

The long-lasting endopeptidase activity of BoNT is a critical biological activity inside the nerve cell, as it prompts proteolysis of the SNARE proteins, involved in the exocytosis of the neurotransmitter acetylcholine. Thus, the BoNT endopeptidase activity is an appropriate clinical target for designing new small-molecule antidotes against BoNT with the potential to reverse the paralysis syndrome of botulism. In principle, small-molecule inhibitors (SMIs) can gain entry into BoNT-intoxicated cells if they have a suitable octanol-water partition coefficient (log P) value and other favorable characteristics (P. Leeson, Nature 481:455–456, 2012, https://doi.org/10.1038/481455a). Several efforts have been made in the past to develop SMIs, but inhibitors effective under in vitro conditions have not in general been effective in vivo or in cellular models (L. M. Eubanks, M. S. Hixon, W. Jin, S. Hong, et al., Proc Natl Acad Sci U S A 104:2602–2607, 2007, https://doi.org/10.1073/pnas.0611213104). The difference between the in vitro and cellular efficacy presumably results from difficulties experienced by the compounds in crossing the cell membrane, in conjunction with poor bioavailability and high cytotoxicity. The screened nitrophenyl psoralen (NPP) effectively antagonized BoNT/A in both in vitro and ex vivo assays. Importantly, NPP inhibited the BoNT/A light chain but not other general zinc endopeptidases, such as thermolysin, suggesting high selectivity for its target. Small-molecule (nonpeptidic) inhibitors have better oral bioavailability, better stability, and better tissue and cell permeation than antitoxins or peptide inhibitors.
KEYWORDS: botulinum neurotoxin type A, high-throughput screening, IC50, cell-based SNAP-25 assay, phrenic nerve-hemidiaphragm preparation
ABSTRACT
Botulinum neurotoxins (BoNTs), the most poisonous proteins known to humankind, are a family of seven (serotype A to G) immunologically distinct proteins synthesized primarily by different strains of the anaerobic bacterium Clostridium botulinum. Being the causative agents of botulism, the toxins block neurotransmitter release by specifically cleaving one of the three soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) proteins, thereby inducing flaccid paralysis. The development of countermeasures and therapeutics against BoNTs is a high-priority research area for public health because of their extreme toxicity and potential for use as biowarfare agents. Extensive research has focused on designing antagonists that block the catalytic activity of BoNTs. In this study, we screened 300 small natural compounds and their analogues extracted from Indian plants for their activity against BoNT serotype A (BoNT/A) as well as its light chain (LCA) using biochemical and cellular assays. One natural compound, a nitrophenyl psoralen (NPP), was identified to be a specific inhibitor of LCA with an in vitro 50% inhibitory concentration (IC50) value of 4.74 ± 0.03 µM. NPP was able to rescue endogenous synaptosome-associated protein 25 (SNAP-25) from cleavage by BoNT/A in human neuroblastoma cells with an IC50 of 12.2 ± 1.7 µM, as well as to prolong the time to the blocking of neutrally elicited twitch tensions in isolated mouse phrenic nerve-hemidiaphragm preparations.
IMPORTANCE The long-lasting endopeptidase activity of BoNT is a critical biological activity inside the nerve cell, as it prompts proteolysis of the SNARE proteins, involved in the exocytosis of the neurotransmitter acetylcholine. Thus, the BoNT endopeptidase activity is an appropriate clinical target for designing new small-molecule antidotes against BoNT with the potential to reverse the paralysis syndrome of botulism. In principle, small-molecule inhibitors (SMIs) can gain entry into BoNT-intoxicated cells if they have a suitable octanol-water partition coefficient (log P) value and other favorable characteristics (P. Leeson, Nature 481:455–456, 2012, https://doi.org/10.1038/481455a). Several efforts have been made in the past to develop SMIs, but inhibitors effective under in vitro conditions have not in general been effective in vivo or in cellular models (L. M. Eubanks, M. S. Hixon, W. Jin, S. Hong, et al., Proc Natl Acad Sci U S A 104:2602–2607, 2007, https://doi.org/10.1073/pnas.0611213104). The difference between the in vitro and cellular efficacy presumably results from difficulties experienced by the compounds in crossing the cell membrane, in conjunction with poor bioavailability and high cytotoxicity. The screened nitrophenyl psoralen (NPP) effectively antagonized BoNT/A in both in vitro and ex vivo assays. Importantly, NPP inhibited the BoNT/A light chain but not other general zinc endopeptidases, such as thermolysin, suggesting high selectivity for its target. Small-molecule (nonpeptidic) inhibitors have better oral bioavailability, better stability, and better tissue and cell permeation than antitoxins or peptide inhibitors.
INTRODUCTION
Clostridium botulinum is an anaerobic spore-forming bacterium whose different strains produce seven immunologically different serotypes of botulinum neurotoxin (BoNT), serotypes A to G. BoNTs are responsible for the life-threatening disease botulism. Their long-lasting paralytic effect, extreme toxicity, ease of production, and lack of antidotes make BoNTs tier 1 biothreat agents according to the U.S. Centers for Disease Control and Prevention. BoNT serotype A (BoNT/A) is the most potent of all the BoNT serotypes (1), with an estimated human lethal intravenous dose of 1 to 5 ng/kg of body weight (2, 3). In contrast to their lethal properties, very low doses of BoNT/A are widely used for alleviating the symptoms of various disorders, such as cervical dystonia, blepharospasm, strabismus, migraine, neuropathic pain, and severe hyperhidrosis (1). They have also been used for cosmetic applications since the early 1990s (4).
Thus, BoNT is a unique molecule with both toxic and therapeutic capabilities as a biowarfare and potent biothreat agent, on the one hand, and as a therapeutic and a cosmetic agent, on the other. BoNT poses an exceptional challenge for public health management. Because of the toxin’s therapeutic use, mass vaccination against BoNT is impractical and undesirable. The potential of BoNT to cause mass casualties has led to intense efforts to develop effective countermeasures and antidotes (5).
BoNTs consist of a light chain (LC) and a heavy chain (HC), each of which contributes to the toxicity. BoNTs bind and are internalized into nerve terminals via the HC, whose N-terminal region helps translocate the LC into the cytosol. The LC is a zinc endopeptidase that specifically cleaves one of the soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) proteins: synaptosome-associated protein 25 (SNAP-25), synaptobrevin, or syntaxin (6). BoNT/A cleaves SNAP-25, thereby blocking exocytosis of acetylcholine at the neuromuscular junction, leading to flaccid paralysis (7).
Current therapy for botulism involves respiratory supportive care and the administration of antitoxin. The only antitoxin approved for adult human use is a polyclonal heptavalent (serotype A, B, C, D, E, F, G) equine antitoxin designated botulism antitoxin (BAT). To treat infant botulism, BabyBIG, which is derived from plasmapheresis of human serum, has been developed (8). By neutralizing circulating toxins, these antitoxins block further binding of the toxins to the neuronal cells, but they do not reverse the paralysis caused by the toxin inside the cells. To be effective, these antibodies must be administered before there is an appreciable uptake of toxin by neurons. Since the treatment window for botulism with antibody-based therapy is limited, a challenging task of early detection and diagnosis is essential for success.
Apart from antibodies, the only near-term options for antidotes are small-molecule inhibitors (SMIs) or peptide-based inhibitors for targeting various functional domains of BoNT, such as the translocation, binding, or catalytic domain. Small peptide-based drugs and receptor mimics have been developed as therapeutic targets (9–16). Inhibitors and drugs targeted at toxin binding, internalization, and translocation have a very limited therapeutic window and thus have a limited scope. Inhibitors targeting the proteolytic domain of BoNT represent a more feasible approach. Therefore, small-molecule drug candidates (peptidic and nonpeptidic) targeting the catalytic domain are becoming increasingly more important in drug discovery efforts against botulism. The major thrust of this effort has been directed against BoNT/A, since this is the most potent serotype in humans and has the longest half-life in cells (17–19).
Our research is an attempt to identify an antagonist for BoNT/A. In this study, we screened a library of natural compounds and their analogues, made available by researchers at the University of Delhi, against the Zn2+ endopeptidase domain of BoNT/A. Using a progression of biochemical and cellular assays of increasing complexity, we have identified a nitrophenyl psoralen (NPP) derivative (Fig. 1) to be a selective and potent inhibitor of BoNT/A from the natural product library.
FIG 1.

Substrate design for the HTS endopeptidase assays. (A) Schematic of the substrate SNAP-25 (aa 1 to 206) and proteolytic fragments generated following cleavage of SNAP-25 at residues Q197 and R198 by BoNT/A. The residues shown in red were used for FRET peptide substrate development. (B) Sequence of FRET peptide substrate used for HTS of inhibitors. (C) Full-length substrate (GST [26.31 kDa, purple], SNAP25a [23.82 kDa, blue], GFP [26.94 kDa, green], SNAG [77.03 kDa]) for confirmation of inhibition by NPP. SNAG cleaves into 48.73-kDa and 28.33-kDa fragments when incubated with BoNT/A and into 46.78-kDa and 30.27-kDa fragments when incubated with BoNT/E. EGCP, enhanced green fluorescent protein.
RESULTS
Screening of natural compounds.
