

Abstract
Serine/arginine‐rich splicing factor 1 (SRSF1) has been linked to various human cancers including pediatric acute lymphoblastic leukemia (ALL). Our previous study has shown that SRSF1 potentially contributes to leukemogenesis; however, its underlying mechanism remains unclear. In this study, leukemic cells were isolated from pediatric ALL bone marrow samples, followed by immunoprecipitation assays and mass spectrometry analysis specific to SRSF1. Subcellular localization of the SRSF1 protein and its mutants were analyzed by immunofluorescence staining. Cell growth, colony formation, cell apoptosis, and the cell cycle were investigated using stable leukemic cell lines generated with lentivirus‐mediated overexpressed WT or mutant plasmids. Cytotoxicity of the Tie2 kinase inhibitor was also evaluated. Our results showed the phosphorylation of SRSF1 at tyrosine 19 (Tyr‐19) was identified in newly diagnosed ALL samples, but not in complete remission or normal control samples. Compared to the SRSF1 WT cells, the missense mutants of the Tyr‐19 phosphorylation affected the subcellular localization of SRSF1. In addition, the Tyr‐19 phosphorylation of SRSF1 also led to increased cell proliferation and enhanced colony‐forming properties by promoting the cell cycle. Remarkably, we further identified the kinase Tie2 as a potential therapeutic target in leukemia cells. In conclusion, we identify for the first time that the phosphorylation state of SRSF1 is linked to different phases in pediatric ALL. The Tyr‐19 phosphorylation of SRSF1 disrupts its subcellular localization and promotes proliferation in leukemia cells by driving cell‐cycle progression. Inhibitors targeting Tie2 kinase that could catalyze Tyr‐19 phosphorylation of SRSF1 offer a promising therapeutic target for treatment of pediatric ALL.
Keywords: acute lymphoblastic leukemia, childhood, splicing factor SRSF1, Tie2 kinase, tyrosine phosphorylation
Abbreviations
- ALL
acute lymphoblastic leukemia
- BM
bone marrow
- CR
complete remission
- IP
immunoprecipitation
- ITP
immune thrombocytopenia
- ND
newly diagnosed
- NC
negative control
- PI
propidium iodide
- PRMT1
protein arginine methyltransferase 1
- PTM
post‐translational modification
- RRM1/2
RNA recognition motif 1/2
- SRSF1
serine/arginine‐rich splicing factor 1
1. INTRODUCTION
Acute lymphoblastic leukemia is the most common malignancy and a major cause of cancer‐related mortality in children. In the past two decades, remarkable progress has been made in the treatment of pediatric ALL. The long‐term survival rates have approached 85%‐90% in developed countries, which are attributed to accurate diagnostic stratification and optimal risk‐directed polychemotherapy. Despite favorable outcomes for most pediatric ALL, disease relapse still occurs in approximately 10%‐15% of children, leading to therapeutic failure and death.1 Therefore, better understanding of the pathogenesis in pediatric leukemia is urgently needed to break through such a therapeutic bottleneck.
Serine/arginine‐rich splicing factor 1 is a prototypical serine‐rich protein. In addition to constitutive and alternative splicing,2 SRSF1 plays many essential roles in regulating mRNA stability, nuclear export, nonsense‐mediated mRNA decay, translation, and mRNA processing.2, 3, 4, 5 Serine/arginine‐rich splicing factor 1 is also connected to genomic instability, cell‐cycle arrest, and apoptosis.6 These diverse biological functions of SRSF1 are mainly due to its structure, the strict regulation of expression or PTMs.7 Serine/arginine‐rich splicing factor 1 is composed of a classical conserved RNA recognition motif (RRM1), a short linker domain, a second pseudo RNA recognition motif (RRM2), and an RS domain. Multiple lines of experiments have indicated that SRSF1 is a shuttling protein dependent on the phosphorylation state in the RS domain and arginine methylation in the link region between RRM1 and RRM2.8, 9, 10
Owing to its upregulation in various cancers, SRSF1 has been regarded as a proto‐oncoprotein.11, 12, 13 In mice, ectopic expression of SRSF1 is able to drive the oncogenic transformation of fibroblasts and epithelial cells through enhanced proliferation and compromised apoptosis.12, 14 Indeed, our previous study showed that SRSF1 is highly expressed in pediatric ALL; knockdown of SRSF1 in leukemia cells results in an increase in early cell apoptosis and shows sensitivity of leukemia cells to chemotherapeutic drugs, suggesting a potential oncogenic role of SRSF1.15 Interestingly, we discovered a close link between SRSF1 and PRMT1 in leukemogenesis.15 Protein arginine methyltransferase 1 can methylate SRSF1 at arginine 93, 97, and 109 in the G‐Hinge region in vitro. Blocking methylation of 3 arginine results in the cytoplasmic accumulation of SRSF1 and consequent changes of its biological functions in cells.10 Nevertheless, other PTMs such as phosphorylation might be involved in the pathogenesis of ALL as well. Disordered phosphorylation of SRSF1 has a causative effect on proto‐oncogenes. For example, SRSF1 can be activated following hyperphosphorylation at serines 199, 201, 227, and 234, mediating therapeutic resistance of non‐small‐cell lung cancer.16, 17 Another report showed that the phosphorylation of SRSF1 regulates alternative splicing of tumor‐related Rac1b, which is overexpressed in colon tumor and required to maintain tumor cell survival.18 Although many studies have documented the serine phosphorylation of SRSF1,19, 20, 21, 22, 23, 24 there has been a lack of study on tyrosine phosphorylation of SRSF1. Given that protein tyrosine phosphorylation controls a wide variety of cellular events, including cell proliferation and differentiation,25, 26 we then asked whether tyrosine phosphorylation of SRSF1 is involved in leukemogenesis.
Using mass spectrometry analysis, we identified for the first time that the phosphorylation state of SRSF1 is linked to different phases of pediatric ALL. Notably, the phosphorylation of SRSF1 at Tyr‐19 residue controls the subcellular localization of SRSF1 and promotes cell proliferation by accelerating the cell‐cycle progression. These data provide a novel posttranslational marker in pediatric ALL. Furthermore, we identified the tyrosine kinase Tie2 as a potential therapeutic target; loss of function of the Tyr‐19 phosphorylation of SRSF1 shows chemoresistance to the Tie2 kinase inhibitor, suggesting that phosphorylation of SRSF1 at Tyr‐19 residue might be a part of the survival mechanisms for ALL.
2. MATERIALS AND METHODS
2.1. Patient samples
Bone marrow samples from 7 patients were collected randomly, including 4 ND patients and 3 in CR. All patients were diagnosed with ALL using a combination of morphology, immunology, cytogenetics, and molecular biology. Six samples (ND = 3 [patients #1‐3] and CR = 3 [patients #5‐7]) were used for mass spectrometry following IP with an anti‐SRSF1 Ab (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and 1 sample (patient #4) was used for IP with an antiphosphotyrosine Ab (PY20) (Abcam, Cambridge, UK). The characteristics of these patients are described in detail in Table S1. Three immune ITP samples were used as normal controls. Informed consent was obtained from patients, guardians or patients.
