

Summary
Natural products luteolin, ellagic acid and punicic acid as a combination have the potential to inhibit prostate cancer metastasis.
Abstract
Prostate cancer (PCa) is the second cause of cancer deaths in men in the USA. When the cancer recurs, early stages can be controlled with hormone ablation therapy to delay the rate of cancer progression but, over time, the cancer overcomes its hormone dependence, becomes highly aggressive and metastasizes. Clinical trials have shown that pomegranate juice (PJ) inhibits PCa progression. We have previously shown that the PJ components luteolin (L), ellagic acid (E) and punicic acid (P) together inhibit growth of hormone-dependent and -independent PCa cells and inhibit their migration and chemotaxis towards CXCL12, a chemokine that is important in PCa metastasis. On the basis of these findings, we hypothesized that L+E+P inhibit PCa metastasis in vivo. To test this possibility, we used a severe combined immunodeficiency mouse model in which luciferase-expressing human PCa cells were injected subcutaneously near the prostate. Tumor progression was monitored with bioluminescence imaging weekly. We found that L+E+P inhibits PC-3M-luc primary tumor growth, inhibits the CXCL12/CXCR4 axis for metastasis and none of the tumors metastasized. In addition, L+E+P significantly inhibits growth and metastasis of highly invasive Pten−/−;K-rasG12D prostate tumors. Furthermore, L+E+P inhibits angiogenesis in vivo, prevents human endothelial cell (EC) tube formation in culture and disrupts preformed EC tubes, indicating inhibition of EC adhesion to each other. L+E+P also inhibits the angiogenic factors interleukin-8 and vascular endothelial growth factor as well as their induced signaling pathways in ECs. In conclusion, these results show that L+E+P inhibits PCa progression and metastasis.
Introduction
Prostate cancer (PCa) is the most common male malignancy and the second leading cause of cancer-related death among men in America. The American Cancer Society has estimated that a total of 238 590 new cases will be diagnosed and 29 720 men will die of PCa in 2013 (1). To date, there is no real cure for the disease beyond surgery and/or radiation. Early stages can be controlled with hormone ablation therapy that suppresses the rate of PCa growth. However, over time, the cancer overcomes its hormone dependence, becomes castration-resistant PCa and metastasizes to the lung, liver and bone (2).
Chemotherapy is available but the chemotherapeutic drugs are aggressive and have many negative side effects. As a result, researchers are looking for novel strategies to treat PCa. Food and Drug Administration–approved sipuleucel-T (Provenge®) is an autologous cellular immunotherapy to treat metastatic PCa. The median overall survival rate of patients who received sipuleucel-T was only improved by 4.5 months and treatment is costly but some patients survived much longer (3). Novel androgen receptor antagonists, such as enzalutamide (Xtandi®), and androgen biosynthesis inhibitors, such as abiraterone (Zytiga®), have shown great promise as androgen deprivation therapy to prolong overall survival rate among patients with metastatic PCa (4–8). Another novel drug, Cabozantinib, a potent dual inhibitor of the tyrosine kinases MET and VEGFR2, has been shown to reduce or stabilize metastatic bone lesions in patients with castration-resistant PCa (9,10). However, all of these treatments have adverse side effects.
Recently, pomegranate juice (PJ) has been identified as a natural agent to fight PCa. Mounting evidence shows that PJ has great potential to inhibit the growth and reduce the invasiveness of PCa cells both in vitro and in vivo (11–14). In 2006, a 2-year phase II clinical trial of patients with PCa with rising prostrate-specific antigen (PSA) were given 8 oz of PJ by mouth daily. PSA doubling time lengthened with treatment from a mean of 15.6 months at baseline to 54.7 months post-treatment (P < 0.001) (15). More recently in 2013, another phase II clinical trial of patients with PCa with rising PSA received 1 g (comparable to about 8 oz of PJ) or 3 g of pomegranate extract daily for up to 18 months. PSA doubling time lengthened more than 6 months from 11.9 to 18.5 months (P < 0.001) with no significant difference between dose groups (16). The statistically significant prolongation of PSA doubling time and the lack of metastatic progression in any of the patients strongly suggest a potential of PJ for treatment of PCa.
We have previously shown that PJ inhibits the migratory and metastatic properties of hormone refractory PCa cells by stimulating cell adhesion and inhibiting cell migration/chemotaxis (17). However, PJ contains many components and as a whole it is difficult to determine how to best maximize its use in treating PCa. A way to overcome this challenge is to identify components of PJ that are responsible for the antimetastatic effect of the whole juice. The juice contains a rich complement of polyphenolic compounds such as delphinidin, punicalin, punicalagin, quercetin and luteolin (12). Pomegranate pericarp is rich in tannates of gallic acid and ellagic acid, which are strongly antioxidant (18). Pomegranate seed oil is rich in steroids and sterols (19). Remarkably, the oil is composed of 80% punicic acid, which is a rare C18 octadecatrienoic fatty acid. Many of these compounds have been shown to have anticancer properties (20–23). Nevertheless, the specific components of PJ that have antimetastatic effects against PCa are largely unknown. Recently, we showed that a combination of a polyphenolic compound (luteolin/L), an antioxidant (ellagic acid/E) and a seed oil component (punicic acid/P), when used in cultured PCa cells individually and in combination, stimulate cell adhesion and inhibit cell migration/chemotaxis, processes critical for metastasis. We also found that the combination of L+E+P is additively more potent than L, E or P individually (24). They stimulate cell adhesion, inhibit cell migration and inhibit chemotaxis of the PCa cells via CXCL12/CXCR4, a chemokine axis that is important in metastasis of PCa cells (24). We have also shown similar antimetastatic effects of L+E+P on breast cancer cells (25). However, there is no direct evidence yet showing that L+E+P inhibits PCa metastasis in vivo. Therefore, in the current study, we tested the effect of L+E+P on metastasis of PCa in a mouse tumor model. We found that L+E+P administration inhibits growth and metastasis of luciferase expressing human PCa (PC-3M-luc) xenograft tumors in severe combined immunodeficiency (SCID) mice. L+E+P treatment also inhibits growth and metastasis of allograft tumors of the highly invasive mouse PCa cells with PTEN deletion and K-Ras activation. In addition, L+E+P treatment inhibits tumor angiogenesis and angiogenesis-related molecular properties of endothelial cells. Our findings strongly suggest that L+E+P can potentially be used to deter metastasis of PCa.
