

The discovery of complete ammonia oxidizers (comammox), which oxidize ammonia to nitrate via nitrite, refutes the century-old paradigm that nitrification requires the activity of two types of microbes and redefines a key process in the biogeochemical nitrogen cycle. Understanding the functional relationships between comammox and other nitrifiers is important for ecological studies on the nitrogen cycle. Therefore, the diversity and contribution of comammox should be considered during ecological analyses of nitrifying microorganisms. In this study, a ubiquitous and highly diverse distribution of comammox was observed in various environmental samples, similar to the distribution of canonical ammonia-oxidizing bacteria. The proportion of comammox was relatively high in coastal water and sediment samples, whereas it was nearly undetectable in open-ocean samples. The ubiquitous distribution and high diversity of comammox indicate that these microorganisms might be important contributors to nitrification.
KEYWORDS: AOA, AOB, comammox, diversity, metagenome, partial nested PCR
ABSTRACT
The discovery of complete ammonia oxidizers (comammox) refutes the century-old paradigm that nitrification requires the activity of two types of microbes. Determining the distribution and abundance of comammox in various environments is important for revealing the ecology of microbial nitrification within the global nitrogen cycle. In this study, the ubiquity and diversity of comammox were analyzed for samples from different types of environments, including soil, sediment, sludge, and water. The results of a two-step PCR using highly degenerate primers (THDP-PCR) and quantitative real-time PCR (qPCR) supported the relatively high abundance of comammox in nearly half of all samples tested, sometimes even outnumbering canonical ammonia-oxidizing bacteria (AOB). In addition, a relatively high proportion of comammox in tap and coastal water samples was confirmed via analysis of metagenomic data sets in public databases. The diversity of comammox was estimated by comammox-specific partial nested PCR amplification of the ammonia monooxygenase subunit A (amoA) gene, and phylogenetic analysis of comammox AmoA clearly showed a split of clade A into clades A.1 and A.2, with the proportions of clades A.1, A.2, and B differing among the various environmental samples. Moreover, compared to the amoA genes of AOB and ammonia-oxidizing archaea (AOA), the comammox amoA gene exhibited higher diversity indices. The ubiquitous distribution and high diversity of comammox indicate that they are likely overlooked contributors to nitrification in various ecosystems.
IMPORTANCE The discovery of complete ammonia oxidizers (comammox), which oxidize ammonia to nitrate via nitrite, refutes the century-old paradigm that nitrification requires the activity of two types of microbes and redefines a key process in the biogeochemical nitrogen cycle. Understanding the functional relationships between comammox and other nitrifiers is important for ecological studies on the nitrogen cycle. Therefore, the diversity and contribution of comammox should be considered during ecological analyses of nitrifying microorganisms. In this study, a ubiquitous and highly diverse distribution of comammox was observed in various environmental samples, similar to the distribution of canonical ammonia-oxidizing bacteria. The proportion of comammox was relatively high in coastal water and sediment samples, whereas it was nearly undetectable in open-ocean samples. The ubiquitous distribution and high diversity of comammox indicate that these microorganisms might be important contributors to nitrification.
INTRODUCTION
Nitrification is a central part of the nitrogen cycle and the only oxidative biological process linking nitrogen fixation and denitrification in natural ecosystems (1). For more than a century, nitrification was thought to be a two-step process (2) that involves ammonia oxidation to nitrite by chemolithoautotrophic ammonia-oxidizing microorganisms and nitrite oxidation to nitrate by nitrite-oxidizing bacteria (NOB). Autotrophic ammonia oxidation has been described for specific groups of microorganisms, including canonical ammonia-oxidizing bacteria (AOB) within Betaproteobacteria and Gammaproteobacteria (3) and ammonia-oxidizing archaea (AOA) within Thaumarchaea (4).
However, the discovery of complete ammonia oxidizers (comammox), which oxidize ammonia to nitrate via nitrite, refutes this century-old paradigm that nitrification requires the activity of two types of microbes, and it redefines a key process of the biogeochemical nitrogen cycle (5, 6). Theoretical analyses of kinetics and thermodynamics predict that comammox have a high growth yield and a low specific growth rate (7), which has recently been supported by experimental observations (8). Comammox were initially identified through a combination of enrichment and metagenomics from a trickling filter connected to an aquaculture system (9) and a biofilm on the walls of a pipe under the flow of hot water raised from a deep oil exploration well (10). All known comammox are members of the genus Nitrospira and possess the full genetic complement for both ammonia and nitrite oxidation (9, 10). Metagenomic evidence of the presence of comammox in drinking water distribution systems (11, 12), groundwater-fed rapid sand filters (13–15), wastewater treatment processes (16–19), and agriculture soils (20) has also been reported.
The functional relationships of comammox with other nitrifiers, such as AOB, AOA, NOB, and anaerobic ammonia oxidizers may depend on whether the main activity of the comammox in the specific environment is ammonia oxidation or nitrite oxidation. Therefore, the diversity and contribution of comammox are important topics for ecological studies of the nitrogen cycle and of ammonia- or nitrite-oxidizing microorganisms.
The widespread distribution of comammox in both manmade environments and a variety of natural terrestrial ecosystems has been analyzed by data mining of metagenomic data sets (9, 10). Some researchers have used the 16S rRNA (21) or nxrB (nitrite-oxidoreductase subunit B) (22) genes for population analyses of comammox communities. However, these genes should not be used to identify comammox because these bacteria are not distinguishable via phylogenetic analyses of these genes from strict NOB in the genus Nitrospira (23).
The genomes of all identified comammox contain the ammonia monooxygenase subunit A (amoA) gene, which has been widely employed as a functional and phylogenetic marker for population analyses of AOB (3) and AOA (24). Phylogenetic analyses have revealed that known comammox form a monophyletic group with AOB in Betaproteobacteria as a sister group, and AmoA of these two groups can be phylogenetically separated from particulate methane monooxygenase subunit A (PmoA), an enzyme homologous to AmoA, of methane-oxidizing bacteria (MOB) (9). The three comammox species Nitrospira inopinata, “Candidatus Nitrospira nitrificans,” and “Candidatus Nitrospira nitrosa” form a cluster (named clade A) in a phylogenetic tree for AmoA (9, 10). Another cluster (named clade B) was proposed based on the amoA gene sequences from groundwater metagenome bins that contain both ammonia and nitrite oxidation-related genes (10). Putative comammox amoA gene sequences were previously identified as an “unusual” pmoA gene of the methanotroph genus Crenothrix in Gammaproteobacteria (for clade A) (25) or presumed a methanotroph from Alphaproteobacteria (for clade B) (26) and excluded from ecological analyses of nitrifiers.