2.2. Cell culture
Nalm‐6, a pre‐B ALL cell line, was cultured in RPMI‐1640 medium (Gibco, Waltham, MA, USA). Nalm‐6 stable cell lines were generated with lentivirus‐mediated overexpressed WT or mutant SRSF1 with 0.5% puromycin selection (Sigma‐Aldrich, St. Louis, MO, USA). 293T and HeLa cell lines were cultured in DMEM (Gibco). All mediums were supplemented with 10% (v/v) FBS (Gibco), and cells were incubated at 37°C in a humidified incubator containing 5% CO2.
2.3. Western blot analysis
Western blot analyses were carried out as described previously.15 Proteins were probed using an anti‐SRSF1 Ab overnight at 4°C. After washing with TBS‐T, the membranes were incubated with a secondary Ab (1:5000; Pierce, Rockford, IL, USA) for 45 minutes at room temperature. The proteins were visualized using an enhanced chemiluminescence kit (Amersham, Arlington Heights, IL, USA).
2.4. Immunoprecipitation assays
Mononuclear cells of BM samples were isolated by Ficoll gradient centrifugation (TBD, Tianjin, China). Cells were washed with PBS and then incubated on ice for 30 minutes in RIPA buffer (20 mmol/L Tris, 150 mmol/L NaCl, 2 mmol/L Na3VO4, 10 mmol/L NaF, 1 mmol/L EDTA, 0.1% Triton X‐100, Proteinase Inhibitor Cocktail [Roche, Branchburg, NJ, USA], PhosSTOP [Roche], and 10 mmol/L PMSF). Then 1.0 μg anti‐SRSF1 Ab or anti‐PY20 was used for binding overnight at 4°C to undertake IP using 1.0 mg cell lysates, as described previously.15 The IP was evaluated by standard western blot analysis as described above.
2.5. Coomassie brilliant blue staining and mass spectrometry
The samples from IP were separated by SDS‐PAGE, and protein bands were visualized by staining with Coomassie blue. Stained bands were excised and digested in gel with trypsin, and the tryptic peptides were analyzed by an LTQ Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) coupled online to an Easy‐nLC 1000 (Thermo Fisher Scientific) in the data‐dependent mode. Phosphopeptides were identified by searching the Homo sapiens database from UniProt using the software Proteome Discoverer (version 1.4; Thermo Fisher Scientific, Waltham, MA, USA).
2.6. Plasmid construction
SRSF1‐WT encoding human WT SRSF1 was subcloned into the EcoRI and SalI sites of the pEGFP‐C2 vector to construct the pEGFP‐C2‐SRSF1‐WT plasmid, which was further used as a template to construct pEGFP‐C2‐SRSF1‐Y19D and pEGFP‐C2‐SRSF1‐Y19F by site‐directed mutagenesis. The Tyr‐19 of SRSF1 was altered to aspartic acid (D) or phenylalanine (F) using the primers as follows: 5′aacgattgccgcatcgacgtgggtaactt3′ and 5′gtcgatgcggcaatcgttgttccctgcgg3′; or 5′aacgattgccgcatcttcgtgggtaactt 3′ and 5′gaagatgcggcaatcgttgttccctgcgg3′.
The PCR was carried out with the following cycling parameters: initial denaturation at 95°C for 30 seconds, 18 cycles of denaturation at 95°C for 30 seconds, annealing at 55°C for 1 minute, and extension at 68°C for 5 minutes. The PCR amplification products were digested with the DpnI (Takara, Otsu, Japan) (2.5 μL 10× buffer, 0.5 μL DpnI and 100 ng in 22 μL DNA) at 37°C for 3 hours to destroy parental and hemiparental plasmids. The plasmids were purified using an Endo‐free Plasmid Mini Kit II (Omega, Atlanta, GA, USA) according to the manufacturer's instructions. We replaced the EcoRI‐BamHI fragment in pCDH‐EF1‐CMV‐T2A‐puro vector with the corresponding fragment from pEGFP‐C2‐SRSF1‐WT, pEGFP‐C2‐SRSF1‐Y19D, and pEGFP‐C2‐SRSF1‐Y19F to construct pCDH‐EF1‐MCS‐T2A‐puro‐SRSF1‐WT, pCDH‐EF1‐MCS‐T2A‐puro‐SRSF1‐Y19D, and pCDH‐EF1‐MCS‐T2A‐puro‐SRSF1‐Y19F, respectively. The plasmids were verified by sequencing.
2.7. Transfection and infection assays
The pEGFP‐C2‐SRSF1‐WT, pEGFP‐C2‐SRSF1‐Y19D, or pEGFP‐C2‐SRSF1‐Y19F plasmid was transiently transfected in HeLa cells. The empty vector was used as a negative control using LipoMax (Sudgen, Beijing, China) according to the manufacturer's instructions.
PCDH‐EF1‐MCS‐T2A‐puro‐SRSF1‐WT or its mutants were used to cotransfect 293T cells together with two packaging vectors, PAX and PMD. Viral supernatants were collected 48 and 72 hours after transfection and purified with the 0.45‐μm filter prior to infection. Nalm‐6 cells (2 × 104) were plated in 6‐well plates and transduced by spinoculation (1500 g, 90 minutes, 32°C) with virus‐containing supernatants for 2 rounds. The cells were cultured with medium containing 0.5% puromycin 48 hours after the infection to selected stable cell lines. Stable clones were assessed with western blot analysis 4 weeks after selection.
2.8. Immunofluorescence staining
Cells were washed with PBS 36 hours after transfection. They were fixed with 4% paraformaldehyde for 20 minutes at 4°C and DAPI (1:1000; Beyotime, Shanghai, China) was applied to stain nuclei. Immunofluorescence analysis of localization was carried out using a confocal microscope.
2.9. Cell viability assays
Nalm‐6 stable cells overexpressing WT or mutant SRSF1 were plated at a density of 2 × 103 cells per 100 μL in a 96‐well plate in triplicate and cultured for 0, 24, 48, 72, or 96 hours. Viable cells were tested using the MTS assay (Cell Titer 96 Aqueous One Solution reagent; Promega, Madison, WI, USA). The relative survival rate was normalized to the values at 0 hour. For cytotoxicity assays, cells (2 × 103) were treated with different doses (0‐200 μmol/L) of Tie2 kinase inhibitor (Selleck, Houston, TX, USA) for 72 hours, and cells with only media were used as negative controls. Viable cells were tested using the MTS assay referred to above. Blank media only readouts were used as baseline blank controls. The relative cell viabilities were calculated by the following formula: cell viability = dosing/negative control × 100%.
2.10. Colony formation assays
To undertake colony‐forming assays, 1 mL complete medium containing 1.2% agarose was added to each well of 12‐well plates. Five hundred Nalm‐6 stable cells were suspended in 1 mL complete medium containing 0.7% agarose and were plated over the layer of the agarose‐containing medium. To explore the inhibition of clone formation, medium was supplemented with Tie2 kinase inhibitor (10 μmol/L). These plates were kept at 4°C for 10 minutes to allow solidification and then incubated at 37°C in a humidified atmosphere containing 5% CO2. After 2 weeks, the colonies were fixed in methanol, stained with 0.1% crystal violet (Sigma‐Aldrich), and the colonies consisting of ≥50 cells were counted under an inverted phase‐contrast microscope.