Materials and methods
PCa cell injection and monitoring of tumor size
Experiments were approved by the Institutional Animal Care and Use Committee of the University of California, Riverside. Male SCID mice (5–6 weeks old) were anesthetized and were injected ventrally under the skin near the region of the prostate with 2 × 106 PC-3M-luc2 cells or 1 × 106Pten−/−;K-rasG12D cells suspended in 100 µl Dulbecco’s phosphate-buffered saline. One day after the injection of the tumor cells, mice were randomly divided into two groups (7 mice/group). One group of mice received L+E+P treatment (64 µg/component/day) once a day, 5 days/week for 8 weeks and the other received only vehicle (Dulbecco’s phosphate-buffered saline) for the same period of time, via intraperitoneal (IP) injection. For the weekly imaging, mice were anesthetized and were given the substrate D-luciferin by IP injection at 150 mg/kg in Dulbecco’s phosphate-buffered saline 5–10min before imaging. Bioluminescence imaging (BLI) was performed with a bioluminescent imaging system (ONYX, Stanford Photonics) composed of a highly sensitive cooled CCD camera mounted in a light-tight specimen box. Images and measurements of BLI were acquired and analyzed using WinView software. To quantify the BLI intensity, the tumor of each mouse was manually outlined using Matlab’s built-in function ‘roipoly’. The mean values of the gray scale intensities inside the tumor were then calculated for quantitative analysis. The tumor diameters were measured and the volumes were calculated by the formula: Volume = (width)2 × length/2 (26).
PCa patient samples
Human prostate tumor samples were obtained through an institutional review board–approved protocol. Matched normal and tumor tissues from a single patient were obtained at the time of prostatectomy. The fragments used were shown by frozen section to contain or not contain tumor. Two patient-derived xenograft tumors were also used. Tissue was removed from subcutaneous xenografts in SCID mice, at sixth passage level.
Tube formation assay
Fifty microliters of Matrigel (BD Biosciences, San Jose, CA) was applied to the center of 35-mm cell culture dishes to evenly coat a 1.5-cm diameter area. The coated plates were incubated for 30min at 37°C. A total of 3 × 104 human microvascular endothelial cells (HMVECs) suspended in 200 µl Dulbecco’s modified Eagle’s medium were plated onto the Matrigel-coated area. To study the effect of PJ components on tube formation, HMVECs were treated after the cells adhered for 1 h and then observed for tube formation. To determine whether the PJ components disrupt preformed tubes, HMVECs were treated after the cells had formed tubes and observed for tube disruption. The tubes were observed with an inverted phase contrast microscope intermittently for 12 h. The number of formed tubes was recorded.
In vivo angiogenesis
C57BL mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All experimental protocols were approved by the UCR Institutional Animal Care and Use Committee. The mice were treated with L+E+P (64 µg of each) by IP injection daily for 2 weeks. The hair was then removed from the back of the mice using Nair hair remover (Madera, CA) and they were injected with interleukin (IL)-8 (100 ng/20 μl saline) or vascular endothelial growth factor (VEGF; 200 ng/20 μl saline) using an insulin syringe. IL-8 and VEGF were each injected in two sites symmetrically located on the back of each mouse every 24 h for 4 days. Injection sites were labeled using a permanent marker to ensure that repeated injections for the same site all took place in the same spot; on each animal, the pair of injection sites for IL-8 and the pair for VEGF were on opposite sides of the backbone and at least 2cm apart to avoid possible cross-over effects. Skin samples from the injected areas were collected and observed at day 5.
Immunolabeling
Frozen tissue sections were immunolabeled with Ki67 or αSMA antibodies, mounted with Vectashield, and viewed using Nikon Microphot-FXA fluorescence microscope with a Nikon DS-Fi1 digital camera and Nikon NIS-Elements software.
Statistical analysis
Data analysis was performed using the Student’s t-test or one-way analysis of variance on raw data with GraphPad Instat software (GraphPad Software, La Jolla, CA).
Results
L+E+P inhibits tumor growth, metastasis and angiogenesis of prostate xenograft and allograft tumors
To study the effects of L+E+P on the metastasis of PCa in vivo, male SCID mice were injected ventrally under the skin with luciferase-expressing human PCa cells (PC-3M-luc). Tumor growth and progression to metastasis of both treated and vehicle groups were monitored weekly by BLI (Figure 1A and B). Without treatment, the tumors grew to the maximum size allowed by the Institutional Animal Care and Use Committee in 8 weeks (Figure 1A).
Fig. 1.
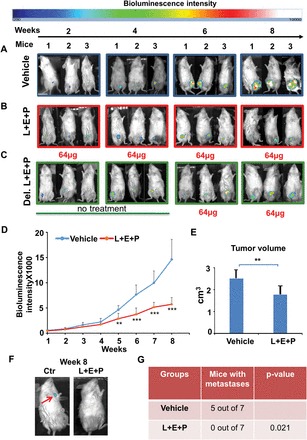
L+E+P inhibits growth and metastasis of PC-3M-luc xenograft tumors in SCID mice. (A and B) 2×106 PC-3M-luc cells were injected into SCID mice subcutaneously; 1 day after injection of the tumor cells, mice in the treated group received 64 µg each of L+E+P via IP once a day, 5 days/week for 8 weeks and the control mice received only vehicle (Dulbecco’s phosphate-buffered saline). (C) In a group of mice receiving delayed L+E+P treatment (Del. L+E+P), 64 µg L+E+P treatment started at week 5 (Vehicle group n = 7, L+E+P group n = 7, Delayed L+E+P group n = 5). Tumor growth and progression in Vehicle, L+E+P and Delayed L+E+P groups of mice were monitored by BLI weekly. Representative images at week 2, 4, 6 and 8 are shown (A–C). (D) The weekly BLI intensities of mice in Vehicle and L+E+P group were quantified and the mean intensities were plotted. (E) Mice were euthanized at the end of the 8th week and tumor volume was determined using the formula: Volume = (width)2 × length/2 (P = 0.0052). (F) Tumors from mice in the Vehicle group metastasized by week 8 as shown by the BLI when the bioluminescence signals from primary tumors were blocked. Arrow indicates a metastasis. (G) Comparison of number of mice showing metastases between Vehicle and L+E+P groups was quantified using the Fisher’s exact test (P = 0.021). Bars represent standard error of the mean. **P < 0.01, ***P < 0.001.
