

Abstract
While the extensive hunt for therapeutics combating Alzheimer’s disease (AD) has fallen short of delivering effective treatments, breakthroughs towards understanding the disease mechanisms and identifying areas for future research have nevertheless been enabled. The majority of clinical trials with β- and γ-secretase modulators have been suspended from additional studies or terminated due to toxicity issues and health concerns. The lack of progress in developing innovative AD therapies has also prompted a resurgence of interest in more traditional symptomatic treatments with cholinesterase inhibitors and N-methyl-d-aspartate receptor antagonists, as well as in the research of immune response modulators. Recently, evidence has emerged showing that inhibitors of arginine metabolism and in particular blockers of arginase, an enzyme that catalyzes the breakdown of l-arginine, could present an effective therapeutic candidate for halting the progression of AD and boosting cognition and memory. In this commentary, we present a brief overview of reports on arginase inhibitors in AD mouse models and discuss emerging advantages and areas for careful consideration on the road to clinical translation.
Electronic supplementary material
The online version of this article (10.1007/s13311-018-0668-6) contains supplementary material, which is available to authorized users.
KeyWords: l-Norvaline, Arginine metabolism, Nitric oxide, Amyloid beta, Cognitive enhancers, Alzheimer’s disease.
Alzheimer’s disease (AD) is a chronic neurodegenerative condition characterized by progressive loss of higher brain functions, deposition of intraneuronal neurofibrillary tangles, and accumulation of extracellular amyloid plaques in the brain. The principal constituent of tangles is the hyper-phosphorylated protein tau, while plaques are comprised of fibrils of the β-amyloid (Aβ) peptide. Both biomolecules in physiological amounts play multiple roles in neuronal biology and brain functions, but when in excess and misfolded, turn toxic to affect a range of processes [1, 2]. Soluble Aβ oligomers in particular are thought to interfere with a wide range of brain functions, including synaptic transmission and plasticity, dendritic integrity and spine formation, axonal trafficking, and receptors and ion channels as well as the biology of glial cells and the immune response [3, 4].
Therefore, targeting amyloid pathology and restoring functional impairments due to amyloidosis in the brain have been key priorities in the search for disease-modifying therapy (DMT). In this pursuit, modulation of amyloid precursor protein (APP) processing and Aβ metabolism have been identified as principal strategic directions. Extensive research with the development of a variety of β-, and γ-secretase (BACE and GACE) inhibitors, with their therapeutic applicability, has been of major interest to academic laboratories and pharmaceutical companies, which enabled major advances in the research of their pharmacology and toxicology [5–8]. Despite overwhelmingly promising results from in vitro cell culture and animal experiments in vivo, the outcome of human clinical trials has turned out to be highly unfavorable. Pharmaceutical giants such as Eli Lilly, AstraZeneca, Boehringer Ingelheim, Vitae Pharmaceuticals, and Merck as well as many research laboratories, after investing vast resources and manpower, have put on hold or turned down many research programs, due to toxicity concerns of BACE and GACE inhibitors in humans [9–12]. While extremely disappointing, the toxicity of drug candidates targeting these key enzymes is hardly surprising given their critical involvement in a wide range of complex neurobiological processes in the brain. Nevertheless, one of the hard lessons which have emerged from these studies is that future reports of disease-modifying effects of BACE and GACE modulators in AD models should be treated with caution and verified independently before drawing conclusions and their advancement into clinical trials.
Given the rapidly increasing prevalence of AD and the urgent need for effective therapies, more traditional themes of therapeutic research, such as development and optimization of cholinesterase inhibitors and N-methyl-d-aspartate receptor antagonists, have recently become the subject of renewed interest [13–15]. Further in-depth research along with the development of safer means and methods for immunotherapies to suppress the neuro-inflammatory response and facilitate Aβ clearance with reduction in its toxic effects on neural mechanisms and functions is another AD research area undergoing revival. Among these, a recent study by Kan and co-workers showed that in CVN-AD mice (which are mNos2-deficient and transgenic for the Swedish K670N/M671L vasculotropic, Dutch/Iowa E693Q/D694N mutant APP) characterized by extensive amyloidosis and neurodegeneration in regions affected by Aβ deposits, the local immune response is strongly suppressed, possibly making a contribution towards the AD-like pathological process [16]. Indeed, the hippocampus and part of the cortex of these mice characterized with the most extensive neuronal death and highest amyloid load show enrichment of immune-suppressive CD11c+ microglia. The same brain regions also show a significant rise in the level of arginase, an enzyme which catalyzes the breakdown of l-arginine, causing significant reduction in its level in the affected brain tissue. Remarkably, pharmacological inhibition of arginase activity by eflornithine (known with the trade name Vaniqa) leads to amelioration of the AD-like pathology in CVN-AD model mice with suppression of CD11c+ expression in amyloid-affected areas. While the authors conclude that the principal mechanisms of the therapeutic-like effects of eflornithine are mediated through modulation of microglial activity downstream to increased levels of l-arginine in the brain, eflornithine is also a potent and irreversible inhibitor of another key enzyme—ornithine decarboxylase [16–18], which is a major regulator of the growth and functions of endothelial cells and vascular smooth muscles.
The use of other more selective inhibitors could provide a better elucidation of the role of l-arginine in the AD-like pathological process in mouse models, with the potential for developing selective therapies. In this issue of Neurotherapeutics, Polis et al. address this topic using a potent arginase inhibitor, l-norvaline, in a 3xTg-AD model [19]. Homozygous mice harboring APP KM670/671NL, a human mutant PS1 (M146V) knock-in, and tau (P301L), show a spectrum of neuropathological changes closely replicating those documented in the human AD brain [20, 21]. Importantly, unlike synthetic DMT candidates targeting APP cleaving proteases, l-norvaline has been used for many years as an anti-inflammatory compound, which is attributed to its potent inhibition of arginase activity in endothelial cells. l-Norvaline is also an active ingredient of supplements taken by athletes to improve muscle growth, an effect ascribed to its stimulating actions on tissue metabolism and perfusion [22, 23]. The latter appears to result partly from the fact that by inhibiting arginase activity, l-norvaline blocks the breakdown of l-arginine to l-ornithine and urea and by doing so favors the processing of l-arginine via the l-citrulline and nitric oxide (NO) route [24, 25] (Fig. 1). Increased bioavailability of NO in turn causes strong dilation of blood vessels, stimulating nutrient and oxygen supply and reducing oxidative stress as well as modulating the activity of calcium channels. The boosting effect of l-norvaline on tissue metabolism and associated with it modulation of the NO response are seemingly well tolerated and are not limited to the peripheral vasculature, but extend into the functions of neural tissue and the central nervous system.
Fig. 1.
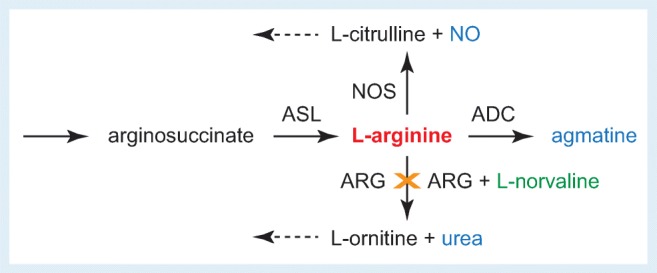
Schematic of l-arginine metabolism pathways and inhibition of arginase (ARG) by l-norvaline. NOS = nitric oxide synthase; ASL = argininosuccinate lysate; ADC = arginine decarboxylase; NO = nitric oxide. Broken arrows indicate directions of additional processing of metabolites
In their study, Polis et al. treated young adult wild-type and 3xTg-AD mice with l-norvaline (supplemented in the drinking water) over 10 weeks, followed by carefully carried out tests for cognitive and memory functions, with subsequent wide range of biochemical, molecular, and histochemical tests and microscopic studies of brain tissue from l-norvaline treated 3xTg-AD mice compared with vehicle-treated 3xTg-AD group, as well as wild-type controls [19]. As expected, l-norvaline was tolerated well by both wild-type and 3xTg-AD mice, causing no notable changes in their well-being or routine physiological activities. In Y-maze and in hidden platform swimming tests, 3xTg-AD mice showed improved cognitive performance as compared to vehicle-treated group. Boosting effects of l-norvaline on cognition were related to a reduction in the load of prefibrillar and fibrillary Aβ in the brain, as revealed by immunoreactivity assays in hippocampal lysates, as well as decreases in amyloid plaques in the cerebral cortex as evident from histochemical tests and microscopic studies. Using high-resolution light microscopy and analysis of the density of dendritic spines in cortical and hippocampal neurons, a notable increase in the number of dendritic spines was also detected in 3xTg-AD treated with the arginase inhibitor, an observation that correlated with the reads of proteomic assays, showing higher levels of presynaptic and postsynaptic proteins, as compared to vehicle-only-treated groups. These findings suggest the potent ameliorative effects of N-norvaline on synaptic homeostasis and integrity of synaptic connections in 3xTg-AD mice, which otherwise display an elaborate AD-like synaptic pathology and dendritic spine loss [20, 21]. The reduction in plaque load and neuroprotective effects of l-norvaline correlated with modulation of microglial activity in the 3xTg-AD mouse brain, as evident from reduced Iba1 immune reactivity, as well as increased viability of astrocytes in the hippocampal and cortical slices. Overall, these novel and converging results suggest that l-norvaline counters neurodegeneration and significantly slows down AD-like pathology in 3xTg-AD mice, boosting their cognitive and memory functions (Fig. 2).
Fig. 2.
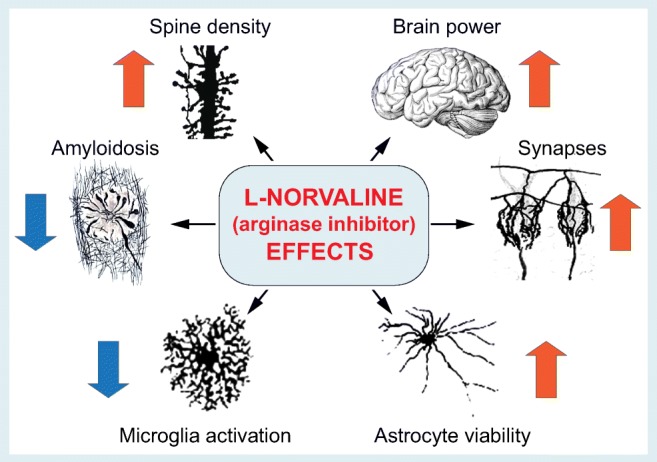
Assessment of the effects of l-norvaline on neuronal and glial cell integrity, viability, and brain functions in 3xTg-AD mice. Arrows indicate the directionality of change. Insert images used with permission from [26, 27]
From a basic neurotherapeutics standpoint, the deceleration of the pathological process and the boosting effects of l-norvaline on cognition along with excellent tolerability present a highly advantageous development that calls for future in-depth research and independent verification. From a neuropathological standpoint, the principal novelty is that these studies demonstrate that the constitutive activity of arginase and its pharmacological modulation could play a fundamental role in the pathobiology of AD, and hence present a potential therapeutic target. In this context, it is worth noting that the augmented expression of arginase has been suggested previously as a risk factor for developing AD [28, 29]. Upregulation of arginase has also been related to several other acute and chronic neurological conditions, which include multiple sclerosis, stroke, and traumatic brain injury as well as retinal diseases [29, 30]. Dysregulation of arginase activity has also been implicated in other neurodegenerative disorders such as Huntington’s [31] and Parkinson’s diseases [29, 30]. Although the report by Polis and colleagues makes a strong case for the potential benefits of the arginase inhibitor l-norvaline in 3xTg-AD mice, at this stage, careful assessment of the effects of this amino acid in other AD models is necessary, with further research required into its potential subtle side effects on brain function as well as on overall animal physiology. As noted earlier, the excellent tolerability of l-norvaline with overall ameliorative effects makes it a superb drug candidate, facilitating its use in future preclinical studies and clinical translation.
With the stakes getting increasingly higher for discovering an effective DMT against such a complex disease as AD, expecting a simple solution is outwardly unrealistic and premature. The positive outcome from the studies discussed above implies that targeting arginine metabolism, one of the most fundamental biochemical processes, might yield a multifaceted (yet not fully understood) solution to a complex disease such as AD. And this should be viewed in a promising light. In the end, what is urgently needed in the fight against AD is finding an effective treatment that will reduce human suffering and improve quality of life. With a better understanding of underlying mechanisms for beneficial effects, we can look forward to better times to come.
Electronic supplementary material
(PDF 1224 kb)
Acknowledgments
Authors would like to thank their colleagues and students for insightful discussion and for their enthusiasm towards neuroscience research.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Abbreviations
- AD
Alzheimer’s disease
- NMDA
N-Methyl-d-aspartate
- Aβ
β-Amyloid
- DMT
Disease-modifying therapies
- APP
Amyloid precursor protein
- BACE
β-Secretase
- GACE
γ-Secretase
- NO
Nitric oxide
- 3xTg-AD
Triple transgenic AD mouse
- ARG
Arginase
- NOS
Nitric oxide synthase
- ASL
Argininosuccinate lysate
- ADC
Arginine decarboxylase
References
- 1.Ovsepian SV, et al. Synaptic vesicle cycle and amyloid beta: Biting the hand that feeds. Alzheimers Dement. 2018;14(4):502–513. doi: 10.1016/j.jalz.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 2.Spires-Jones TL, Hyman BT. The Intersection of Amyloid Beta and Tau at Synapses in Alzheimer’s Disease. Neuron. 2014;82(4):756–771. doi: 10.1016/j.neuron.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nature Reviews Molecular Cell Biology. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 4.Walsh DM, Selkoe DJ. Oligomers in the brain: The emerging role of soluble protein aggregates in neurodegeneration. Protein and Peptide Letters. 2004;11(3):213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- 5.Haass C. Take five--BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 2004;23(3):483–8. doi: 10.1038/sj.emboj.7600061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vassar R, et al. The beta-secretase enzyme BACE in health and Alzheimer’s disease: regulation, cell biology, function, and therapeutic potential. J Neurosci. 2009;29(41):12787–94. doi: 10.1523/JNEUROSCI.3657-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imbimbo BP, Giardina GA. gamma-secretase inhibitors and modulators for the treatment of Alzheimer’s disease: disappointments and hopes. Curr Top Med Chem. 2011;11(12):1555–70. doi: 10.2174/156802611795860942. [DOI] [PubMed] [Google Scholar]
- 8.Kumar D, et al. Secretase inhibitors for the treatment of Alzheimer’s disease: Long road ahead. Eur J Med Chem. 2018;148:436–452. doi: 10.1016/j.ejmech.2018.02.035. [DOI] [PubMed] [Google Scholar]
- 9.Egan MF, et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N Engl J Med. 2018;378(18):1691–1703. doi: 10.1056/NEJMoa1706441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mullard A. BACE inhibitor bust in Alzheimer trial. Nat Rev Drug Discov. 2017;16(3):155. doi: 10.1038/nrd.2017.43. [DOI] [PubMed] [Google Scholar]
- 11.Mullard A. BACE failures lower AD expectations, again. Nat Rev Drug Discov. 2018;17(6):385. doi: 10.1038/nrd.2018.94. [DOI] [PubMed] [Google Scholar]
- 12.De Strooper B. Lessons from a failed gamma-secretase Alzheimer trial. Cell. 2014;159(4):721–6. doi: 10.1016/j.cell.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 13.Anand, R., K.D. Gill, and A.A. Mahdi, Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology, 2014. 76 Pt A: p. 27–50. [DOI] [PubMed]
- 14.Ovsepian SV, O’Leary VB, Zaborszky L. Cholinergic Mechanisms in the Cerebral Cortex: Beyond Synaptic Transmission. Neuroscientist. 2016;22(3):238–51. doi: 10.1177/1073858415588264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rafii MS, Aisen PS. Recent developments in Alzheimer’s disease therapeutics. BMC Med. 2009;7:7. doi: 10.1186/1741-7015-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kan MJ, et al. Arginine Deprivation and Immune Suppression in a Mouse Model of Alzheimer’s Disease. Journal of Neuroscience. 2015;35(15):5969–5982. doi: 10.1523/JNEUROSCI.4668-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abeloff MD, et al. Phase I trial and pharmacokinetic studies of alpha-difluoromethylornithine--an inhibitor of polyamine biosynthesis. J Clin Oncol. 1984;2(2):124–30. doi: 10.1200/JCO.1984.2.2.124. [DOI] [PubMed] [Google Scholar]
- 18.Pepin J, et al. Difluoromethylornithine for arseno-resistant Trypanosoma brucei gambiense sleeping sickness. Lancet. 1987;2(8573):1431–3. doi: 10.1016/S0140-6736(87)91131-7. [DOI] [PubMed] [Google Scholar]
- 19.Polis, P., et al., L-norvaline Reverses Cognitive Decline and Synaptic Loss in a Murine Model of Alzheimer’s Disease. Neurotherapeutics, 2018. 10.1101/354290. [DOI] [PMC free article] [PubMed]
- 20.Oddo S, et al. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiology of Aging. 2003;24(8):1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 21.Oddo S, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular A beta and synaptic dysfunction. Neuron. 2003;39(3):409–421. doi: 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 22.Mitchell WK, et al. Supplementing essential amino acids with the nitric oxide precursor, l-arginine, enhances skeletal muscle perfusion without impacting anabolism in older men. Clin Nutr. 2017;36(6):1573–1579. doi: 10.1016/j.clnu.2016.09.031. [DOI] [PubMed] [Google Scholar]
- 23.Ohta F, et al. Low-dose L-arginine administration increases microperfusion of hindlimb muscle without affecting blood pressure in rats. Proc Natl Acad Sci U S A. 2007;104(4):1407–11. doi: 10.1073/pnas.0610207104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris SM. Arginine metabolism: Boundaries of our knowledge. Journal of Nutrition. 2007;137(6):1602s–1609s. doi: 10.1093/jn/137.6.1602S. [DOI] [PubMed] [Google Scholar]
- 25.Wu GY, Morris SM. Arginine metabolism: nitric oxide and beyond. Biochemical Journal. 1998;336:1–17. doi: 10.1042/bj3360001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeFelipe, J. and S. L., The Beautiful Brain: The Drawings of Ramon y Cajal. 2017, New York: Abrams & Chronicle Books.
- 27.Zhang Y, Barres BA. Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr Opin Neurobiol. 2010;20(5):588–94. doi: 10.1016/j.conb.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Hansmannel F, et al. Is the Urea Cycle Involved in Alzheimer’s Disease? Journal of Alzheimers Disease. 2010;21(3):1013–1021. doi: 10.3233/JAD-2010-100630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caldwell RB, et al. Arginase: an old enzyme with new tricks. Trends Pharmacol Sci. 2015;36(6):395–405. doi: 10.1016/j.tips.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shosha E, et al. Arginase 2 promotes neurovascular degeneration during ischemia/reperfusion injury. Cell Death Dis. 2016;7(11):e2483. doi: 10.1038/cddis.2016.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patassini S, et al. Identification of elevated urea as a severe, ubiquitous metabolic defect in the brain of patients with Huntington’s disease. Biochemical and Biophysical Research Communications. 2015;468(1–2):161–166. doi: 10.1016/j.bbrc.2015.10.140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1224 kb)