In our previous study, we improved a protocol for community analysis of AOB and MOB using a two-step PCR technique with highly degenerate primers (THDP-PCR) that target the copper-containing membrane-bound monooxygenase (CuMMO) gene family, which includes the amoA and pmoA genes (27). With this method, a relatively high proportion of comammox was observed in some environmental samples (27).
In the present study, THDP-PCR, quantitative real-time PCR (qPCR), and data mining of metagenomic/metatranscriptomic data sets were applied to compare the proportions of comammox with those of AOB in various environmental samples. In addition, the population structure and diversity of comammox in these samples were analyzed by partial nested PCR amplification using newly designed comammox-specific primer sets.
RESULTS
Ubiquity of comammox.
Communities of comammox, AOB, and MOB in various soil, sediment, sludge, water, and leaf surface samples were analyzed by THDP-PCR amplification targeting of the CuMMO gene family. HiSeq and MiSeq sequencing generated averages of 39,129 and 3,600 reads per sample, respectively. Figure 1A shows the proportions of comammox and AOB in different types of environmental samples, with the two sequencing methods (HiSeq and MiSeq) yielding similar fractions for all samples. The proportions of comammox were higher than those of AOB in 12 of the 21 samples, and the minimal comammox to AOB ratio observed in all 21 samples was approximately 1:5. In nearly all samples, the proportions of AOB in the genus Nitrosospira were higher than those of AOB in the genus Nitrosomonas. The very low proportion of comammox clade B members may be related to the limited coverage of the highly degenerate primers for THDP-PCR, as described below.
FIG 1.

Proportions or abundances of the amoA gene from comammox and AOB in 21 different environmental samples, assessed by two-step PCR with highly degenerate primers (THDP-PCR) (A) or by quantitative real-time PCR, compared with the abundance of the 16S rRNA gene from total bacteria (TB) (B). In panel A, the positive error bars indicate differences between the two sequencing methods (Illumina HiSeq and MiSeq). In panel B, the positive error bars indicate the standard deviation of three replicates, and the units of the results are copies per gram for soil/sediment/sludge samples, copies per liter for water samples and copies per square meter for leaf surface. An asterisk (*) indicates that the results were under the detection limit. For detailed information about the primer sets used in this analysis, please see Table 1.
Although the comammox-specific qPCR primers cover only comammox clade A, the qPCR analysis of these environmental samples also supported the relatively high abundances of comammox, sometimes even outnumbering AOB, in 9 of 21 samples (Fig. 1B). The fractions of comammox among total bacteria were highest in the tap water (0.5%) and coastal water (0.3%) samples, exceeding these fractions of AOB and corroborating the results obtained by THDP-PCR. The contents of comammox and AOB were similar in river and lake sediment samples, while AOB were much more abundant in river and lake water samples.
The proportions of comammox and other ammonia oxidizers (AOB and AOA) were also analyzed using 275 metagenomic data sets and 175 metatranscriptomic data sets of different environmental samples in the NCBI Sequence Read Archive (SRA) database (Table S1). According to the data from 111 metagenomic data sets with at least 30 amoA gene sequences, the proportions of comammox among total aerobic ammonia oxidizers were high in freshwater (52.3 ± 37.5%), urban waterway sediments (45.8 ± 19.5%), drinking water systems (35.9 ± 33.2%), coastal and estuary water (35.4 ± 20.7%), and paddy soils (26.8 ± 11.5%) (Fig. 2). In some drinking water influent, river water, ground water, and urban waterway sediment samples, the proportion of comammox among the total ammonia-oxidizing microorganism population was higher than 70%. However, the proportion of comammox in the metatranscriptomic data sets was much lower for most of the environmental samples (Fig. S1 and Table S1), including all wastewater treatment systems (2.4 ± 1.2%) and estuary and coastal (2.5 ± 1.1%) samples. Comammox-related reads were nearly absent from the metagenome and metatranscriptomic data sets from open ocean samples, and this result was confirmed by data in the Community Cyberinfrastructure for Advanced Marine Microbial Ecology Research and Analysis (CAMERA) database (Table S2).
FIG 2.
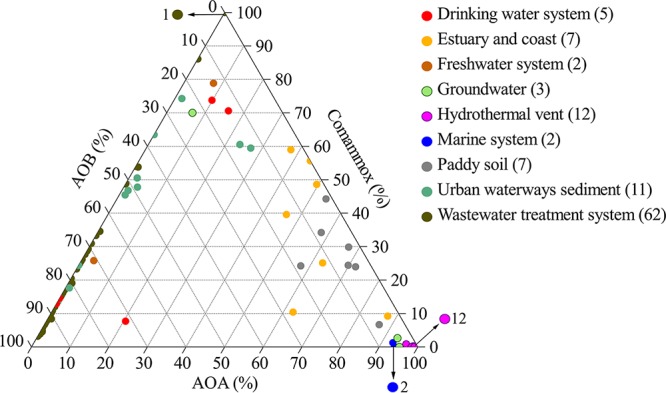
A ternary plot of the proportions of comammox, AOB, and AOA in 111 metagenomic data sets of different environmental samples from the NCBI SRA database with amoA gene sequence read numbers of at least 30. The numbers directed by arrows in the figure indicate the numbers of overlapping specific color circles. The numbers in parentheses denote the numbers of data sets used in this analysis for specific environmental types. For detailed information for each data set, please see Table S1.
Diversity of comammox.
To determine the diversity of comammox in various environments, the comammox-specific primer sets A189Y/C576r (for first-round PCR) and CA209f/C576r (for second-round PCR) (Table 1), covering both clades A and B, were designed and applied in partial nested PCR. Using this approach resulted in successful amplification with relatively high specificity for the comammox amoA gene (>70% of the sequences in 13/18 samples) for all but three of the samples (hot spring sediment, glacier soil, and wetland soil samples) (Table S3). Compared with the results obtained using recently reported primer sets separately targeting clades A and B (28), the results from our approach with partial nested PCR showed much higher amplification efficiency for most of the samples (Fig. S2), with similar coverage of the comammox population (Fig. S3).
TABLE 1.