We had previously determined in vitro that the most effective combination of L+E+P was when the components were used at equal doses in the mixture (24). Therefore, we tested increasing doses of the mixture in a ratio of 1:1:1 starting with 16 µg per component, which was the most effective dose in in vitro assays (24). We tested L+E+P at 16, 32, 64 and 128 µg of each component and found that 16 µg of each was ineffective in deterring tumor growth, 32 µg had a small effect and 128 µg killed the mice; 64 µg was most effective. Therefore, we used 64 µg of each component for all experiments. During the 8 weeks of treatment, no apparent toxicity was observed with 64 µg/component/day in any of the L+E+P-treated mice, whereas this concentration significantly inhibited tumor growth as indicated by the decreased intensity of BLI (Figure 1B). We also found that L+E+P was effective in deterring tumor growth when treatment was started at week 5 of tumor development (Figure 1C). The weekly tumor BLI intensities of mice in vehicle and L+E+P group were quantified and the mean intensities plotted. L+E+P significantly reduced the BLI intensity as compared with the vehicle group (Figure 1D). At week 8, all the mice were euthanized and the volume of the tumors was measured after excision. L+E+P treatment significantly decreased tumor volume compared with mice treated with the vehicle (Figure 1E), and it completely inhibited metastatic incidence in PC-3M-luc xenograft tumors as shown by BLI when the signal from the primary tumor was blocked (Figure 1F). Among the vehicle-treated group, tumors in five of seven mice metastasized by week 8. However, none of the tumors in seven mice treated with L+E+P metastasized by week 8 (Figure 1G).
Our previous results in vitro showed that the treatment of L+E+P is more effective than L, E or P individually. To test this result in vivo, we treated the mice with PC3M-luc tumors with L, E or P individually and compared the effectiveness to L+E+P (Supplementary Figure S1, available at Carcinogenesis Online). We found that the combination of L+E+P was additively more effective than L, E or P individually as indicated by BLI intensities and the tumor volumes (Supplementary Figure S1A–Supplementary Data, available at Carcinogenesis Online). Consistent with our in vitro findings, L is more potent than E or P individually.
Because we saw a decrease in tumor growth rate, we used a marker for Ki67 to determine whether L+E+P affected tumor cell proliferation and observed significant inhibition of proliferation (Figure 2A and B). To determine whether L+E+P treatment affects angiogenesis in the tumor, sections from the control and treated groups were stained with Masson’s trichrome to highlight the collagen surrounding the blood vessels. We found that the number of blood vessels in treated mice was significantly lower than in the vehicle-treated mice (Figure 2C). The effects of L+E+P on tumor biology are summarized in Figure 2D.
Fig. 2.
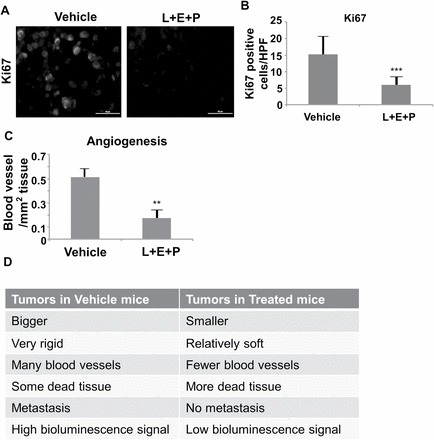
L+E+P inhibits angiogenesis of PC-3M-luc xenograft tumors in SCID mice. Tumors collected from the Vehicle and L+E+P treated groups were fixed and sectioned. (A) Ki67 immunolabeling of tumors from Vehicle and L+E+P groups. (B) The numbers of Ki67 positively stained cells were averaged in 10 high-power (40×) fields. (C) Quantification of blood vessels in each section. The total number of blood vessels were counted/mm2 of tumor tissue. (D) Summary of the observed differences between tumors from Vehicle and L+E+P-treated groups. Bars represent standard error of the mean. **P < 0.01, ***P < 0.001.
Because not all xenograft tumors from PC-3M-luc cells in SCID mice metastasized, we injected the highly invasive mouse cancer cell line with PTEN deletion and K-RAS activation (Pten−/−;K-rasG12D) to evaluate the effects of L+E+P on metastasis of this aggressive PCa cell line (27). To initiate these studies, we tested the effect of L+E+P on cultured cells and found that L+E+P increases cell adhesion, decreases cell migration and decreases chemotaxis towards CXCL12 much like it did on PC3 cells (Supplementary Figure S2A–Supplementary Data, available at Carcinogenesis Online).
For the experiments in vivo, SCID mice were injected with Pten−/−;K-rasG12D cells subcutaneously in the region of the prostate, and then divided into two groups. One group of mice received L+E+P treatment (64 µg/component/day 5 days a week) and the other group received only the vehicle. Because of the tumor aggressiveness, all mice were treated for only 4 weeks and euthanized at the end of the fourth week. We found that L+E+P significantly decreased the size of the tumors compared with the vehicle-treated group (Figure 3A). By the end of week 4, all tumors in the vehicle-treated group had metastasized and the major sites of metastasis were the lung and liver. The number of metastatic lesions was counted macroscopically; L+E+P treatment significantly inhibited the number of metastatic lesions in to both the lung (Figure 3B and C) and the liver (Figure 3B and D). We summarize and compare lung and liver metastases between control and L+E+P group in Figure 3E. In addition, we found that L+E+P reduced the number of blood vessels in the Pten−/−;K-rasG12D tumors (Supplementary Figure S3A, available at Carcinogenesis Online). L+E+P also inhibited tumor cell proliferation as shown by reduced Ki67-positive cells (Supplementary Figure S3B, available at Carcinogenesis Online). These findings show that L+E+P effectively inhibits metastasis in both the PC-3M-luc tumors and the highly invasive Pten−/−;K-rasG12D tumors.