2.11. Cell apoptosis assay
Cells (2 × 105) were treated with 50 μmol/L of cytarabine (Pharmacia & Upjohn, Kalamazoo, MI, USA), or 50 μmol/L normal saline as the negtive control (NC). Cells were harvested at 24 hours after treatment. Apoptosis was detected using an FITC Annexin V/PI Kit (BD, San Jose, CA, USA), and cells were analyzed immediately by flow cytometry (FACS Aria II; BD). Annexin V+/PI− cells identified early apoptosis.
2.12. Cell cycle analysis
Cells were washed with PBS and then fixed with ice‐cold 70% ethanol at 4°C for 24 hours. Samples were then stained with PI (50 μg/mL; Sigma‐Aldrich) containing RNase (100 μg/mL; Sigma‐Aldrich) for 30 minutes at 37°C, followed by flow cytometry analysis.
2.13. RNA sequencing
Total RNA was extracted from the Nalm‐6 stable cells overexpressing WT SRSF1, mutants (SRSF1‐Y19D or SRSF1‐Y19F) or the empty vector. Sequencing libraries were generated using a NEBNext Ultra RNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA) and sequenced on an Illumina HiSeq 4000 platform (Illumina Inc., San Diego, CA, USA). The 150‐bp paired‐end reads were generated and aligned to the reference genome (Homo_sapiens.GRCh38.87.chr) using Hierarchical Indexing for Spliced Alignment of Transcripts 2 (2.1.0; http://ccb.jhu.edu/software/hisat2/index.shtml). High‐throughput sequencing (0.6.0; https://htseq.readthedocs.io/en/release_0.9.1/overview.html#paper) was used to count the read numbers, then the fragments per kilobase million value of each gene was calculated based on the length of the gene and read count mapped to this gene. Differential expression analysis was carried out using Identify Differentially Expressed Genes from RNA‐seq data (1.18.0; http://www.bioconductor.org/packages/release/bioc/html/DEGseq.html). Alternative splicing was analyzed by Asprofile (b‐1.0.4; http://ccb.jhu.edu/software/ASprofile/). The full‐sequence dataset was deposited in the GEO database (accession no. GSE118727).
2.14. Statistical analysis
All experiments were repeated at least 3 times. Statistical analyses were undertaken using GraphPad Prism version 6 (GraphPad Software Inc., San Diego, CA, USA). Data are presented as the mean ± SEM. Data were analyzed using an unpaired t test for comparisons of two cohorts. One‐way ANOVA was used to analyze three or more group comparisons. P < .05 was considered as significant.
3. RESULTS
3.1. Serine/arginine‐rich splicing factor 1 is phosphorylated at Tyr‐19 residue in newly diagnosed pediatric ALL samples
To determine the clinical relevance of our findings, we first completed a comprehensive analysis using leukemic cells isolated from primary pediatric ALL BM samples. The percentage of blast cells in the BM is at least 85% when diagnosed. We accumulated SRSF1 protein by IP with an anti‐SRSF1 Ab in ND ALL, CR, and ITP samples. The protein bands were visualized by staining with Coomassie blue (Figure 1A). Stained bands were further excised for mass spectrometry analysis. As shown in Figure 1B, the phosphorylation of Tyr‐19 was further identified in 2 ND samples, but not in CR or ITP samples (Figure 1B). To confirm this finding, we examined the tyrosine phosphorylation state of SRSF1 accumulated by IP with an antiphosphotyrosine Ab (PY20) in cells from 1 ND sample. The result showed SRSF1 is phosphorylated at the tyrosine residues, supporting our mass spectrometry analysis (Figure 1C).
Figure 1.
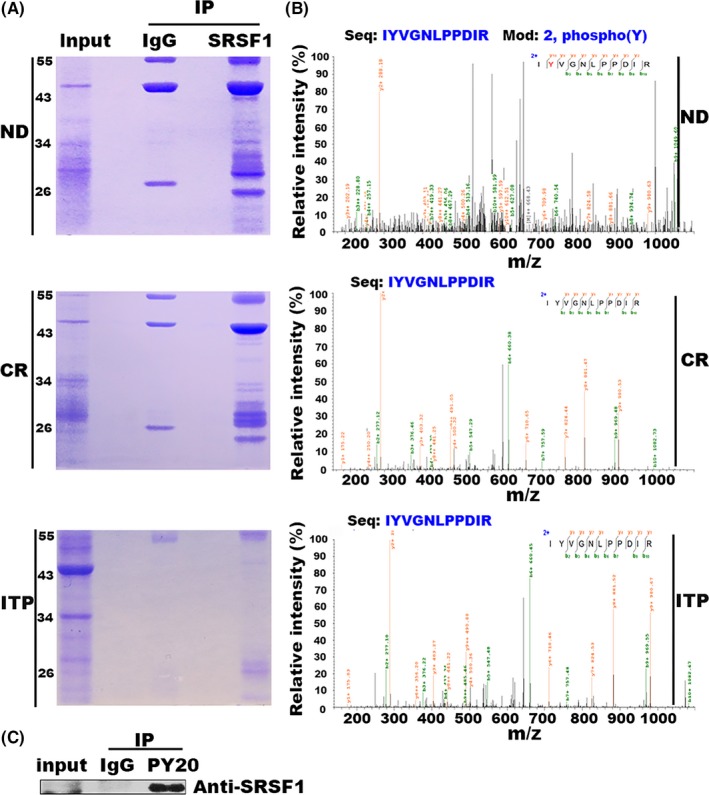
Identification of tyrosine 19 (Tyr‐19) phosphorylation of serine/arginine‐rich splicing factor 1 (SRSF1). A, SRSF1 protein cumulated by immunoprecipitation (IP) from acute lymphoblastic leukemia samples (newly diagnosed [ND] or complete remission [CR]) or immune thrombocytopenia (ITP) samples was separated by SDS‐PAGE, followed by Coomassie brilliant blue staining. ITP was used as a normal control. Anti‐IgG Ab was used as a negative control. B, Tyr‐19 phosphorylation of SRSF1 was identified in ND samples, but not in CR or ITP samples by mass spectrometry. C, IP was carried out in an acute lymphoblastic leukemia ND sample with an antiphosphotyrosine Ab (PY20), and an anti‐IgG Ab was used as a negative control. The result indicates that endogenous SRSF1 was phosphorylated at the tyrosine residues
3.2. Phosphorylation at Tyr‐19 residue affects the subcellular localization of SRSF1
As the biological function of SRSF1 is tightly controlled by its subcellular localization, we then asked whether Tyr‐19 phosphorylation of SRSF1 influences its localization and induces subsequent effects. To address this issue, we constructed a WT plasmid for SRSF1 (SRSF1‐WT). Next, we mutated the Tyr‐19 (Y19) of SRSF1 into the phenylalanine (F) (SRSF1‐Y19F) to prevent phosphorylation, and into the aspartic acid (D) (SRSF1‐Y19D) to retain the phosphorylation (Figure 2A).27, 28, 29 The subcellular localization of SRSF1‐WT and the missense mutants were detected using indirect immunofluorescence and a GFP‐tag after the transient transfection in HeLa cells. As shown in Figure 2B, SRSF1‐WT and mutant proteins shuttled between the nucleus and cytoplasm. Compared to SRSF1‐WT, the percentage of pancellular distribution was increased in the cells transfected with the SRSF1‐Y19D plasmid, but decreased in the cells transfected with the SRSF1‐Y19F (Figure 2C), suggesting that the Tyr‐19 phosphorylation, to some extent, promotes SRSF1 to locate both in the nucleus and cytoplasm.
Figure 2.
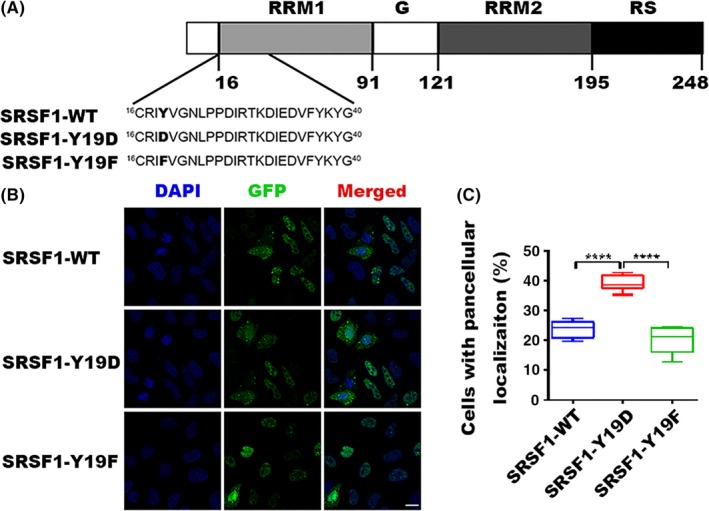
Localization of WT and mutant serine/arginine‐rich splicing factor 1 (SRSF1). A, Modular structure of SRSF1 and mutants in RNA recognition motif 1 (RRM1). The tyrosine 19 residue (Y19, shown in in boldface type) is phosphorylated, and we mutated it to aspartic acid (D) or phenylalanine (F). B, Indirect immunofluorescence of HeLa cells transfected WT or mutant GFP‐tagged SRSF1. Panels in the right column show the merged images of GFP and DAPI signals. C, Statistics for cells with pancellular (both nucleus and cytoplasm) localization of SRSF1. Three independent experiments were carried out and 3 views were chosen randomly in each experiment. Bar represents SEM. Total number of GFP‐positive cells is 5000 for GFP‐tagged WT or mutant SRSF1. ****P < .0001
3.3. Tyrosine 19 phosphorylation of SRSF1 promotes cell proliferation in Nalm‐6 cells
We next questioned the implication of phosphorylated SRSF1 at Tyr‐19 in leukemia cells. We first generated lentivirus‐mediated SRSF1‐WT and mutant stable cell lines using a pre‐B ALL cell line, Nalm‐6. The expression of SRSF1 in these cell lines was evaluated by western blot (Figure 3A). We then utilized WT and mutant leukemic cells to evaluate the effects of SRSF1 on cell growth. The empty vector‐transfected leukemic cells were used as controls. Enhanced SRSF1, including SRSF1‐WT and the mutants, significantly increased cell viability in Nalm‐6 compared to the control cells. Further analysis showed that SRSF1‐Y19D played a more intensive role in promoting cell growth than other types (Figure 3B). In addition, SRSF1‐Y19D also led to enhanced colony‐forming properties compared to other stable cell lines. (Figure 3C,D), suggesting that phosphorylated SRSF1 at Tyr‐19 contributes to the oncogenic mechanisms, such as proliferation, in ALL.
Figure 3.
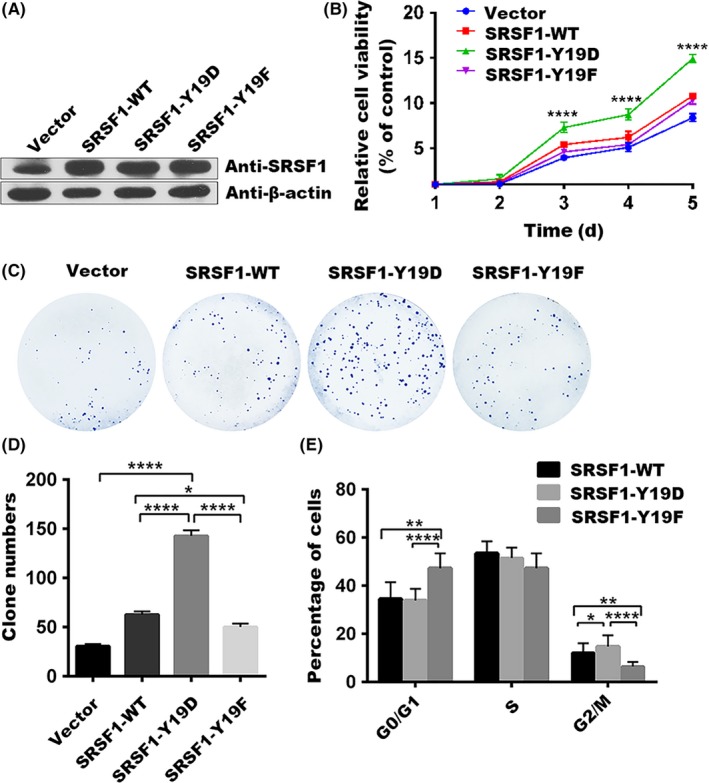
Tyrosine 19 phosphorylation of serine/arginine‐rich splicing factor 1 (SRSF1) promotes proliferation of Nalm‐6 cells by accelerating the cell cycle. A, Nalm‐6 stable cells generated with lentivirus‐mediated overexpressed WT or mutant SRSF1 were assessed with western blotting. B, Cell proliferation abilities were compared among the Nalm‐6 stable cells overexpressing the empty vector, SRSF1‐WT, or its mutants with MTS assays. Point, mean of three independent experiments; bar represents SEM. ****P < .0001. C,D, Colony formation abilities were compared among Nalm‐6 stable cells overexpressing the empty vector, SRSF1‐WT, or its mutants with clone formation assays, and the number of clones were counted. Column, mean of 3 independent experiments; bar represents SEM. Each experiment performed in triplicate. ****P < .0001. E, Proportion of cells at G0/G1, G2/M, and S phase were assessed with flow cytometry and plotted for quantification. The percentage of Nalm‐6 stable cells overexpressing SRSF1‐WT or its mutants in each stage of the cell cycle is shown. Column, mean of 3 independent experiments; bar represents SEM. Each experiment performed in triplicate. *P < .05, **P < .01, ****P < .0001
3.4. Tyrosine 19 phosphorylation of SRSF1 accelerates cell cycle in Nalm‐6 cells
To explore the mechanism of how Tyr‐19 phosphorylation of SRSF1 regulates the proliferation of Nalm‐6 cells, we examined the cell‐cycle distribution by flow cytometry. The results showed that SRSF1‐Y19F resulted in cell‐cycle arrest in the G0/G1 phase, whereas SRSF1‐Y19D led to a significant increase of cells in G2/M phase (Figure 3E), indicating that Tyr‐19 phosphorylation of SRSF1 enhances cell‐cycle progression, thereby leading to dominant growth of leukemic cells.
3.5. Tyrosine 19 phosphorylation of SRSF1 has no effect on cell apoptosis
In our previous study, we reported that SRSF1 plays an antiapoptotic role in Nalm‐6 cells following chemotherapy.15 This feature allowed us to investigate whether Tyr‐19 phosphorylation of SRSF1 leads to cell apoptosis following treatment. We introduced cytarabine or NS into the stable cell lines. Cellular apoptosis was evaluated at 24 hours after cytarabine or NS treatment. The FACS analyses based on annexin V/PI staining revealed a significant cytarabine‐induced apoptosis in the control cells, whereas SRSF1‐WT cells showed a decreased apoptosis, consistent with our previous work. However, there was no difference in cell apoptosis between SRSF1‐WT and other phosphorylated types (Figure 4). These data suggest that cell apoptosis upon chemotherapies in Nalm‐6 cells may not due to the Tyr‐19 phosphorylation of SRSF1.
Figure 4.
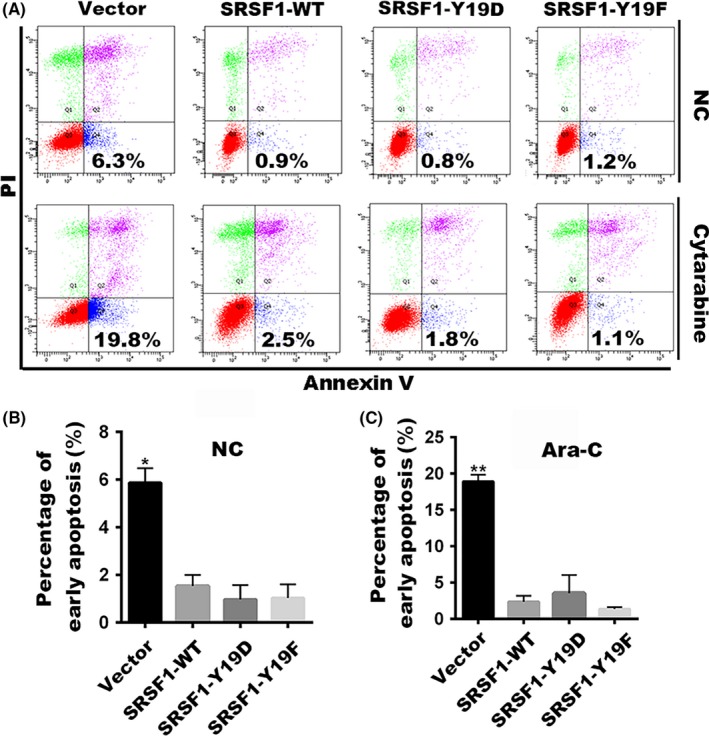
Tyrosine 19 phosphorylation has no influence on cell apoptosis. A, The percentage of early apoptotic cells (annexin V+/propidium iodide−) with cytarabine or the negative control (NC) treatment was shown. B,C, Differences in the percentages of early apoptosis cells were compared among Nalm‐6 stable cells overexpressing the empty vector, SRSF1‐WT, or its mutants treated with NS or cytarabine. Column, mean of 3 independent experiments were repeated; bar represents SEM. Each experiment performed in triplicate. *P < .05, **P < .01
3.6. Tyrosine kinase Tie2 could be a potential therapeutic target in leukemic cells
Mounting evidence has suggested that nearly 50% of the 90 human tyrosine kinases contribute to human cancers.30 Imatinib and gefitinib are the successful examples of tyrosine kinase inhibitor therapies for cancers.31, 32, 33 In light of the findings above, we sought to predict potential specific tyrosine kinases using three websites: PPSP (http://ppsp.biocuckoo.org/), NetPhos (http://www.cbs.dtu.dk/services/NetPhos/), and GPS (http://gps.biocuckoo.org/). Interestingly, the tyrosine kinase Tie2 was highlighted twice with a strong possibility to phosphorylate the Tyr‐19 residue.
To confirm this novel target, we next undertook MTS assays in stable cell lines using a specific Tie2 kinase inhibitor. As shown in Figure 5A, Tie2 kinase inhibitor triggered cell death in leukemic cells generated with empty vectors, SRSF1‐WT or SRSF1‐Y19F in a dose‐dependent manner. Among them, SRSF1‐WT transfected cells showed more sensitivity to Tie2 kinase inhibition (Figure 5B), and SRSF1‐Y19F cells showed resistant to the Tie2 kinase inhibitor compared to the empty vector‐transfected cells. The same is true in the colony formation assay, which showed more intensive effect of the Tie2 kinase inhibitor on the colony‐forming properties of SRSF1‐WT cells (Figure 5C,D). These results suggest that the tyrosine phosphorylation, especially the phosphorylation of SRSF1 at Tyr‐19, plays an important role in the dominant growth and survival of leukemic cells.
Figure 5.
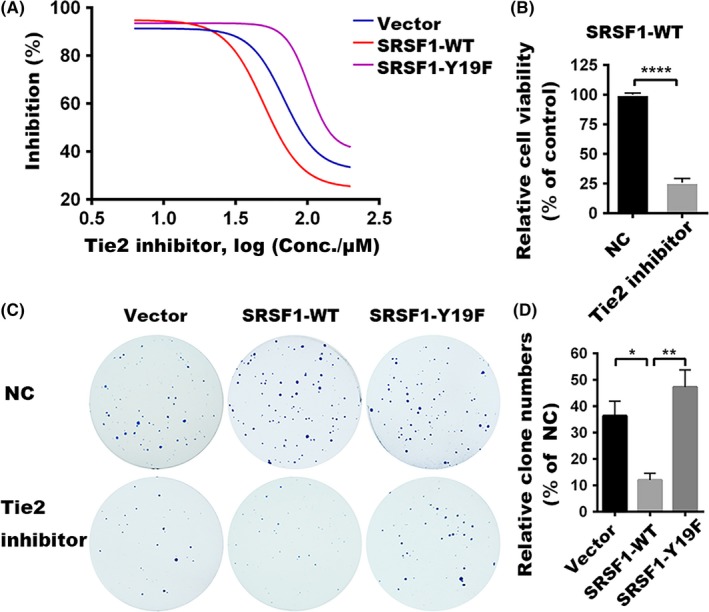
Cytotoxicity of Tie2 kinase inhibitor. A, Cytotoxicity assays were undertaken with varying concentrations (0‐200 μmol/L) of Tie2 kinase inhibitor. Relative cell viabilities were plotted in GraphPad Prism 6 using a variable slope logistic curve. B, Viabilities of Nalm‐6 stable cells overexpressing SRSF1‐WT were compared between cells treated with Tie2 inhibitor (200 μmol/L) and the negative control (NC). C, Colony formation assays were undertaken with the treated Tie2 inhibitor (10 μmol/L), cells treated with DMSO were used as NC. D, Number of clones was counted. Inhibition rates of colony formation between Tie2 inhibitor and NC groups for each stable cell were compared. Bar represents SEM. Each experiment performed in triplicate. *P < .05, **P < .01
4. DISCUSSION
Although Tyr phosphorylation represents <1% of the phosphoproteome,34 perturbations in Tyr phosphorylation underlie many human diseases, especially in cancers.26 In the current study, we, for the first time, identified the phosphorylation of SRSF1 at Tyr‐19 residue in ND samples with pediatric ALL, implying that this phosphorylation site is very likely related to disease progression. The Tyr‐19 residue presents in the RRM1 domain that is required for oncogenic activity of SRSF1, and the RRM2 and RS domains are dispensable for this activity.35
Serine/arginine‐rich splicing factor 1 is a shuttling protein that shows predominant nuclear localization in the steady state.36 The movement of SRSF1 among cellular compartments is tightly controlled. The phosphorylation state of SRSF1 at the RS domain is crucial for the normal function of SRSF1 by modulating the subcellular localization.8, 9 Here, we identified a novel posttranslational signal that affects the subcellular localization of SRSF1. As shown, the Tyr‐19 phosphorylation of SRSF1 promotes itself localized both in the nucleus and in the cytoplasm, which is quite different from the SRSF1‐WT that is predominantly localized in the nucleus. This dysregulation suggests that Tyr‐19 phosphorylation could disturb the cellular distribution of SRSF1 in both the cytosolic and nuclear compartments.
In the nucleus, the splicing function of SRSF1 has been extensively characterized. For example, cell apoptotic regulators (bridging integrator 1, BCL2 like 11, intracellular adhesion protein D, and BCL2 family apoptosis regulator),14, 37, 38 and the factors involving cell proliferation and cell‐cycle progression (ribosomal protein S6 kinase B1, MAP kinase interacting serine/threonine kinase 2, cyclin D1, and macrophage stimulating 1 receptor) have been identified to participate in SRSF1‐regulated alternative splicing events in tumorigenesis.12, 38, 39, 40 In addition, the relative concentration of SRSF1 in the nucleus is another important determinant in alternative splicing regulation.41, 42, 43 Thus, the disordered distribution of SRSF1 induced by Tyr‐19 phosphorylation is very likely to affect the alternative splicing profiles of its target gene transcripts, thus leading to leukemogenesis. The RNA sequencing analysis of the stable leukemic cell lines overexpressing SRSF1‐WT or mutants showed changes in the alternative splicing of certain target genes (Table S2). For example, alteration of MST1R (Ron) splicing is involved. A Ron isoform, called ΔRon, has been identified in certain tumors and correlates with the metastatic phenotype that might promote the proliferation.39, 44 The antiapoptotic isoforms of cell apoptotic regulators including CASP8‐L, CASP9‐b, and Rac1b were also upregulated following Tyr‐19 phosphorylation.18, 45, 46 However, our result showed that Tyr‐19 phosphorylation of SRSF1 has no effect on cell apoptosis; other mechanisms are likely to reverse the antiapoptosis effect.
In the cytoplasm, SRSF1 is reported to activate mTOR signaling, which is a major contributor to tumor growth and survival.47 The hyperactive mTORC1 pathway promotes the phosphorylation of its substrates S6K and 4E‐BP1 to enhance translation,48, 49, 50, 51 which further contributes to tumorigenesis by elevating transcription factors with a high turnover, such as the oncoprotein c‐myc and others.52, 53, 54 Moreover, SRSF1‐mediated oncogenesis is attributed to the activation of the Raf‐MEK‐ERK signaling pathway, which impinges on all the functional hallmarks of cancer cells. A nuclear‐retained SRSF1 chimeric protein fails to activate these signaling pathways, and it plays few roles in tumor formation, suggesting that its activation is mainly attributed to the cytosolic part of SRSF1.35 In addition, a large number of genes are increased by the Try‐19 phosphorylation of SRSF1 (Table S3): chemokine (C‐X‐C) ligand 1 (CXCL1), activating transcription factor 5 (ATF5), and ATP/GTP binding protein‐like 2 (AGBL2). Studies showed elevated CXCL1 is associated with tumor progression and poor prognosis in cancers;55, 56 and the overexpression of ATF5 or AGBL2 enhances malignancy in cancer cells.57, 58 The mechanisms of SRSF1 within different cellular compartments need to be further studied.
Dysregulation of tyrosine phosphorylation of proteins leads to disordered signaling pathways and contributes to oncogenic malignancies.59 Using the MTS and colony formation assays, we found that enhanced SRSF1, especially the Tyr‐19 phosphorylation of SRSF1, promotes cell growth and proliferation in Nalm‐6 cells by accelerating the cell cycle, suggesting the Tyr‐19 phosphorylation is most likely to activate oncogenic pathways.
Based on our discoveries of Tyr‐19 phosphorylation in SRSF1, it is logical to question its implication in ALL therapy. In the present study, Tie2 was predicted with a strong possibility to phosphorylate the Tyr‐19 residue using the online tools. Tie2, a receptor tyrosine kinase, is aberrantly expressed in various human cancers.60, 61, 62, 63, 64, 65 Tie2 induces cell proliferation and migration in human papillary thyroid carcinoma through the PI3K/AKT pathway.66 Tie2 inhibition elicits angiosarcoma growth delay through enhanced apoptosis of tumor cells.67 It has also been correlated with poor prognosis in myelodysplastic syndromes.68, 69 A phase I study with ARRY‐614, a dual inhibitor of p38 MAPK and Tie2, is underway in patients with myelodysplastic syndromes.70 Based on our results, the phosphorylation of SRSF1 at Tyr‐19 markedly increases the proliferation and enhances colony‐forming properties of Nalm‐6 cells, which can be reversed by the Tie2 kinase inhibitors. Supported by this evidence, targeting Tie2 kinase could offer a promising therapeutic strategy for the treatment of ALL.
Taken together, we identified for the first time that the phosphorylation state of SRSF1 is linked to different phases in ALL. Our results underscore that phosphorylation of SRSF1 at the Tyr‐19 residue disrupts subcellular localization of SRSF1 and promotes cell proliferation in leukemic cells by accelerating cell‐cycle progression. The Tie2 kinase is able to catalyze Tyr‐19 phosphorylation of SRSF1 and offers a promising therapeutic target for the treatment of pediatric ALL. These findings undoubtedly add a new layer in understanding of how posttranslational mechanisms support leukemia progression. Future work exploring the mechanisms involved in this process will likely reveal more connections between PTMs and the pathogenesis of pediatric ALL.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
This work was supported by the Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Grants (No. ZY201404), the Beijing Municipal Administration of Hospitals DengFeng Program (No. DFL20151101), and the Capital Health and Development of Special Grant (No. 2016‐1‐2091). We would like to thank the families and each of the pediatric ALL patients who participated in this study. We also thank Ting Li for flow cytometry experiments, and members of the Bao's laboratory for helpful discussions.
Xu L, Zhang H, Mei M, et al. Phosphorylation of serine/arginine‐rich splicing factor 1 at tyrosine 19 promotes cell proliferation in pediatric acute lymphoblastic leukemia. Cancer Sci. 2018;109:3805–3815. 10.1111/cas.13834
Contributor Information
Shilai Bao, Email: slbao@genetics.ac.cn.
Huyong Zheng, Email: zhenghuyong@bch.com.cn.
REFERENCES
- 1. Pui CH, Yang JJ, Hunger SP, et al. Childhood acute lymphoblastic leukemia: progress through collaboration. J Clin Oncol. 2015;33:2938‐2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285‐298. [DOI] [PubMed] [Google Scholar]
- 3. Zhang Z, Krainer AR. Involvement of SR proteins in mRNA surveillance. Mol Cell. 2004;16:597‐607. [DOI] [PubMed] [Google Scholar]
- 4. Sanford JR, Gray NK, Beckmann K, Caceres JF. A novel role for shuttling SR proteins in mRNA translation. Genes Dev. 2004;18:755‐768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang Y, Gattoni R, Stevenin J, Steitz JA. SR splicing factors serve as adapter proteins for TAP‐dependent mRNA export. Mol Cell. 2003;11:837‐843. [DOI] [PubMed] [Google Scholar]
- 6. Das S, Krainer AR. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol Cancer Res. 2014;12:1195‐1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gonçalves V, Jordan P. Posttranscriptional regulation of splicing factor SRSF1 and its role in cancer cell biology. Biomed Res Int. 2015;2015:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blaustein M, Pelisch F, Tanos T, et al. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat Struct Mol Biol. 2005;12:1037‐1044. [DOI] [PubMed] [Google Scholar]
- 9. Cazalla D, Zhu J, Manche L, Huber E, Krainer AR, Caceres JF. Nuclear export and retention signals in the RS domain of SR proteins. Mol Cell Biol. 2002;22:6871‐6882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sinha R, Allemand E, Zhang Z, Karni R, Myers MP, Krainer AR. Arginine methylation controls the subcellular localization and functions of the oncoprotein splicing factor SF2/ASF▿†. Mol Cell Biol. 2010;30:2762‐2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gout S, Brambilla E, Boudria A, et al. Abnormal expression of the pre‐mRNA splicing regulators SRSF1, SRSF2, SRPK1 and SRPK2 in non small cell lung carcinoma. PLoS One. 2012;7:e46539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto‐oncogene. Nat Struct Mol Biol. 2007;14:185‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anczuków O, Akerman M, Cléry A, et al. SRSF1‐regulated alternative splicing in breast cancer. Mol Cell. 2015;60:105‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anczuków O, Rosenberg AZ, Akerman M, et al. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol. 2012;19:220‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zou L, Zhang H, Du C, et al. Correlation of SRSF1 and PRMT1 expression with clinical status of pediatric acute lymphoblastic leukemia. J Hematol Oncol. 2012;5:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shultz JC, Goehe RW, Wijesinghe DS, et al. Alternative splicing of caspase 9 is modulated by the phosphoinositide 3‐kinase/Akt pathway via phosphorylation of SRp30a. Can Res. 2010;70:9185‐9196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goehe RW, Shultz JC, Murudkar C, et al. hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase‐9 pre‐mRNA processing. J Clin Investig. 2010;120:3923‐3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gonçalves V, Henriques A, Pereira J, et al. Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor‐related Rac1b in colorectal cells. RNA. 2014;20:474‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hagopian JC, Ma CT, Meade BR, et al. Adaptable molecular interactions guide phosphorylation of the SR protein ASF/SF2 by SRPK1. J Mol Biol. 2008;382:894‐909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huynh N, Ma CT, Giang N, et al. Allosteric interactions direct binding and phosphorylation of ASF/SF2 by SRPK1. Biochemistry. 2009;48:11432‐11440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bullock AN, Das S, Debreczeni JE, et al. Kinase domain insertions define distinct roles of CLK kinases in SR protein phosphorylation. Structure. 2009;17:352‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu S, Kang K, Zhang J, et al. A novel Physarum polycephalum SR protein kinase specifically phosphorylates the RS domain of the human SR protein, ASF/SF2. Acta Biochim Biophys Sin. 2009;41:657‐667. [DOI] [PubMed] [Google Scholar]
- 23. Ma CT, Velazquez‐Dones A, Hagopian JC, Ghosh G, Fu XD, Adams JA. Ordered multi‐site phosphorylation of the splicing factor ASF/SF2 by SRPK1. J Mol Biol. 2008;376:55‐68. [DOI] [PubMed] [Google Scholar]
- 24. Malanga M, Czubaty A, Girstun A, Staron K, Althaus FR. Poly(ADP‐ribose) binds to the splicing factor ASF/SF2 and regulates its phosphorylation by DNA topoisomerase I. J Biol Chem. 2008;283:19991‐19998. [DOI] [PubMed] [Google Scholar]
- 25. Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. 2000;69:373‐398. [DOI] [PubMed] [Google Scholar]
- 26. Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol. 2009;21:140‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lievens PMJ, Kuznetsova T, Kochlamazashvili G, et al. ZDHHC3 tyrosine phosphorylation regulates neural cell adhesion molecule palmitoylation. Mol Cell Biol. 2016;36:2208‐2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kazi JU, Chougule RA, Li T, et al. Tyrosine 842 in the activation loop is required for full transformation by the oncogenic mutant FLT3‐ITD. Cell Mol Life Sci. 2017;74:2679‐2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou Z, Li M, Zhang L, et al. Oncogenic kinase‐induced PKM2 tyrosine 105 phosphorylation converts non‐oncogenic PKM2 to a tumor promoter and induces cancer stem‐like cells. Can Res. 2018;78:2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Del Rosario AM, White FM. Quantifying oncogenic phosphotyrosine signaling networks through systems biology. Curr Opin Genet Dev. 2010;20:23‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR‐ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038‐1042. [DOI] [PubMed] [Google Scholar]
- 32. Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342‐4349. [DOI] [PubMed] [Google Scholar]
- 33. Kim KS, Jeong JY, Kim YC, et al. Predictors of the response to gefitinib in refractory non‐small cell lung cancer. Clin Cancer Res. 2005;11:2244‐2251. [DOI] [PubMed] [Google Scholar]
- 34. Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci USA. 1980;77:1311‐1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shimoni‐Sebag A, Lebenthal‐Loinger I, Zender L, Karni R. RRM1 domain of the splicing oncoprotein SRSF1 is required for MEK1‐MAPK‐ERK activation and cellular transformation. Carcinogenesis. 2013;34:2498‐2504. [DOI] [PubMed] [Google Scholar]
- 36. Caceres JF, Screaton GR, Krainer AR. A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev. 1998;12:55‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li X, Wang J, Manley JL. Loss of splicing factor ASF/SF2 induces G2 cell cycle arrest and apoptosis, but inhibits internucleosomal DNA fragmentation. Genes Dev. 2005;19:2705‐2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gautrey HL, Tyson‐Capper AJ. Regulation of Mcl‐1 by SRSF1 and SRSF5 in cancer cells. PLoS ONE. 2012;7:e51497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ghigna C, Giordano S, Shen H, et al. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell. 2005;20:881‐890. [DOI] [PubMed] [Google Scholar]
- 40. Olshavsky NA, Comstock CE, Schiewer MJ, et al. Identification of ASF/SF2 as a critical, allele‐specific effector of the cyclin D1b oncogene. Can Res. 2010;70:3975‐3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Blanco FJ, Bernabeu C. The splicing factor SRSF1 as a marker for endothelial senescence. Front Physiol. 2012;3:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Leva V, Giuliano S, Bardoni A, et al. Phosphorylation of SRSF1 is modulated by replicational stress. Nucleic Acids Res. 2012;40:1106‐1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kurogi Y, Matsuo Y, Mihara Y, et al. Identification of a chemical inhibitor for nuclear speckle formation: implications for the function of nuclear speckles in regulation of alternative pre‐mRNA splicing. Biochem Biophys Res Comm. 2014;446:119‐124. [DOI] [PubMed] [Google Scholar]
- 44. Ghigna C, De Toledo M, Bonomi S, et al. Pro‐metastatic splicing of Ron proto‐oncogene mRNA can be reversed: therapeutic potential of bifunctional oligonucleotides and indole derivatives. RNA Biol. 2010;7:495‐503. [DOI] [PubMed] [Google Scholar]
- 45. Breckenridge DG, Nguyen M, Kuppig S, Reth M, Shore GC. The procaspase‐8 isoform, procaspase‐8L, recruited to the BAP31 complex at the endoplasmic reticulum. Proc Natl Acad Sci USA. 2002;99:4331‐4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Massiello A, Chalfant CE. SRp30a (ASF/SF2) regulates the alternative splicing of caspase‐9 pre‐mRNA and is required for ceramide‐responsiveness. J Lipid Res. 2006;47:892‐897. [DOI] [PubMed] [Google Scholar]
- 47. Karni R, Hippo Y, Lowe SW, Krainer AR. The splicing‐factor oncoprotein SF2/ASF activates mTORC1. Proc Natl Acad Sci USA. 2008;105:15323‐15327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729‐734. [DOI] [PubMed] [Google Scholar]
- 49. Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006;25:6416‐6422. [DOI] [PubMed] [Google Scholar]
- 50. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424‐430. [DOI] [PubMed] [Google Scholar]
- 51. Ben‐Hur V, Denichenko P, Siegfried Z, et al. S6K1 alternative splicing modulates its oncogenic activity and regulates mTORC1. Cell Rep. 2013;3:103‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Michlewski G, Sanford JR, Caceres JF. The splicing factor SF2/ASF regulates translation initiation by enhancing phosphorylation of 4E‐BP1. Mol Cell. 2008;30:179‐189. [DOI] [PubMed] [Google Scholar]
- 53. De Benedetti A, Harris AL. eIF4E expression in tumors: its possible role in progression of malignancies. Int J Biochem Cell Biol. 1999;31:59‐72. [DOI] [PubMed] [Google Scholar]
- 54. Holland EC, Sonenberg N, Pandolfi PP, Thomas G. Signaling control of mRNA translation in cancer pathogenesis. Oncogene. 2004;23:3138‐3144. [DOI] [PubMed] [Google Scholar]
- 55. Zou A, Lambert D, Yeh H, et al. Elevated CXCL1 expression in breast cancer stroma predicts poor prognosis and is inversely associated with expression of TGF‐β signaling proteins. BMC Cancer. 2014;14:781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cao Z, Fu B, Deng B, Zeng Y, Wan X, Qu L. Overexpression of Chemokine (C‐X‐C) ligand 1 (CXCL1) associated with tumor progression and poor prognosis in hepatocellular carcinoma. Cancer Cell Int. 2014;14:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sears TK, Angelastro JM. The transcription factor ATF5: role in cellular differentiation, stress responses, and cancer. Oncotarget. 2017;8:84595‐84609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang H, Ren Y, Pang D, Liu C. Clinical implications of AGBL2 expression and its inhibitor latexin in breast cancer. World J Surg Oncol. 2014;12:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Blume‐Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355‐365. [DOI] [PubMed] [Google Scholar]
- 60. Kitajima D, Kasamatsu A, Nakashima D, et al. Tie2 regulates tumor metastasis of oral squamous cell carcinomas. J Cancer. 2016;7:600‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kukk E, Wartiovaara U, Gunji Y, et al. Analysis of Tie receptor tyrosine kinase in haemopoietic progenitor and leukaemia cells. Br J Haematol. 1997;98:195‐203. [DOI] [PubMed] [Google Scholar]
- 62. Mitsutake N, Namba H, Takahara K, et al. Tie‐2 and angiopoietin‐1 expression in human thyroid tumors. Thyroid. 2002;12:95‐99. [DOI] [PubMed] [Google Scholar]
- 63. Peters KG, Kontos CD, Lin PC, et al. Functional significance of Tie2 signaling in the adult vasculature. Recent Prog Horm Res. 2004;59:51‐71. [DOI] [PubMed] [Google Scholar]
- 64. Shirakawa K, Tsuda H, Heike Y, et al. Absence of endothelial cells, central necrosis, and fibrosis are associated with aggressive inflammatory breast cancer. Can Res. 2001;61:445‐451. [PubMed] [Google Scholar]
- 65. Wang J, Wu K, Zhang D, et al. Expressions and clinical significances of angiopoietin‐1, ‐2 and Tie2 in human gastric cancer. Biochem Biophys Res Comm. 2005;337:386‐393. [DOI] [PubMed] [Google Scholar]
- 66. Ye K, Li J, Li X, Chang S, Zhang Z. Ang1/Tie2 induces cell proliferation and migration in human papillary thyroid carcinoma via the PI3K/AKT pathway. Oncol Lett. 2018;15:1313‐1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hasenstein JR, Kasmerchak K, Buehler D, et al. Efficacy of Tie2 receptor antagonism in angiosarcoma. Neoplasia. 2012;14:131‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cheng CL, Hou HA, Jhuang JY, et al. High bone marrow angiopoietin‐1 expression is an independent poor prognostic factor for survival in patients with myelodysplastic syndromes. Br J Cancer. 2011;105:975‐982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Keith T, Araki Y, Ohyagi M, et al. Regulation of angiogenesis in the bone marrow of myelodysplastic syndromes transforming to overt leukaemia. Br J Haematol. 2007;137:206‐215. [DOI] [PubMed] [Google Scholar]
- 70. Garcia‐Manero G, Khoury HJ, Jabbour E, et al. A phase I study of oral ARRY‐614, a p38 MAPK/Tie2 dual inhibitor, in patients with low or intermediate‐1 risk myelodysplastic syndromes. Clin Cancer Res. 2015;21:985‐994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials