus far only been employed for the synthesis of carbo-substituted quinazolines from the corresponding halogenated precursors. The Negishi cross-coupling between the 4-substituted 2-chloro-6,7-dimethoxyquinazolines 11 and CH3ZnCl reagent generated in situ from methyl lithium and zinc(II) chloride in the presence of tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) as a Pd(0) catalyst source in dioxane, for example, previously afforded compounds 12 (Scheme 2) [17].
Scheme 2.
Negishi cross-coupling of 11 with MeZnCl.

Reagents and conditions: (i) MeLi, ZnCl2, Pd(PPh3)4, dioxane.
Negishi cross-coupling of 13 with 5-(1-N-methylpyrazole)zinc chloride 14 under palladium catalysis afforded triazoloquinazoline 15 in 40% yield (Scheme 3) [23].
Scheme 3.
Negishi cross-coupling of 13 with 14.

Reagents and conditions: (i) Pd(PPh3)4, DMF, 50 °C.
(2-Pyridyl)zinc chloride prepared, in turn, from 2-bromopyridine by halogen-metal exchange with isopropylmagnesium chloride followed by addition of zinc chloride [24], was reacted with 2-chloro-5-iodo-6,7-dimethoxyquinazolin-4-amine 16 in the presence of palladium acetate-triphenylphosphine catalyst complex in THF under reflux to afford a mixture of the cross-coupled product 17 and the reduced derivative 18 (Scheme 4) [25]. The latter was formed as a sole product by treatment of 16 with 2-iodopyridine in the presence of activated zinc and Pd(OAc)2–PPh3 mixture in DMF.
Scheme 4.
Negishi cross-coupling of 2-chloro-5-iodo-6,7-dimethoxyquinazolin-4-amine 16.

Reagents and conditions: (i) 2-PyZnCl, Pd(OAC)2, PPh3, THF, reflux.
Despite high sensitivity of organozinc reagents to air and moisture, their high compatibility with various functional groups make the Negishi cross-coupling reaction a powerful tool for the formation of carbon-carbon bonds.
3.3. Application of Stille Cross-Coupling in the Synthesis of Polysubstituted Quinazolines
The Stille cross-coupling which involves the reaction between organostannanes and organic halides or tosylates in the presence of Pd catalyst [26] has thus far only been employed for the synthesis of polysubstituted quinazolines from the corresponding halogenated precursors. 5-Chlorotriazoloquinazoline, for example, was subjected to Stille cross-coupling with heterarylstannanes using Pd(PPh3)4-CuI catalyst mixture in DMF at 70 °C to afford a series of 5-heteraryl-substituted triazoloquinazolines in 20%–78% yields [23]. Under the same reaction conditions, the analogous 5-tosyltriazoloquinazoline 19 reacted with heterarylstannanes 20 to afford the corresponding 5-heteraryl–substituted derivatives 21 in reasonable yields (Scheme 5) [23].
Scheme 5.
Stille cross-coupling of 5-tosyltriazoloquinazoline 19 with heterarylstannanes.

| Het | %Yield Condition (i) | %Yield Condition (ii) | |
|---|---|---|---|
| 21a | 2-thienyl | 91 | 78 |
| 21b | 2-furyl | 73 | 87 |
| 21c | 2-(N-methylimidazole) | - | 52 |
| 21d | 5-(N-methyl-[1,2,4]-triazole) | - | 69 |
Reagents and conditions: (i) Pd(PPh3)4, DMF, 70 °C, 8 h; (ii) Pd(PPh3)4, CuI, DMF, 70 °C, 1 h.
A modification of the Stille cross-coupling involving the use of 6-bromo-2,4-dichloroquinazoline 22 as substrate and trimethylalane (1.2 equiv.) as coupling partner in the presence of Pd(PPh3)4 as a source of active Pd(0) catalyst to afford a mixture of the C-4 substituted 23 (47%) and C-6 cross-coupled product 24 (16%) has been described before (Scheme 6) [16]. The preponderance of the C-4 cross-coupled product is a consequence of the increased reactivity of the C(4)–Cl bond due to the α-effect and strong coordination of Pd(0) with N-3 lone pair electrons in the oxidative-addition step. The 2-position of the pyrimidine ring, on the other hand, is generally known to be less reactive to oxidative addition of Pd than the 4-position [27].
Scheme 6.
Regioselective alkylation of 6-bromo-2,4-dichloroquinazoline 22.

Reagents and Conditions: (i) Al(CH3)3, Pd(PPh3)4, THF, reflux.
3.4. Application of Sonogashira Cross-Coupling in the Synthesis of Polysubstituted Quinazolinones and Quinzoline Derivatives
The Sonogashira cross-coupling of aryl halides or tosylates with terminal acetylenes in the presence of Pd(0) catalyst source and copper(I) iodide (CuI) as co-catalyst provides an effective route for Csp2-Csp bond formation to afford arylalkynes and conjugated enynes [28]. A combination of palladium and copper as catalysts results in the increased reactivity of the reagents and as a result, Sonogashira cross-coupling can also be carried out at room temperature [28]. This reaction has been employed extensively in the synthesis of alkynylated quinazolinones and quinazolines with potential application in medicinal and material chemistry. Initial Pd/C-PPh3-CuI catalyzed Sonogashira cross-coupling of 2-amino-5-iodobenzamide 25 with terminal acetylenes in the presence of triethylamine as a base in ethanol under reflux afforded the 5-alkynyl-2-aminobenzamides 26 (Scheme 7) [29]. The latter were, in turn, subjected to cyclocondensation with cycloalkanones 27 using Amberlyst-15 as a catalyst in acetonitrile under ultrasonic irradiation at room temperature for 2–3 min. to afford a series of spiro 6-alkynyl-2,3-dihydroquinazolin-4(1H)-ones 28.
Scheme 7.
Sonogashira cross-coupling of 25 and subsequent cyclocondensation of 26 and 27.

| n | R | %Yield | |
|---|---|---|---|
| 28a | 1 | Ph | 98 |
| 28b | 2 | Ph | 98 |
| 28c | 3 | Ph | 95 |
| 28d | 2 | -CH2CH2I | 97 |
Reagents and conditions: (i) R-C≡CH, 10% Pd/C, PPh3, CuI, NEt3, EtOH, 70 °C, 4 h; (ii) Amberlyst-15, ultrasound irradiation (40 KHz), CH3CN, r.t., 2–3 min.
The Sonogashira cross-coupling of 6-iodoquinazolinedione 29 or its NCH3-4(3H)-oxo derivative with ethyl 2-(1-butanesulfonamido)pent-4-yn-l-oate 30 in the presence of Pd(PPh3)4-CuI catalyst mixture previously afforded the cross-coupled products 31 as potent fibrinogen receptor antagonists (Scheme 8) [30].
Scheme 8.
Sonogashira cross-coupling of 29 with 30.

Reagents and conditions: (i) Pd(PPh3)4, Cul, HNEt2, 40 °C.
Sardon et al. subjected the 6-iodo-N-isopropylquinazolin-4-amines 32 to Sonogashira cross-coupling with propargyl alcohol in the presence of PdCl2(PPh3)2-1,1-bis(diphenylphosphino)ferrocene catalyst complex, CuI as co-catalyst and NEt3 in DMF at 50 °C to afford the corresponding 6-alkynylated quinazoline-4-amines 33a and 33b with selective inhibition of AurA vs. AurB (Scheme 9) [4]. Under the same reaction conditions, N-(3-fluorophenyl)-6-iodoquinazolin-4-amine also coupled with 2-methylbut-3-en-2-ol to yield the 6-alkynylated derivative in 73% yield [4].
Scheme 9.
Sonogashira cross-coupling of 32 with propargyl alcohol.

