

Abstract
Tripartite motif 36 (TRIM36) belongs to the TRIM family, most members of which are involved in ubiquitination and degradation of target proteins by functioning as E3 ubiquitin ligases. The function of TRIM36 has not been well documented, therefore, we investigated the clinical significance and function of TRIM36 in human prostate cancer (PC). Multivariate logistic regression analysis showed that TRIM36 immunoreactivity was an independent predictor of cancer‐specific survival of PC patients. Gain‐of‐function study revealed that overexpression of TRIM36 suppressed cell proliferation and migration of LNCaP, 22Rv1, and DU145 cells. Moreover, TRIM36 knockdown using siRNA suppressed apoptosis and promoted cell proliferation and migration in LNCaP and 22Rv1 cells. Furthermore, our microarray analysis revealed that the apoptosis‐related pathway was significantly upregulated by TRIM36 overexpression. The TUNEL assay showed that apoptosis promoted by docetaxel treatment was alleviated in siTRIM36‐treated LNCaP and 22Rv1 cells. Taken together, these results suggest that high expression of TRIM36 is associated with favorable prognosis and that TRIM36 plays a tumor‐suppressive role by inhibiting cell proliferation and migration as well as promoting apoptosis in PC.
Keywords: immunohistochemistry, microarray, prostate cancer, TRIM family, tripartite motif
Abbreviations
- AR
androgen receptor
- COS
cos‐box
- CRPC
castration‐resistant prostate cancer
- DHT
dihydrotestosterone
- DTX
docetaxel
- IR
immunoreactivity
- MTS
3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium
- PC
prostate cancer
- PS
Aprostate specific antigen
- qRT‐PCR
quantitative RT‐PCR
- TNFSF10
tumor necrosis factor superfamily member 10
- TRIM
tripartite motif
1. INTRODUCTION
Prostate cancer is one of the most common cancers diagnosed in developed countries.1, 2 Despite an increased ability to detect PC and the presence of various types of treatment, including androgen deprivation therapy,3 PC could still be lethal after it progresses to CRPC.4, 5 Multiple mechanisms are proposed for AR involvement and the development of CRPC.5 Nevertheless, in men with metastatic disease, the prognosis is poor with an average overall survival of 24‐48 months.1 Therefore, further investigation is required to discover therapies with new targets involving the AR signaling pathway that can effectively treat or prevent the progression of disease in PC patients.
Tripartite motif family proteins possess three key structures such as the RING finger, B‐box, and coiled‐coil domains. It is also known that these unique proteins could function as E3 ubiquitin ligases that are crucial in the process of ubiquitination.6, 7 In addition, these proteins are considered to regulate biological functions such as cell proliferation,8, 9, 10, 11 DNA repair,12, 13, 14 signal transduction,15 and apoptosis.16 There are many reports suggesting its association with various cancers.7, 8, 9, 10, 11, 13, 14, 16, 17
Tripartite motif 36 was first reported as a novel haploid germ cell‐specific RING finger protein that is involved in the acrosome reaction.18 Later, Balint et al19 identified TRIM36 from the tumor suppressor gene region at chromosome 5q22.3. After being identified as an androgen‐responsive gene,20 subsequent reports revealed its association with the microtubule‐binding process,21 which affects the cell cycle.22 In the present study, we investigated the clinical impact and tumor‐suppressive role of TRIM36 on carcinogenesis of PC.
2. MATERIALS AND METHODS
2.1. Patient characteristics and tissue preparation
Ninety‐two prostatectomy specimens were obtained from open radical prostatectomy undertaken between April 1987 and December 2001. Staging was carried out according to the AJCC TNM staging system (https://www.cancer.org/cancer/prostate‐cancer/detection‐diagnosis‐staging/staging.html). This study was approved by the institutional ethical committee (#2283), and is in accordance with the Helsinki Declaration. Each patient provided written informed consent.
2.2. Immunostaining and immunohistochemical assessment
Immunohistochemistry for TRIM36 expression was carried out using the streptavidin‐biotin method as previously described.11 We used 1:200 diluted rabbit polyclonal Ab to TRIM36 for the primary Ab. Sections were well washed in Tris‐buffered saline with Tween‐20 after applying primary antibody overnight at 4°C. Sections were incubated with CSII (Dako, Carpinteria, CA, USA). For negative controls, normal rabbit IgG was used. All sections were counterstained using Carracci's hematoxylin.
Immunostained slides were evaluated for IR scores as described previously.23 Briefly, IR was evaluated by the sum of intensity and area score of immunostaining. Intensity score was rated from 0 to 3+ (0, none; 1, weak; 2, moderate; 3, strong), and area score was rated from 0 to 5 (0, none; 1, <1/100; 2, 1/100‐1/10; 3, 1/10‐1/3; 4, 1/3‐2/3; 5, >2/3 of the total area). The TRIM36 IR was considered “high” when the IR sum score was 4+ or higher. A score of 4+ was considered the cut‐off point, as the median value of the sum score was 4+, and the mean score was 3.49. The optimal cut‐off value in the receiver operating characteristic curve analysis for predicting cancer‐specific survival was IR sum 3+ in PC patients. Two observers (YY and NK) evaluated the slides, and a third observer (TF) estimated the scores of the slides in case of disagreement between the 2 observers.
2.3. Cell culture and reagents
293T cells were grown in DMEM supplemented with 10% FBS, 50 U/mL penicillin, and 50 μg/mL streptomycin. LNCaP, 22Rv1, and DU145 cells were grown in RPMI medium supplemented with 10% FBS, 50 U/mL penicillin, and 50 μg/mL streptomycin. All prostate cancer cell lines were validated as the expected cell type by short tandem repeat analyses in 2015. The antibodies used in this study were anti‐TRIM36 from Thermo Fisher Scientific (cat#PA5‐28401; Tokyo, Japan), anti‐AR (H‐280) from Santa Cruz Biotechnology (Santa Cruz, CA, USA), β‐actin from Sigma (St. Louis, MO, USA), anti‐TNFSF10 (sc‐8440; Santa Cruz Biotechnology), and anti‐BAX (sc‐493; Santa Cruz Biotechnology). The following reagents were purchased from the indicated companies: DHT (Wako, Saitama, Japan), bicalutamide (Sigma Aldrich Japan, Tokyo, Japan), crystal violet (Nacalai Tesque, Tokyo, Japan).