Three hundred compounds extracted from Indian medicinal plants were screened for their activity against the light chain of BoNT/A (LCA). The modern pharmaceutical industry was born from botanical medicine due to the structural diversity of natural compounds (20). A rapid, robust, and accurate fluorescence-based assay was used to screen the inhibitor candidates in the natural compound library.
A quantitative fluorescence resonance energy transfer (FRET) assay was used as a high-throughput screening assay (HTS) for the initial evaluation of LCA inhibitors (11, 12). In brief, the peptide substrate was a 14-amino-acid (aa) SNAP-25 reporter that included the LCA cleavage site (Q197-R198). The fluorophore fluorescein isothiocyanate (FITC) was linked to the N terminus of the reporter, while the quencher 4-{[4-(dimethylamino)phenyl]azo}benzoic acid (DABCYL) was attached to the C terminus (Fig. 1B). As the BoNT/A endopeptidase cleaves the peptide substrate, the quenching group is removed and the fluorescence signal increases. In the presence of an effective inhibitor of LCA, the protease is unable to cleave the reporter peptide and no fluorescence is recorded.
Out of 300 compounds, 4 were selected as hits, having >95% inhibition of the endopeptidase activity at a concentration of 25 µg/ml (the criterion for efficacy). NPP (Fig. 2) was found to inhibit the protease activity of LCA, and it was identified to be the most effective inhibitor during the initial HTS. The 3 other hits were not pursued further due to the lower level of purity of those compounds.
FIG 2.
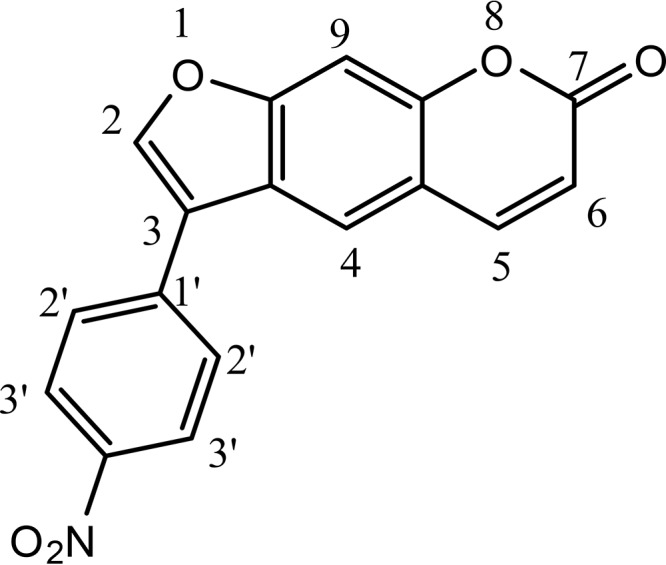
Structure of 3-(4-nitrophenyl)-7H-furo[3,2-g]chromen-7-one, which is a nitrophenyl psoralen (NPP).
The structure of the most potent inhibitor of the 4 hits was confirmed to be 3-(4-nitrophenyl)-7H-furo[3,2-g]chromen-7-one, which is a nitrophenyl psoralen (NPP) derivative (Fig. 1), by its 1H and 13C nuclear magnetic resonance (NMR) spectra and mass spectrum. Hydrogen 1 and carbon 13 NMR parameters were recorded as follows. 1H NMR (400 MHz, dimethyl sulfoxide [DMSO]) showed the following parameters: δ ppm 6.43 (1 H, d, J = 9.52, C-6H), 7.09 (1 H, s, C-4H), 7.71 (1 H, s, C-9H), 8.04 (2 H, d, J = 6.4 Hz, C-2ʹ and C-6ʹH), 8.09 (1 H, s, C-2H), 8.15 (2 H, d, J = 6.6 Hz, C-3ʹ and C-5ʹH), 8.32 (1 H, d, J = 8.8 Hz, C-5H) (NMR descriptions, s, singlet; d, doublet; m, multiple; for C and H numbers, see Fig. 1). 13C NMR (100 MHz, CDCl3) showed the following parameters: δ ppm 109.3 (C-3); 114.3, 114.7, 121.5, 123.9, 125.1, 129.8, 137.4, 144.0, and 144.5 (aromatic carbons); 150.8 (C-2); 152.0 (C-4ʹ); 158.4 (C-7).
High-resolution mass spectrometric analysis revealed [M + H]+ to be 308.0569, consistent with the molecular formula of NPP (C17H9NO5).
Following this initial screen, a similar FRET assay using full-length SNAP-25 (aa 1 to 206) was performed to confirm the designated hit and to eliminate false-positive results due to the potential fluorescence quenching in the HTS assay.
Determination of IC50 value of NPP using the peptide-based assay.
The inhibitory activity of NPP was further investigated using the 14-aa peptide substrate. NPP produced a concentration-dependent reduction of the endopeptidase activity of LCA, reaching 90% inhibition at 50 μM NPP (Fig. 3). The 50% inhibitory concentration (IC50) value for NPP was determined to be 4.74 ± 0.03 µM (Fig. 3).
FIG 3.

Concentration-response curves to determine the inhibition of LCA by NPP using a 14-aa SNAP-25 reporter peptide as a substrate. Various concentrations of NPP were incubated with LCA and fluorescent peptide substrate. Percent inhibition was determined by measuring the fluorescence using an excitation wavelength of 490 nm and an emission wavelength of 523 nm. The experiments were performed in 10 mM HEPES buffer, pH 7.4, containing 150 mM NaCl, 1.25 mM freshly prepared dithiothreitol (DTT), and 0.05% Tween 20 at 37°C. The symbols represent the mean ± SD for data obtained from three replicate samples. The curve was fit by nonlinear regression analysis and yielded an IC50 of 4.74 ± 0.03 µM. [C], concentration.
Confirmation of endopeptidase inhibition using full-length SNAP-25.
Although use of the SNAP-25 peptide reporter is convenient for HTS studies, it is not a true representation of the SNAP-25 (1 to 206 aa) substrate. SNAP-25 cleavage by BoNT/A is a multistep process, and it involves a number of structural changes near the toxin’s catalytic pocket upon substrate binding (21, 22), as well as interactions of full-length SNAP-25 with α- and β-exosites on LCA which are not possible with the truncated 14-mer peptide. Xue et al. (2014) demonstrated that α-exosite binding can enhance both the catalytic activity and stability of LCA (23). The full-length substrate with an N-terminal glutathione S-transferase (GST) tag and a C-terminal green fluorescent protein (GFP) tag (designated SNAG) was used as the substrate for LCA in these studies. The fluorescent tags do not interfere with the cleavage by LCA and yet provide cleavage products with large size differences, making the detection of cleaved SNAP-25 and its inhibition more accurate. LCA at 2 nM was sufficient to cleave the 1.5 µM SNAG substrate. Cleavage or inhibition was determined after subtracting the background intensity of SNAG in the absence of LCA (control lane) to obtain the actual enzymatic activity of LCA (Fig. 4).
FIG 4.
Confirmation of inhibitory activity of NPP using the SNAG construct (GST–SNAP-25–GFP). SNAG is labeled as the negative control (NC; lane 1). SNAG incubated with the 2 nM LCA served as the positive control (PC; lane 2). LCA was incubated with serially diluted (2×) inhibitor (80, 40, 20, 10, 5, 2.5, and 1.25 μM; lanes 4 to 10). Percent inhibition was determined using densitometry analysis with Bio-Rad Image Lab (v.5.2.1) software. SNAP-25 cleavage by NPP was reduced in a concentration-dependent manner. The IC50 was determined to be 6.3 ± 2.02 µM (mean ± SD, n =3). The gel is representative of the gels from 3 independent experiments.
Different concentrations of inhibitor with a given concentration of enzyme were used to evaluate NPP’s effective inhibition. NPP showed a concentration-dependent inhibition of LCA, inhibiting 50% of SNAG cleavage at 6.3 ± 2.0 µM (Fig. 4). The IC50 of NPP for the cleavage of the SNAG substrate (6.3 µM) was similar to that for the cleavage of the 14-mer peptide-based substrate (4.7 µM).
Analysis of inhibitor specificity.
Thermolysin is a 34.6-kDa thermostable metalloprotease secreted by Bacillus thermoproteolyticus. Thermolysin preferentially cleaves sites with bulky and aromatic residues (Ile, Leu, Val, Ala, Met, Phe) in the substrate. Since thermolysin shares the same zinc-binding motif (HEXXH) as LCA (24–27), this enzyme was used as a model to examine the specificity of NPP. As indicated in Fig. 5, two different concentrations of thermolysin (50 nM and 500 nM) were used with SNAG as the substrate. The results demonstrated that NPP did not inhibit the cleavage of SNAG by thermolysin (Fig. 5). Thermolysin can cleave SNAG at 176 different sites, which results in multiple low-molecular-weight bands. This suggests that NPP lacks any general protease promiscuity. Furthermore, NPP showed substantially lower levels of inhibition of the endopeptidase activity of the light chain of BoNT/B (LCB) (IC50, 39.6 ± 16.4 µM) and the light chain of BoNT/E (LCE) (IC50, >80 µM) (see Fig. S1 and S2 in the supplemental material) than LCA (IC50, 4.7 µM), which suggests a high specificity for LCA. This feature of NPP makes it a good candidate for therapeutics development.
FIG 5.
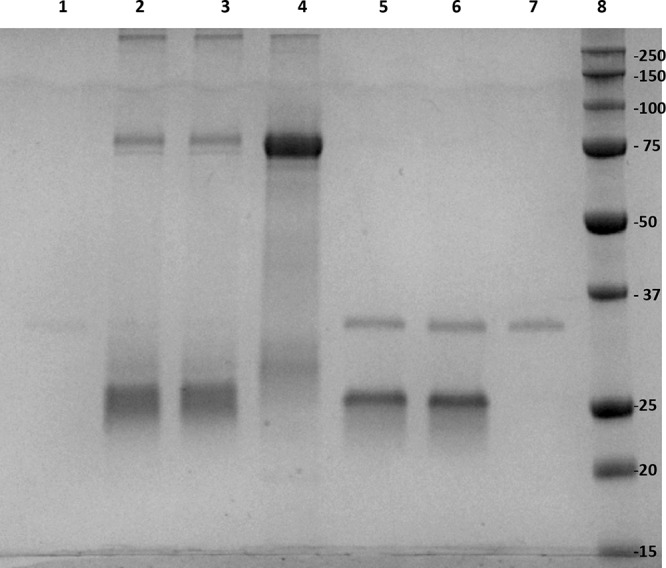
NPP inhibition specificity analysis using thermolysin as an enzyme. Two different concentrations of thermolysin (50 nM and 500 nM) were preincubated with 6.5 µM NPP (lane 2 and lane 5, respectively), followed by incubation with SNAG for comparison with the thermolysin reaction in the absence of NPP (lane 3 and lane 6, respectively). SNAG at 1.5 µM (lane 4), thermolysin at 50 nM (lane 1), and thermolysin at 500 nM (lane 7) were used as controls. Lanes 1 and 7 had thermolysin in the absence of a substrate or inhibitor. Lane 8, SDS-PAGE molecular weight standard (the numbers on the right are in kilodaltons).
Inhibition of BoNT/A endopeptidase activity in M17 cells.
To be effective as therapeutic agents against BoNT/A, small molecules must cross the plasma membrane of intoxicated neurons and inhibit the LC in the cytosol. Moreover, while the crystal structure of LCA under in vitro conditions is well-known, the nature of the LC in intoxicated neurons remains elusive, as it undergoes unfolding and refolding during its escape from the endosome, with the possibility of enzymatic modifications and strong interactions with the cell membrane. Furthermore, in vivo assays are expensive, time-consuming, and more variable. Thus, cellular models based on neuronal cell cultures mimicking in vivo intoxication are essential for more advanced compound screening. To further confirm the inhibitory effects of our selected lead inhibitor at the cellular level, NPP was first incubated with 30 nM BoNT/A, and then the mixture was used to treat M17 neuroblastoma cells. NPP showed inhibition with an IC50 of 12.2 ± 1.7 µM (Fig. 6).
FIG 6.
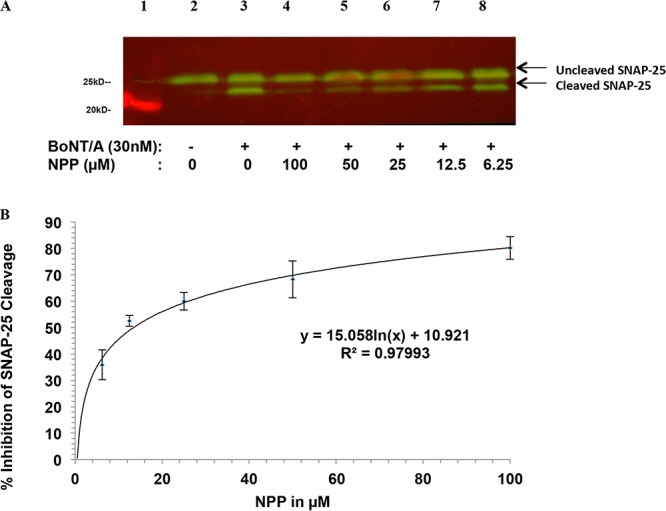
Inhibition by NPP of BoNT/A-mediated cleavage of SNAP-25 in M17 neuroblastoma cells, determined using Western blot analysis. (A) Lane 1, marker lane; lane 2, control cells without BoNT/A as a negative control; lane 3, cells incubated with 30 nM BoNT/A as a positive control; lanes 4 to 8, 30 nM BoNT/A was mixed with 1 of 5 concentrations of NPP for 30 min, respectively, and the cells were incubated with the toxin-inhibitor mixture for 24 h prior to assay. (B) Percent inhibition was determined by densitometry. The IC50 was calculated using nonlinear regression analysis. Symbols represent the mean ± SD from three independent experiments.
Effect of NPP on BoNT/A-mediated muscle paralysis.
Twitch tensions were elicited at a frequency of 0.03 Hz by supramaximal stimulation of the phrenic nerve in isolated mouse diaphragm muscles. To examine the protective action of NPP on diaphragmatic tensions, muscles were first exposed to BoNT/A at room temperature (22°C) for 30 min, which resulted in the nearly irreversible binding of the toxin to the nerve terminal. At this low temperature, no impairment of muscle tension occurs because little or no toxin internalization takes place (28, 29). Excess toxin was removed by three washes with control Tyrode’s solution, followed by addition of 0.12% DMSO (vehicle) or 60 µM NPP to the bathing solution. The bath temperature was raised to 37°C to promote toxin internalization, and muscles were monitored for the development of BoNT/A-mediated paralysis. The results of a typical experiment illustrating the protection provided by NPP on diaphragmatic tension are shown in Fig. 7.
FIG 7.

Time course of BoNT/A-mediated muscle inhibition of twitch tension elicited at a frequency of 0.03 Hz in isolated mouse hemidiaphragm muscles. Muscles were exposed to 5 pM BoNT/A at zero time for 30 min at 22°C (first arrowhead). At the end of this incubation period, the muscles were washed to remove unbound BoNT/A to limit entry of the toxin during the recording period and to minimize interactions of BoNT/A and NPP in the extracellular environment. The temperature was increased to 37°C to initiate muscle paralysis. Subsequently, 60 µM NPP dissolved in DMSO or vehicle (0.12% DMSO) was added from concentrated stock solutions to determine the effects of NPP on BoNT/A-mediated paralysis (second arrowhead). Additions were made slowly (30 to 60 s) to minimize the precipitation of NPP and to prevent the generation of muscle fasciculations from transient formation of high concentrations of the solvent prior to its equilibration in the bath. Other than muscle fasciculations (which were observed in one of six experiments), DMSO had no effect on muscle tension at the concentration examined.
In the absence of BoNT/A, muscle tensions were well maintained, undergoing a <10% reduction in amplitude over a 4-h recording period. Exposure of muscles to 5 pM BoNT/A followed 30 min later by exposure to NPP or 0.12% DMSO led to a progressive decline in twitch tension after a brief latent period (Fig. 7). Data from 7 experiments in 0.12% DMSO disclosed a latency of 58.3 ± 13.8 min and a half-paralysis time (HPT) of 116.9 ± 17.3 min (mean ± standard deviation [SD]); latencies and HPTs are referenced to the time after the bath temperature was increased to 37°C (Fig. 7). Addition of 60 µM NPP after the washout of unbound BoNT/A increased the latency to 76.4 ± 14.5 min and extended the HPT to 147.1 ± 16.5 min (n = 6). Both changes were statistically significant by a two-tailed t test (P < 0.05 for the former and P < 0.01 for the latter).
Cytotoxicity of NPP.
To determine the cytotoxicity profile of NPP, M17 cells were incubated with the compound for 24 h, and cytotoxicity was determined using the 3-(4,5-dimethylthizol-2-yl)-2,5-diphenyltetrazolium (MTT) assay. As shown in Fig. 8, NPP caused little or no cytotoxicity at any of the concentrations tested (0.1 to 310 µM).
FIG 8.
The cellular toxicity of NPP was determined using the MTT assay. At 80% confluence, cells were incubated with different concentrations of inhibitors for 24 h. Six wells with 0.5% DMSO without inhibitors were used as a positive control. Six empty wells without any cells were also used as a negative control to remove the background signal. SD is a representation of 6 wells.
Drug-like properties.
The drug-like properties of NPP at physiological pH were determined using preADMET software from the Bioinformatics and Molecular Design Research Center (BMDRC), Seoul, South Korea. NPP’s drug-like properties were analyzed by using the most well-known drug-like rule, which is normally referred to as the “Pfizer rule” or the “rule of five,” which is also known as Lipinski’s rule (30), and another comprehensive medicinal chemistry (CMC)-like drug rule (31, 32). NPP followed all the criteria for both of these classical rules. Some of NPP’s drug-like properties were a molecular weight of 307 (according to the criteria, the molecular weight should be <500), an octanol-water partition coefficient (log P) value of 3.6 (according to the criteria, the value should be 2 to 4), a distribution constant at a pH of 7.4 (log D) value of 3.5 (according to the criteria, the value should be <5), a polar surface area (PSA) of 85 (according to the criteria, the PSA should be <140), and 0 hydrogen bond donors (according to the criteria, there should be <5 hydrogen bond donors).
DISCUSSION
Currently, there is no treatment to reverse the intoxication caused by BoNTs. The only therapy for botulism is an equine polyclonal heptavalent botulism antitoxin (BAT), which is generally used in conjunction with intensive care and mechanical ventilation. Even though new monoclonal antibodies targeting circulating BoNTs have been evaluated in clinical trials (33), their effectiveness, like that of BAT, is restricted by a narrow window for treatment. Efforts to discover drugs based on SMIs have intensified. To be an effective inhibitor of BoNT/A toxicity, the compound must possess the following five characteristics: (i) high selectivity, (ii) high target affinity, (iii) low toxicity, (iv) high cell permeation, and (v) a favorable absorption, distribution, metabolism, and excretion (ADME) profile.
Several research groups have screened and developed synthetic small molecules inhibiting the proteolytic activity of BoNT/A LC over a concentration range of 0.1 to 10 µM using various approaches (34–37). SMIs containing quinolinol derivatives (16, 38–40) and hydroxamates (41–43) have been examined extensively. The quinolinols that were found to be effective in the mouse hemidiaphragm assay have been reported to have poor solubility, high toxicity, and low bioavailability (16, 38). New-generation hydroxamate-based SMIs were shown to be effective inhibitors with IC50 values in the nanomolar to low-micromolar range in in vitro assays (41, 44–46). However, because of their general binding to zinc, these compounds are known to suffer from significant off-target effects, including interaction with the hERG cardiac potassium channel, and mutagenic effects (47). Additionally, these hydroxamate compounds are reported to have poor bioavailability, short metabolic half-lives, and high cytotoxicity (48, 49). Our group also identified 30-kDa RNA aptamers with IC50s in the nanomolar range, but it has been a challenging task to deliver aptamers into cells (11).
In this study, we have successfully identified a serotype-specific natural product-based molecule active against the endopeptidase activity of LCA. Natural compounds have more diverse structural scaffolds than synthetic compounds, have low toxicity with a potentially better ADME profile (50–52), and, thus, can provide a promising starting point for the development of varied therapeutic agents. Of all the drugs discovered, ∼60% of anticancer agents were reported to be either natural products or natural product-based drugs (53). During the period from 2005 to 2010, 7 natural products and 12 natural product derivatives were approved for use in clinical practice (54–56). As previously reported, the antibotulinum natural compound toosendanin (57) was effective in antagonizing BoNT in the low-nanomolar range and acted by interfering with the HC domain of BoNT during the translocation step. Toosendanin was not effective against the proteolytic domain of BoNT. Buforin I, a natural antimicrobial peptide, was found to inhibit BoNT/B with an IC50 of 1 µM (58). A study of a library of 527 natural compounds has disclosed serotype-specific inhibition of BoNT by 35 compounds/extracts from different plants, demonstrating that natural products are a rich source of BoNT inhibitors (59). Based on knowledge from ayurvedic medicine (a system of medicine with historical roots in the Indian subcontinent) (60), several natural products have been identified, using an in vitro assay, to be effective against BoNT/A. However, such studies have not shown further evidence for the effectiveness of these compounds in a cellular assay. Two natural products, d-chicoric acid and lomofungin, were effective against BoNT/A by interacting with α- and β-exosites, respectively (61). Furthermore, lomofungin was reported to be effective in the cellular assay, albeit with a very high IC50 (131 μM) (61). Indeed, these studies show that natural compounds have the potential to be effective botulism inhibitors.
In this study, we screened a library of 300 natural compounds and analogs for their activity against the endopeptidase activity of BoNT/A and successfully identified the natural product-based molecule NPP to be an effective inhibitor of the proteolytic activity of BoNT/A with an IC50 in the low-micromolar range (4.74 µM with peptide substrate). Even though the assay with a peptide-based substrate was sensitive, rapid, and robust, it was necessary to confirm this finding using the full-length SNAP-25 reporter SNAG because the peptide is unable to interact with important exosites on the BoNT/A LC. As shown in Fig. 4, the efficacy of NPP was also evident using SNAG, as revealed by a similar IC50 (6.3 µM) (22). The finding that NPP inhibits interactions of LCA with both the peptide substrate containing only the active site and the full-length SNAP-25, containing the α-exosite recognition site (which is distant from the LCA cleavage site) (22), suggests that NPP interacts directly with the active site of LCA. To understand the detailed mechanism of action, further experiments, such as enzyme inhibitor kinetic studies, molecular dynamic (MD) studies, or inhibitor enzyme crystallography studies, can be performed.
In addition to the in vitro assay, we tested NPP in a cellular model, using M17 neuroblastoma cells. Our results showed that the IC50 value of NPP in the cellular model is 12.2 µM. This value is much lower than that of leading compounds reported in the literature (11, 62, 63). The cellular efficacy of NPP was further corroborated in the isolated mouse phrenic nerve-hemidiaphragm preparation. These studies revealed that NPP is able to extend BoNT/A-mediated muscle paralysis by a factor of 1.4 (Fig. 7), which is comparable to that found with the most effective mercaptoacetamide metalloprotease inhibitor tested by Jacobson et al. (64). In addition to its high efficacy, the cytotoxicity of NPP was remarkably low, and even at 310 µM, >90% of M17 cells remained viable after 24 h (Fig. 3). Moreover, at a concentration of 60 µM, NPP produced no apparent toxicity in diaphragm muscle, as evidenced by the absence of contractures or the loss of muscle tension.
We have also investigated the specificity of NPP, and results suggested that it is highly specific to BoNT/A, with inhibition of other serotypes, viz., BoNT/B and BoNT/E, occurring at substantially higher concentrations (IC50s, over 8-fold and 17-fold higher, respectively). This is possibly due to the specific molecular interaction between NPP and LCA. Even though the active-site residues HEXXH are conserved in all the BoNT serotypes, BoNT LCs are extremely selective for the three SNARE proteins, with exclusive cleavage (65). There are multiple reports published for the inhibitors of BoNT serotype A (11, 16, 41, 42, 62), but there are only 3 reports published on SMIs of BoNT/B (66–68) and only 2 reports published on SMIs of BoNT/E (68, 69). Additional studies of BoNT/A/B/E and NPP interactions using molecular dynamics or/and crystallography will shed light on the specific mechanism of action of NPP toward BoNT/A. Furthermore, the results also suggest that the NPP is not a general zinc chelator, as the compound did not inhibit thermolysin, which has a zinc-binding motif similar to that of LCA. X-ray crystallography or molecular dynamic (MD) simulation in solution phase can reveal the specific features of the interaction between NPP and its target, LCA, and help identify the core pharmacophore involved in the inhibition of the endopeptidase activity of BoNT/A. This will help to optimize the design of NPP analogs for improved efficacy in future studies.
NPP is a derivative of psoralen, a naturally occurring main ingredient in the plant Psoralea corylifolia, which has been used in the treatment of alopecia areata, vitiligo, and tinea (70). Psoralen exerts other biological activities, such as prevention of the proliferation of mucoepidermoid carcinoma, mammary cancer cells, and bladder carcinoma (71). Psoralen is a photosensitizing substance that enhances skin sensitivity by enhancing melanin production. Therefore, it is widely used in psoralen plus UV type A (PUVA) treatment for psoriasis, eczema, etc., when administered orally as well topically (70). Thus, the psoralen-derived compound NPP will have better drug-like properties with an approved clinical application. Our preliminary ADMET analysis of NPP has already shown that it has favorable pharmaceutical properties when Lipinski’s and CMC-like rules for drug candidates are followed (30–32, 72).
In summary, we have screened a natural product library and applied a sequential assay approach for the development of antidotes against BoNT/A. Using this approach, we have identified a potent natural product-based inhibitor, NPP, active against BoNT/A. Further optimization of its structure through structural analysis and improvement of its ADME properties through analysis of the structure-function relationship (SAR) are expected to lead to the development of NPP-based antidotes against botulism in preclinical and clinical studies.
MATERIALS AND METHODS
Materials.
The natural product inhibitor library was obtained from the Chemistry Department of the University of Delhi, Delhi, India. The synthetic peptide substrate for HTS was synthesized with a purity of >95% and was from New England Peptide (Gardner, MA). BoNT/A LC (LCA), BoNT/B LC (LCB), and BoNT/E LC (LCE) were purified in our laboratories at the University of Massachusetts Dartmouth and the Institute of Advanced Sciences (73). A 3-(4,5-dimethylthizol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell cytotoxicity assay kit was obtained from ATCC (Manassas, VA). Cell culture media were procured from ATCC (Manassas, VA). Antibiotics for the cell culture (kanamycin, penicillin, and streptomycin) were purchased from Life Technologies (Grand Island, NY). Rabbit anti-SNAP-25 antibody and alkaline phosphatase-conjugated anti-rabbit IgG were purchased from Sigma-Aldrich (St. Louis, MO). BCIP (5-bromo-4-chloroindolyl phosphatase) was acquired from Thermo Fisher Scientific (Waltham, MA).
Purification of BoNT/A, BoNT/B, and BoNT/E LCs.
All LCs were produced and purified as described previously using Ni2+ columns (73, 74). LCA, LCB, and LCE were purified in phosphate buffer (10 mM sodium phosphate, pH 8.0, containing 300 mM NaCl). Each LC was dialyzed against 10 mM phosphate buffer, pH 7.3, containing 150 mM NaCl. Purified LCs were stored in 20% aqueous glycerol at −80°C. The measured concentrations of the enzymes were as follows: for LCA, 0.63 mg/ml; for LCB, 1.53 mg/ml; and for LCE, 0.78 mg/ml.
FRET substrate peptide.
A 14-amino acid SNAP-25-derived FRET peptide was used to assay LCA endopeptidase activity as published previously (11). To more efficiently label the peptide with the fluorophore FITC, β-alanine was added to the N terminus. The sequence of the peptide is FITC-β(Ala)-Thr-(d-Arg)-Ile-Asp-Gln-Ala-Asn-Gln-Arg-Ala-Thr-Lys(DABCYL)-norleucine-CONH2. The 4-{[4-(dimethylamino)phenyl]azo}benzoic acid (DABCYL) component served as the FITC quencher (Fig. 1B). The peptide cleavage site is between the Gln and Arg residues, which are underlined in the peptide sequence above. The FRET reporter was synthesized by New England Peptide (Gardener, MA) and had a purity of >95%.
HTS.
The library of 300 natural compounds from Indian medicinal plants and their selective synthetic analogs was a gift from the University of Delhi, Delhi, India; these were screened for their activity against the endopeptidase activity of LCA, using the FRET peptide as a substrate. To carry out the screening, 100 nM LCA was preincubated with 50 µg/ml of the test compound in a 96-well clear-bottom microtiter plate (Corning, NY) at 37°C for 30 min, after which 50 µl of a 10 µM peptide reporter was added to the reaction mixture. The final working concentrations of the FRET peptide, LCA, and inhibitor candidates from the 300-compound library were 5 μM, 50 nM, and 25 µg/ml, respectively. All compounds were dissolved in DMSO; the final DMSO concentration in each well was <0.5% (vol/vol). Each plate contained at least 8 wells for the positive control and 8 wells for the negative control. The plates were incubated at 37°C for 30 min to allow the endopeptidase reaction to proceed. The positive-control well contained LCA without any library compound but had the same concentration of DMSO as the wells containing an inhibitor. The negative controls were peptide substrate without LCA incubated in the presence and absence of a library compound. The plates were read using a Spectra Max M5 fluorescence microplate reader (Molecular Devices, CA) with an excitation wavelength of 490 nm, an emission wavelength of 523 nm, and a cutoff of 495 nm.
The high-throughput screening (HTS) assays require not only a high-throughput capability and robustness but also adequate sensitivity, reproducibility, and accuracy in order to identify hits among a large number of compounds. The Zʹ factor (statistical effect size factor) (equation 1) has been used to accurately and rapidly evaluate HTS assays for the identification of hits from large chemical libraries. An ideal HTS assay would possess a Zʹ factor of 1, which indicates that there is no variation in the assay (the SD is 0) or that the data dynamic range is indefinite. In practical terms, for an assay with a Zʹ factor of between 0.5 and 1, the separation band between positive and negative controls is large and, therefore, the assay is suitable for HTS (75). In contrast, for an HTS assay with a Zʹ factor of between 0.5 and 0, the separation of the controls is small and the assay yields doubtful results. Furthermore, when the Zʹ factor is less than 0, there is no separation band between the positive and negative controls and the determination of hits is not possible.
To evaluate the HTS assay in our experiments, 18 individual plates with 8 positive and 8 negative controls were examined independently for each plate. The average Zʹ factor was calculated using equation 1.
| (1) |
where SD+ is the standard deviation for the positive controls, SD− is the standard deviation for the negative controls, Ave+ is the average for the positive control, and Ave− is the average for the negative control. The average value for Zʹ was determined to be 0.82 ± 0.08; the Zʹ factors of >0.8 for the 18 independent plates indicated the suitability of the assay protocol for HTS.
For accuracy and precision, we employed eight negative and eight positive controls per plate. Inhibition of the endopeptidase activity of LCA by the compounds was evaluated using the standard deviation for triplicate samples. The percent inhibition of LCA, after prior incubation with compounds from the library, was calculated using equation 2.
| (2) |
where IC is the value for inhibitor control, NC is the value for the negative control, and PC is the value for the positive control.
IC50 determination.
The concentration-response curve for NPP was performed using the peptide-based substrate. The 50% inhibitory concentration (IC50) value was interpolated from the concentration-response curve using a nonlinear polynomial regression. Eight concentrations of inhibitor were serially diluted in triplicate to generate the concentration-response curves.
Confirmation of inhibition using full-length recombinant SNAP-25.
An N-terminal glutathione S-transferase (GST) tag and a C-terminal green fluorescent protein (GFP) were attached to full-length, 24-kDa recombinant SNAP-25 to obtain GST–SNAP-25–GFP (SNAG). SNAG was used as a substrate to evaluate the inhibition of LCA endopeptidase activity (Fig. 1C). The 77-kDa SNAG fusion protein was purified using a glutathione affinity column. In the absence of the N- and C-terminal tags, cleavage of SNAP-25 by LCA yields a truncated SNAP-25 (from cleavage of the last 9 C-terminal residues) that differs from the parent compound by only 1 kDa. The 77-kDa SNAG is cleaved by LCA into 28.3-kDa and 48.7-kDa products with a molecular weight difference of 20 kDa, which is more clearly discerned on SDS-PAGE gels.
The endopeptidase reaction was carried out by incubating 1.5 μM SNAG substrate with 2 nM recombinant LCA at 37°C for 30 min. The reaction buffer used was 10 mM HEPES, pH 7.4, containing 150 mM NaCl and 1.25 mM freshly prepared dithiothreitol (DTT) and 0.05% Tween 20. Serially diluted NPP inhibitor was preincubated for 30 min at 37°C with LCA. After the addition of SNAG, the reaction mixture was incubated for an additional 30 min, and then the reaction was stopped with the addition of SDS-PAGE sample buffer followed by boiling for 3 min in a water bath. The reaction of 2 nM LCA with 1.5 µM SNAG in the absence of inhibitor was used as a positive control, and 1.5 µM SNAG in the absence of both inhibitor and LCA was used as a negative control. The reaction results were examined and viewed by running a precast mini-SDS-PAGE gel (4% to 20% Tris HCl; 10 wells; Bio-Rad Laboratories). Densitometric analysis of three independent gels was performed using Bio-Rad Image Lab (v.5.2.1) software. Percent inhibition was calculated using equation 2.
Analysis of inhibitor specificity.
Thermolysin (Sigma-Aldrich, St. Louis, MO), a zinc endopeptidase, was used to examine the selectivity of the natural compounds against LCA. NPP was first incubated with two different concentrations of thermolysin (100 nM or 1,000 nM) at 37°C for 30 min. Three micromolar SNAG was added to each thermolysin sample, and the contents were incubated at 37°C for 30 min. The reaction mixtures were then analyzed by SDS-PAGE (4% to 20% polyacrylamide gel). The final concentration of SNAG in the reaction mixture was 1.5 μM, the final concentration of inhibitor was kept near its IC50 value (6.3 µM), and the final concentration of thermolysin was either 50 nM or 500 nM. Densitometry analysis was performed using Bio-Rad Image Lab (v.5.2.1) software.
Serotype specificity of selected inhibitors.
To determine the selectivity of the inhibitors toward different BoNT serotypes, the effects of NPP on LCB and LCE were also examined. The FRET peptide substrate for LCB, o-Abz-Dnp (ortho-aminobenzoic acid-2,4-dinitrophenyl) (VAMPtide; List Biological Laboratories, CA), has an excitation wavelength of 321 nm and an emission wavelength of 418 nm. Inhibition of the endopeptidase activity of LCB was determined by preincubating the LC with or without NPP for 30 min at 37°C, followed by the addition of 5 µM OBZ-DNP in the assay buffer (10 mM HEPES buffer, 150 mM NaCl, 1.25 mM DTT, 0.05% Tween 20, pH 7.4). Inhibition was measured using a SpectraMax M5 plate reader.
LCA and LCE cleave different residues in SNAP-25; LCE cleaves between amino acids R180 and I181, whereas LCA cleaves between amino acids Q197 and R198 (Fig. 2). LCE was assayed for inhibition of endopeptidase activity using the SNAG substrate. The assay was performed using a protocol similar to that described above for the SNAG-based assay of LCA in the section “Confirmation of inhibition using full-length recombinant SNAP-25.”
Neuroblastoma cell-based assay.
M17 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS). Cells were seeded in 12-well plates with 2.5 × 105 cells per well and maintained in a humidified atmosphere of 5% CO2 at 37°C. The medium was changed after 24 h, and the cells were allowed to grow to 90% confluence. Varied concentrations of inhibitor and 30 nM BoNT/A were preincubated at 37°C for 30 min; the positive control was BoNT/A (without addition of the inhibitor). M17 cells were incubated for 36 h with the BoNT/A-inhibitor mixtures, washed with phosphate-buffered saline (PBS), and harvested into preweighed Eppendorf screw-cap vials. The cells were then pelleted by centrifugation (6,500 × g for 3 min). The supernatant was carefully removed, and the pellet was incubated with 10 µl of mammalian protein extraction reagent (M-PER; Thermo Fisher Scientific, USA) plus 10 μl of SDS-PAGE sample buffer and boiled for 10 min to inactivate the residual toxin. The samples were then subjected to immunoblot analysis.
Immunoblot analysis.
Aliquots of protein were electrophoresed through 15% polyacrylamide gels. Proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore Corp., Bedford, MA) and processed for immunoblot analysis. Blotting was performed with 5% nonfat dry milk in TBST (10 mM Tris, 150 mM NaCl, 0.1% [vol/vol] Tween 20, pH 7.4) for 1 h at room temperature, and the blots were probed with rabbit anti-SNAP-25 antibody (1:2,500 dilution; Sigma-Aldrich, St. Louis, MO) in 1% nonfat dry milk in TBST for 1 h at room temperature. Following three washes with TBST, secondary antibody incubation was performed using goat anti-rabbit IgG conjugated to IRdye 800 (1:15,000 dilution; Abcam, Cambridge, MA) in 1% nonfat dry milk in TBST. After incubation for 1 h at room temperature, the blots were washed and visualized with a LI-COR Odyssey detection system (LI-COR Biotechnology, Lincoln, NE, USA). The percent inhibition of SNAP-25 cleavage intensity was determined via quantitative densitometry (Odyssey Imager; LI-COR Biotechnology).
Mouse phrenic nerve-hemidiaphragm assay.
Adult male CD-1 mice (weight, 25 to 33 g) were used in all experiments (Charles River Laboratories, Wilmington, MA). The animals were maintained under an AAALAC-accredited animal care and use program, with food and water provided ad libitum. Mice were euthanized by exposure to excess isoflurane followed by decapitation, and diaphragms with attached phrenic nerves were rapidly excised and dissected into left and right hemidiaphragm preparations. All animal experiments were carried out under a protocol approved by the Animal Care and Use Committee at the United States Army Medical Research Institute of Chemical Defense (USAMRICD).
For measurement of isometric twitch tension, hemidiaphragm pairs were transferred to 20-ml twitch baths containing oxygenated (95% O2, 5% CO2) Tyrode’s solution, and the phrenic nerve was stimulated by supramaximal pulses of a 0.1-ms duration at 0.033 Hz to elicit twitch tensions. Tyrode’s solution contained 135 mM NaCl, 5.0 mM KCl, 1.0 mM MgCl2, 2.0 mM CaCl2, 15.0 mM NaHCO3, 1.0 mM Na2HPO4, and 11.0 mM glucose (pH 7.3 to 7.4). Resting tensions were maintained at 1 g to obtain optimal active tensions. Tensions were transduced to electrical signals using Grass FT03 force-displacement transducers (Natus Medical Inc., San Carlos, CA), digitized, and analyzed offline using pClamp software (v.10.1; Molecular Devices, Sunnyvale, CA).
To assess the efficacy of NPP, both hemidiaphragms were first exposed to 5 pM BoNT/A at room temperature for 30 min, followed by three washes to remove excess BoNT/A. Under these conditions, BoNT binds to its receptor without undergoing significant internalization. After the last wash, NPP was added to one hemidiaphragm and an equivalent volume of DMSO was added to the other to serve as a vehicle control. The temperature was then increased to 37°C to initiate internalization, and the time for half paralysis was determined. The above-described order of toxin and NPP additions ensures that the drug does not act by inactivating BoNT in solution prior to its binding to the nerve terminal. Statistical analyses were performed using a two-tailed t test (GraphPad InStat software, v.3.10). P values of <0.05 were considered significant.
MTT cytotoxicity assay.
The cytotoxicity of the inhibitors was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell viability assay. M17 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; ATCC, Manassas, VA) containing 10% fetal bovine serum (FBS) and the antibiotics kanamycin (100 µg/ml), penicillin (100 units/ml), and streptomycin (100 µg/ml) (Life Technologies, Grand Island, NY) and maintained in a humidified incubator supplied with 5% CO2 at 37°C in 96-well l-lysine-coated plates. Upon reaching 80% confluence, the medium was washed and replenished with fresh serum-free DMEM without antibiotics. The cells were then incubated with different concentrations of inhibitors for 24 h. For the positive control, six wells without inhibitors were used. Six empty wells without any cells were also used for the negative control to allow the subsequent removal of the background signal. Cell viability was measured after 24 h using the MTT assay (ATCC, Manassas, VA) in triplicate, as recommended by the manufacturer’s protocol. The absorbance at 570 nm was recorded with a Molecular Devices SpectraMax M5 microplate reader. The relative viability of the cells was normalized using the absorbance from the positive and negative controls.
Drug-like properties.
The drug-like properties of NPP were determined using preADMET software (v.2.0; Bioinformatics and Molecular Design Research Center [BMDRC], Seoul, South Korea). preADMET is a web-based tool for predicting ADME and the drug-like properties of small molecules.
Supplementary Material
ACKNOWLEDGMENTS
We sincerely acknowledge the financial support provided by the United States Department of Defense (DoD) Consortium (W81XWH-14-C-0070) and by the Maryada Foundation.
We appreciate Suraj K. Singh and Shashwat Malhotra for kindly providing NMR data for the NPP compound used in the study.
The views expressed in this article are those of the authors and do not reflect the official policy of the Department of the Army, Department of Defense, or the U.S. Government.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01280-18.
REFERENCES
- 1.Kukreja R, Chang TW, Cai S, Lindo P, Riding S, Zhou Y, Ravichandran E, Singh BR. 2009. Immunological characterization of the subunits of type A botulinum neurotoxin and different components of its associated proteins. Toxicon 53:616–624. doi: 10.1016/j.toxicon.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 2.Cai S, Singh BR, Sharma S. 2007. Botulism diagnostics: from clinical symptoms to in vitro assays. Crit Rev Microbiol 33:109–125. doi: 10.1080/10408410701364562. [DOI] [PubMed] [Google Scholar]
- 3.Breidenbach MA, Brunger AT. 2005. New insights into clostridial neurotoxin-SNARE interactions. Trends Mol Med 11:377–381. doi: 10.1016/j.molmed.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 4.Salti G, Ghersetich I. 2008. Advanced botulinum toxin techniques against wrinkles in the upper face. Clin Dermatol 26:182–191. doi: 10.1016/j.clindermatol.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 5.Patel K, Cai S, Singh BR. 2014. Current strategies for designing antidotes against botulinum neurotoxins. Expert Opin Drug Discov 9:319–333. doi: 10.1517/17460441.2014.884066. [DOI] [PubMed] [Google Scholar]
- 6.Dong M, Yeh F, Tepp WH, Dean C, Johnson EA, Janz R, Chapman ER. 2006. SV2 is the protein receptor for botulinum neurotoxin A. Science 312:592–596. doi: 10.1126/science.1123654. [DOI] [PubMed] [Google Scholar]
- 7.Schiavo G, Matteoli M, Montecucco C. 2000. Neurotoxins affecting neuroexocytosis. Physiol Rev 80:717–766. doi: 10.1152/physrev.2000.80.2.717. [DOI] [PubMed] [Google Scholar]
- 8.Francisco AM, Arnon SS. 2007. Clinical mimics of infant botulism. Pediatrics 119:826–828. doi: 10.1542/peds.2006-0645. [DOI] [PubMed] [Google Scholar]
- 9.Fan Y, Barash JR, Lou J, Conrad F, Marks JD, Arnon SS. 2016. Immunological characterization and neutralizing ability of monoclonal antibodies directed against botulinum neurotoxin type H. J Infect Dis 213:1606–1614. doi: 10.1093/infdis/jiv770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng LW, Stanker LH, Henderson TD II, Lou J, Marks JD. 2009. Antibody protection against botulinum neurotoxin intoxication in mice. Infect Immun 77:4305–4313. doi: 10.1128/IAI.00405-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai S, Lindo P, Park JB, Vasa K, Singh BR. 2010. The identification and biochemical characterization of drug-like compounds that inhibit botulinum neurotoxin serotype A endopeptidase activity. Toxicon 55:818–826. doi: 10.1016/j.toxicon.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang TW, Blank M, Janardhanan P, Singh BR, Mello C, Blind M, Cai S. 2010. In vitro selection of RNA aptamers that inhibit the activity of type A botulinum neurotoxin. Biochem Biophys Res Commun 396:854–860. doi: 10.1016/j.bbrc.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ravichandran E, Janardhanan P, Patel K, Riding S, Cai S, Singh BR. 2016. In vivo toxicity and immunological characterization of detoxified recombinant botulinum neurotoxin type A. Pharm Res 33:639–652. doi: 10.1007/s11095-015-1816-x. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt JJ, Stafford RG. 2002. A high-affinity competitive inhibitor of type A botulinum neurotoxin protease activity. FEBS Lett 532:423–426. doi: 10.1016/S0014-5793(02)03738-9. [DOI] [PubMed] [Google Scholar]
- 15.Burnett JC, Li B, Pai R, Cardinale SC, Butler MM, Peet NP, Moir D, Bavari S, Bowlin T. 2010. Analysis of botulinum neurotoxin serotype A metalloprotease inhibitors: analogs of a chemotype for therapeutic development in the context of a three-zone pharmacophore. Open Access Bioinformatics 2010:11–18. doi: 10.2147/OAB.S7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roxas-Duncan V, Enyedy I, Montgomery VA, Eccard VS, Carrington MA, Lai H, Gul N, Yang DC, Smith LA. 2009. Identification and biochemical characterization of small-molecule inhibitors of Clostridium botulinum neurotoxin serotype A. Antimicrob Agents Chemother 53:3478–3486. doi: 10.1128/AAC.00141-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keller JE, Neale EA, Oyler G, Adler M. 1999. Persistence of botulinum neurotoxin action in cultured spinal cord cells. FEBS Lett 456:137–142. doi: 10.1016/S0014-5793(99)00948-5. [DOI] [PubMed] [Google Scholar]
- 18.Keller JE, Cai F, Neale EA. 2004. Uptake of botulinum neurotoxin into cultured neurons. Biochemistry 43:526–532. doi: 10.1021/bi0356698. [DOI] [PubMed] [Google Scholar]
- 19.Pellett S, Bradshaw M, Tepp WH, Pier CL, Whitemarsh RCM, Chen C, Barbieri JT, Johnson EA. 2018. The light chain defines the duration of action of botulinum toxin serotype a subtypes. mBio 9:e00089-18. doi: 10.1128/mBio.00089-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Momtaz K, Fitzpatrick TB. 1998. The benefits and risks of long-term PUVA photochemotherapy. Dermatol Clin 16:227–234. doi: 10.1016/S0733-8635(05)70005-X. [DOI] [PubMed] [Google Scholar]
- 21.Chen S, Kim JJ, Barbieri JT. 2007. Mechanism of substrate recognition by botulinum neurotoxin serotype A. J Biol Chem 282:9621–9627. doi: 10.1074/jbc.M611211200. [DOI] [PubMed] [Google Scholar]
- 22.Breidenbach MA, Brunger AT. 2004. Substrate recognition strategy for botulinum neurotoxin serotype A. Nature 432:925–929. doi: 10.1038/nature03123. [DOI] [PubMed] [Google Scholar]
- 23.Xue S, Javor S, Hixon MS, Janda KD. 2014. Probing BoNT/A protease exosites: implications for inhibitor design and light chain longevity. Biochemistry 53:6820–6824. doi: 10.1021/bi500950x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh BR. 2000. Intimate details of the most poisonous poison. Nat Struct Biol 7:617–619. doi: 10.1038/77900. [DOI] [PubMed] [Google Scholar]
- 25.Singh BR. 2006. Botulinum neurotoxin structure, engineering, and novel cellular trafficking and targeting. Neurotox Res 9:73–92. doi: 10.1007/BF03033925. [DOI] [PubMed] [Google Scholar]
- 26.Zuniga JE, Schmidt JJ, Fenn T, Burnett JC, Arac D, Gussio R, Stafford RG, Badie SS, Bavari S, Brunger AT. 2008. A potent peptidomimetic inhibitor of botulinum neurotoxin serotype A has a very different conformation than SNAP-25 substrate. Structure 16:1588–1597. doi: 10.1016/j.str.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montecucco C, Schiavo G. 1994. Mechanism of action of tetanus and botulinum neurotoxins. Mol Microbiol 13:1–8. doi: 10.1111/j.1365-2958.1994.tb00396.x. [DOI] [PubMed] [Google Scholar]
- 28.Simpson LL. 1980. Kinetic studies on the interaction between botulinum toxin type A and the cholinergic neuromuscular junction. J Pharmacol Exp Ther 212:16–21. [PubMed] [Google Scholar]
- 29.Adler M, Deshpande SS, Apland JP, Murray B, Borrell A. 2012. Reversal of BoNT/A-mediated inhibition of muscle paralysis by 3,4-diaminopyridine and roscovitine in mouse phrenic nerve-hemidiaphragm preparations. Neurochem Int 61:866–873. doi: 10.1016/j.neuint.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 30.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26. doi: 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 31.Ajay 2002. Predicting drug-likeness. Why and how? Curr Top Med Chem 2:1273–1286. doi: 10.2174/1568026023392968. [DOI] [PubMed] [Google Scholar]
- 32.Ajay A, Walters WP, Murcko MA. 1998. Can we learn to distinguish between “drug-like” and “nondrug-like” molecules? J Med Chem 41:3314–3324. doi: 10.1021/jm970666c. [DOI] [PubMed] [Google Scholar]
- 33.Nayak SU, Griffiss JM, McKenzie R, Fuchs EJ, Jurao RA, An AT, Ahene A, Tomic M, Hendrix CW, Zenilman JM. 2014. Safety and pharmacokinetics of XOMA 3AB, a novel mixture of three monoclonal antibodies against botulinum toxin A. Antimicrob Agents Chemother 58:5047–5053. doi: 10.1128/AAC.02830-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Opsenica I, Filipovic V, Nuss JE, Gomba LM, Opsenica D, Burnett JC, Gussio R, Solaja BA, Bavari S. 2012. The synthesis of 2,5-bis(4-amidinophenyl)thiophene derivatives providing submicromolar-range inhibition of the botulinum neurotoxin serotype A metalloprotease. Eur J Med Chem 53:374–379. doi: 10.1016/j.ejmech.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Šilhár P, Alakurtti S, Čapková K, Xiaochuan F, Shoemaker CB, Yli-Kauhaluoma J, Janda KD. 2011. Synthesis and evaluation of library of betulin derivatives against the botulinum neurotoxin A protease. Bioorg Med Chem Lett 21:2229–2231. doi: 10.1016/j.bmcl.2011.02.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cardellina JH, Roxas-Duncan VI, Montgomery V, Eccard V, Campbell Y, Hu X, Khavrutskii I, Tawa GJ, Wallqvist A, Gloer JB, Phatak NL, Höller U, Soman AG, Joshi BK, Hein SM, Wicklow DT, Smith LA. 2012. Fungal bis-naphthopyrones as inhibitors of botulinum neurotoxin serotype A. ACS Med Chem Lett 3:387–391. doi: 10.1021/ml200312s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Videnovic M, Opsenica DM, Burnett JC, Gomba L, Nuss JE, Selakovic Z, Konstantinovic J, Krstic M, Segan S, Zlatovic M, Sciotti RJ, Bavari S, Solaja BA. 2014. Second generation steroidal 4-aminoquinolines are potent, dual-target inhibitors of the botulinum neurotoxin serotype A metalloprotease and P. falciparum malaria. J Med Chem 57:4134–4153. doi: 10.1021/jm500033r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caglic D, Krutein MC, Bompiani KM, Barlow DJ, Benoni G, Pelletier JC, Reitz AB, Lairson LL, Houseknecht KL, Smith GR, Dickerson TJ. 2014. Identification of clinically viable quinolinol inhibitors of botulinum neurotoxin A light chain. J Med Chem 57:669–676. doi: 10.1021/jm4012164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harrell WA Jr, Vieira RC, Ensel SM, Montgomery V, Guernieri R, Eccard VS, Campbell Y, Roxas-Duncan V, Cardellina JH II, Webb RP, Smith LA. 2016. A matrix-focused structure-activity and binding site flexibility study of quinolinol inhibitors of botulinum neurotoxin serotype A. Bioorg Med Chem Lett doi: 10.1016/j.bmcl.2016.11.019. [DOI] [PubMed] [Google Scholar]
- 40.Lai H, Feng M, Roxas-Duncan V, Dakshanamurthy S, Smith LA, Yang DC. 2009. Quinolinol and peptide inhibitors of zinc protease in botulinum neurotoxin A: effects of zinc ion and peptides on inhibition. Arch Biochem Biophys 491:75–84. doi: 10.1016/j.abb.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 41.Eubanks LM, Hixon MS, Jin W, Hong S, Clancy CM, Tepp WH, Baldwin MR, Malizio CJ, Goodnough MC, Barbieri JT, Johnson EA, Boger DL, Dickerson TJ, Janda KD. 2007. An in vitro and in vivo disconnect uncovered through high-throughput identification of botulinum neurotoxin A antagonists. Proc Natl Acad Sci U S A 104:2602–2607. doi: 10.1073/pnas.0611213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boldt GE, Eubanks LM, Janda KD. 2006. Identification of a botulinum neurotoxin A protease inhibitor displaying efficacy in a cellular model. Chem Commun (Camb) 2006:3063–3065. [DOI] [PubMed] [Google Scholar]
- 43.Pang YP, Davis J, Wang S, Park JG, Nambiar MP, Schmidt JJ, Millard CB. 2010. Small molecules showing significant protection of mice against botulinum neurotoxin serotype A. PLoS One 5:e10129. doi: 10.1371/journal.pone.0010129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Malley S, Sareth S, Jiao G-S, Kim S, Thai A, Cregar-Hernandez L, McKasson L, Margosiak SA, Johnson AT. 2013. Virtual medicinal chemistry: in silico pre-docking functional group transformation for discovery of novel inhibitors of botulinum toxin serotype A light chain. Bioorg Med Chem Lett 23:2505–2511. doi: 10.1016/j.bmcl.2013.03.030. [DOI] [PubMed] [Google Scholar]
- 45.Thompson AA, Jiao G-S, Kim S, Thai A, Cregar-Hernandez L, Margosiak SA, Johnson AT, Han GW, O’Malley S, Stevens RC. 2011. Structural characterization of three novel hydroxamate-based zinc chelating inhibitors of the Clostridium botulinum serotype A neurotoxin light chain metalloprotease reveals a compact binding site resulting from 60/70 loop flexibility. Biochemistry 50:4019–4028. doi: 10.1021/bi2001483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Čapková K, Hixon MS, Pellett S, Barbieri JT, Johnson EA, Janda KD. 2010. Benzylidene cyclopentenediones: first irreversible inhibitors against botulinum neurotoxin A’s zinc endopeptidase. Bioorg Med Chem Lett 20:206–208. doi: 10.1016/j.bmcl.2009.10.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goracci L, Deschamps N, Randazzo GM, Petit C, Dos Santos Passos C, Carrupt P-A, Simões-Pires C, Nurisso A. 2016. A rational approach for the identification of non-hydroxamate HDAC6-selective inhibitors. Sci Rep 6:29086. doi: 10.1038/srep29086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Capek P, Zhang Y, Barlow DJ, Houseknecht KL, Smith GR, Dickerson TJ. 2011. Enhancing the pharmacokinetic properties of botulinum neurotoxin serotype A protease inhibitors through rational design. ACS Chem Neurosci 2:288–293. doi: 10.1021/cn200021q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen S, Kozikowski AP. 2016. Why hydroxamates may not be the best histone deacetylase inhibitors—what some may have forgotten or would rather forget? ChemMedChem 11:15–21. doi: 10.1002/cmdc.201500486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu CP, Ohnuma S, Ambudkar SV. 2011. Discovering natural product modulators to overcome multidrug resistance in cancer chemotherapy. Curr Pharm Biotechnol 12:609–620. doi: 10.2174/138920111795163887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waltenberger B, Mocan A, Šmejkal K, Heiss EH, Atanasov AG. 2016. Natural products to counteract the epidemic of cardiovascular and metabolic disorders. Molecules 21:e807. doi: 10.3390/molecules21060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Atanasov AG, Waltenberger B, Pferschy-Wenzig EM, Linder T, Wawrosch C, Uhrin P, Temml V, Wang L, Schwaiger S, Heiss EH, Rollinger JM, Schuster D, Breuss JM, Bochkov V, Mihovilovic MD, Kopp B, Bauer R, Dirsch VM, Stuppner H. 2015. Discovery and resupply of pharmacologically active plant-derived natural products: a review. Biotechnol Adv 33:1582–1614. doi: 10.1016/j.biotechadv.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Newman DJ, Cragg GM, Snader KM. 2003. Natural products as sources of new drugs over the period 1981-2002. J Nat Prod 66:1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- 54.Clardy J, Walsh C. 2004. Lessons from natural molecules. Nature 432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- 55.Newman DJ. 2008. Natural products as leads to potential drugs: an old process or the new hope for drug discovery? J Med Chem 51:2589–2599. doi: 10.1021/jm0704090. [DOI] [PubMed] [Google Scholar]
- 56.Newman DJ, Cragg GM. 2007. Natural products as sources of new drugs over the last 25 years. J Nat Prod 70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 57.Li MF, Shi YL. 2006. Toosendanin interferes with pore formation of botulinum toxin type A in PC12 cell membrane. Acta Pharmacol Sin 27:66–70. doi: 10.1111/j.1745-7254.2006.00236.x. [DOI] [PubMed] [Google Scholar]
- 58.Garcia GE, Moorad DR, Gordon RK. 1999. Buforin I, a natural peptide, inhibits botulinum neurotoxin B activity in vitro. J Appl Toxicol 19:S19–S22. doi:. [DOI] [PubMed] [Google Scholar]
- 59.Hines HB, Kim AD, Stafford RG, Badie SS, Brueggeman EE, Newman DJ, Schmidt JJ. 2008. Use of a recombinant fluorescent substrate with cleavage sites for all botulinum neurotoxins in high-throughput screening of natural product extracts for inhibitors of serotypes A, B, and E. Appl Environ Microbiol 74:653–659. doi: 10.1128/AEM.01690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yalamanchili C, Manda VK, Chittiboyina AG, Guernieri RL, Harrell WA Jr, Webb RP, Smith LA, Khan IA. 2016. Utilizing ayurvedic literature for the identification of novel phytochemical inhibitors of botulinum neurotoxin A. J Ethnopharmacol 197:211–217. [DOI] [PubMed] [Google Scholar]
- 61.Eubanks LM, Silhar P, Salzameda NT, Zakhari JS, Xiaochuan F, Barbieri JT, Shoemaker CB, Hixon MS, Janda KD. 2010. Identification of a natural product antagonist against the botulinum neurotoxin light chain protease. ACS Med Chem Lett 1:268–272. doi: 10.1021/ml100074s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burnett JC, Ruthel G, Stegmann CM, Panchal RG, Nguyen TL, Hermone AR, Stafford RG, Lane DJ, Kenny TA, McGrath CF, Wipf P, Stahl AM, Schmidt JJ, Gussio R, Brunger AT, Bavari S. 2007. Inhibition of metalloprotease botulinum serotype A from a pseudo-peptide binding mode to a small molecule that is active in primary neurons. J Biol Chem 282:5004–5014. doi: 10.1074/jbc.M608166200. [DOI] [PubMed] [Google Scholar]
- 63.Boldt GE, Kennedy JP, Janda KD. 2006. Identification of a potent botulinum neurotoxin a protease inhibitor using in situ lead identification chemistry. Org Lett 8:1729–1732. doi: 10.1021/ol0603211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jacobson AR, Adler M, Silvaggi NR, Allen KN, Smith GM, Fredenburg RA, Stein RL, Park JB, Feng X, Shoemaker CB, Deshpande SS, Goodnough MC, Malizio CJ, Johnson EA, Pellett S, Tepp WH, Tzipori S. 2017. Small molecule metalloprotease inhibitor with in vitro, ex vivo and in vivo efficacy against botulinum neurotoxin serotype A. Toxicon 137:36–47. doi: 10.1016/j.toxicon.2017.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pirazzini M, Rossetto O, Eleopra R, Montecucco C. 2017. Botulinum neurotoxins: biology, pharmacology, and toxicology. Pharmacol Rev 69:200–235. doi: 10.1124/pr.116.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hanson MA, Oost TK, Sukonpan C, Rich DH, Stevens RC. 2000. Structural basis for BABIM inhibition of botulinum neurotoxin type B protease. J Am Chem Soc 122:11268–11269. doi: 10.1021/ja005533m. [DOI] [Google Scholar]
- 67.Adler M, Nicholson JD, Cornille F, Hackley BE Jr. 1998. Efficacy of a novel metalloprotease inhibitor on botulinum neurotoxin B activity. FEBS Lett 429:234–238. doi: 10.1016/S0014-5793(98)00492-X. [DOI] [PubMed] [Google Scholar]
- 68.Montgomery VA, Ahmed SA, Olson MA, Mizanur RM, Stafford RG, Roxas-Duncan VI, Smith LA. 2015. Ex vivo inhibition of Clostridium botulinum neurotoxin types B, C, E, and F by small molecular weight inhibitors. Toxicon 98:12–19. doi: 10.1016/j.toxicon.2015.02.012. [DOI] [PubMed] [Google Scholar]
- 69.Kumar G, Agarwal R, Swaminathan S. 2012. Discovery of a fluorene class of compounds as inhibitors of botulinum neurotoxin serotype E by virtual screening. Chem Commun (Camb) 48:2412–2414. doi: 10.1039/c2cc17158a. [DOI] [PubMed] [Google Scholar]
- 70.da Silva VB, Kawano DF, Carvalho I, da Conceicao EC, de Freitas O, da Silva CH. 2009. Psoralen and bergapten: in silico metabolism and toxicophoric analysis of drugs used to treat vitiligo. J Pharm Pharm Sci 12:378–387. doi: 10.18433/J3W01D. [DOI] [PubMed] [Google Scholar]
- 71.Liu Y, Zhang G, Liao Y, Wang Y. 2015. Binding characteristics of psoralen with trypsin: insights from spectroscopic and molecular modeling studies. Spectrochim Acta A Mol Biomol Spectrosc 151:498–505. doi: 10.1016/j.saa.2015.07.018. [DOI] [PubMed] [Google Scholar]
- 72.Leeson P. 2012. Drug discovery: chemical beauty contest. Nature 481:455–456. doi: 10.1038/481455a. [DOI] [PubMed] [Google Scholar]
- 73.Li L, Singh BR. 1999. High-level expression, purification, and characterization of recombinant type A botulinum neurotoxin light chain. Protein Expr Purif 17:339–344. doi: 10.1006/prep.1999.1138. [DOI] [PubMed] [Google Scholar]
- 74.Kumar R, Kukreja RV, Cai S, Singh BR. 2014. Differential role of molten globule and protein folding in distinguishing unique features of botulinum neurotoxin. Biochim Biophys Acta 1844:1145–1152. doi: 10.1016/j.bbapap.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 75.Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.