Reagents and conditions: (i) PdCl2(PPh3)2, 1,1-bis(diphenylphosphino)ferrocene, CuI, NEt3, DMF, 50 °C.
Alkynylation of 4-chloro-6,7-dimethoxyquinazoline 34 with various terminal alkynes 35 (1.5 equiv) in the presence of palladium catalyst, CuI as a co-catalyst and NEt3 as a base in DMF afforded the 4-alkynylquinazolines 36 as scaffolds for potent epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (Scheme 10) [7].
Scheme 10.
Sonogashira cross-coupling of 6,7-disubtituted-4-chloroquinazoline 34.

| X | EGFR TK, IC50 (nM) | |
|---|---|---|
| 36a | 0 | 5600 |
| 36b | -(CH2)2- | 14 |
| 36c | -CH2O- | 15 |
| 36d | -C(Me2)CH2- | 13 |
| 36e | -C(Me2)N(Me)- | 80 |
| 36f | -(CH2)3- | 3 |
Reagents and conditions: (i) Pd(PPh3)4 or PdCl2(PPh3)2, CuI, NEt3, DMF, r.t.
Sonogashira cross-coupling of the 2-substituted 4-chloroquinazolines 37 with terminal alkynes 38 using Pd(PPh3)4 and CuI in the presence of cesium carbonate (Cs2CO3) in dry DMF at room temperature afforded a series of the corresponding 4-alkynylquinazolines 39 in high yields (Scheme 11) [31].
Scheme 11.
Sonogashira cross-coupling of 37.

| R' | R'' | %Yield | |
|---|---|---|---|
| 39a | -CH3 | Ph | 90 |
| 39b | -CH3 | Cyclopropyl | 98 |
| 39c | H | Ph | 95 |
| 39d | -CF3 | Ph | 75 |
| 39e | -CF3 | Cyclopropyl | 60 |
| 39f | H | Cyclopropyl | 72 |
Reagents and conditions: (i) Pd(PPh4)3, CuI, Cs2CO3, DMF, r.t., 3 h.
Attempted Sonogashira cross-coupling of 4-chloro-2-trichloromethylquinazoline 40 with phenylacetylene 41 using triethylamine as a base, Pd(PPh3)4 as a source of the reactive Pd(0) species and CuI as a co-catalyst in THF did not afford the expected cross-coupled product [31]. The use of Cs2CO3 as a base, Pd(OAc)2 as a catalyst in DMF, on the other hand, afforded the cross-coupled product 42 in low yield (15%) along with other undesirable products 43–45 (Scheme 12). The presence of trichloromethyl group at position 2 of 40 was found to complicate the outcome of this reaction. Likewise, 4-bromo- and 4-iodo-2-trichloromethylquinazolines prepared, in turn, from 2-trichloromethyl quinazolin-4(3H)-ones using tetrabutylammonium bromide (TBABr) and tetrabutylammonium iodide (TBAI) in the presence of P2O5 in toluene under reflux were cross-coupled with cyclopropylacetylene and phenylacetylene to afford the corresponding 4-alkynyl-2-trichloromethylquinazolines in 15% and 9%, respectively [31]. These poor yields indicate that the 4-chloroquinazoline moiety is preferred over the 4-bromo- or 4-iodoquinazoline framework in the cross-coupling reactions.
Scheme 12.
Sonogashira cross-coupling of 4-chloro-2-trichloromethylquinazoline 40.

Reagents and conditions: (i) Cs2CO3, Pd(OAc)2, CuI, DMF, r.t., 3 h.
6-Tosyl-[1,2,3]-triazoloquinazoline derivative has been cross-coupled with trimethylsilylacetylene in the presence of PdCl2(PPh3)2 and CuI in DMF at 60 °C to afford the 5-trimethylsilanylethynyl-[1,2,3]-triazoloquinazoline in 88% yield [23]. Sonogashira cross-coupling of the analogous 2-arylquinazoline-4-tosylates 46 with terminal alkynes 47 (R = aryl, cyclopropyl, n-butyl or cyclohexen-1-yl) using N-heterocyclic carbenes (NHC) as ligands in the presence of Pd(0)–Cu catalyst complex recently afforded a series of 4-alkynyl-2-arylquinazolines 48 in 58%–95% yields (Scheme 13) [32]. The NHC ligands have been found to exhibit similar electronic properties to phosphines, being strongly σ-donating and weakly π-acidic and also to offer very high catalytic activity combined with stability and longevity in comparison with phosphine ligands [33].
Scheme 13.

Sonogashira cross-coupling of quinazoline-4-tosylates 46.
Sonogashira cross-coupling of 2,4-dichloroquinazoline [15] and 6-bromo-2,4-dichloroquinazoline [34] with stoichiometric amount of terminal alkynes previously led to exclusive replacement of the 4-chloro atom. Selective mono-alkynylation of 6-bromo-2,4-dichloroquinazoline 22 with tert-butyl acetylene 49 to afford the C-4 substituted product 50 in 67% yield was achieved in the presence of PdCl2(PPh3)2-CuI catalyst mixture and triethylamine as a base in THF at room temperature (Scheme 14) [17]. Likewise, Sonogashira cross-coupling of 2,4-dichloroquinazoline with 4-dimethylaminophenylacetylene (2.5 equiv.) in the presence of PdCl2(PPh3)2-CuI catalyst mixture in diisopropylamine at 70 °C for 15 h afforded the 4-alkynylated derivative, exclusively [35].
Scheme 14.
Site-selective C-4 alkynylation of 6-bromo-2,4-dichloroquinazoline 22.

Reagents and conditions: (i) NEt3, PdCl2(PPh3)2, CuI, THF, r.t., 20 h.
Ripin et al., previously subjected 4-chloro-6-iodoquinazoline 51 to BOC-protected propargylamine 52 in the presence of PdCl2(PPh3)2-CuI catalyst mixture and i-Pr2NH in THF at r.t. for 3 h followed by amination of the incipient 6-alkynylated-4-chloroquinazoline 53 to afford the alkynylated anilinoquinazoline derivative 54 (Scheme 15) [5]. However, the propargylamine used and the reaction conditions outlined in the scheme caused the target compound to differ from those described in the Experimental Section. This example, nevertheless, represents the first and only reported site-selective alkynylation involving a chloro-iodo quinazoline derivative as a substrate. This is because most of the site selectivity involving di- or trihalogeno quinazolines focus on the reactivity of the chloro-chloro or chloro-bromo substituted derivatives. Sonogashira cross-coupling of 2,4-diamino-4-iodoquinazoline with methyl 4-ethynylbenzoate catalysed by Pd(OAc)2-tris(2-tolyl)phosphine-CuI catalyst mixture in the presence of NEt3 in DMF at 60 °C for 24 h also afforded the 6-alkynylated derivative in 95% yield [36].
Scheme 15.
Site-selective alkynylation of 51 and subsequent amination of 53.

Reagents and conditions: (i) PdCl2(PPh3)2, CuI, i-Pr2NH, THF, r.t., 3 h; (ii) 3-(6-methylpyridin-3-yloxy)-2-methyl benzenamine, t-BuOH, dichloroethane, 80 °C, 1 h.
The 2-aryl-6,8-dibromo-4-chloroquinazolines 55 derived from the corresponding 2-aryl-6,8-dibromoquinazolin-4(3H)-one precursors have recently been found to undergo Sonogashira cross-coupling with terminal acetylenes (propargyl alcohol, phenylacetylene and 2-pyridinylacetylene) through exclusive replacement of the 4-chloro atom to afford the 4-alkynyl-6,7-dibromoquinazolines 56 in 53%–72% yield (Scheme 16) [37]. The increased reactivity of the C(4)-Cl bond observed for the bromo-chloro substituted quinazolines 22 and 55 is attributed to α-nitrogen effect, which makes the C-4 position highly activated than the other positions bearing Cl or Br [15,34].
Scheme 16.
Site-selective Sonogashira cross-coupling of 55.