Fig. 3.
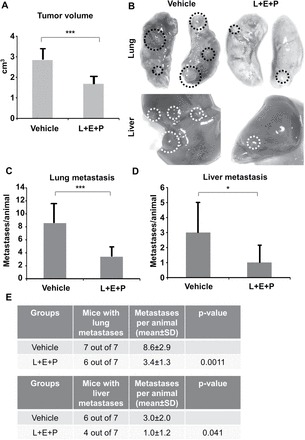
L+E+P inhibits metastasis of Pten−/−;K-rasG12D mouse cancer cell allograft tumors. A total of 1×106Pten−/−;K-rasG12D mouse cancer cells were injected into SCID mice subcutaneously near the prostate region. One day after the injection of the tumor cells, the L+E+P group of mice received 64 µg each of L+E+P once a day, 5 days/week for 4 weeks, and the control group received only the vehicle (Dulbecco’s phosphate-buffered saline) (Vehicle group n = 7, L+E+P group n = 7). (A) Mice were euthanized at the end of the 4th week and tumor volume was determined using the formula: Volume = (width)2 × length/2 (P = 0.0002). (B) Lung and liver metastases in mice from Vehicle and L+E+P group. Circles indicate the metastatic lesions. (C and D) The number of metastatic lesions in lung (C) (P = 0.0011) and liver (D) (P = 0.041) were counted macroscopically in each animal. (E) Summary of the number of metastases in lung and liver. Bars represent standard error of the mean. *P < 0.05, ***P < 0.001.
Effects of L+E+P involve inhibiting the CXCL12/CXCR4 and AKT signaling axis
It has been well established that the CXCL12/CXCR4 signaling axis plays a critical role in cancer metastasis. We have previously shown that L+E+P reduces CXCR4 levels and inhibits the downstream signaling pathways of CXCL12 in human endothelial cells in vitro (24). To investigate the effect of L+E+P on CXCL12/CXCR4 axis in vivo, we examined the protein levels of CXCR4 in the L+E+P-treated PC-3M-luc tumors and found that treatment with these components significantly decreased the protein levels of CXCR4. Furthermore, we found that Gα13, PI3K and p-AKT proteins involved in the signaling downstream of CXCR4 were also decreased (Figure 4A and B). To investigate whether the inhibitory effect of L+E+P on tumor growth is mediated by reducing the level of activated AKT, we treated the mice with PC3M-luc tumors with a small molecule AKT activator, SC79 (28) and examined whether AKT activation could reverse the effect of L+E+P. We found that SC79 treatment at least partially reversed the effect of L+E+P on tumor growth as indicated by comparison of BLI intensities (Figure 4C and D). SC79 treatment also reversed the effect of L+E+P on reducing the level of p-AKT in the tumors (Figure 4E). Because Akt is a key molecule in the signaling pathway for CXCL12/CXCR4, these findings show that L+E+P inhibits the CXCL12/CXCR4/AKT chemotactic axis in the tumor and are consistent with the observed L+E+P inhibition of metastasis.
Fig. 4.
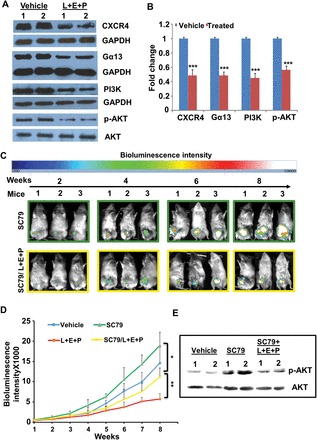
The effects of L+E+P involve inhibiting CXCL12/CXCR4 and AKT signaling axis. (A) PC3M-luc tumors from Vehicle and L+E+P groups of mice were collected and total protein was extracted and analyzed by immunoblotting for CXCR4, Gα13, PI3K and p-AKT (S473). (B) Densitometry of the bands. (C) A total of 2×106 PC-3M-luc cells were injected into SCID mice subcutaneously. One group of mice (group SC79, n = 3) received AKT activator SC79 treatment weekly (0.04mg/g) via IP and the other group (group SC79/L+E+P, n = 3) received the SC79 treatment weekly plus L+E+P treatment once a day, 5 days/week. Tumor growth was monitored by BLI weekly. Representative images at week 2, 4, 6 and 8 are shown. (D) The weekly BLI intensities of mice in SC79 and SC79/L+E+P group were quantified and the mean intensities were plotted. (E) Tumors from SC79 and SC79/L+E+P groups of mice were collected and total protein was extracted and analyzed by immunoblotting for p-AKT (S473). Bars represent standard error of the mean. **P < 0.01.
L+E+P alters the angiogenic properties of HMVECs
To study the molecular mechanisms of inhibition of angiogenesis by L+E+P as observed in the tumors, we performed a variety of cellular and molecular assays in vitro using HMVEC. These cells were treated with L+E+P at 2 or 4 µg/ml (higher concentrations were detrimental to these cells) and performed adhesion assays, much like we did for the tumor cells, using a gelatin-coated substrate and measured the trypsinization time required to completely detach all cells from the culture dish as an indicator of cell adhesion strength (17,24,25). We tested for adhesion to the substrate at 12 and 24 h after initiation of treatment by recording the time it took for trypsinization to remove all of the cells from the dish. Control represents no treatment. Treatment with L+E+P decreased the ability of HMVECs to adhere to the substrate (Figure 5A). The reason for not presenting statistical significance is because the loss of adhesion is similar from culture to culture and it occurs rapidly when the cells begin to detach. In addition, using the scratch wound assay, we found that L+E+P treatment significantly decreased cell migration of HMVECs in a dose-dependent manner (Figure 5B).