2.4. Plasmid construction and transfection
Human TRIM36 cDNA was amplified by PCR. The generated amplicon was subcloned into pCDNA3 (Invitrogen, St. Louis, MO, USA) with an N‐terminal His tag to generate mammalian expression plasmid. Cells were cultured in 6‐well plates 24 hours before transfection. Transfection of expression vector containing TRIM36 cDNA or empty vector (control) was carried out using X‐tremeGENE (Sigma Aldrich Japan), according to the manufacturer's protocol. The cell extracts were analyzed after 72 hours by western blotting.
2.5. Western blot analysis
Western blot analysis was undertaken as previously described.11 293T cells transfected with TRIM36 and empty vector (without tagged TRIM36) were used as controls. Membranes were incubated with the appropriate dilutions of specific primary Abs at 4°C overnight (anti‐TRIM36 1:200, anti‐AR 1:2000, anti‐BAX 1:100, anti‐TNFSF10 1:100, and anti‐β‐actin 1:2000). Blots were detected with either anti‐rabbit IgG or anti‐mouse IgG.
2.6. RNA extraction and qRT‐PCR
Total RNA extraction was carried out using ISOGEN reagent (Nippon Gene, Tokyo, Japan), and first‐strand cDNA was generated by using PrimeScript (Takara, Kyoto, Japan). The generated cDNA was subjected to real‐time PCR using an Applied Biosystems Step one plus real‐time PCR system based on SYBR Greenfluorescence (NIPPON Genetics, Tokyo, Japan). mRNA expression levels were normalized by GAPDH, and qRT‐PCR was undertaken as previously described.24 The following PCR primer sequences were used for qRT‐PCR: GAPDH forward, 5′‐ GGTGGTCTCCTCTGACTTCAACA; GAPDH reverse, 5′‐ GTGGTCGTTGAGGGCAATG; TRIM36 forward, 5′‐ AGTTCTGGAAGAGAGGAAATC‐3′; TRIM36 reverse, 5′‐ TCCATTTGAGTCTGAAATTG‐3′; BAX forward, 5′‐GTCGCCCTTTTCTACTTTGC‐3′; BAX reverse, 5′‐CTCAGCCCATCTTCTTCCAG‐3′; TNFSF10 forward, 5′‐AGACCTGCGTGCTGATGATCGTG‐3′; and TNFSF10 reverse, 5′‐CAAGCAATGCCACTTTTGCA‐3′.
2.7. Small interfering RNA transfection
Knockdown of TRIM36 and AR was carried out using siRNA transfection. Two specific siRNAs targeting TRIM36, and 1 non‐targeting siRNA (siRNA control) were purchased from Invitrogen. These siRNAs were transfected to PC cells (LNCaP and 22Rv1) by using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. Effects of knockdown of TRIM36 and AR were confirmed by qRT‐PCR and western blot analysis. Small interfering RNA targeting TRIM36 (cat# 1299001, #1 HSS 183097, and #2 HSS 183098) and siRNAs targeting AR (#1 S1539 and #2 S1538) were purchased from Thermo Fisher Scientific.
2.8. Cell proliferation assay
LNCaP and 22RV1 cells were seeded in 96‐well plates 24 hours before transfection. The MTS assay was carried out using the Cell Titer 96 Aqueous One Solution Cell Proliferation Assay (Promega, Osaka, Japan) according to the manufacturer's instructions after transfection. Assays were carried out in 6 wells, and data are presented as mean value ± SD.
2.9. Analysis of cell viability
LNCaP cells were seeded in 24‐well plates 24 hours before transfection. Cell viability was measured using crystal violet staining. The numbers of stained cells were counted under a microscope. Assays were carried out in quintuplicate, and data are presented as mean value ± SD.
2.10. Cell migration assay
The migration assay was carried using a cell culture insert attached with an 8.0‐μm‐pore sized PET filter (Becton Dickinson, Franklin Lakes, NJ, USA). Then RPMI medium with 10% FBS including 300 cells of LNCaP, 22Rv1, or DU145 cells were added to the lower chamber, and 700 μL specific medium was added in the insert. The cells on the upper surface of the filter were carefully removed 48 hours after transfection and were wiped with a cotton swab. The filter was dipped in methanol for 30 minutes, washed with fresh PBS, and stained with Giemsa for 30 seconds. After washing three times with fresh PBS, filters were mounted on glass slides. The cells migrated on the lower surface were counted in three randomly selected fields under a microscope at a magnification of ×200. Data are presented as mean value ± SD.
2.11. Cell apoptosis assay
We carried out the TUNEL assay using the DEADEND Fluorometric TUNEL System (Promega, Madison, WI, USA) to evaluate effect of TRIM36 knockdown for apoptosis in PC cells. Prostate cancer cells (3.0 × 104 per well) were seeded in 6‐well culture plates and incubated for 24 hours. Cells were transfected with siRNAs as described elsewhere. After incubating for 24 hours, control cells were treated with DTX (1 nmol/L) and replated to poly‐l‐lysine coated glass (Matsunami Glass Industries, Osaka, Japan) inside a 24‐well culture plate. Seventy‐two hours after transfection, cells were treated with TUNEL staining according to the manufacturer's protocol. Nuclear staining was carried out using DAPI. Signals were captured using a digital microscope (Fluoview FV10i; Olympus, Tokyo, Japan). Percentages of apoptotic cells were evaluated in 3 randomly selected fields (×100), and data are presented as mean value ± SD.
2.12. Microarray analysis
In order to evaluate the target pathway and regulated genes of TRIM36, we undertook a microarray analysis. LNCaP cells were treated with either TRIM36 overexpression or TRIM36 knockdown and compared with the matched controls. Total RNAs from siTRIM36‐treated LNCaP, TRIM36‐overexpressed LNCaP, and controls were extracted by using the Qiagen RNAeasy micro kit according to the manufacturer's instructions. RNA integrity number values were above 9.0 in all RNA samples. Clarioum S array was (Affymetrix, Thermo Fisher Scientific) used according to the manufacturer's protocol. By integrating the data of TRIM36 overexpression and TRIM36 knockdown results, we found signals and pathways specifically regulated by TRIM36. To identify upregulated genes by TRIM36, we selected downregulated genes by TRIM36 knockdown (downregulate <0.95‐fc) from top 450 genes that were upregulated by TRIM36 overexpression. Similarly, to identify downregulated genes by TRIM36, we selected upregulated genes by TRIM36‐knockdown (upregulate more than 1.05‐fc) from the top 450 genes that were downregulated by TRIM36 overexpression. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis were carried out using the Database for Annotation, Visualization and Integrated Discovery (DAVID 6.8, http://david.ncifcrf.gov/).