Reagents and conditions: (i) R'C≡CH, Pd(PPh3)4, CuI, Cs2CO3, THF, r.t., 24 h.
3.5. Application of Heck Cross-Coupling in the Synthesis of Polysubstituted Quinazolinones and Quinazolines
The Heck reaction which involves Pd-catalyzed [Pd(PPh3)4, PdCl2(PPh3)2, or Pd(OAc)2] carbon-carbon bond formation through inter- or intramolecular cross-coupling reaction between organohalides or triflates with alkenes is a powerful tool for the construction of alkenyl- and aryl-substituted alkenes. 2-Substituted 6-iodoquinazolin-4(3H)-one 57 was previously reacted with unprotected allyl amidines or guanidines 58 in the presence of palladium acetate as active Pd(0) source, tri-(o-tolyl)phosphine as ligand and triethylamine in acetonitrile at 80 °C to afford a series of the 6-vinyl substituted products 59 as potential vitronectin receptor (ανβ, integrin) antagonists (Scheme 17) [38]. To our knowledge, this represents the only example of the application of the Heck cross-coupling reactions on halogenated quinazolinones to afford alkenyl-substituted derivatives.
Scheme 17.
Heck cross-coupling of 57 with allyl amidines or guanidines.

| Y | %Yield | |
|---|---|---|
| 59a | 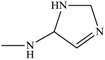 |
77 |
| 59b | 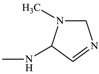 |
79 |
| 59c | 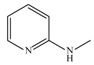 |
81 |
| 59d | 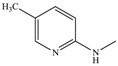 |
56 |
| 59e | 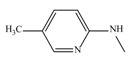 |
62 |
| 59f | 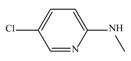 |
54 |
| 59g | 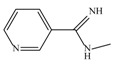 |
66 |
| 59h | 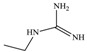 |
78 |
Reagents and conditions: (i) Pd(OAc)2, (o-tolyl)3P, NEt3, CH3CN, 80 °C.
The Heck cross-coupling reaction has, however, been applied extensively on the synthesis of poly-carbosubstituted derivatives from the corresponding halogenated quinazoline precursors. For example, initial hydro-zirconation of the terminal alkynes 60 followed by a Heck-type cross-coupling involving the reaction of the incipient alkene-like intermediate with 6,7-dialkoxy-4-iodoquinazolines 61 in THF at room temperature afforded the analogous 4-alkenyl-6,7-dialkoxyquinazolines 62 (Scheme 18) [7]. Of interest is that compounds 62 serve as epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors [7].
Scheme 18.
Heck-type cross-coupling of incipient hydro-zirconated 60 with 61.

| R | X | EGFR TK, IC50 (nM) | |
|---|---|---|---|
| 62a | -Me | -(CH2)2- | 4 |
| 62b | -Me | -C(Me)N(Et2) | 30 |
| 62c | -Me | 0 | 15 |
| 62d | -Et | -(CH2)2- | 5 |
| 62e | -Et | -(CH2)3- | 140 |
Reagents and conditions: (i) Cp2Zr(H)Cl, Pd(PPh3)4, THF, r.t.
The Heck cross-coupling of 2-(furan-2-yl)-6-iodo-N-isopropylquinazolin-4-amine 63 with tert-butyl acrylate or 2-methylbut-3-en-2-ol in the presence of Pd(OAc)2–tri-o-tolylphosphine catalyst complex and NEt3 in acetonitrile at 100 °C afforded the 6-alkenyl-4-(isopropylamino)quinazolin-6-yl)acrylates 64a and 64b in 63% and 86% yield, respectively (Scheme 19) [4]. Under the same reaction conditions, N-(3-fluorophenyl)-6-iodoquinazolin-4-amine reacted with 2-methylbut-3-en-2-ol to afford the 6-alkenylated derivative in 73% yield [4].
Scheme 19.
Heck cross-coupling of 6-iodo-4-(isopropylamino)quinazolin-6-yl)acrylates 63.

Reagents and conditions: (i) alkene, NEt3, Pd(OAc)2, (o-Tol)3P, acetonitrile, 100 °C, 48–72 h.
6-Iodo-N-phenylquinazolin-4-amine 65 was recently cross-coupled with styrene or phenylbutadiene derivatives in the presence of Pd(OAc)2-PPh3 catalyst complex and piperidine in THF under reflux to yield the corresponding 6-carbosubstituted N-phenylquinazolin-4-amines 66 albeit in low yields (Scheme 20) [38]. (E)-6-(4-Dimethylaminostyryl)-N-phenylquinazolin-4-amine 66a was found to exhibit ERBB2-induced fluorescence thus demonstrating its utility as a turn-on fluorescence kinase inhibitor in MCF7 cells [39].
Scheme 20.
Heck cross-coupling of 65 with styrene or phenylbutadiene.

| n | R | % Yield | |
|---|---|---|---|
| 66a | 1 | -N(CH3)2 | 16 |
| 66b | 1 | -OMe | 25 |
| 66c | 1 | -H | 16 |
| 66d | 1 | -CN | 20 |
| 66e | 1 | -NO2 | 24 |
| 66f | 2 | -N(CH3)2 | 16 |
| 66g | 2 | -OMe | 13 |
| 66h | 2 | -H | 14 |
| 66i | 2 | -CN | 25 |
| 66j | 2 | -NO2 | 20 |
3.6. Application of Suzuki-Miyaura Cross-Coupling in the Synthesis of Polysubstituted Quinazolinones and Quinazolines
Palladium-catalyzed Suzuki-Miyaura cross-coupling reaction between organoboron compounds and organic halides or triflates and tosylates is a general and efficient method for the formation of Csp2–Csp2 bonds [40]. This cross-coupling reaction has several advantages including functional group compatibility, low toxicity of reagents and intermediates, easy availability of boron derivatives, high thermal stability and good tolerance toward oxygen and aqueous solvents [40]. Suzuki-Miyaura cross-coupling reaction is an area of intense research for the synthesis of aryl- and arylvinyl-substituted quinazolinones and quinazolines with potential application in material and medicinal chemistry. Suzuki cross-coupling of the 2-aryl-6,8-dibromo-2,3-dihydroquinazolin-4(1H)-ones 67 with arylboronic acids yielded the corresponding 2,6,8-triaryl-2,3-dihydroquinazolin-4(1H)-ones 68 (77%–91%), which were in turn, dehydrogenated using iodine (2 equiv.) in ethanol under reflux to afford the potentially tautomeric 2,6,8-triarylquinazolin-4(3H)-ones 69 in 71%–96% yields (Scheme 21) [41]. A three layered greenish-yellow electroluminescent device based on the analogous 2-[4ꞌ-(N,N-dimethylaminophenyl)]-2,3-dihydroquinazolin-4(1H)-one as an emissive layer sandwiched between a hole transporting layer, N,N′-diphenyl-N,N′-bis(3-methylphenyl)-1,1-biphenyl-4,4'-diamine (TPD), and an electron transporting layer, tris(8-hydroxyquinolinato)aluminium (Alq3) {ITO/TPD/MAPQ/Alq3/Al} has been developed [42]. Likewise, quinazolin-4(3H)-one–based compounds exhibit interesting photophysical (electronic absorption and emission) properties [43]. The electronic absorption and emission spectra of 2-(4-methoxystyryl)-3-phenylquinazolin-4(3H)-one acquired in acetonitrile at 25 °C, for example, revealed that this compound absorbs in the ultra-violet region (λab 348 nm) and emits in the visible region at λem 485 nm [44].
Scheme 21.
Suzuki-Miyaura cross-coupling of 67 and subsequent dehydrogenation of 68.

Reagents and conditions: (i) ArB(OH)2 (2.2 equiv.), PdCl2(PPh3)2, Xphos, K2CO3, DMF-water (4/1, v/v), 120 °C, 2 h; (ii) I2 (2 equiv.), ethanol, reflux, 2 h.
The Suzuki-Miyaura cross-coupling of tetrabromobis-heterocycles 70 with arylboronic acids (4.5 equiv.) in the presence of PdCl2(PPh3)2-tricyclohexylphosphine (PCy3) catalyst complex in dioxane-water mixture (3/1, v/v) in the presence of K2CO3 under reflux afforded the corresponding tetraarylbis-heterocycles 71 in 60%–81% yield, exclusively (Scheme 22) [45]. Lack of selectivity in the cross-coupling is the consequence of equal Csp2–Br bond dissociation energies [15]. The electronic absorption and emission properties of these bis-heterocycles in dimethylsulfoxide (DMSO) and acetic acid in conjunction with quantum chemical methods showed a strong correlation with the substituents on the aryl groups and the size of the alkyl substituent on the 2-position of the quinazolin-4(1H)-one moiety as well as the substitution pattern on the propyl chain [45].
Scheme 22.
Suzuki-Miyaura cross-coupling of bis-heterocycles 70 with arylboronic acids.