Fig. 5.
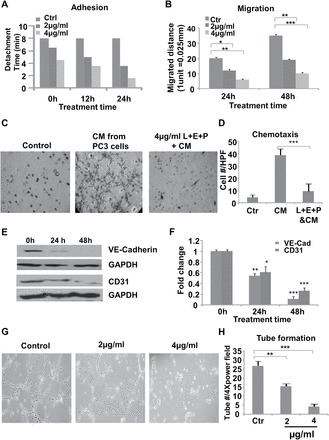
L+E+P changes angiogenesis-related properties of HMVEC cells. (A) HMVEC cells were plated onto gelatin-coated plates and were treated with L+E+P at 2 or 4 µg/ml for 12 or 24h. Time required to completely trypsinize all cells from the plate was recorded. (B) Confluent HMVEC-1 cultures were scratch wounded and were treated with L+E+P at 2 or 4 µg/ml for 24 or 48h. The distance migrated from the wounded edge was recorded at indicated time points. (C) HMVECs were seeded onto the collagen-coated upper side of 8 µm pore size membranes of inserts inside transwell units and were treated with L+E+P at 4 µg/ml for 12h. Conditioned medium collected from PC3 culture was introduced into the lower chamber to induce chemotaxis for 4h. L+E+P inhibited chemotaxis of the ECs towards the conditioned media of the cancer cells. (D) The number of cells that migrated through the pores was counted and averaged in 10 high-power (40×) fields. (E) HMVECs were treated with L+E+P at 4 µg/ml for 24 or 48h, and protein extracts were analyzed by immunoblotting for VE-cadherin and CD31. (F) Densitometry of the bands in E. (G) HMVEC-1 cells were plated onto Matrigel-coated plates and allowed to form tubes for 4h. Tubes were then treated with L+E+P at 2 or 4 µg/ml for 4h. L+E+P treatment resulted in dismantling of the endothelial tubes. (H) Tube number per 4× power field. Bars represent standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001.
It is well established that tumor cells can attract endothelial cells through chemotaxis. To investigate the effects of L+E+P on chemotaxis of endothelial cells towards the tumor cells, we performed chemotaxis assays with PC3 conditioned medium and found that L+E+P treatment significantly inhibited the PC3 conditioned medium-induced chemotaxis (Figure 5C and D).
To further investigate the molecular mechanisms of L+E+P on HMVECs, we determined the effect of these components on levels of VE-cadherin and CD31, which are two important proteins in endothelial cell adhesion to each other and in tube formation in blood vessels. Treatment for 24 or 48 h with L+E+P decreased the levels of both proteins and did so in a time-dependent manner (Figure 5E and F). Given these results, we also tested the possibility that L+E+P could have an effect on endothelial cell tube formation. HMVECs were treated with L+E+P after the tubes were allowed to form and found that these components significantly disrupted the formed tubes (Figure 5G and H). When L+E+P treatment was done immediately after the cells had adhered to the substrate, tube formation was prevented (Supplementary Figure S4A and Supplementary Data, available at Carcinogenesis Online). Both effects occurred in a dose-dependent manner. These results suggest that L+E+P inhibits tumor-induced angiogenesis through changing endothelial cell processes that are involved in cell migration, chemotaxis and tube formation.
It is well established that IL-8 and VEGF are potent angiogenesis inducers and that tumor cells in general produce high levels of both. We examined the levels of IL-8 and VEGF in normal human prostate tissues and prostate tumors. Levels of both growth factors are low in the normal prostate tissue but are consistently increased in the human prostate tumors (Figure 6A and B). To determine the effect of L+E+P on IL-8 and VEGF production, we treated PC3 cells with L+E+P and found that these PJ components significantly reduce the IL-8 and VEGF production (Figure 6C and D).
Fig. 6.
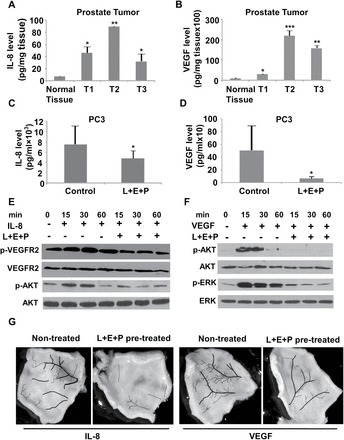
L+E+P inhibits production of proangiogenic factors in tumors and inhibits signaling in HMVEC cells. (A and B) Levels of IL-8 and VEGF in normal human prostate tissue and in three different human prostate tumors. (C and D) In comparison to untreated tumors, IL-8 and, especially, VEGF production by PC3 cells is significantly reduced when treated with 8 µg/ml L+E+P. (E) HMVEC cells were pretreated with 4 µg/ml L+E+P for 12h and then treated with IL-8 at 100ng/ml for 60min in the presence or absence of L+E+P. Protein extracts were analyzed by immunoblotting for p-AKT (S473) and VEGFR2 phosphorylation (Y1054). (F) HMVEC cells were pretreated with 4 µg/ml L+E+P for 12h and then treated with VEGF at 200ng/ml for 60min in the presence or absence of L+E+P. Protein extracts were analyzed by immunoblotting for p-AKT (S473) and p-ERK (T202/Y204). (G) C57BL mice were treated with 64 μg of L+E+P for 2 weeks and then injected under the skin on the back (after hair removal) with IL-8 (100ng/20 μl saline) or VEGF (200ng/20 μl saline) at symmetric sites (see text) every 24h for 4 days. Skin samples from the injected areas were collected at day 5 and photographed. The blood vessels were manually highlighted. Bars represent standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001.
We have previously shown that IL-8-induced angiogenesis occurs via transactivation/phosphorylation of VEGFR2 (29). To determine whether L+E+P inhibits IL-8-induced angiogenesis via this signaling mechanism, we examined whether IL-8 induces the phosphorylation/activation of VEGFR2 and subsequent activation of AKT. Treatment with L+E+P strongly inhibits IL-8-induced phosphorylation of VEGFR2 and phosphorylation of AKT at 15–60min (Figure 6E). VEGF is known to promote endothelial cell proliferation through activation of the PI3K/AKT and MEK/ERK signaling pathways (30). L+E+P inhibits VEGF-induced phosphorylation of ERK and AKT (Figure 6F), suggesting that L+E+P inhibits angiogenesis by inhibiting the response of endothelial cells to the proangiogenic factors IL-8 and VEGF in addition to affecting the endothelial cells themselves.
To study whether L+E+P inhibits IL-8- or VEGF-induced angiogenesis in vivo, C57BL mice were injected for 2 weeks with 64 µg each of L+E+P once/day. IL-8 or VEGF was injected under the skin once/day for 4 consecutive days to examine whether the angiogenic effects were inhibited by systemically pretreating the animals with L+E+P. The skin samples were collected, imaged and the blood vessels were manually highlighted. We found that angiogenesis induced by IL-8 or VEGF was inhibited by L+E+P as shown by reduced number of blood vessels in L+E+P-treated skin samples (Figure 6G and Supplementary Figure S5A and Supplementary Data, available at Carcinogenesis Online).
Discussion
PCa that has become metastasized has a poor prognosis and remains a significant therapeutic challenge. In this study, we have tested the hypothesis that L+E+P has antimetastatic effects in vivo and have found that these PJ components (i) inhibit growth of primary tumors; (ii) inhibit cellular and molecular processes critical for metastasis; (iii) inhibit tumor angiogenesis and change angiogenesis-related properties of human endothelial cells; (iv) inhibit the CXCL12/CXCR4 chemotactic signaling axis, which is known to be important in cancer metastasis.
Using SCID mice and a luciferase-expressing human PCa cell line to obtain xenograft tumors, we investigated the potential for tumor metastasis. The advantage of this model is that tumor growth and metastasis can be monitored in real time. Treatment with L+E+P significantly suppressed tumor growth but, more importantly, it completely eliminated metastasis of these tumors. However, the fact that in control tumors, progression to the metastatic stage did not occur 100% of the time limits the power of this result. This led us to test the effects of these components on a more aggressive tumor.
It has been shown that inhibition of the PTEN/PI3K pathway combined with activation of the Ras/MAPK pathway promotes PCa metastasis (27). As a result, we also investigated a highly invasive mouse PCa cell line which has PTEN deletion and K-ras activation (Pten−/−;K-rasG12D) to generate allograft tumors. In this model, all control mice metastasized by the end of week 4, and we observed abundant metastases in lung and liver. In the corresponding mice treated with L+E+P by IP injection, tumor growth was again significantly suppressed, but in this case, metastasis to lung and liver also occurred but the overall number of metastases was significantly reduced. We are currently performing studies to determine how long past 8 weeks treatment with L + E+ P prevents metastasis.
Detectable ellagic acid and metabolites can be identified briefly and in low quantity in plasma and urine within an hour of both oral and IP administration. When given orally, most ellagic acid seems to be converted by intestinal microorganisms to urolithins and their glucuronides (31–33). These urolithin glucuronides are then absorbed and concentrated in prostate tissue, where they may exert antiproliferative and antioxidant effects. Thus, ellagic acid itself does not seem to be a major, biologically effective component of orally administered PJ. However, when injected IP, ellagic acid itself ends up in the plasma and in the prostate of mice (31). Much less data exist to describe the bioavailability and activity of luteolin or punicic acid after oral ingestion by humans. Our data suggest that a combination of PJ constituents may have antitumor activity when administered parenterally at a nontoxic dose. Although not as convenient as oral administration, parenteral dosing of PJ components may have novel biologic effects by bypassing intestinal metabolism. Comparative studies of the biologic activities of L, E and P by both routes will be necessary to define the optimum methods to use these compounds in clinical cancer care.
There is a limit to how large a tumor can grow without angiogenesis (34). Indeed, angiogenesis plays a crucial role in the survival, proliferation and metastatic potential of PCa tumors through providing nutrients and oxygen (35). Therefore, angiogenesis is an attractive treatment target in many types of solid tumors, including PCa tumors. To date, the most successful antiangiogenic agent is bevacizumab, a monoclonal antibody against VEGF (36). Natural agents such as pomegranate extract have been shown to inhibit angiogenesis in PCa (37) but the active components responsible for the antiangiogenic effects are largely unknown. We have shown here that L+E+P inhibits angiogenesis by reducing the number of blood vessels in the tumors. New blood vessels are formed in response to interaction between tumor cells and endothelial cells, growth factors and extracellular matrix components (38). In cultured endothelial cells, we show that L+E+P significantly, and in a dose-dependent manner, decreases endothelial cell adhesion, migration and tube formation, all of which are important processes in angiogenesis. In addition, VE-cadherin and CD31, two of the important endothelial-specific adhesion proteins that maintain the integrity of blood vessels at adherens junctions (39), are completely (VE-cadherin) or significantly (CD31) inhibited by L+E+P treatment. These findings suggest that the antiangiogenic effects of L+E+P are at least partially due to directly changing the cellular and molecular properties of endothelial cells.
It is well established that tumor-secreted factors can chemoattract local stromal cells, such as fibroblasts and macrophages, and distant cells such as endothelial cells (40). We show that L+E+P inhibits chemotaxis of endothelial cells towards PC3-conditioned medium, suggesting that these components inhibit angiogenesis also through perturbing the communication between tumor and endothelial cells. Many studies have demonstrated that tumor cells secrete growth factors such as IL-8 and VEGF to stimulate the migration and proliferation of endothelial cells (41). IL-8 and VEGF are known as potent promoters of angiogenesis and the level of IL-8 and VEGF are in general increased in various tumors (42). IL-8 has been shown to stimulate cell migration via the PI3K/AKT signaling pathway (43), and VEGF is known to promote endothelial cell proliferation through activation of the PI3K/AKT and MEK/ERK signaling pathways (30). We show that L+E+P treatment significantly reduces the production of IL-8 and VEGF in PC3 cells and inhibits the endothelial cell response to IL-8 and VEGF.
It has been well established that PCa cells develop ways to bypass the need for testosterone and then the cancer progresses rapidly. The CXCL12 receptor CXCR4 is widely expressed in various tumor cells and is responsible for metastasis to the most common destinations such as lung and liver (44,45). As a result, the CXCL12/CXCR4 signaling axis has become an attractive therapeutic target for metastasis. Neutralizing CXCR4 function with an antagonist or an antibody has been shown to inhibit prostate and breast cancer metastasis (44,46). We show that L+E+P reduces the level of CXCR4 in prostate tumors and inhibits the CXCR4 downstream signaling Gα13, which is the G protein α subunit involved in CXCL12-induced chemotaxis (47). Together with the finding that the effect of L+E+P on tumor growth was partially reversed by AKT activation, these results strongly suggest that L+E+P inhibits PCa metastasis also through targeting the CXCL12/CXCR4/AKT signaling axis.
It is well know that PCa significantly metastasizes to the bone. We could not test the effects of L+E+P on bone metastasis because our SCID mouse model is limited by how long the mice live and prostate metastasis to the bone takes a long time to develop. However, because we find that L+E+P affects cellular and molecular processes important for metastasis, we expect L+E+P also has the potential to inhibit bone metastasis. Furthermore, on the basis of our previous findings (24), L+E+P also inhibits cell growth and downregulates genes involved in anti-apoptosis and cell cycle progression. Therefore, these natural products have multiple molecular targets in the cell and the observed antimetastatic effect of L+E+P is potentially a result of multiple combined actions such as inhibiting cell growth, inducing apoptosis, inhibiting chemotaxis and tumor angiogenesis. We show that L+E+P inhibits cell proliferation as detected with staining for Ki67, a protein active during the cell cycle but absent when cells are in G0. Because we find virtually no staining for Ki67 in tumors of animals treated with L+E+P, this suggests that the cells are arrested in G0.
In conclusion, L+E+P can be used in combination to prevent PCa growth and metastasis and because these are natural products they easily could be used in humans in the near future. Furthermore, it may be possible to develop them into novel drugs that can be made more effective than the natural products in preventing cancer progression.
Supplementary material
Supplementary Figures 1–5 can be found at Supplementary Data
Funding
The research was supported by a gift from a private donor.
Supplementary Material
Acknowledgements
We would like to thank Dr D.Binder and M.Hsu for making the cryomicrotome available to us for tissue sectioning and H.W.Green for constructive discussions.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations:
- BLI
bioluminescence imaging
- E
ellagic acid
- EC
endothelial cell
- HMVEC
human microvascular endothelial cell
- IP
intraperitoneal
- L
luteolin
- P
punicic acid
- PCa
prostate cancer
- PJ
pomegranate juice
- PSA
prostrate-specific antigen
- SCID
severe combined immunodeficiency
- VEGF
vascular endothelial growth factor.
References
- 1. American Cancer Society. http://www.cancer.org/ (15 December 2013, date last accessed).
- 2. Bubendorf L., et al. (2000)Metastatic patterns of prostate cancer: an autopsy study of 1,589 patients. Hum. Pathol., 31, 578–583. [DOI] [PubMed] [Google Scholar]
- 3. Higano C.S., et al. (2010)Sipuleucel-T. Nat. Rev. Drug Discov., 9, 513–514. [DOI] [PubMed] [Google Scholar]
- 4. Scher H.I., et al. (2010)Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet, 375, 1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sartor O., et al. (2013)Abiraterone and its place in the treatment of metastatic CRPC. Nat. Rev. Clin. Oncol., 10, 6–8. [DOI] [PubMed] [Google Scholar]
- 6. Pal S.K., et al. (2013)Enzalutamide for the treatment of prostate cancer. Expert Opin. Pharmacother., 14, 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ryan C.J., et al. (2013)Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med., 368, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stein M.N., et al. (2012)Abiraterone in prostate cancer: a new angle to an old problem. Clin. Cancer Res., 18, 1848–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yakes F.M., et al. (2011)Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther., 10, 2298–2308. [DOI] [PubMed] [Google Scholar]
- 10. Smith D.C., et al. (2013)Cabozantinib in patients with advanced prostate cancer: results of a phase II randomized discontinuation trial. J. Clin. Oncol., 31, 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Albrecht M., et al. (2004)Pomegranate extracts potently suppress proliferation, xenograft growth, and invasion of human prostate cancer cells. J. Med. Food, 7, 274–283. [DOI] [PubMed] [Google Scholar]
- 12. Syed D.N., et al. (2008)Dietary agents for chemoprevention of prostate cancer. Cancer Lett., 265, 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rettig M.B., et al. (2008)Pomegranate extract inhibits androgen-independent prostate cancer growth through a nuclear factor-kappa B-dependent mechanism. Mol. Cancer Ther., 7, 2662–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Malik A., et al. (2005)Pomegranate fruit juice for chemoprevention and chemotherapy of prostate cancer. Proc. Nat. Acad. Sci. USA, 102, 14813–14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pantuck A.J., et al. (2006)Phase II study of pomegranate juice for men with rising prostate-specific antigen following surgery or radiation for prostate cancer. Clin. Cancer Res., 12, 4018–4026. [DOI] [PubMed] [Google Scholar]
- 16. Paller C.J., et al. (2013)A randomized phase II study of pomegranate extract for men with rising PSA following initial therapy for localized prostate cancer. Prostate Cancer Prostatic Dis., 16, 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang L., et al. (2011)Cellular and molecular mechanisms of pomegranate juice-induced anti-metastatic effect on prostate cancer cells. Integr. Biol. (Camb)., 3, 742–754. [DOI] [PubMed] [Google Scholar]
- 18. Gil M.I., et al. (2000)Antioxidant activity of pomegranate juice and its relationship with phenolic composition and processing. J. Agric. Food Chem., 48, 4581–4589. [DOI] [PubMed] [Google Scholar]
- 19. El Kar C., et al. (2011)Pomegranate (Punica granatum) juices: chemical composition, micronutrient cations, and antioxidant capacity. J. Food Sci., 76, C795–C800. [DOI] [PubMed] [Google Scholar]
- 20. Hora J.J., et al. (2003)Chemopreventive effects of pomegranate seed oil on skin tumor development in CD1 mice. J Med. Food, 6, 157–161. [DOI] [PubMed] [Google Scholar]
- 21. Lansky E.P., et al. (2005)Pomegranate (Punica granatum) pure chemicals show possible synergistic inhibition of human PC-3 prostate cancer cell invasion across Matrigel. Invest. New Drugs, 23, 121–122. [DOI] [PubMed] [Google Scholar]
- 22. Zhou Q., et al. (2009)Luteolin inhibits invasion of prostate cancer PC3 cells through E-cadherin. Mol. Cancer Ther., 8, 1684–1691. [DOI] [PubMed] [Google Scholar]
- 23. Gasmi J., et al. (2010)Growth inhibitory, antiandrogenic, and pro-apoptotic effects of punicic acid in LNCaP human prostate cancer cells. J Agric. Food Chem., 58, 12149–12156. [DOI] [PubMed] [Google Scholar]
- 24. Wang L., et al. (2012)Specific pomegranate juice components as potential inhibitors of prostate cancer metastasis. Transl. Oncol., 5, 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rocha A., et al. (2012)Pomegranate juice and specific components inhibit cell and molecular processes critical for metastasis of breast cancer. Breast Cancer Res. Treat., 136, 647–658. [DOI] [PubMed] [Google Scholar]
- 26. Euhus D.M., et al. (1986)Tumor measurement in the nude mouse. J. Surg. Oncol., 31, 229–234. [DOI] [PubMed] [Google Scholar]
- 27. Mulholland D.J., et al. (2012)Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res., 72, 1878–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jo H., et al. (2012)Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc. Nat. Acad. Sci. USA, 109, 10581–10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petreaca M.L., et al. (2007)Transactivation of vascular endothelial growth factor receptor-2 by interleukin-8 (IL-8/CXCL8) is required for IL-8/CXCL8-induced endothelial permeability. Mol. Biol. Cell, 18, 5014–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marshall C.J. (1995)Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell, 80, 179–185. [DOI] [PubMed] [Google Scholar]
- 31. Seeram N.P., et al. (2007)Pomegranate ellagitannin-derived metabolites inhibit prostate cancer growth and localize to the mouse prostate gland. J. Agric. Food Chem., 55, 7732–7737. [DOI] [PubMed] [Google Scholar]
- 32. González-Sarrías A., et al. (2010)Occurrence of urolithins, gut microbiota ellagic acid metabolites and proliferation markers expression response in the human prostate gland upon consumption of walnuts and pomegranate juice. Mol. Nutr. Food Res., 54, 311–322. [DOI] [PubMed] [Google Scholar]
- 33. Vicinanza R., et al. (2013)Pomegranate juice metabolites, ellagic acid and urolithin a, synergistically inhibit androgen-independent prostate cancer cell growth via distinct effects on cell cycle control and apoptosis. Evid. Based. Complement. Alternat. Med., 2013, 247504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carmeliet P., et al. (2000)Angiogenesis in cancer and other diseases. Nature (London), 407, 249–257. [DOI] [PubMed] [Google Scholar]
- 35. Weis S.M., et al. (2011)Tumor angiogenesis: molecular pathways and therapeutic targets. Nat. Med., 17, 1359–1370. [DOI] [PubMed] [Google Scholar]
- 36. Aragon-Ching J.B., et al. (2008)The role of angiogenesis inhibitors in prostate cancer. Cancer J., 14, 20–25. [DOI] [PubMed] [Google Scholar]
- 37. Sartippour M.R., et al. (2008)Ellagitannin-rich pomegranate extract inhibits angiogenesis in prostate cancer in vitro and in vivo. Int. J. Oncol., 32, 475–480. [PubMed] [Google Scholar]
- 38. Jahroudi N., et al. (1995)The role of endothelial cells in tumor invasion and metastasis. J. Neurooncol., 23, 99–108. [DOI] [PubMed] [Google Scholar]
- 39. Vestweber D., et al. (2009)Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol., 19, 8–15. [DOI] [PubMed] [Google Scholar]
- 40. Wels J., et al. (2008)Migratory neighbors and distant invaders: tumor-associated niche cells. Genes Dev., 22, 559–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ahmad S.A., et al. (2002)The role of the microenvironment and intercellular cross-talk in tumor angiogenesis. Semin. Cancer Biol., 12, 105–112. [DOI] [PubMed] [Google Scholar]
- 42. Waugh D.J., et al. (2008)The interleukin-8 pathway in cancer. Clin. Cancer Res., 14, 6735–6741. [DOI] [PubMed] [Google Scholar]
- 43. Lai Y., et al. (2011)Interleukin-8 induces the endothelial cell migration through the activation of phosphoinositide 3-kinase-Rac1/RhoA pathway. Int. J. Biol. Sci., 7, 782–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taichman R.S., et al. (2002)Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res., 62, 1832–1837. [PubMed] [Google Scholar]
- 45. Zlotnik A., et al. (2011)Homeostatic chemokine receptors and organ-specific metastasis. Nat. Rev. Immunol., 11, 597–606. [DOI] [PubMed] [Google Scholar]
- 46. Huang E.H., et al. (2009)A CXCR4 antagonist CTCE-9908 inhibits primary tumor growth and metastasis of breast cancer. J. Surg. Res., 155, 231–236. [DOI] [PubMed] [Google Scholar]
- 47. Tan W., et al. (2006)The Galpha13-Rho signaling axis is required for SDF-1-induced migration through CXCR4. J. Biol. Chem., 281, 39542–39549. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.