2.13. Statistical analyses
We used the statistical software JMP Pro version 11.0.2 (2010; SAS Institute Japan, Tokyo, Japan) for data analysis. Pearson's test and Fisher's test were used (when frequency was <5) for analysis of association of TRIM36 with clinicopathological parameters. Student's t test was used in the analysis of qRT‐PCR, MTS assay, TUNEL assay, and migration assay. Log‐rank test was carried out to analyze the statistical difference of progression‐free survival and cancer‐specific survival. Univariate and multiple logistic regression models were used to evaluate independent predictors of cancer‐specific survival in PC patients. P values < .05 were considered to be statistically significant.
3. RESULTS
3.1. Tripartite motif 36 IR is significantly associated with Gleason score, pT stage, progression‐free survival, and cancer‐specific survival
Representative immunostaining pictures of TRIM36 and controls are shown in Figure 1A‐F. Tripartite motif 36 immunostaining was observed both in the nuclei and cytoplasm. The rate of high TRIM36 IR (sum ≥4) was significantly higher in patients with lower Gleason score and pT stage (P = .0003 and P = .028, respectively, as shown in Table 1). Kaplan‐Meier curves were analyzed for difference between patients with low and high TRIM36 IR. Patients with low TRIM36 IR had significantly poor progression‐free survival and cancer‐specific survival compared to those with high TRIM36 IR (P = .045 and P = .0035, respectively, as shown in Figure 1G,H). Moreover, multivariate analysis using a logistic regression model revealed that TRIM36 IR was an independent predictor of cancer‐specific survival in patients with PC (P = .014; Table 2).
Figure 1.

Immunohistochemistry of tripartite motif 36 (TRIM36) in prostate cancer (PC). A‐F, Representative images of immunohistochemistry. A, Positive control (testis specimen immunostained with anti‐TRIM36 antibody). B, Negative control (testis specimen immunostained with rabbit IgG antibody). C, Anti‐TRIM36 in benign prostate, intensity score 3+. D, Anti‐TRIM36 in PC with Gleason score (GS) 3+3, intensity score 2+. E, Anti‐TRIM36 in PC with GS 4+5, intensity score 1+. F, Anti‐TRIM36 in PC with GS 4+5, intensity score 0+. Scale bar = 100 μm. G, Progression‐free survival of PC patients. H, Cancer‐specific survival of PC patients. IR, immunoreactivity
Table 1.
Relationships between tripartite motif 36 (TRIM36) immunoreactivity (IR) score and clinicopathological findings in prostate cancer
| TRIM36 IR score (N = 92) | |||
|---|---|---|---|
| Sum <4 (N = 45) | Sum ≥4 (N = 47) | P value | |
| Serum PSA level | |||
| PSA <10 ng/mL | 24 | 22 | .5300 |
| PSA ≥10 ng/mL | 20 | 25 | |
| Gleason score (GS) | |||
| GS < 8 | 17 | 36 | .0003 |
| GS ≥ 8 | 28 | 11 | |
| T stage | |||
| pT < 3b | 24 | 36 | .0280 |
| pT ≥ 3b | 21 | 11 | |
| N stage | |||
| pN = 0 | 36 | 44 | .0600 |
| pN ≥ 1 | 9 | 3 | |
Immunoreactivity score was evaluated by using the sum of intensity (0, none; 1, weak; 2, moderate; 3, strong) and area score (0, none; 1, <1/100; 2, 1/100‐1/10; 3, 1/10‐1/3; 4, 1/3‐2/3; 5, >2/3 of the total area).PSA, prostate‐specific antigen. High GS (GS ≥ 8) and high pTstage (pT ≥ 3) were significantly associated with low IR of TRIM36 are indicated in bold.
Table 2.
Univariate and multivariate analyses for cancer‐specific survival in prostate cancer patients
| Parameters | Univariate | Multivariate | ||
|---|---|---|---|---|
| OR (95% CI) | P value | OR (95% CI) | P value | |
| PSA (PSA <10 ng/mL vs ≥10 ng/mL) | 1.3 (0.4‐4.5) | .5900 | ||
| Gleason score (GS) (GS <8 vs ≥8) | 0.03 (0.002‐0.20) | <.0001 | 0.110 (0.005‐0.800) | .0270 |
| T stage (T <3b vs ≥3b) | 0.10 (0.02‐0.35) | .0002 | 0.28 (0.04‐1.30) | .110 |
| N stage (N0 vs ≥N1) | 0.17 (0.04‐0.7) | .0150 | 0.60 (0.11‐3.20) | .550 |
| TRIM36 IR score (sum ≥ 4 vs <4) | 0.05 (0.002‐0.28) | .0001 | 0.100 (0.005‐0.660) | .014 |
Logistic regression models were used for univariate and multivariate analyses. P value of <.05 was considered to be statistically significant.
CI, confidence interval; IR score; immunoreactivity score (sum of intensity and area scores); OR, odds ratio; PSA, prostate‐specific antigen; TRIM36, tripartite motif 36. Multivariate analysis showed that GS and TRIM36 IR score were significantly associated with cancer‐specific survival are indicated in bold.
3.2. Tripartite motif 36 is induced by androgen stimulation in LNCaP and 22Rv1 cells
We observed that the mRNA level of TRIM36 was elevated after androgen stimulation with DHT in LNCaP and 22Rv1 cells (Figures 2A and S1A). mRNA level of TRIM36 was repressed by bicalutamide treatment in LNCaP cells (Figure 2B). mRNA level of TRIM36 was maximally elevated at 12 hours after androgen stimulation in LNCaP and 22Rv1 cells (Figures 2C and S1B). Protein level of TRIM36 was increased by androgen treatment in LNCaP and 22Rv1 cells (Figures 2D and S1C). Protein level of AR was also repressed after AR knockdown in the presence of DHT (Figures 2E and S1D). mRNA level of TRIM36 was repressed after AR knockdown in LNCaP and 22Rv1 cells (Figures 2F and S1E).