Primers used in this study
| Application | Primer name | Sequence (5′ to 3′)a | Degeneracy | Annealing temperature (°C) | Amplicon length (bp) | Source and/or reference(s) |
|---|---|---|---|---|---|---|
| THDP-PCR for amoA/pmoA gene of comammox, AOB, and MOB | A189Y-tag | GCCGGAGCTCTGCAGATATCGGNGACTGGGAYTTYTGGb | 16 | 59 | 513 | 27; this study |
| HD616N-tag (first step) | GCCGGAGCTCTGCAGATATCAYCWKVCKNAYRTAYTCNGGb | 6,144 | ||||
| tag-barcode (second step) | XXXXXXXXGCCGGAGCTCTGCAGATATCb,c | 1 | 59 | |||
| Partial nested PCR for the amoA gene of comammox | A189Y | GGNGACTGGGAYTTYTGG | 16 | 52 | 415 | This study |
| C576r (first step) | GAAGCCCATRTARTCNGCC | |||||
| CA209f | GAYTGGAARGAYCGNCA | 32 | 50 | |||
| C576r-barcode (second step) | XXXXXXXXXXXXGAAGCCCATRTARTCNGCCc | 16 | ||||
| PCR for amoA gene of AOB | amoA-1F-barcode | XXXXXXXXXXXXGGGGTTTCTACTGGTGGTc | 1 | 57 | 491 | 31 |
| amoA-2R | CCCCTCKGSAAAGCCTTCTTC | 4 | ||||
| PCR for amoA gene of AOA | Arch-amoAF-barcode | XXXXXXXXXXXXSTAATGGTCTGGCTTAGACGc | 2 | 53 | 635 | 24 |
| Arch-amoAR | GCGGCCATCCATCTGTATGT | 1 | ||||
| qPCR for total bacterial 16S rRNA gene | 338F | ACTCCTACGGGAGGCAGC | 1 | 55 | 200 | 39, 40 |
| 536R | GTATTACCGCGGCKGCTG | 2 | ||||
| qPCR for amoA gene of AOB | amoA-1F | GGGGTTTCTACTGGTGGT | 1 | 57 | 491 | 31 |
| amoA-2R | CCCCTCKGSAAAGCCTTCTTC | 4 | ||||
| qPCR for amoA gene of comammox clade A | A378f | TGGTGGTGGTGGTCNAAYTAT | 8 | 55 | 278 | This study |
| C616r | ATCATCCGRATGTACTCHGG | 6 | ||||
| THDP-PCR for amoAB gene of comammox | A378f-tag | GCCGGAGCTCTGCAGATATCTGGTGGTGGTGGTCNAAYTATb | 8 | 50 | 683 | This study |
| HDamo/pmoB (first step) | GCCGGAGCTCTGCAGATATCCKCATBCKNADRAAYGGYTCb | 1,152 | ||||
| tag-barcode (second step) | XXXXXXXXGCCGGAGCTCTGCAGATATCb,c | 1 | 59 |
Degenerate bases: R, A/G; Y, C/T; K, G/T; S, G/C; W, A/T; H, A/T/C; V, A/G/C; B, G/C/T; and N, A/T/C/G.
The underlined bases indicate the sequences of the tag.
XXXXXXXX and XXXXXXXXXXXX indicate the presence of 8- and 12-base barcodes, respectively, used to differentiate samples in high-throughput sequencing mixtures.
A phylogenetic tree (Fig. 3) of comammox amoA genes based on sequences from these 18 environmental samples clearly showed separation of clades A and B, with clade A being further divided into two monophyletic groups, clades A.1 and A.2. The split of clades A.1 and A.2 was supported by high bootstrap values at nodes of comammox clades A.1 (bootstrap value = 73) and A.2 (bootstrap value = 87) in a phylogenetic tree constructed using the maximum-likelihood method. The maximum-parsimony and nearest-neighbor phylogenetic trees also supported the division of Clade A into two monophyletic groups (Fig. 3). A similarity matrix of different operational taxonomic units (OTUs) with a 90% threshold also supported the separation of clades A and B as well as that of clades A.1 and A.2 (Table S5). Comammox in clade B were relatively dominant in forest soil (96% of the total comammox), plateau soil, paddy soil, and river sediment (>40%). This result means that the low proportion of comammox clade B in these samples, as determined by the THDP-PCR method, may be caused by limited coverage of the primer set. Regardless, comammox in clade A were dominant in all other samples (>95%) (Fig. 4). The proportions of clades A.1 and A.2 differed in various environmental samples; clade A.1 was dominant in activated sludge, anaerobic sludge, lake sediment, lake water, tidal flat sediment, and coastal water samples (>80%), whereas clade A.2 was dominant in coal mine soil, dry land soil, and tap water samples (>80%) (Fig. 4).
FIG 3.

Maximum-likelihood phylogenetic tree of comammox AmoA based on 30 major OTUs (no less than 0.7%) with a 90% similarity cutoff. The data in parentheses following the OTU numbers indicate the average content of each OTU in the 18 different environmental samples amplified by comammox-specific partial nested PCR (amplification failed for the other three of the 21 samples). The data in parentheses following the bootstrap values indicate the bootstrap values in the neighbor-joining and maximum-parsimony trees. The details of the content of each OTU for each environmental sample are listed in Table S4.
FIG 4.

Relative proportions of the different clades of comammox in 18 different environmental samples (amplification failed for the other three of the 21 samples) analyzed using partial nested PCR with comammox-specific primer sets. For the detailed read numbers of different types of ammonia oxidizers, please see Table S3.
Overall, 1,052 and 198 unique OTUs were identified based on 97% and 90% similarity cutoff thresholds, respectively. Both the observed and extrapolated comammox richness varied among different environmental samples. At a 90% clustering threshold, the Chao1 richness estimates ranged from 34 (for tap water) to 114 (for the leaf surface); at a 97% clustering threshold, the Chao1 richness estimates ranged from 75 (for tap water) to 243 (for river water) (Table 2). In addition, plateau soil and tidal flat sediment samples exhibited relatively high Shannon indices. To compare diversity between the different comammox clades and AOB and AOA, the same number (1,900) of amoA gene sequences of comammox, AOB, and AOA were randomly selected from each of 12 samples that had been efficiently amplified for all three types of ammonia oxidizers (Fig. 5). When a 97% or 90% similarity cutoff was used, the whole-sample amoA gene diversity indices of comammox were much higher than those of AOA and AOB, with higher diversity for clade A.1 than for clades A.2 and B (Fig. 5). In addition, the diversity indices for each of the 12 samples showed the same tendency (Table S6). A diversity index comparison based on the same regions of the amoA gene in comammox and AOB or in comammox and AOA also supported the diversity of the comammox amoA gene being higher than the diversity of the AOB and AOA amoA genes (Fig. S4).
TABLE 2.