Reagents and conditions: (i) ArB(OH)2 (4.5 equiv.), PdCl2(PPh3)2, PCy3, K2CO3, dioxane–water (3/1, v/v), reflux, 5 h.
Previously, the Suzuki-Miyaura cross-coupling of furfurylboronic acid and 6-iodoquinazolin-4(3H)-one catalysed by Pd(OAc)2–tri-t-butylphosphine catalyst complex afforded 5-(4-oxo-3,4-dihydroquinazolin-6-yl)furan-2-carbaldehyde as a precursor for the synthesis of Lapatinib 3 [5]. Initial Suzuki-Miyaura cross-coupling of 2-(2-furyl)-6-iodoquinazolin-4(3H)-one with 4-methoxyphenylboronic in the presence of Pd(OAc)2-K2CO3 mixture in dioxane-water (3:1, v/v) and subsequent oxidative-aromatization into the 4-chloroquinazoline followed by amination has also been described in the literature [4]. The Suzuki-Miyaura cross-coupling of 6-bromo/chloro-2-cyclopropyl-3-((pyridin-3-yl)methyl)quinazolin-4(3H)-one 72 with aryl-, heteroaryl- and alkylboronic acids previously afforded 2-cyclopropyl-6-phenyl-3-((pyridin-3-yl)methyl)quinazolin-4(3H)-one 73 as potential α-glucosidase inhibitors (Scheme 23) [46]. Several palladium catalysts were used as sources of active Pd(0) species in the presence of K3PO4 in tetrahydropyran (THP) at room temperature and among them, bis-(dicyclohexylphosphino)ferrocene]dichloropalladium(II) [PdCl2(dcpf)] and bis(di-tert-butylphosphino) ferrocene]dichloropalladium(II) [PdCl2(dtbpf)] were found to be better catalysts that produced the desired products 73 in high yields. Although THP was found to be a superior solvent than dioxane at room temperature, it involved prolonged reaction time (36 h) and some of the arylboronic acids failed to react with the halogenated substrates. Complete transformation and improved yields were observed within 8 h when dioxane was used as solvent under reflux. Likewise, the use of 6-bromoquinazoline as substrate was found to be superior to 6-chloroquinazoline in terms of yields.
Scheme 23.
Suzuki cross-coupling of 72 with ArB(OH)2.

Reagents and conditions: (i) arylboronic acid, PdCl2(dcpf), K3PO4, dioxane, r.t.
Napier et al. previously reacted the bromoquinazolinone derivative 74 with bis(pinacolato)diboron in the presence of PdCl2(dppf) to afford the boronate intermediate 75, which upon Suzuki cross-coupling with bromobenzaldehyde derivatives afforded the 2,3,6-trisubstituted quinazolin-4(3H)-ones 76 (Scheme 24) [2]. The latter were, in turn, subjected to reductive amination to afford the corresponding 2-(6-aminomethylaryl-2-aryl-4-oxoquinazolin-(3(4H)-yl)acetamides with low nanomolar affinity for the vazsopressin V1b and good selectivity with respect to related receptors V1a, V2 and OT [2].
Scheme 24.
Suzuki-Miyaura cross-coupling of 74 with bis(pinacolato)diboron.

Reagents and conditions: (i) bis(pinacolato)diboron, PdCl2(dppf)KOAc, DMF, 80 °C; (ii) bromobenzaldehydes, Pd(PPh3)4, K3PO4, DMF, 80 °C.
4-(Dimethyl/phenylaminophenyl)quinazolines were recently prepared via the Suzuki-Miyaura cross-coupling of 4-chloroquinazoline with arylboronic acids and the compounds were found to exhibit absorption bands in the UV region and to emit green light upon irradiation [35]. Kieffer et al. also subjected 4-chloro-2-trichloromethylquinazoline 40 to Suzuki-Miyaura cross-coupling with arylboronic acids (2.5 equiv.) in the presence of Pd(OAc)2 in DMF to afford the corresponding 4-aryl-2-trichloromethylquinazolines 77 (50%–65%) with antiplasmodial properties (Scheme 25) [31].
Scheme 25.
Suzuki-Miyaura cross-coupling of 4-chloro-2-trichloromethylquinazoline 40.

Reagents and Conditions: (i) ArB(OH)2 (2.5 equiv), Pd(OAc)2, Cs2CO3, DMF, reflux, 2 h.
The Suzuki-Miyaura cross-coupling of 4,7-dichloro-2-(2-methylprop-1-enyl)-6-nitroquinazoline 78 with arylboronic acids (1.5 equiv) in the presence of Na2CO3 and Pd(PPh3)4 in dimethoxyethane-ethanol mixture (9:1, v/v) under microwave irradiation afforded the C-4 substituted products 79 (Ar: 3-NO2Ph-, 5-Me-2-thienyl-, 4-ClPh-, 3-CF3Ph-) in 43%–78% yield, exclusively (Scheme 26) [47]. In the case of 4-methoxyphenyl derivative, the C-4 substituted product 79 (Ar = 4-MeOC6H4-) was only obtained in 68% yield using 1.2 equivalent of the 4-methoxyphenylboronic acid whereas the use of 1.5 equivalent afforded a mixture of 79 and the disubstituted product 80 in 45% and 15% yield, respectively. The disubstituted product 80 (Ar, Ar′ = 4-MeOC6H4-) on the other hand, was formed as the sole product in 70% yield when an excess of the 4-methoxyphenylboronic acid (4 equiv.) was used as a coupling partner. Under the same reaction conditions, 78 reacted with phenyl-, 4-fluorophenyl- and 2-tolylboronic acids to afford products 80 in 85, 71 and 65% yield, respectively (Scheme 26). Cross-coupling of 79 to afford the C-7 substituted product 80 was generally achieved using arylboronic acid (2 equiv.) in the presence of Pd(PPh3)4 and Na2CO3 in DMF/ethanol mixture (9:1, v/v) under microwave conditions for 3 h.
Scheme 26.
Suzuki-Miyaura cross-coupling of 4,7-dichloroquinazolines 78.

Reagents and Conditions: (i) ArB(OH)2, Na2CO3, Pd(PPh3)4 , DME/ethanol (9:1, v/v), MW (300 W), 80 °C, 3 h.
Although Sonogashira cross-coupling of 2,4-dichloroquinazoline with terminal acetylenes occurs exclusively at the C-4 position, these substrates were found to undergo Pd(PPh3)4 catalyzed diarylation with arylboronic acids under reflux for 24 h to afford novel (E,E)-2,4-bis(dimethyl/ phenylaminophenyl)quinazolines with absorption bands in the UV region and they emit green light upon irradiation [35]. Hitherto, regioselective Pd-catalyzed Suzuki-Miyaura cross-coupling reaction of 2,4,7-trichloroquinazoline 81 with aryl- and heterarylboronic acids was reported to favor coupling at C-4 position albeit in low yield due to competitive hydrolysis at this site [48]. The authors, in turn, temporarily deactivated the C-4 position as thioether 82 to effect regioselective Suzuki-Miyaura cross-coupling with arylboronic acid derivatives at the C-2 position to yield 83 (Scheme 27). A second Suzuki-Miyaura cross-coupling with arylboronic acids using Pd(PPh3)4–copper(I) thiophene-2-carboxylate (CuTC) catalyst complex in THF under reflux afforded the C-4 arylated product 84, which was in turn arylated at the C-7 position to afford trisubstituted quinazolines 85 [48].
Scheme 27.
Sequential site-selective Suzuki-Miyaura cross-coupling of 81.

Reagents and conditions: (i) propane-2-thiol, NaH, degassed THF, r.t.; (ii) Ar'B(OH)2, Pd(OAc)2, PPh3, Na2CO3, DME and H2O (10:1, v/v), 75 °C, N2; (iii) Ar''B(OH)2, Pd(PPh3)4, CuTC, THF, 50 °C, N2 atmosphere; (iv) Ar'''B(OH)2, Pd(OAc)2, PPh3, Na2CO3, DME:H2O (10:1, v/v), reflux.
Amination of 4-chloro-2-(furan-2-yl)-6-iodoquinazoline with isopropylamine in propan-2-ol followed by Suzuki-Miyaura cross-coupling of 2-(furan-2-yl)-6-iodo-N-isopropylquinazoline with arylboronic acids in the presence of palladium acetate and potassium carbonate in aqueous dioxane under reflux previously afforded a series of 4-amino-6-arylquinazoline [4]. An in situ C–OH activation of the 2-substituted quinazolin-4(3H)-ones 86 as tosylates followed by Suzuki-Miyaura cross-coupling with arylboronic acids previously afforded a series of 2,4-diarylquinazolines 87 in a single-pot operation (Scheme 28) [49].
Scheme 28.
Suzuki-Miyaura cross-coupling of tosylates generated in situ from 86.

| R | Ar | %Yield | |
|---|---|---|---|
| 87a | C6H5 | -C6H5 | 85 |
| 87b | -C6H5 | 4-MeC6H4- | 88 |
| 87c | -C6H5 | 4-MeOC6H4- | 90 |
| 87d | -C6H5 | 4-ClC6H4- | 79 |
| 87e | 4-MeC6H4- | -C6H5 | 83 |
| 87f | 4-MeC6H4- | 4-MeC6H4- | 85 |
| 87g | 4-MeC6H4- | 4-MeOC6H4- | 85 |
| 87h | 4-MeC6H4- | 4-ClC6H4- | 79 |
| 87i | 4-MeOC6H4- | -C6H5 | 81 |
| 87j | 4-MeOC6H4- | 4-ClC6H4- | 75 |
| 87k | 4-MeOC6H4- | 4-MeC6H4- | 83 |
| 87l | 4-ClC6H4- | -C6H5 | 85 |
| 87m | 4-ClC6H4- | 4-ClC6H4- | 80 |
| 87n | 4-ClC6H4- | 4-MeC6H4- | 90 |
| 87o | 4-ClC6H4- | 4-MeOC6H4- | 75 |
| 87p | -CH2CH2CH3 | -C6H5 | 63 |
| 87q | -CH(CH3)2 | -C6H5 | 43 |
Reagents and Conditions: (i) ArB(OH)2, K2CO3, PdCl2(PPh3)2, TsCl, THF–H2O (20:1, v/v), 60 °C.
The Suzuki-Miyaura cross-coupling of N-(3-fluorophenyl)-6-iodoquinazolin-4-amine 88 with 4-methoxyphenylboronic acid 89 using Pd(OAc)2 as a catalyst in the presence of Cs2CO3 in aqueous dioxane afforded the N-(3-fluorophenyl)-6-(4-methoxyphenyl)quinazolin-4-amine 90 in 70% yield (Scheme 29) [4]. Cross-coupling of the analogous 6-iodo-N-phenylquinazolin-4-amine with arylboronic acids in the presence of PdCl2(PPh3)2-PPh3 catalyst complex, Cs2CO3 as a base in DMF under reflux for 24 h afforded the corresponding 6-aryl-N-phenylquinazolin-4-amines albeit in low yields [39]. These compounds were found to exhibit ERBB2-induced fluorescence and to represent a turn-on probe that can report binding to the kinase domain in organic solutions [39].
Scheme 29.
The Suzuki cross-coupling reaction of N-(3-fluorophenyl)-6-iodoquinazolin-4-amine.

Reagents and Conditions: (i) Cs2CO3, Pd(OAc)2, dioxane–water (3:1, v/v), reflux, 30 min.
Initial Suzuki-Miyaura cross-coupling of 4-amino-6-iodoquinazoline 91 and furfurylboronic acid in the presence of 10% Pd/C and triethylamine in ethanol under reflux (i) or through C–H activation using PdCl2-KOAc mixture (ii) previously afforded the 4-anilino-6-furfurylquinazoline 92, a precursor for the synthesis of Lapatinib 3 (Scheme 30) [5].
Scheme 30.
Pd/C catalysed Suzuki-Miyaura cross-coupling of 91.

Reagents and conditions: (i) 10% Pd/C, NEt3, EtOH, 60 °C; (ii) PdCl2, KOAc.
A series of 2-aryl-6,8-dibromo-4-(arylethynyl)quinazolines 56 was, in turn, subjected to the Suzuki-Miyaura cross-coupling with arylboronic acids in the presence of PdCl2(PCy3)2 and K2CO3 in dioxane under reflux for 4 h to afford the corresponding 2,6,8-triaryl-4-(2-phenylethynyl)quinazolines 93 with potential photophysical properties (Scheme 31) [37]. Lack of selectivity for the cross-coupling was attributed to comparable Csp2–Br bond strengths based on the literature precedents on the analogous 2-aryl-6,8-dibromo-4-methoxyquinolines [50] and tribromoquinoline [51].
Scheme 31.
Suzuki-Miyaura cross-coupling of 56 with arylvinylboronic acid.

Reagents and conditions: (i) ArB(OH)2, PdCl2(PCy3)2, K2CO3, dioxane, reflux, 4 h.
Microwave-assisted consecutive bis-SNAr/bis-Suzuki cross-coupling reaction involving initial amination of 6,8-dibromo-2,4-dichloroquinazoline 94 afforded 6,8-dibromo-N2,N4-bis(3-chlorophenyl) quinazoline-2,4-diamine 95, which upon cross-coupling with arylboronic acids (2 equiv.) yielded the corresponding 2,4,6,8-tetrasubstituted quinazolines 96 without selectivity (Scheme 32) [52]. Moreover, the authors performed these consecutive steps in a single-pot operation to afford products 96, exclusively.
Scheme 32.
Single-pot amination and Suzuki-Miyaura cross-coupling of 94.

Reagents and conditions: (i) 3-ClC6H4NH2, DME,-EtOH (9:1, v/v), MW, 100 °C; (ii) ArB(OH)2, PdCl2(PPh3)2, K2CO3, H2O, MW, 100 °C, 1 h.
3.7. Application of Buchwald-Hartwig Cross-Coupling in the Synthesis of Polyheteroatom-Substituted Quinazolinones
The Buchwald-Hartwig involves the direct C–N or C–O bond formation between aryl- or heteroaryl halides and amines or alcohols in the presence of stoichiometric amount of a base and palladium as a catalyst. This reaction was previously applied on 6-bromo- and 6-chloro-2-cyclopropyl-3-(pyridyl-3-methyl)quinazolin-4(3H)-ones 97 using a variety of aryl, heteraryl and alkyl amines as coupling partners in the presence of Pd2(dba)3–DavePhos catalyst complex and t-BuONa in 1,4-dioxane at 100 °C to afford the corresponding 6-aminated derivatives 98 in moderate to high yields (Scheme 33) [53].
Scheme 33.
Palladium catalyzed amination of 97.

Reagents and conditions: (i) Pd2(dba)3, DavePhos, t-BuONa, dioxane, 100 °C, 8 h.
Buchwald-Hartwig cross-coupling of 6-bromoquinazolin-4(3H)-one 99 with morpholine or 1-(2-fluorophenyl)piperizine in the presence palladium acetate-xantphos catalyst complex and potassium tert-butoxide in dioxane under reflux recently afforded the 6-(morpholin-4-yl)quinazolin-4(3H)-ones 100a (X = O) and 100b (X = 2-FC6H4N-) in 89% and 88% yield, respectively (Scheme 34) [54]. Under the same reaction conditions, the 3-(4-methoxybenzyl)- and 3-benzyl-6-bromobenzo[h]quinazolin-4(3H)-one derivatives afforded the corresponding 6-(morpholin-4-yl)benzo[h]quinazolin-4(3H)-one derivatives in 45% and 40% yield, respectively. The 3-(4-methoxybenzyl)-6-(morpholin-4-yl)benzo[h]quinazolin-4(3H)-one was found to be highly toxic against HT29 cancer cells (IC50 = 4.12 μM) [54].
Scheme 34.
Buchwald-Hartwig amination of quinazolin-4(3H)-one 102.

Reagents and conditions: (i) morpholine or 1-(2-fluorophenyl)piperizine, Pd(OAc)2, t-BuOK, toluene, 100 °C, 20 h.
Baarlaam et al. performed the amination of 3-(p-methoxybenzyl) protected 8-bromo-6-fluoro-2-methylquinazolino-4(3H)-one 101 with primary and secondary amines without the use of transition metal catalyst and isolated 6-amino substituted products 102 as major or sole products (Scheme 35) [55]. A halogen atom (X = Br, I) meta to the fluorine leaving group is envisioned to exert a sufficient inductive effect to permit the substitution under standard laboratory conditions. However, this reaction could not be extended to the alkoxides due to deprotonation and deactivation of the quinazoline ring towards substitution [55].
Scheme 35.
Regioselective C-6 amination of 101.

| R', R'' | %Yield 102 | %Yield 103 | |
|---|---|---|---|
| a | 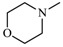 |
68 | 12 |
| b | 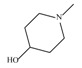 |
79 | 9 |
| c | 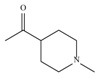 |
68 | 7 |
| d | 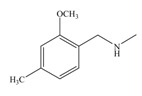 |
45 | - |
| e | 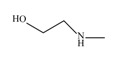 |
41 | - |
| f | 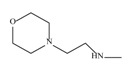 |
43 | - |
| g | 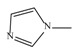 |
39 | - |
Reagents and conditions: (i) R'R''NH, 140 °C, 3 days.
3.8. Palladium Catalyzed Cyanation in the Synthesis of Polysubstituted Quinazolines
Cyanation of aromatic halides in the presence of palladium(0) catalyst source has proven to be superior to the Rosemund-Von Braun reaction which employs stoichiometric amount of CuCN under high temperature conditions accompanied by tedious work-up [56]. An excess of the strong σ-donor cyanide ion which has high affinity for palladium tend to poison the catalyst and, in turn, retard the progress of reaction [56]. A microwave assisted cyanation of 8-bromo-6-fluoro-2-methylquinazolino-4(3H)-one 104 using zinc cyanide in the presence of tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3)–xanthphos catalyst complex afforded the C8–CN substituted product 105, exclusively (Scheme 36) [55]. Zn(CN)2 in this case serves as a source of low concentration of cyanide ion during the reaction to avoid poisoning the catalyst.
Scheme 36.
Palladium catalyzed C-8 cyanation of 104 with zinc cyanide.

Reagents and conditions: (i) Zn(CN)2, Pd2(dba)3, xanthphos, DMA, 160 °C MW, 10 min.
A series of 2-substituted 4-tosylquinazolines 106 was recently subjected to copper(I) cyanide (2 equiv.) in the presence of palladium(II) acetate–bis(diphenylphosphino)ferrocene (DPPF) catalyst complex and cesium carbonate in toluene under reflux to yield the corresponding 4-cyanoquinazoline 107 in moderate to high yields (Scheme 37) [57]. Under the same conditions, 4-chloro-2-(4-methoxyphenyl)quinazoline was found to be less reactive and to afford the corresponding 4-cyanoquinazoline in 11% yield.
Scheme 37.
Palladium catalyzed cyanation of 4-tosylquinazolines with CuCN.

Reagents and conditions: (i) CuCN, Pd(OAc)2, DPPF, Cs2CO3, toluene, 80 °C, 8–12 h.
The nitrile group offers a useful functionality for subsequent manipulations to important functional groups such as hydrolysis into acids, hydration into amides, reduction into amines or aldehydes, and cycloaddition into various heterocycles.
4. Conclusions and Perspective
The observed reactivity of the dihalogenated quinazolinones in metal catalyzed C-C bond formation follows a similar trend to that observed for the other aryl/heteroaryl halides (C-I > C-Br >> C-Cl), whereby selective coupling occurs with the more intrinsically reactive iodides or bromides in the presence of chlorides. The C(4)-Cl bond of quinazoline derivatives, on the other hand, is highly activated than the other positions bearing Cl or Br due to α-nitrogen effect and in general, the oxidative addition of Pd(0) to a C(4)-Cl bond occurs easily at room temperature without the use of specialized and expensive ligands. Moreover, the 4-chloroquinazoline moiety is preferred over the 4-bromo- or 4-iodoquinazoline framework in the cross-coupling reactions [31]. Despite the successes in site-selective or sequential metal-catalyzed halogen substitution reactions, the application of this strategy towards the synthesis of polysubstituted quinazolinones and quinazolines from the corresponding di- or trihalogenated precursors in a single-pot operation remains a challenge. In our view, this could be realized through the use of di- or trihalogenated quinazolinones or quinazolines bearing mixed halogen atoms to increase the diversity of substitution on the heterocyclic scaffold/s to afford compounds with interesting biological or photophysical properties. Heterocyclic compounds with intramolecular charge transfer properties continue to attract considerable attention for potential applications in organic electroluminescent diodes, organic solar cells, polarity probes and nonlinear optics. Polycarbo-substituted quinazolinones and quinazoline derivatives comprise electron-deficient heterocyclic scaffolds (quinazolinone or quinazoline framework) as electron-acceptors linked directly to the aryl ring or through π-conjugated bridge/s to comprise donor-π-acceptor systems with intramolecular charge transfer properties. Given the need for efficient methods for the incorporation of quinazolinone and quinazoline moieties in pharmaceutical compounds or materials, it can be expected that growth and exciting new advances will continue in this important subdomain of metal-catalyzed cross-coupling research.
Acknowledgments
Financial support from to the University of South Africa and the National Research Foundation (NRF) in SA is gratefully acknowledged.
Author Contributions
Authors contributed equally.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Rudolph J., Esler W.P., O’Connor S., Coish P.D.G., Wickens P.L., Brands M., Bierer D.E., Bloomquist B.T., Bondar G., Chen L., et al. Quinazolinone derivatives as orally available Ghrelin receptor antagonists for the treatment of diabetes and obesity. J. Med. Chem. 2007;50:5202–5216. doi: 10.1021/jm070071+. [DOI] [PubMed] [Google Scholar]
- 2.Napier S.E., Letourneau J.J., Ansari N., Auld D.S., Baker J., Best S., Campbell-Wan L., Chan R., Craighead M., Desai H., et al. Synthesis and SAR studies of novel 2-(6-aminomethylaryl-2-aryl-4-oxo-quinazolin-3(4H)-yl)acetamide Vasopressin V1b receptor antagonists. Bioorg. Med. Chem. Lett. 2011;21:3813–3817. doi: 10.1016/j.bmcl.2011.04.022. [DOI] [PubMed] [Google Scholar]
- 3.Zhu X., Wu L., Qiao H., Han T., Chen S., Liu X., Jiang R., Wei Y., Feng D., Zhang Y., et al. Autophagy stimulates apoptosis in HER2-overexpressing breast cancers treated by lapatinib. J. Cell. Biochem. 2013;114:2643–2653. doi: 10.1002/jcb.24611. [DOI] [PubMed] [Google Scholar]
- 4.Sardon T., Cottin T., Xu J., Giannis A., Vernos I. Development and biological evaluation of a novel aurora A kinase inhibitor. ChemBioChem. 2009;10:464–478. doi: 10.1002/cbic.200800600. [DOI] [PubMed] [Google Scholar]
- 5.Ripin D.H.B., Bourassa D.E., Brandt T., Castaldi M.J., Frost H.N., Hawkins J., Johnson P.J., Masset S.S., Neumann K., Phillips J., et al. Evaluation of kilogram-scale Sonagashira, Suzuki, and Heck coupling routes to Oncology candidate CP-724,714. Org. Process Res. Dev. 2005;9:440–450. doi: 10.1021/op050039u. [DOI] [Google Scholar]
- 6.Bernotas R.C., Ullrich J.W., Travins J.M., Wrobel J.E., Unwalla R.J. Preparation of quinazoline compounds as modulators of Liver X receptors (LXRs) WO2009020683 A2. 2009 Feb 12;
- 7.Kitano Y., Suzuki T., Kawahara E., Yamazaki T. Synthesis and inhibitory activity of 4-alkynyl and 4-alkenylquinazolines: Identification of new scaffolds for potent EGFR tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2007;17:5863–5867. doi: 10.1016/j.bmcl.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 8.De Vries J.G. Palladium-catalysed coupling reactions. Top. Organomet. Chem. 2012;42:1–34. [Google Scholar]
- 9.Connelly D.J., Cusack D., O’Sullivan T.P., Guiry P.J. Synthesis of quinazolinones and quinazolines. Tetrahedron. 2005;61:10153–10202. doi: 10.1016/j.tet.2005.07.010. [DOI] [Google Scholar]
- 10.Besson T., Chosson E. Microwave-assisted synthesis of bioactive quinazolines and quinazolinones. Comb. Chem. High Throughput Screen. 2007;10:903–917. doi: 10.2174/138620707783220356. [DOI] [PubMed] [Google Scholar]
- 11.Wang D., Gao F. Quinazoline derivatives. Synthesis and bioactivities. Chem. Cent. J. 2013;7:95. doi: 10.1186/1752-153X-7-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patil D.A., Patil P.O., Deshmukh P.K., Patil G.B., Shewale B.D., Patil D.D., Gattani S.G. Synthesis of 2,3-disubstituted quinazolin-4(3H)-ones. Res. J. Pharm. Technol. 2010;3:979–1003. [Google Scholar]
- 13.Grushin V.V., Alper H. Transformation of chloroarenes, catalysed by transition metal complexes. Chem. Rev. 1994;94:1047–1062. doi: 10.1021/cr00028a008. [DOI] [Google Scholar]
- 14.Hassan J., Sévignon M., Gozzi C., Schulz E., Lemaire M. Aryl-aryl bond formation one century after the discovery of the Ullmann reaction. Chem. Rev. 2002;102:1359–1469. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- 15.Garcia Y., Schoenebeck F., Legault C.Y., Merlic C.A., Houk K.N. Theoretical bond dissociation energies of halo-heterocycles: Trends and relationships to regioselectivity in palladium-catalyzed cross-coupling reactions. J. Am. Chem. Soc. 2009;131:6632–6639. doi: 10.1021/ja9004927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mangalagiu I., Benneche T., Undheim K. Trialkalalanes in palladium-catalyzed chemo- and regioselective alkylations. Tetrahedron Lett. 1996;37:1309–1312. doi: 10.1016/0040-4039(95)02381-X. [DOI] [Google Scholar]
- 17.Charpiot B., Brun J., Donze I., Naef R., Stefani M., Mueller T. Quinazolines: Combined type 3 and 4 phosphodiesterase inhibitors. Bioorg. Med. Chem. Lett. 1998;8:2891–2896. doi: 10.1016/S0960-894X(98)00508-3. [DOI] [PubMed] [Google Scholar]
- 18.Hintermann L., Xiao L., Labonne A. A general and selective copper-catalyzed cross-coupling of tertiary Grignard reagents with azacyclic electrophiles. Angew. Chem. Int. Ed. 2008;47:8246–8250. doi: 10.1002/anie.200803312. [DOI] [PubMed] [Google Scholar]
- 19.Rueping M., Ieawsuwan W. A manganese-catalyzed cross-coupling reaction. Synlett. 2007;2:247–250. doi: 10.1055/s-2007-968013. [DOI] [Google Scholar]
- 20.Negishi E. Palladium- or nickel-catalyzed cross coupling. A new selective method for carbon-carbon bond formation. Acc. Chem. Res. 1982;15:340–348. doi: 10.1021/ar00083a001. [DOI] [Google Scholar]
- 21.Knochel P., Singer R.D. Preparation and reactions of polyfunctional organozinc reagents in organic synthesis. Chem. Rev. 1993;93:2117–2188. doi: 10.1021/cr00022a008. [DOI] [Google Scholar]
- 22.Legros J.-Y., Primault G., Fiaud J.-C. Syntheses of acetylquinolines and acetylisoquinolines via palladium-catalyzed coupling reactions. Tetrahedron. 2001;57:2507–2514. doi: 10.1016/S0040-4020(01)00076-X. [DOI] [Google Scholar]
- 23.Jones P., Chambers M. Rapid analogue synthesis of C-5 substituted 1,2,3-triazolo[1,5-a]quinazolines. Tetrahedron. 2002;58:9973–9981. doi: 10.1016/S0040-4020(02)01351-0. [DOI] [Google Scholar]
- 24.Boulton L.T., Fox M.E., Hodgson P.B., Lennon I.C. Zinc-mediated intramolecular acyl and imino transfer reactions of aryl iodides. Tetrahedron Lett. 2005;46:983–986. doi: 10.1016/j.tetlet.2004.12.034. [DOI] [Google Scholar]
- 25.Krasovsky A., Knochel P.A. A LiCl-mediated Br/Mg exchange reaction for the preparation of functionalized aryl- and heteroarylmagnesium compounds from organic bromides. Angew. Chem. Int. Ed. 2004;43:3333–3336. doi: 10.1002/anie.200454084. [DOI] [PubMed] [Google Scholar]
- 26.Stille J.K. The Palladium-catalyzed cross-coupling reactions of organotin reagents with organic electrophiles. Angew. Chem. Int. Ed. 1986;25:508–524. doi: 10.1002/anie.198605081. [DOI] [Google Scholar]
- 27.Achelle S., Plé N., Kreher D., Attias A.-J., Arfaoui L., Charra F. Star-shaped ethynylpyrimidine with long alkoxyl side chains: Synthesis, fluorescence and 2D self-assembling. Tetrahedron Lett. 2009;50:7055–7058. doi: 10.1016/j.tetlet.2009.09.169. [DOI] [Google Scholar]
- 28.Sonogashira K. Development of Pd-Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides. J. Organomet. Chem. 2002;653:46–59. doi: 10.1016/S0022-328X(02)01158-0. [DOI] [Google Scholar]
- 29.Rambabu D., Kumar S.K., Sreenivas B.Y., Sandra S., Kandale A., Misra P., Rao M.V., Pal M. Ultrasound-based approach to spiro-2,3-dihydroquinazolin-4(1H)-ones: Their in vitro evaluation against chorismate mutase. Tetrahedron Lett. 2013;54:495–501. doi: 10.1016/j.tetlet.2012.11.057. [DOI] [Google Scholar]
- 30.Liverton N.J., Armstrong D.J., Claremon D.A., Remy D.C., Baldwin J.J., Lynch R.J., Zhang G., Gould R.J. Nonpeptide glycoprotein IIb/IIIa inhibitors: Substituted quinazolinediones and quinazolinones as potent fibrinogen receptor antagonists. Bioorg. Med. Chem. Lett. 1998;8:483–186. doi: 10.1016/S0960-894X(98)00047-X. [DOI] [PubMed] [Google Scholar]
- 31.Kieffer C., Verhaeghe P., Primas N., Castera-Ducros C., Gellis A., Rosas R., Rault S., Rathelot P., Vanelle P. Sonogashira cross-coupling reaction in 4-chloro-2-trichloromethylquinazoline series is possible despite a side dimerization reaction. Tetrahedron. 2013;69:2987–2995. doi: 10.1016/j.tet.2013.01.094. [DOI] [Google Scholar]
- 32.Peng Y., Huang P., Wang Y., Zhou Y., Yuan J., Yang Q., Jiang X., Deng Z., Xu J. Synthesis of 4-alkynylquinazolines: Pd-Cu-cocatalyzed coupling of quinazoline-4-tosylates with terminal alkynes using N-heterocyclic carbenes as ligands. Org. Biomol. Chem. 2014;12:5922–5927. doi: 10.1039/C4OB00700J. [DOI] [PubMed] [Google Scholar]
- 33.Kantchev E.A.B., O’Brien C.J., Organ M.G. Palladium complexes of N-heterocyclic carbenes as catalysts for cross-coupling reactions—A synthetic chemist’s perspective. Angew. Chem. Int. Ed. 2007;46:2768–2813. doi: 10.1002/anie.200601663. [DOI] [PubMed] [Google Scholar]
- 34.Mangalagiu I., Benneche T., Undheim K. Ethenylation and alkynylation in palladium-catalyzed carbosubstitution in heteroazines. Acta Chem. Scand. 1996;50:914–917. doi: 10.3891/acta.chem.scand.50-0914. [DOI] [Google Scholar]
- 35.Achelle S., Rodríguez-López J., Robin-le Guen F. Synthesis and photophysical studies of a series of quinazoline chromophores. J. Org. Chem. 2014;79:7564–7571. doi: 10.1021/jo501305h. [DOI] [PubMed] [Google Scholar]
- 36.Harris N.V., Smith C., Bowden K. A simple synthesis of 5,8,10-trideazaminopterin analogs. Synlett. 1990:577–578. [Google Scholar]
- 37.Mphahlele M.J., Paumo H.K., El-Nahas A.M., El-Hendawy M.M. Synthesis and photophysical property studies of the 2,6,8-triaryl-4-(phenylethynyl)quinazolines. Molecules. 2014;19:795–818. doi: 10.3390/molecules19010795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lawson E.C., Kinney W.A., Luci D.K., Yabut S.C., Wisnoski D., Maryanoff B.E. The Heck reaction with unprotected allylic amidines and guanidines. Tetrahedron Lett. 2002;43:1951–1953. doi: 10.1016/S0040-4039(02)00182-X. [DOI] [Google Scholar]
- 39.Dhuguru J., Liu W., Gonzalez W.G., Babinchak W.M., Miksovska J., Landgraf R., Wilson J.N. Emission tuning of fluorescent kinase inhibitors: Conjugation length and substituent effects. J. Org. Chem. 2014;79:4940–4947. doi: 10.1021/jo500520x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyaura N. Organoboron compounds. Top. Curr. Chem. 2002;219:11–59. [Google Scholar]
- 41.Mphahlele M.J., Maluleka M.M., Khoza T.A. Suzuki cross-coupling of the 2-aryl-6,8-dibromo-2,3-dihydroquinazolin-4(1H)-ones and transformation of the resulting 2,6,8-triaryl-2,3-dihydroquinazolin-4(1H)-ones. Bull. Chem. Soc. Ethiop. 2014;28:81–90. [Google Scholar]
- 42.Wang S., Lou H., Liu Y., Yu G., Lu P., Zhu D. Greenish-yellow electroluminescent devices using a novel dihydroquinazolinone derivative as emitting layer. J. Mater. Chem. 2001;11:2971–2973. doi: 10.1039/b105102b. [DOI] [Google Scholar]
- 43.Bakalova S.M., Santos A.G., Timcheva I., Kaneti J., Filipova I.L., Dobrikov G.M., Dimitrov V.D. Electronic absorption and emission spectra and computational studies of some 2-aryl, 2-styryl, and 2-(4′-aryl)butadienyl quinazolin-4-ones. J. Mol. Struct. Theochem. 2004;710:229–234. doi: 10.1016/j.theochem.2004.07.037. [DOI] [Google Scholar]
- 44.Nosova E.V., Stupina T.V., Lipunova G.N., Valova M.S., Slepukhin P.A., Charushin V.N. 3-Phenyl/pyridinyl derivatives of trans-2-(aryl/heteryl)vinyl-3H-quinazolin-4-ones: Synthesis and fluorescent properties. Int. J. Org. Chem. 2012;2:56–53. doi: 10.4236/ijoc.2012.21010. [DOI] [Google Scholar]
- 45.Mmonwa M.M., Mphahlele M.J., El-Hendawy M.M., El-Nahas A.M., Koga N. Synthesis and photophysical properties of the 2-(3-(2-alkyl-6,8-diaryl-4-oxo-1,2,3,4-tetrahydroquinazolin-2-yl)propyl)-6,8-diarylquinazolin-4(3H)-ones. Molecules. 2014;19:9712–9735. doi: 10.3390/molecules19079712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garlapati R., Pottabathini N., Gurram V., Kasani K.S., Gundla R., Thulluri C., Machiraju P.K., Chaudhary A.B., Addepally U., Dayam R., et al. Development if α-glucosidase inhibitors by room temperature C–C cross couplings of quinazolinones. Org. Biomol. Chem. 2013;11:4778–4791. doi: 10.1039/c3ob40636a. [DOI] [PubMed] [Google Scholar]
- 47.Kabri Y., Verhaeghe P., Gellis A., Vanelle P. Regioselective Suzuki-Miyaura reaction: Application to the microwave-promoted synthesis of 4,7-diarylquinazoline. Molecules. 2010;15:2949–2961. doi: 10.3390/molecules15052949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wipf P., George K.M. Regioselective palladium-catalyzed cross-coupling reactions of 2,4,7-trichloroquinazoline. Synlett. 2010:644–648. doi: 10.1055/s-0029-1219391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qiu G.Q., Huang P., Yang Q., Lu H., Xu J., Deng Z., Zhang M., Peng Y. Synthesis of 4-arylquinazolines by arylation of quinazolin-4-ones under mild conditions. Synthesis. 2013;45:3131–3136. doi: 10.1055/s-0033-1339848. [DOI] [Google Scholar]
- 50.Khoza T.A., Maluleka M.M., Mama N., Mphahlele M.J. Synthesis and photophysical properties of the 2-aryl-6,8-bis(arylethenyl)-4-methoxyquinolines. Molecules. 2012;17:14186–14204. doi: 10.3390/molecules171214186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akrawi O.A., Mohammed H.H., Langer P. Synthesis and Suzuki-Miyaura reaction of 3,6,8-tribromoquinoline: A structural revision. Synlett. 2013;24:1121–1124. doi: 10.1055/s-0032-1316904. [DOI] [Google Scholar]
- 52.Kabri Y., Crozet D., Redon S., Vanelle P. One-pot chemoselective synthesis of 2,4,6,8-tetrasubstituted quinazolines via microwave-assiusted consecutive bis-SNAr/bis-Suzuki–Miyaura cross-coupling reactions. Synthesis. 2014;46:1613–1620. doi: 10.1055/s-0033-1341105. [DOI] [Google Scholar]
- 53.Garlapati R., Pottabathini N., Gurram V., Chaudhary A.B., Chunduri V.R., Patro B. Pd catalyzed amination of 6-halo-2-cyclopropyl-3-(pyridyl-ylmethyl)quinazolin-4(3H)-one. Tetrahedron Lett. 2012;53:5162–5166. doi: 10.1016/j.tetlet.2012.07.061. [DOI] [Google Scholar]
- 54.Nowak M., Malinowski Z., Jozwiak A., Fornal E., Blaszczyk A., Kontek R. Substituted benzoquinazolinones. Part 1: Synthesis of 6-aminobenzo[h]quinazolinones via Buchwald-Hartwig amination from 6-bromobenzo[h]quinazolinones. Tetrahedron. 2014;70:5153–5160. doi: 10.1016/j.tet.2014.05.117. [DOI] [Google Scholar]
- 55.Barlaam B., Haris C.S., Lecoq J., Nguyen H.T.H. Preparation of 6-aminoquinazolin-4(3H)-ones via direct SNAr on the quinazoline ring. Tetrahedron. 2012;68:534–543. doi: 10.1016/j.tet.2011.11.008. [DOI] [Google Scholar]
- 56.Wen Q., Jin J., Zhang L., Luo Y., Lu P., Wang Y. Copper-mediated cyanation reactions. Tetrahedron Lett. 2014;55:1271–1280. doi: 10.1016/j.tetlet.2014.01.032. [DOI] [Google Scholar]
- 57.Zhao X., Zhou Y., Yang Q., Xie Y., Ding Q., Deng Z., Zhang M., Xu J., Peng Y. Palladium-catalyzed cyanation of quinazoline-4-tosylates for access to 4-CN–functionalized quinazolines. Synthesis. 2013;45:3245–3250. doi: 10.1055/s-0033-1339918. [DOI] [Google Scholar]