Figure 2.

Tripartite motif 36 (TRIM36) is induced by androgen stimulation in LNCaP cells. A, mRNA level of TRIM36 was determined by quantitative RT‐PCR in LNCaP cells that were treated with dihydrotestosterone (DHT; 1, 10, and 100 nmol/L) or ethanol (n = 3, bar = SD; *P < .05, **P < .001, Student's t test). B, Quantitative RT‐PCR was carried out to determine mRNA levels of TRIM36 in LNCaP cells treated with DHT (10 nmol/L) and/or bicalutamide (Bic; 5, 10, and 20 μmol/L) (n = 3, bar = SD; *P < .05, **P < .001, ***P < .0001, Student's t test). C, Change in mRNA levels of TRIM36 after DHT (10 nmol/L) stimulation in LNCaP cells. D, Protein levels of TRIM36 were determined by western blotting after DHT (10 nmol/L) stimulation. IB, immunoblot. E, Protein levels of androgen receptor (AR) were determined by western blotting after treating LNCaP cells with siAR (5 nmol/L). F, mRNA levels of TRIM36 in LNCaP cells after siAR (5 nmol/L) transfection along with DHT (10 nmol/L) stimulation (n = 3, bar = SD; *P < .05, Student's t test)
3.3. Overexpression of TRIM36 promoted proliferation and migration of PC cells
LNCaP, 22Rv1, and DU145 cells were transiently transfected with TRIM36 (Figures 3A,B, S2A,B, and S3A,B). LNCaP, 22Rv1, and DU145 cells overexpressing TRIM36 showed decreased cell growth and motility compared to control cells (Figures 3C‐E, S2C‐E, and S3C‐E). Next, loss‐of‐function study of TRIM36 was carried out using siTRIM36 transfection in PC cells. Two TRIM36‐specific siRNAs (siTRIM36 #1 and siTRIM36 #2) were used for TRIM36 knockdown (Figures 3F,G and S2F,G). Cell growth was significantly increased in LNCaP and 22Rv1 cells (Figures 3H and S2H). We further carried out cell migration assays to determine the effect on motility of PC cells. The number of migrated cells was significantly increased in siTRIM36‐treated cells than in siControl‐treated cells (Figures 3I,J and S2I,J).
Figure 3.

Cell proliferation and migration assays of LNCaP cells in which tripartite motif 36 (TRIM36) is overexpressed or knocked down. A, mRNA levels of TRIM36 were determined by quantitative RT‐PCR in TRIM36‐transfected LNCaP cells (n = 3, bar = SD; **P < .001, Student's t test). B, Protein levels of TRIM36 were analyzed in TRIM36‐transfected LNCaP cells by western blotting. C, MTS assay of TRIM36‐transfected LNCaP cells (bar = SD; *P < .05, Student's t test). D, Representative pictures of migration assay. TRIM36‐transfected and control‐treated LNCaP cells are shown. Scale bar = 10 μm. E, Quantification of migrated cells of TRIM36‐transfected and control‐treated LNCaP cells (bar = SD; **P < .001, Student's t test). F, mRNA levels of TRIM36 were determined by quantitative RT‐PCR in siTRIM36‐transfected LNCaP cells (n = 3, bar = SD; **P < .001, Student's t test). G, Protein levels of TRIM36 were analyzed in siTRIM36‐transfected LNCaP cells by western blotting. H, MTS assay of siTRIM36‐transfected LNCaP cells (bar = SD; *P < .05, Student's t test). I, Representative pictures of migration assay. siTRIM36‐transfected and control‐treated LNCaP cells are shown. Scale bar = 10 μm. J, Quantification of migrated cells of siTRIM36‐transfected and control‐treated LNCaP cells (bar = SD; *P < .05, Student's t test). IB, immunoblot
3.4. Tripartite motif 36 regulates the apoptosis‐related pathway and promotes apoptosis in PC cells
Furthermore, we investigated target genes and pathways that are potentially regulated by TRIM36 overexpression and knockdown in LNCaP cells using microarray analysis. With microarray data for TRIM36 overexpression, we identified 197 genes that were upregulated by TRIM36 (fold change 1.7‐6.1) as well as 185 genes downregulated (fold change 0.1‐0.5). Interestingly, pathway analysis of these TRIM36‐regulated genes indicated that the apoptosis‐related pathway was significantly upregulated by TRIM36 (Figure 4A). The T‐cell receptor signaling pathway was significantly downregulated by TRIM36 (Figure 4B).
Figure 4.
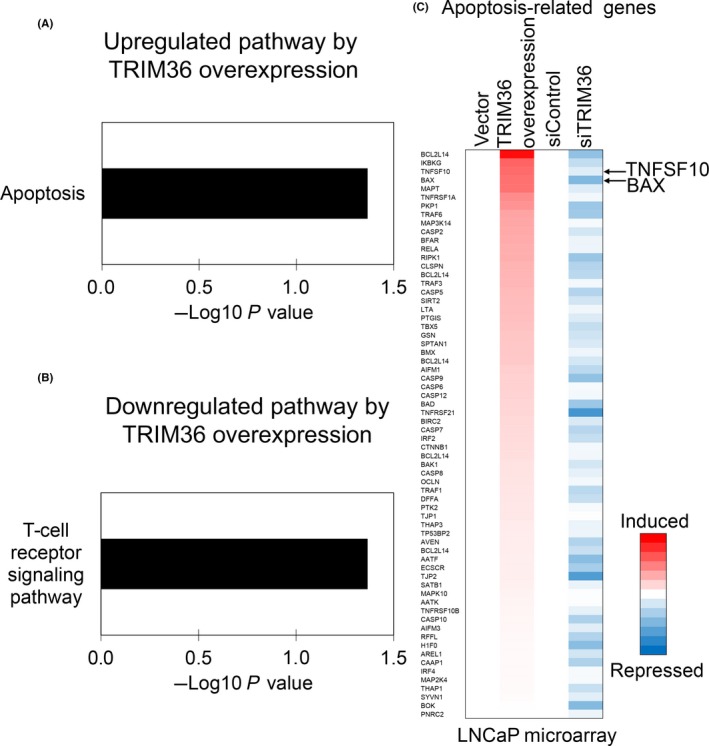
Identification of tripartite motif 36 (TRIM36)‐regulated genes by microarray analysis. A, Representative upregulated pathway in TRIM36‐transfected LNCaP cells. B, Representative downregulated pathway in TRIM36‐transfected LNCaP cells. C, TRIM36 induces expression of apoptosis‐related genes. Heat map shows the mRNA expression levels of apoptosis‐related genes compared with control samples in microarray analysis
As shown in Figure 4C, we identified BAX and TNFSF10 as representative TRIM36‐regulated genes in the apoptosis‐related pathway in LNCaP cells. Quantitative RT‐PCR analysis confirmed that TRIM36 overexpression increased mRNA and protein levels of BAX and TNFSF10 (Figures 5A‐C, S4A,B and S5A,B). Furthermore, TRIM36 knockdown decreased mRNA levels of BAX and TNFSF10 (Figures 5D,E and S4C). We also undertook TUNEL assays to evaluate apoptosis in TRIM36‐overexpressing LNCaP, 22Rv1, and DU145 cells. Apoptosis was induced in TRIM36‐overexpressing LNCaP cells (Figures 5F,G, S4D,E and S5C,D). Next, increased mRNA levels of BAX and TNFSF10 by DTX supplementation were attenuated after TRIM36 knockdown (Figures 6A,B and S6A,B). Accordingly, cell viability was in line with enhanced cell survival by TRIM36 knockdown despite DTX treatment (Figures 6C,D and S6C). Moreover, by TUNEL assay, we observed that apoptosis induced by DTX treatment was inhibited by TRIM36 knockdown (Figures 6E,F and S6D,E).