Diversity indices with 97% or 90% similarity cutoffs for comammox amoA gene sequencesa
| Sample name | Cutoff |
|||||
|---|---|---|---|---|---|---|
| 97% |
90% |
|||||
| No. of OTUs | Chao1 | Shannon | No. of OTUs | Chao1 | Shannon | |
| Dry land soil | 82 | 160 | 3.00 | 36 | 66 | 2.43 |
| Coal mine soil | 64 | 99 | 3.17 | 41 | 59 | 2.08 |
| Landfill soil | 141 | 224 | 4.44 | 69 | 81 | 3.79 |
| Paddy soil | 85 | 139 | 3.89 | 37 | 46 | 2.96 |
| Plateau soil | 144 | 187 | 5.33 | 50 | 60 | 4.18 |
| Forest soil | 56 | 99 | 2.54 | 30 | 93 | 2.04 |
| Tidal flat sediment | 128 | 184 | 5.03 | 64 | 84 | 4.20 |
| Saltmarsh sediment | 117 | 148 | 4.25 | 66 | 73 | 3.83 |
| Coastal sediment | 76 | 144 | 3.55 | 41 | 53 | 3.06 |
| River sediment | 139 | 225 | 4.76 | 73 | 90 | 3.83 |
| Lake sediment | 111 | 211 | 2.81 | 49 | 65 | 2.22 |
| Anaerobic sludge | 51 | 181 | 1.93 | 26 | 59 | 1.66 |
| Tap water | 50 | 75 | 2.44 | 27 | 34 | 1.48 |
| Coastal water | 50 | 107 | 2.78 | 28 | 43 | 2.22 |
| River water | 162 | 243 | 4.87 | 61 | 84 | 3.86 |
| Lake water | 54 | 90 | 2.69 | 31 | 61 | 1.97 |
| Leaf surface | 78 | 152 | 3.40 | 38 | 114 | 2.84 |
Amplified with partial nested PCR in different environmental samples after the read numbers were rarified to 1,900 for each sample.
FIG 5.

Rarefaction curves of the number of OTUs, the Chao1 estimator, and the Shannon index using OTU similarity thresholds of 97% (A) and 90% (B) for the amoA gene among different comammox clades, AOB, and AOA, based on a combination of 1,900 randomly selected reads from each of the 12 different environmental samples from which comammox, AOB, and AOA amoA genes were efficiently amplified. For diversity index comparison of amoA genes of comammox, AOB, and AOA for each sample, please see Table S6.
Coverage evaluation of comammox-targeting primers using metagenomic and metatranscriptomic data sets.
Our diversity analyses based on sequences obtained using PCR approaches might be prone to bias due to the coverage limitation of the primers. The identified comammox amoA gene sequences from the 450 public metagenomic and metatranscriptomic data sets described above were also utilized to evaluate the coverage of the designed primers (Table 3). The newly modified primer A189Y, which has two degenerate positions more than the normally used primer, A189 (29), greatly increased the coverages for comammox clade A and clade B, from 65% to 99% and from 85% to 98%, respectively. The highly degenerate primer HD616N showed high coverage for comammox clade A (97%) but not for clade B (67%), which would limit the amplification of comammox clade B by the THDP-PCR method. The primer sets A189Y/C576r and CA209/C576r, which were designed in this study for comammox-specific diversity analysis, showed high coverage (>90%) for clades A.1, A.2, and B. The common reverse primer used for partial nested PCR amplification, C576r, was mismatched with the relevant sequences in all members of AOB, the sister group of comammox. The primer set A378f/C616r, used for qPCR analysis of comammox clade A, did not match the corresponding sequences in AOB, and coverage of clade B was also very low (0 and 2%, respectively).
TABLE 3.
Coverage evaluation of primers based on the amoA gene sequencesa
| Primer | Clade (% [no./total]) |
AOB (% [no./total]) | |||
|---|---|---|---|---|---|
| A.1 | A.2 | A | B | ||
| A189Y | 99 (282/286) | 97 (672/690) | 98 (954/976) | 94 (50/53) | 98 (1,944/1,977) |
| HD616N | 97 (267/274) | 96 (76/79) | 97 (343/353) | 67 (31/46) | 51 (2,531/4,955) |
| CA209f | 98 (278/285) | 86 (506/586) | 90 (784/871) | 98 (63/64) | 98 (2,429/2,506) |
| C576r | 98 (277/283) | 87 (72/83) | 95 (349/366) | 91 (30/33) | 0 (0/5,051) |
| A378f | 78 (188/241) | 73 (66/91) | 77 (254/332) | 0 (0/36) | 0 (0/4,298) |
| C616r | 84 (231/274) | 58 (46/79) | 78 (277/353) | 2 (1/46) | 0 (0/4,951) |
| A189 | 76 (217/286) | 60 (415/690) | 65 (632/976) | 85 (45/53) | 53 (1,045/1,977) |
| A682 | 67 (130/194) | 26 (24/92) | 54 (154/286) | 73 (35/48) | 66 (412/626) |
Extracted from metagenomic or metatranscriptomic data sets in public databases, as described in Table S1. A sequence was considered to be covered by this primer if it had no mismatches or only one mismatch that was not located in the last four positions near the 3′ end. The numbers in parentheses denote the number of sequences matched with each primer and the number of extracted sequences that cover the region of this primer.
Although amoA gene sequences of comammox in clades A.1 and B were detected in previous diversity analyses of methane/ammonia-oxidizing bacteria using the primer set A189/A682 (29), clade A.2-related amoA gene sequences were not efficiently detected with this primer set (according to the NCBI database; data not shown). This difference may be due to the low clade A.2 coverage (26%) of primer A682, as evaluated by in silico analysis (Table 3), and one mismatch of the 3′ end of primer A682 with the comammox clade A.2 sequences was observed in this study (Fig. S5).
DISCUSSION
Determining the diversity and proportion of comammox species, a newly discovered type of ammonia-oxidizing bacteria, in various environments is important for understanding their contributions to the global nitrogen cycle.
The THDP-PCR method greatly enhances the coverage of the amoA/pmoA gene (27). Based on the wide coverage of PCR primers, this approach can be used for simultaneous analysis of the community composition of both AOB and comammox. The results of the THDP-PCR analysis in this study showed that the proportions of comammox were higher than or similar to those of AOB in many of the environmental samples assayed, even though this method provided limited coverage of comammox clade B. The high proportions of comammox in various environments, based on the qPCR (Fig. 1B) and public metagenomic data set analyses (Fig. 2), support the ubiquitous distribution and relative abundance of comammox. It is interesting that there were low proportions of comammox amoA in metatranscriptomic data sets, even for wastewater treatment systems, which were shown to have a high proportion of comammox in metagenomic data sets. This needs further research.
As reported previously (10), comammox amoA gene sequences have rarely been detected in open ocean metagenomic/metatranscriptomic data (Table S1 and S2). We also failed to amplify comammox amoA genes from open-ocean water and sediment samples (data not shown). However, the THDP-PCR, qPCR (Fig. 1), and public metagenomic data set analysis (Fig. 2 and Table S1) results support the high proportions of comammox in estuary and coastal environments. Whether environmental parameters such as the concentration of ammonia and salinity affects the distribution of comammox warrants further research.
Comammox were found to be distributed in various environments. Several primer sets have been developed by the specific amplification of all comammox clades from various environments. Although primer sets that are separately specific for comammox clade A or clade B amoA have been reported for quantitative real-time PCR and diversity analyses (28), they resulted in lower amplification efficiency than our partial nested PCR approach (Fig. S2). Another designed primer set, covering both clades A and B together, had two mismatches with most of the sequences in clades A and B, as previously described (14). The high-throughput sequencing and evaluation of the primer sets for coverage and specificity clearly demonstrated that the partial nested PCR amplification applied in this study can be used for efficient amplification of amoA of most types of comammox from various environments.
According to the results of partial nested PCR, the proportion of clade A among the total comammox population was higher than 95% in most of the samples in this study (14/18) (Fig. 4), while the dominant comammox type in the forest soil sample was clade B, with clade A as a minor type, as reported recently (28). Furthermore, the relative abundance of comammox clade A was shown to be higher than that of AOB in the forest soil sample by THDP-PCR and qPCR analyses (Fig. 1), which have limitations in amplifying comammox clade B. These results indicate that the content of comammox clade B in the forest sample may be much higher than that of AOB, and that comammox might be important contributors to the nitrogen cycle in forest soils.
The phylogenetic tree of comammox AmoA indicated clear separation of comammox in clade A into two groups, clades A.1 and A.2. The amoA gene of comammox in clade A.2 has not been detected in previous studies of community analyses of AOB or MOB, and the low coverage of commonly used primers (e.g., A189/682) may be one of the reasons (Table 3 and Fig. S5). The proportions of clades A.1 and A.2 varied depending on the environment (Fig. 4), suggesting differential niche adaptation for these two clades.
The diversity of comammox varied substantially in different environments. Among all analyzed samples, the tap water sample showed the lowest diversity and highest comammox proportion among total bacteria, which suggests that specific types of comammox in clade A.2 are adapted to the tap water system. Analysis of comammox amoA gene sequences from metagenomic/metatranscriptomic data sets in public databases (Fig. 2, Fig. S1, and Table S1) and identification of comammox in metagenomic bins from analysis of drinking water systems (11, 12) also support the high proportion of comammox in drinking water.
Unexpectedly, the amoA gene diversity of comammox was much higher than those of AOB and AOA in many of the compared samples in this study, although all identified comammox groups are related to Nitrospira sublineage II (23). The high diversity of the comammox amoA gene might be associated with the niche adaptation potential and thus with their ubiquitous distribution, although further research is needed in this respect.
Although comammox have been observed in various natural and manmade environments, comammox activity has been confirmed in only three enriched strains (9, 10), which, according to the phylogenetic tree, are affiliated with clade A.1 (Fig. 3). Members of clades B and A.2 have been identified as comammox only through the binning of metagenomic data sets from ground water (10) and drinking water (12), respectively. Therefore, the complete nitrification activity of clades B and A.2 should be investigated in future studies. We should also consider that the clustering of comammox based on the amoA gene may not completely match with clustering based on their genomic or physiological characteristics. It is clear from this study that the community composition and diversity of comammox are distinct in different environments. However, the limited number of types of samples may limit the representation of the comammox distribution characteristics in different habitats in this study. Overall, the relationship between comammox distribution and environmental parameters requires further research. To understand the nitrification potential of different environments, the primary environmental factors affecting the abundance of comammox and their composition should be addressed. Furthermore, the contribution of comammox to nitrification relative to the contributions of conventional AOB and AOA should be demonstrated, because numerical dominance does not always coincide with high biochemical activity (30).
MATERIALS AND METHODS
Description of environmental samples and DNA extraction.
Twenty-one various environmental samples were collected, including seven soil samples, seven sediment samples, two sludge samples, four water samples, and one plant leaf surface sample. The soil, sediment, and sludge samples were used directly for DNA extraction. For the water samples, 4 to 5 liters of water were filtered through 0.22-μm nitrocellulose filters (Merck Millipore, Billerica, MA), which were then used for genomic DNA extraction. For the leaf surface sample, 3 pieces of lotus leaves were washed with 1 liter of sterile water and then treated as a water sample. Detailed information for all of the samples is shown in Table S7. The environmental samples were stored at −20°C before DNA extraction. Total genomic DNA was extracted using a DNeasy PowerSoil kit (Qiagen, Dusseldorf, Germany) following the manufacturer’s instructions.
THDP-PCR amplification and high-throughput sequencing.
Amplifications using DNA extracted from the 21 environmental samples were performed using a modified THDP-PCR method (27). Briefly, the first round of genomic DNA PCR amplification was performed using the primer A189Y-tag (degeneracy = 16) and the highly degenerate primer HD616N-tag (degeneracy = 6,144), which provide good coverage of most bacterial amoA and pmoA genes, as well as the newly discovered comammox amoA gene sequences. Genomic DNA from each sample was amplified individually, and the PCR products were purified using an AxyPrep PCR clean-up kit (Axygen, Tewksbury, MA). Tagged primers with different barcodes and 2 μl of purified first-round PCR product were then used for the second round of amplification. The primers employed in the current study, with their targets and corresponding annealing temperatures, are listed in Table 1. The amplification reaction and thermal programs were the same as described previously (27). Appropriately sized PCR product fragments were separated by agarose gel electrophoresis and purified with an AxyPrep PCR clean-up kit (Axygen, Tewksbury, MA). Purified PCR products were assessed with a Qubit 2.0 fluorometer (Thermo Fisher, Waltham, MA) to determine their DNA concentrations and were then pooled at equal concentrations for sequencing using an Illumina HiSeq or MiSeq system according to the manufacturer’s protocols.
PCR amplification and high-throughput sequencing of comammox amoA genes.
For partial nested PCR amplification of the comammox amoA gene, the first round of nested PCR was performed using an automated thermal cycler (Eastwin, Beijing, China) with the primers A189Y (degeneracy = 16) and C576r in a total volume of 50 μl, containing 25 μl of Premix Ex Taq (TaKaRa, Tokyo, Japan), 1 μl of each primer (10 μM), 2 μl of bovine serum albumin (BSA; 20 mg ml−1), and template DNA (1 to 10 ng). The thermal program used was as follows: an initial step at 94°C for 5 min, followed by 20 cycles of 94°C for 1 min, 52°C for 50 s, and 72°C for 50 s, with a final step at 72°C for 10 min. Approximately 2 to 4 μl of the first-round PCR product was used as a template for the second round of partial nested PCR with 2 μl of each of the primers (10 μM) CA209f and C576r-barcode in a total volume of 50 μl and the following thermal program: an initial step at 94°C for 5 min, followed by 30 to 35 cycles of 94°C for 1 min, 50°C for 50 s, and 72°C for 50 s, with a final step at 72°C for 10 min. To compare PCR amplification efficiency and coverage for different comammox types, the primer sets reported by Pjevac et al. (28) were applied as described by the authors. Purified PCR products were pooled at equal concentrations for sequencing using an Illumina HiSeq system according to the manufacturer’s protocols. For amplification of the comammox amoA-amoB gene region with the THDP-PCR method, the tagged highly degenerate primer HDamoB-tag, targeting the amoB and/or pmoB gene, and the tagged comammox-specific primer A378f-tag were used. The two-step PCR amplification process was the same as that described above for the THDP-PCR method, except that the annealing temperature of the first PCR step was 50°C.
PCR amplification and high-throughput sequencing of amoA genes from AOA and AOB.
Amplification of the amoA gene was performed with the primers amoA1F-barcode/amoA2R (31) and Arch-amoAF-barcode/Arch-amoAR (24) for AOB and AOA, respectively, using an automated thermal cycler (Eastwin, Beijing, China). To differentiate samples, a 12-bp-long barcode was added to both forward primers. The PCR conditions were the same as those described by Rotthauwe et al. (31) and Francis et al. (24). Purified PCR products were pooled at equal concentrations for sequencing using an Illumina HiSeq System according to the manufacturer’s protocols.
qPCR.
Copy numbers of the amoA gene from comammox and AOB and copy numbers of the 16S rRNA gene from total bacteria (TB) were quantified by qPCR. The specific primer set A378f/C616r was designed for quantitation of comammox clade A, using sequences of the comammox amoA gene. The specificity of this primer set was confirmed using clone libraries. The primer sets used for quantification of the amoA gene from comammox and AOB and quantification of the 16S rRNA gene from TB and their corresponding annealing temperatures are listed in Table 1. During analysis of the fraction of comammox and AOB among TB, the bias caused by differences in the gene copy numbers of the 16S rRNA and amoA genes in the bacterial genomes was not considered. The reactions were performed in triplicate for each sample, and the 20-μl total reaction volume contained 10 μl of SYBR Premix Ex Taq (TaKaRa, Tokyo, Japan), 2 μl of template DNA (5 to 10 ng), 0.4 μl of each primer (10 μM), 0.2 μl of BSA (20 mg ml−1), and 0.4 μl of ROX reference dye (50×). Plasmid DNAs containing the corresponding gene fragments were serially diluted to generate external standard curves. qPCR was performed using a Stratagene Mx3000P system (Thermo Fisher, Waltham, MA). The thermal cycling programs were as follows: 95°C for 5 min, followed by 40 cycles of 95°C for 30 s, annealing for 30 s (see Table 1 for annealing temperature), and 72°C for 30 s. Melting curves were analyzed to detect the presence of primer dimers. The qPCR results were analyzed using MxPro qPCR software version 3.0 (Thermo Fisher, Waltham, MA). Consistency in the results was confirmed by the strong linear relationship between the threshold cycle and the log value of the gene copy number (for the 16S rRNA gene from TB and the amoA gene from comammox and AOB, R2 values were 0.997, 0.998, and 0.973, and amplification efficiency values were 104.5, 99.6, and 98.2%, respectively).
High-throughput sequencing data analyses.
The quality of the postsequencing data was visualized using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Sequences were primarily processed using the QIIME software package (32). The barcode sequence region (8 bases for THDP-PCR amplicons and 12 bases for partial nested PCR and AOB/AOA amoA gene amplicons) was removed, and the amplicon sequences were filtered based on their Phred quality scores (Q = 20). For THDP-PCR and AOB/AOA amoA amplicons, only one sequence direction was used for further analyses, because assembly of both of the sequence directions was difficult due to the relative length of the PCR products (Table 1 for detailed information); however, for the partial nested PCR amplicons, quality-controlled sequences from both directions were assembled into one relatively long sequence. Next, chosen THDP-PCR amplicons with the same direction and the assembled nested PCR amplicons were assigned to individual samples according to their barcodes and then chimera-checked by USEARCH61 (33) using the expanded CuMMO-related gene database constructed by Wang et al. (27).
For THDP-PCR amplicons, the sequences were first prefiltered using the expanded CuMMO-related protein database with 40% identity using USEARCH61 (33) and then assigned to OTUs based on a 90% identity threshold for nucleic acid sequences using the expanded CuMMO-related gene database via pick_open_reference_otus.py with QIIME (32). Sequences shorter than 200 bp and singletons were removed, and the longest sequence in each OTU was chosen as its representative sequence. Taxonomic identification of the OTU representatives was carried out using MEGAN with the lowest common ancestor, as previously described (27).
Based on the partial nested PCR amplicon data, sequences were assigned to OTUs according to a 97% identity threshold, as described above. Sequences shorter than 250 bp were removed. The set of representative sequences was then translated into protein sequences using FrameBot (34) and filtered using the criteria of amino acid length of ≥126 and OTUs with read numbers of ≥5. Sequences were also reassigned to another series of OTUs based on a 90% identity threshold, through assignment of the representative sequence in each OTU with a 97% identity threshold. The protein sequences of the OTU representatives were employed to construct phylogenic trees using the neighbor-joining, maximum-likelihood, and minimum-parsimony methods. The taxonomy of each OTU was obtained from the phylogenic position of each representative in these trees. To compare diversity indices of different samples with the same sequence numbers, samples with more than 1,900 reads identified as comammox were rarified for diversity index analyses (number of OTUs, Chao1 estimator, and Shannon index), based on OTU thresholds of 97% or 90%, using the single_rarefaction.py script in the QIIME package. The analyses of the AOB/AOA amoA gene data were similar to those of the partial nested PCR amplicon data, except that the length filter criterion was changed to 200 bp due to the relatively shorter length of the nonassembled AOB/AOA amoA sequences. For comparison of the diversity indices of comammox with those of AOB and AOA, the same amoA gene regions of assembled sequences of comammox and forward reads (read 1) of AOB (from 169 to 306 nucleic acid region of the full-length amoA gene of Nitrospira inopinata) and assembled sequences of comammox and reverse reads (read 2) of AOA (from 331 to 561 nucleic acid region of the full-length amoA gene of Nitrospira inopinata) were selected for each sequence and used for the diversity analysis as described above.
Distribution of comammox in public metagenomic and metatranscriptomic data sets.
To confirm the ubiquity and diversity of comammox, 275 metagenomic and 175 metatranscriptomic data sets were downloaded from NCBI SRA (35) through screening with keywords (e.g., metagenome, metatranscriptome, water, soil, waste, and marine). The extracted data were trimmed to remove sequences shorter than 60 bp and low-quality bases (Q = 20) using Sickle version 1.33 (https://github.com/najoshi/sickle). Next, the trimmed paired-end sequences were assembled using fastq-join in ea-utils version 1.1.2-806 (36). For read pairs that could not be assembled, only one read remained to maintain an accurate count. Finally, all of the retained sequences were combined into one file. Based on Functional Gene Pipeline/Repository (37) data and identified comammox AmoA sequences, a reference data subset with 381 AmoA/PmoA representative protein sequences that covered most AOB, AOA, MOB, and comammox was constructed. This AmoA/PmoA reference data subset was used in tBLASTn queries to search against metagenomic or metatranscriptomic data sets with an e-value cutoff of 10. The extracted shotgun sequences were translated (minimum amino acid length, 30) and classified using FrameBot (34) as described above, and the classification was manually checked once again. Potential nonspecific sequences were removed if the sequence could not be translated during FrameBot analysis. For marine-related environments, 55 metagenomic and 25 metatranscriptomic data sets in the CAMERA database (41) were also searched using the same methods described above.
The identified comammox amoA gene sequences were also utilized to evaluate comammox amoA gene-related primers using a previously described pipeline (38). These gene sequences were translated, aligned, and classified using RDP Tools (https://github.com/rdpstaff/RDPTools) after processing in the FunGene pipeline (https://github.com/rdpstaff/fungene_pipeline).
Accession number(s).
The amoA/pmoA gene sequences of comammox, AOB and MOB obtained using the THDP-PCR method and the amoA gene sequences of comammox obtained using the partial nested PCR method were deposited in NCBI GenBank Sequence Read Archive (SRA) under accession number SRP115447. The amoA gene sequences of comammox (using the method reported by Pjevac et al. in reference 28), AOA and AOB for comparison were deposited in NCBI GenBank SRA under accession numbers SRR7279855 to SRR7279858. The sequences inserted in the standard plasmids used in the qPCR analyses were deposited in NCBI GenBank under accession numbers MH454492, MH444515, and MH444516.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (NSFC) (grant numbers 91752207 and 31470222).
We declare no conflict of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01390-18.
REFERENCES
- 1.Gruber N, Galloway JN. 2008. An earth-system perspective of the global nitrogen cycle. Nature 451:293–296. doi: 10.1038/nature06592. [DOI] [PubMed] [Google Scholar]
- 2.Winogradsky S. 1890. Contributions à la morphologie des organismes de la nitrification. Ann Inst Pasteur 1890:213–231. [Google Scholar]
- 3.Purkhold U, Pommerening-Roser A, Juretschko S, Schmid MC, Koops HP, Wagner M. 2000. Phylogeny of all recognized species of ammonia oxidizers based on comparative 16S rRNA and amoA sequence analysis: implications for molecular diversity surveys. Appl Environ Microbiol 66:5368–5382. doi: 10.1128/AEM.66.12.5368-5382.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spang A, Hatzenpichler R, Brochier-Armanet C, Rattei T, Tischler P, Spieck E, Streit W, Stahl DA, Wagner M, Schleper C. 2010. Distinct gene set in two different lineages of ammonia-oxidizing archaea supports the phylum Thaumarchaeota. Trends Microbiol 18:331–340. doi: 10.1016/j.tim.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 5.Kuypers MMM. 2015. A division of labour combined. Nature 528:487–488. doi: 10.1038/528487a. [DOI] [PubMed] [Google Scholar]
- 6.Lawson CE, Lucker S. 2018. Complete ammonia oxidation: an important control on nitrification in engineered ecosystems? Curr Opin Biotechnol 50:158–165. doi: 10.1016/j.copbio.2018.01.015. [DOI] [PubMed] [Google Scholar]
- 7.Costa E, Perez J, Kreft JU. 2006. Why is metabolic labour divided in nitrification? Trends Microbiol 14:213–219. doi: 10.1016/j.tim.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Kits KD, Sedlacek CJ, Lebedeva EV, Han P, Bulaev A, Pjevac P, Daebeler A, Romano S, Albertsen M, Stein LY, Daims H, Wagner M. 2017. Kinetic analysis of a complete nitrifier reveals an oligotrophic lifestyle. Nature 549:269–272. doi: 10.1038/nature23679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Kessel MAHJ, Speth DR, Albertsen M, Nielsen PH, Op den Camp HJM, Kartal B, Jetten MSM, Lücker S. 2015. Complete nitrification by a single microorganism. Nature 528:555–559. doi: 10.1038/nature16459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daims H, Lebedeva EV, Pjevac P, Han P, Herbold C, Albertsen M, Jehmlich N, Palatinszky M, Vierheilig J, Bulaev A, Kirkegaard RH, von Bergen M, Rattei T, Bendinger B, Nielsen PH, Wagner M. 2015. Complete nitrification by Nitrospira bacteria. Nature 528:504–509. doi: 10.1038/nature16461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinto AJ, Marcus DN, Ijaz UZ, Santos QMBD, Dick GJ, Raskin L. 2016. Metagenomic evidence for the presence of comammox Nitrospira-like bacteria in a drinking water system. mSphere 1:e00054-15. doi: 10.1128/mSphere.00054-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang YL, Ma LP, Mao YP, Jiang XT, Xia Y, Yu K, Li B, Zhang T. 2017. Comammox in drinking water systems. Water Res 116:332–341. doi: 10.1016/j.watres.2017.03.042. [DOI] [PubMed] [Google Scholar]
- 13.Bartelme RP, McLellan SL, Newton RJ. 2017. Freshwater recirculating aquaculture system operations drive biofilter bacterial community shifts around a stable nitrifying consortium of ammonia-oxidizing archaea and comammox Nitrospira. Front Microbiol 8:101. doi: 10.3389/fmicb.2017.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fowler SJ, Palomo A, Dechesne A, Mines PD, Smets BF. 2018. Comammox Nitrospira are abundant ammonia oxidizers in diverse groundwater-fed rapid sand filter communities. Environ Microbiol 20:1002–1015. doi: 10.1111/1462-2920.14033. [DOI] [PubMed] [Google Scholar]
- 15.Palomo A, Fowler SJ, Gulay A, Rasmussen S, Sicheritz-Ponten T, Smets BF. 2016. Metagenomic analysis of rapid gravity sand filter microbial communities suggests novel physiology of Nitrospira spp. ISME J 10:2569–2581. doi: 10.1038/ismej.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Annavajhala MK, Kapoor V, Santo-Domingo J, Chandran K. 2018. Comammox functionality identified in diverse engineered biological wastewater treatment systems. Environ Sci Technol Lett 5:110–116. doi: 10.1021/acs.estlett.7b00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Camejo PY, Domingo JS, McMahon KD, Noguera DR. 2017. Genome-enabled insights into the ecophysiology of the comammox bacterium “Candidatus Nitrospira nitrosa.” mSystems 2:e00059-17. doi: 10.1128/mSystems.00059-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan XY, Gao JF, Pan KL, Li DC, Dai HH. 2017. Temporal dynamics of bacterial communities and predicted nitrogen metabolism genes in a full-scale wastewater treatment plant. RSC Adv 7:56317–56327. doi: 10.1039/C7RA10704H. [DOI] [Google Scholar]
- 19.Chao Y, Mao Y, Yu K, Zhang T. 2016. Novel nitrifiers and comammox in a full-scale hybrid biofilm and activated sludge reactor revealed by metagenomic approach. Appl Microbiol Biotechnol 100:8225–8237. doi: 10.1007/s00253-016-7655-9. [DOI] [PubMed] [Google Scholar]
- 20.Orellana LH, Chee-Sanford JC, Sanford RA, Loffler FE, Konstantinidis KT. 2018. Year-round shotgun metagenomes reveal stable microbial communities in agricultural soils and novel ammonia oxidizers responding to fertilization. Appl Environ Microbiol 84:e01646. doi: 10.1128/AEM.01646-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzalez-Martinez A, Rodriguez-Sanchez A, van Loosdrecht MCM, Gonzalez-Lopez J, Vahala R. 2016. Detection of comammox bacteria in full-scale wastewater treatment bioreactors using tag-454-pyrosequencing. Environ Sci Pollut Res 23:25501–25511. doi: 10.1007/s11356-016-7914-4. [DOI] [PubMed] [Google Scholar]
- 22.Tatari K, Musovic S, Gulay A, Dechesne A, Albrechtsen HJ, Smets BF. 2017. Density and distribution of nitrifying guilds in rapid sand filters for drinking water production: dominance of Nitrospira spp. Water Res 127:239–248. doi: 10.1016/j.watres.2017.10.023. [DOI] [PubMed] [Google Scholar]
- 23.Daims H, Lucker S, Wagner M. 2016. A new perspective on microbes formerly known as nitrite-oxidizing bacteria. Trends Microbiol 24:699–712. doi: 10.1016/j.tim.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Francis CA, Roberts KJ, Beman JM, Santoro AE, Oakley BB. 2005. Ubiquity and diversity of ammonia-oxidizing archaea in water columns and sediments of the ocean. Proc Natl Acad Sci U S A 102:14683–14688. doi: 10.1073/pnas.0506625102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stoecker K, Bendinger B, Schoning B, Nielsen PH, Nielsen JL, Baranyi C, Toenshoff ER, Daims H, Wagner M. 2006. Cohn's Crenothrix is a filamentous methane oxidizer with an unusual methane monooxygenase. Proc Natl Acad Sci U S A 103:2363–2367. doi: 10.1073/pnas.0506361103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Radajewski S, Webster G, Reay DS, Morris SA, Ineson P, Nedwell DB, Prosser JI, Murrell JC. 2002. Identification of active methylotroph populations in an acidic forest soil by stable isotope probing. Microbiology 148:2331–2342. doi: 10.1099/00221287-148-8-2331. [DOI] [PubMed] [Google Scholar]
- 27.Wang JG, Xia F, Zeleke J, Zou B, Rhee SK, Quan ZX. 2017. An improved protocol with a highly degenerate primer targeting copper-containing membrane-bound monooxygenase genes for community analysis of methane- and ammonia-oxidizing bacteria. FEMS Microbiol Ecol 93:fiw244. doi: 10.1093/femsec/fiw244. [DOI] [PubMed] [Google Scholar]
- 28.Pjevac P, Schauberger C, Poghosyan L, Herbold CW, van Kessel MAHJ, Daebeler A, Steinberger M, Jetten MSM, Lucker S, Wagner M, Daims H. 2017. AmoA-targeted polymerase chain reaction primers for the specific detection and quantification of comammox Nitrospira in the environment. Front Microbiol 8:e1508. doi: 10.3389/fmicb.2017.01508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holmes AJ, Costello A, Lidstrom ME, Murrell JC. 1995. Evidence that particulate methane monooxygenase and ammonia monooxygenase may be evolutionarily related. FEMS Microbiol Lett 132:203–208. doi: 10.1111/j.1574-6968.1995.tb07834.x. [DOI] [PubMed] [Google Scholar]
- 30.Di HJ, Cameron KC, Shen JP, Winefield CS, O’Callaghan M, Bowatte S, He JZ. 2009. Nitrification driven by bacteria and not archaea in nitrogen-rich grassland soils. Nat Geosci 2:621–624. doi: 10.1038/ngeo613. [DOI] [Google Scholar]
- 31.Rotthauwe JH, Witzel KP, Liesack W. 1997. The ammonia monooxygenase structural gene amoA as a functional marker: molecular fine-scale analysis of natural ammonia-oxidizing populations. Appl Environ Microbiol 63:4704–4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 34.Wang Q, Quensen JF III, Fish JA, Lee TK, Sun YN, Tiedje JM, Cole JR. 2013. Ecological patterns of nifH genes in four terrestrial climatic zones explored with targeted metagenomics using FrameBot, a new informatics tool. mBio 4:e00592-13. doi: 10.1128/mBio.00592-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leinonen R, Sugawara H, Shumway M, International Nucleotide Sequence Database Collaboration. 2011. The Sequence Read Archive. Nucleic Acids Res 39:19–21. doi: 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aronesty E. 2013. Comparison of sequencing utility programs. Open Bioinforma J 1:1–8. doi: 10.2174/1875036201307010001. [DOI] [Google Scholar]
- 37.Fish JA, Chai BL, Wang Q, Sun YN, Brown CT, Tiedje JM, Cole JR. 2013. FunGene: the functional gene pipeline and repository. Front Microbiol 4:291. doi: 10.3389/fmicb.2013.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zou B, Li JF, Zhou Q, Quan ZX. 2017. MIPE: A metagenome-based community structure explorer and SSU primer evaluation tool. PLoS One 12:e0174609. doi: 10.1371/journal.pone.0174609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruff-Roberts AL, Kuenen JG, Ward DM. 1994. Distribution of cultivated and uncultivated cyanobacteria and Chloroflexus-like bacteria in hot-spring microbial mats. Appl Environ Microbiol 60:697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whiteley AS, Bailey MJ. 2000. Bacterial community structure and physiological state within an industrial phenol bioremediation system. Appl Environ Microbiol 66:2400–2407. doi: 10.1128/AEM.66.6.2400-2407.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seshadri R, Kravitz SA, Smarr L, Gilna P, Frazier M. 2007. CAMERA: a community resource for metagenomics. PLoS Biol 5:e75. doi: 10.1371/journal.pbio.0050075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.