Figure 5.
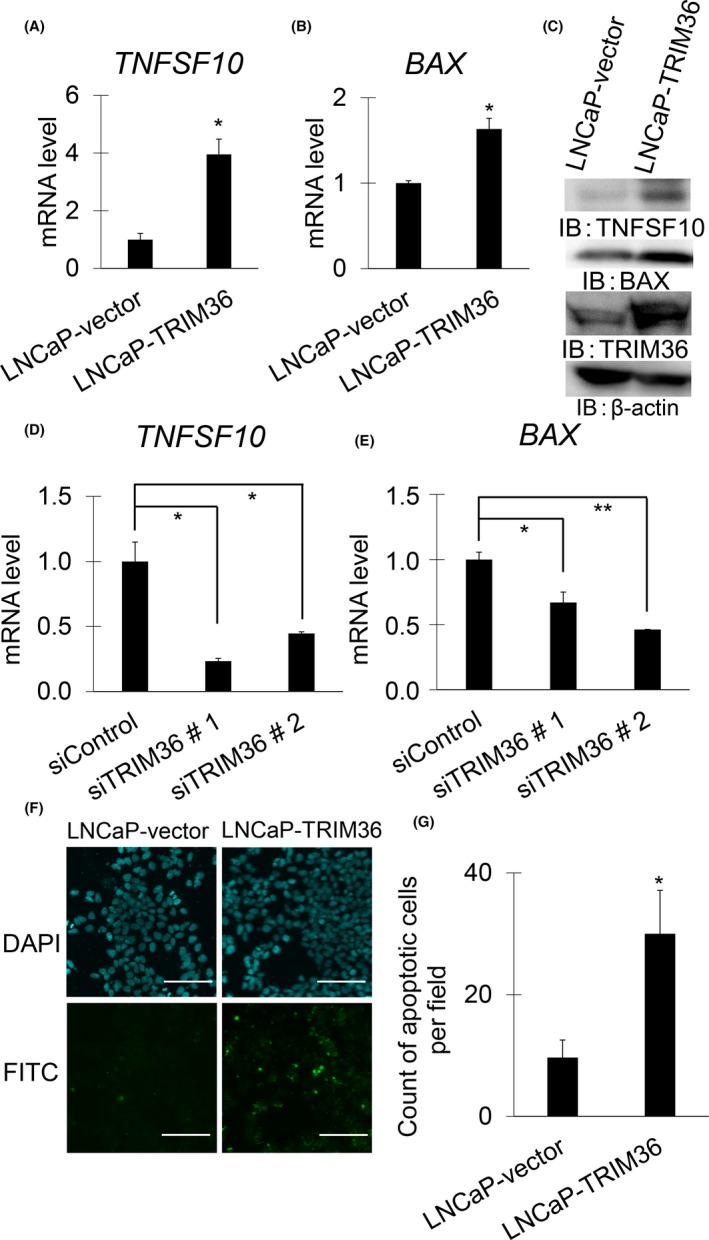
Expression levels of BAX and tumor necrosis factor superfamily member 10 (TNFSF10) determined by quantitative RT‐PCR (qRT‐PCR) and western blot analysis, and TUNEL assay in tripartite motif 36 (TRIM36)‐overexpressed LNCaP cells. A, B, mRNA levels of BAX (A) and TNFSF10 (B) were measured by qRT‐PCR in TRIM36‐transfected LNCaP cells (n = 3, bar = SD; *P < .05, Student's t test). C, Protein levels of BAX and TNFSF10 were analyzed in TRIM36‐transfected LNCaP cells by western blot analysis. IB, immunoblot. D, E, mRNA levels of BAX (D) and TNFSF10 (E) were determined by qRT‐PCR in siTRIM36‐transfected LNCaP cells (n = 3, bar = SD; *P < .05, **P < .001, Student's t test). F, Representative pictures of TUNEL assay in TRIM36‐transfected LNCaP cells. Scale bar = 10 μm. G, Quantification of apoptosis cells (bar = SD; *P < .05, Student's t test)
Figure 6.
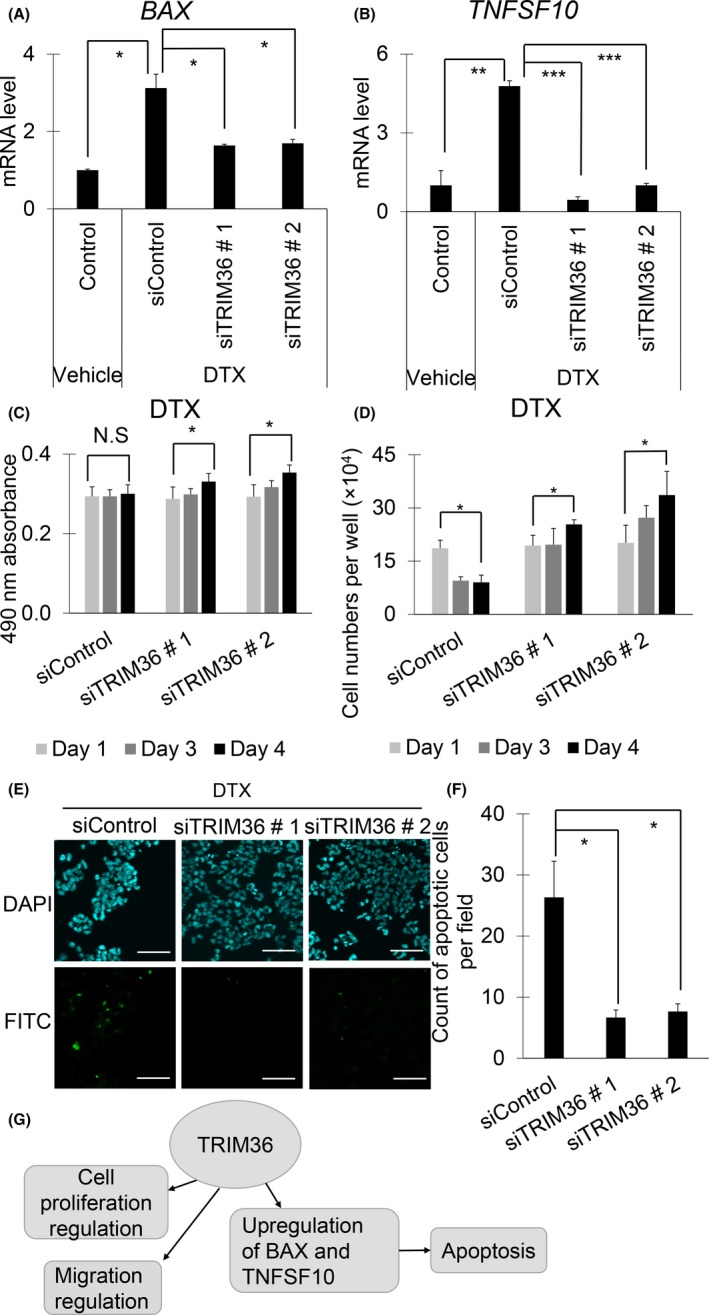
Knockdown of tripartite motif 36 (TRIM36) inhibits mRNAs of BAX and TNFSF10 and also attenuates apoptosis in docetaxel (DTX)‐treated LNCaP cells. A, B, mRNA levels of BAX (A) and TNFSF10 (B) were determined in siTRIM36‐transfected LNCaP cells after DTX (1 nmol/L) treatment by quantitative RT‐PCR (n = 3, bar = SD; *P < .05, **P < .001, ***P < .0001, Student's t test). C, D, Cell growth and viability of siTRIM36‐transfected LNCaP cells after DTX (1 nmol/L) treatment were analyzed by MTS assay (C) and counting viable cells (D) (bar = SD; *P < .05, Student's t test). N.S, not significant. E, Representative pictures of TUNEL assay. Apoptotic cells of siTRIM36‐transfected LNCaP cells after DTX 1 nmol/L treatment are shown. Scale bar = 10 μm. F, Quantification of apoptosis cells in TUNEL assay of siTRIM36‐transfected LNCaP cells after DTX 1 nmol/L treatment (bar = SD; *P < .05, Student's t test). G, Proposed model of TRIM36 functions in prostate cancer. TNFSF10, tumor necrosis factor superfamily member 10
4. DISCUSSION
We previously identified TRIM36 as an androgen‐dependent gene by combining the results of 5′‐cap analysis of gene expression and ChIP analysis in PC cells.20 Further investigation revealed that TRIM36 expression, along with the other 9 signals, correlated with cancer‐specific survival in PC.25 The present study shows that the androgen‐responsive TRIM36 has a significant clinical impact on progression‐free survival and cancer‐specific survival. At a molecular level, TRIM36 overexpression significantly suppressed cell growth and migration, and promoted apoptosis in PC cells. Furthermore, TRIM36 knockdown accelerated cell growth and migration, and suppressed apoptosis. Moreover, we investigated the target pathways of TRIM36 by microarray and found that the apoptosis‐related pathway was significantly regulated by TRIM36. Therefore, TRIM36 might be involved in a tumor‐suppressive role by promoting apoptosis in PC, as summarized in Figure 6G. This finding would be unique, as it is well known that androgen stimulates PC growth, and yet, TRIM36 is also induced by androgen to promote apoptosis. This paradoxical finding might be explained by previously reported in vitro and in vivo studies that showed androgens might repress some types of CRPC cells. Androgen‐repressed human PC cells were repressed by DHT in vitro in a concentration‐dependent manner.26 In addition, a phase II multicohort study by Teply et al27 has shown that bipolar androgen therapy is clinically effective for CRPC. Therefore, it is tempting to speculate that the tumor‐suppressive function of TRIM36 might be involved in the molecular mechanisms of bipolar androgen therapy.
Tripartite motif 36 has two potential molecular functions that are based on two distinct structures. The first structure is the key domains of the TRIM family, containing RING finger, two B‐boxes, and coiled‐coil domains. Possessing the RING finger domain enables the TRIM proteins to function as E3 ubiquitin ligases that are required in the ubiquitination process.6, 28 Tripartite motif 36 has been shown to exhibit ubiquitination activity in the presence of E1, E2, ubiquitin, and ATP, which strongly suggest that TRIM36 is a bona fide E3 ligase.22
As the second structure, TRIM36 possesses COS and fibronectin type III repeat, the most salient characteristic features of subfamily C‐I proteins.29 Notably, other TRIM proteins included in this subgroup are TRIM1, TRIM9, TRIM18, TRIM46, and TRIM67. The COS is considered to be the key domain in microtubule‐binding.21 For instance, the COS domain together with the coiled‐coil domain of TRIM18 can directly bind to microtubules.30 One of the other subfamily C‐I proteins, TRIM46, controls neuronal polarity and axon specification to define the axonal cytoskeletal compartment for regulating microtubule organization.31 Using a bioinformatics approach, Short et al proposed an association between the subfamily C‐I proteins and microtubule binding.22 As for TRIM36, Miyajima et al found that it interacts with the kinetochore protein CENP‐H, and delays cell cycle progression, which may lead to the suppression of cell growth.21 Therefore, it would be postulated that TRIM36 might possess the function of both E3 ubiquitin ligase and microtubule binding.
Our results revealed an association between TRIM36 and the apoptosis‐related pathway. Recently, a different group has reported that TRIM36 enhanced antiandrogen efficacy by inhibiting MAPK/ERK signaling pathways in PC.32 Tripartite motif 36 might have an association with the microtubule‐binding mechanism that leads to decelerating the cell cycle and regulating apoptosis. According to our microarray results, the apoptosis‐related pathway was one of the top TRIM36‐regulated pathways. Genes included in this pathway were BAX and TNFSF10, which are key players of the apoptosis mechanism. BAX is encoded by six exons and has extensive amino acid homology with Bcl‐2.33 BAX promotes apoptosis by heterodimerizing with Bcl‐2, a known inhibitor of apoptotis.34 In PC, several signals induce BAX to enhance apoptosis.35, 36, 37, 38 In the present study, TNFSF10 was also significantly regulated by TRIM36. Additionally, TNFSF10 is known to induce autophagy by regulating TNF receptor‐associated factor 2‐ and receptor‐interacting serine/threonine‐protein kinase 1‐mediated MAPK8/JNK activation.39
Taxanes, especially DTX, have high affinity for microtubules and inhibit microtubular depolymerization to promote apoptotic cell death of PC.40, 41 It is well known that this chemotherapeutic agent is effective in the treatment of PC.42 In our current results, DTX inhibited the cell growth of PC cells. However, DTX did not suppress cell growth of PC cells when it was accompanied with TRIM36 knockdown. Furthermore, mRNA levels of BAX and TNFSF10 were elevated by DTX and countervailed by TRIM36 knockdown. Moreover, cell growth of PC cells was repressed by DTX, whereas cell growth was promoted under TRIM36 knockdown despite DTX supplementation. These findings are in line with the potential function that can be speculated from the molecular features of TRIM36.
In conclusion, TRIM36 has a significant clinical impact on prognosis and plays a tumor‐suppressive role in PC by promoting the apoptosis‐related pathway. Proteomics investigation will be required in future studies in order to elucidate the tumor‐suppressive mechanism of TRIM36 in PC.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
We thank N. Sasaki for technical assistance. This work was supported by grants from P‐CREATE, Japan Agency for Medical Research and Development (to SI), Japan Society for the Promotion of Science (15K15581, 15K15353, and 17H04334 to KT and SI), and Takeda Science Foundation, Japan (to SI and KT).
Kimura N, Yamada Y, Takayama K‐I, et al. Androgen‐responsive tripartite motif 36 enhances tumor‐suppressive effect by regulating apoptosis‐related pathway in prostate cancer. Cancer Sci. 2018;109:3840–3852. 10.1111/cas.13803
Funding information
Takeda Science Foundation, Japan Agency for Medical Research and Development (P‐CREATE), Japan Society for the Promotion of Science (15K15581, 15K15353, and 17H04334).
Naoki Kimura and Yuta Yamada contributed equally to this work.
REFERENCES
- 1. Damber JE, Aus G. Prostate cancer. Lancet. 2008;371(9625):1710‐1721. [DOI] [PubMed] [Google Scholar]
- 2. Grönberg H. Prostate cancer epidemiology. Lancet. 2003;361(9360):859‐864. [DOI] [PubMed] [Google Scholar]
- 3. Fujimura T, Takayama K, Takahashi S, Inoue S. Estrogen and androgen blockade for advanced prostate cancer in the era of precision medicine. Cancers. 2018;10(2):pii: E29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paller CJ, Antonarakis ES. Management of biochemically recurrent prostate cancer after local therapy: evolving standards of care and new directions. Clin Adv Hematol Oncol. 2013;11(1):14‐23. [PMC free article] [PubMed] [Google Scholar]
- 5. Takayama K, Suzuki T, Fujimura T, et al. COBLL1 modulates cell morphology and facilitates androgen receptor genomic binding in advanced prostate cancer. Proc Natl Acad Sci USA. 2018;115(19):4975‐4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ikeda K, Inoue S. TRIM proteins as RING finger E3 ubiquitin ligases. Adv Exp Med Biol. 2012;770:27‐37. [DOI] [PubMed] [Google Scholar]
- 7. Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer. 2011;11:792‐804. [DOI] [PubMed] [Google Scholar]
- 8. Horie‐Inoue K. TRIM proteins as trim tabs for the homeostasis. J Biochem. 2013;154(4):309‐312. [DOI] [PubMed] [Google Scholar]
- 9. Takayama K, Suzuki T, Tanaka T, et al. TRIM25 enhances cell growth and cell survival by modulating p53 signals via interaction with G3BP2 in prostate cancer. Oncogene. 2018;37(16):2165‐2180. [DOI] [PubMed] [Google Scholar]
- 10. Kawabata H, Azuma K, Ikeda K, et al. TRIM44 is a poor prognostic factor for breast cancer patients as a modulator of NF‐kB signaling. Int J Mol Sci. 2017;18(9):pii: E1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yamada Y, Takayama KI, Fujimura T, et al. A novel prognostic factor TRIM44 promotes cell proliferation and migration, and inhibits apoptosis in testicular germ cell tumor. Cancer Sci. 2017;108(1):32‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Masuda Y, Takahashi H, Sato S, et al. TRIM29 regulates the assembly of DNA repair proteins into damaged chromatin. Nat Commun. 2015;6:7299. [DOI] [PubMed] [Google Scholar]
- 13. Regad T, Nellodi C, Nicotera P, Salomoni P. The tumor suppressor Pml regulates cell fate in the developing neocortex. Nat Neurosci. 2009;12:132‐140. [DOI] [PubMed] [Google Scholar]
- 14. Caratozzolo MF, Micale L, Turturo MG, et al. TRIM8 modulates p53 activity to dictate cell cycle arrest. Cell Cycle. 2012;11(3):511‐523. [DOI] [PubMed] [Google Scholar]
- 15. Tan P, He L, Cui J, et al. Assembly of the WHIP‐TRIM14‐PPP6C mitochondrial complex promotes RIG‐mediated antiviral signaling. Mol Cell. 2017;68(2):293‐307. [DOI] [PubMed] [Google Scholar]
- 16. Wang J, Zhu J, Dong M, et al. Knockdown of tripartite motif containing 24 by lentivirus suppresses cell growth and induces apoptosis in human colorectal cancer cells. Oncol Res. 2014;22(1):39‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Urano T, Saito T, Tsukui T, et al. Efp targets 14‐3‐3 sigma for proteolysis and promotes breast tumour growth. Nature. 2002;417(6891):871‐875. [DOI] [PubMed] [Google Scholar]
- 18. Kitamura K, Tanaka H, Nishimune Y. Haprin, a novel haploid germ cell‐specific RING finger protein involved in the acrosome reaction. J Biol Chem. 2003;278(45):44417‐44423. [DOI] [PubMed] [Google Scholar]
- 19. Balint I, Müller A, Nagy A, Kovacs G. Cloning and characterisation of the RBCC728/TRIM36 zinc‐binding protein from the tumor suppressor gene region at chromosome 5q22.3. Gene. 2004;332:45‐50. [DOI] [PubMed] [Google Scholar]
- 20. Takayama K, Tsutsumi S, Katayama S, et al. Integration of cap analysis of gene expression and chromatin immunoprecipitation analysis on array reveals genome‐wide androgen receptor signaling in prostate cancer cells. Oncogene. 2011;30(5):619‐630. [DOI] [PubMed] [Google Scholar]
- 21. Short KM, Cox TC. Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J Biol Chem. 2006;281(13):8970‐8980. [DOI] [PubMed] [Google Scholar]
- 22. Miyajima N, Maruyama S, Nonomura K, et al. TRIM36 interacts with the kinetochore protein CENP‐H and delays cell cycle progression. Biochem Biophys Res Commun. 2009;381(3):383‐387. [DOI] [PubMed] [Google Scholar]
- 23. Ashikari D, Takayama K, Tanaka T, et al. Androgen induces G3BP2 and SUMO‐mediated p53 nuclear export in prostate cancer. Oncogene. 2017;36(45):6272‐6281. [DOI] [PubMed] [Google Scholar]
- 24. Takayama K, Suzuki T, Fujimura T, et al. Dysregulation of spliceosome gene expression in advanced prostate cancer by RNA‐binding protein PSF. Proc Natl Acad Sci USA. 2017;114(39):10461‐10466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujimura T, Takahashi S, Urano T, et al. Expression of androgen and estrogen signaling components and stem cell markers to predict cancer progression and cancer‐specific survival in patients with metastatic prostate cancer. Clin Cancer Res. 2014;20(17):4625‐4635. [DOI] [PubMed] [Google Scholar]
- 26. Zhau HY, Chang SM, Chen BQ, et al. Androgen‐repressed phenotype in human prostate cancer. Proc Natl Acad Sci USA. 1996;93(26):15152‐15157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Teply BA, Wang H, Luber B, et al. Bipolar androgen therapy in men with metastatic castration‐resistant prostate cancer after progression on enzalutamide: an open‐label, phase 2, multicohort study. Lancet Oncol. 2018;19(1):76‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sun Y. Targeting E3 ubiquitin ligases for cancer therapy. Cancer Biol Ther. 2003;2(6):623‐629. [PubMed] [Google Scholar]
- 29. Watanabe M, Hatakeyama S. TRIM proteins and diseases. J Biochem. 2017;161(2):135‐144. [DOI] [PubMed] [Google Scholar]
- 30. Wright KM, Du H, Dagnachew M, Massiah MA. Solution of structure of the microtubule‐targeting COS domain of MID1. FEBS J. 2016;283(16):3089‐3102. [DOI] [PubMed] [Google Scholar]
- 31. Van Beuningen SFB, Will L, Harterink M, et al. TRIM46 controls neuronal polarity and axon specification driving the formation of parallel microtubule arrays. Neuron. 2015;88(6):1208‐1226. [DOI] [PubMed] [Google Scholar]
- 32. Liang C, Wang S, Qin C, et al. TRIM36, a novel androgen‐responsive gene, enhances anti‐androgen efficacy against prostate cancer by inhibiting MAPK/ERK signaling pathways. Cell Death Dis. 2018;9(2):155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl‐2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74(4):609‐619. [DOI] [PubMed] [Google Scholar]
- 34. St Clair EG, Anderson SJ, Oltvai ZN. Bcl‐2 counters apoptosis by Bax heterodimerization‐dependent and independent mechanisms in the T‐cell lineage. J Biol Chem. 1997;272(46):29347‐29355. [DOI] [PubMed] [Google Scholar]
- 35. Ye QF, Zhang YC, Peng XQ, et al. Silencing Notch‐1 induces apoptosis and increases the chemosensitivity of prostate cancer cells to docetaxel through Bcl‐2 and Bax. Oncol let. 2012;3(4):879‐884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maciel‐Silva P, Caldeira I, de Assis Santos I, et al. FAM3B/PANDER inhibits cell death and increases prostate tumor growth by modulating the expression of Bcl‐2 and Bcl‐XL cell survival genes. BMC Cancer. 2018;18(1):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jin X, Zhao W, Zhou P, Niu T. YAP knockdown inhibits proliferation and induces apoptosis of human prostate cancer DU145 cells. Mol Med Rep. 2018;17(3):3783‐3788. [DOI] [PubMed] [Google Scholar]
- 38. Li S, Wu Z, Ma P, et al. Ligand‐dependent EphA7 signaling inhibits prostate tumor growth and progression. Cell Death Dis. 2017;8(10):e3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Voelkel‐Johnson C. TRAIL‐mediated signaling in prostate, bladder and renal cancer. Nat Rev Urol. 2011;8(8):417‐427. [DOI] [PubMed] [Google Scholar]
- 40. Pienta KJ. Preclinical mechanisms of action of docetaxel and docetaxel combinations in prostate cancer. Semin Oncol. 2001;28(4 Suppl 15):3‐7. [DOI] [PubMed] [Google Scholar]
- 41. Stein CA. Mechanisms of action of taxanes in prostate cancer. Semin Oncol. 1999;26(5 Suppl 17):3‐7. [PubMed] [Google Scholar]
- 42. Petrylak DP, Tangen CM, Hussain MHA, et al. Docetaxel and estramustine compared with mitoantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351(15):1513‐1520. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials