

Abstract
Erdheim–Chester disease is a rare histiocytosis with insufficient clinical data. To clarify the clinical features and prognostic factors of Erdheim–Chester disease, we conducted a nationwide survey to collect the detailed data of 44 patients with Erdheim–Chester disease in Japan. The median age of onset of the participants was 51 (range: 23–76) years, and the median number of involved organs per patient was 4 (range: 1–11). The existence of central nervous system disease was correlated with older age (P=0.033), the presence of cardiovascular lesions (P=0.015), and an increased number of involved organs (P=0.0042). The median survival from the onset was 10.4 years, and >3.0 mg/dL C-reactive protein level at onset was associated with worse outcome (median survival, 14.6 vs. 7.4 years; P=0.0016). In a multivariate analysis, age >60 years (hazard ratio, 25.9; 95% confidence interval, 2.82–237; P=0.0040) and the presence of digestive organ involvement (hazard ratio, 4.74; 95% confidence interval, 1.05–21.4; P=0.043) were correlated with worse survival. Fourteen patients had available histological samples of Erdheim– Chester disease lesions. BRAFV600E mutation was detected in 11 patients (78%) by Sanger sequencing. A correlation between BRAF mutation status and clinical factors was not observed. Our study revealed that age and digestive organ involvement influence the outcome of Erdheim–Chester disease patients, and an inflammatory marker, such as C-reactive protein, might reflect the activity of this inflammatory myeloid neoplasm.
Introduction
Erdheim–Chester disease (ECD) is a rare non-Langerhans histiocytosis that was first reported by Jakob Erdheim and William Chester in 1930.1 The number of reports has drastically increased recently, perhaps due to the increased recognition of the disease, and approximately 650–1000 cases have been reported.2–4 ECD typically develops among middle-aged males, and bilateral cortical osteosclerosis occurs in more than 95% of ECD patients.5 Furthermore, some patients experience involvements of the central nervous system (CNS), cardiovascular system, and various other organs.6,7
The pathogenesis of ECD is still unclear, and whether this condition is a type of neoplasia or inflammation is a topic of debate. The high prevalence of mutations in BRAF, NRAS, and various genes involved in the MAPK pathway and the dramatic efficacy of BRAF inhibitors suggest the important role played by the BRAF and MAPK pathways in ECD development.8–11 Recently, Haroche et al. speculated that ECD could be redefined as “inflammatory myeloid neoplasia”, similar to Langerhans cell histiocytosis (LCH).2,12 A mouse model of LCH showed that genetic mutations in hematopoietic stem cells or immature myeloid progenitors induce misguided differentiation, and increase in pathological dendritic cells, which recruit and activate additional inflammatory cells (so-called “innocent bystanders”).13 Cavalli et al. suggested the significance of oncogene-induced senescence (OIS), a protective reaction against oncogenesis, in the recruitment of circulating normal leukocytes in ECD lesions.14,15 In the context of ECD, the local production of pro-inflammatory chemokines and Th1-associated cytokines by BRAFV600E mutated cells attracts circulating normal leukocytes to the sites and contributes to the inflammatory activation and formation of ECD. Further basic studies are warranted to investigate the exact mechanisms of ECD.
The clinical course of ECD is quite heterogeneous, but most cases are progressive and become fatal within a few years. Little is known about the prognostic factors of ECD except that of the involvement of CNS.16 ECD diagnosis is mainly dependent on pathological findings, such as CD68+CD1a− foamy histiocyte infiltration, but the consensus guidelines also demand the clinical and radiological contexts for an appropriate diagnosis of the disease.5 However, our knowledge about the clinical profiles of ECD is unsatisfactory, and more information to consolidate this “context” is required.
Herein, we conducted a nationwide survey on ECD in Japan and clarified some clinical features of this disease. Strikingly, the poor outcome observed among elderly patients and the prognostic impact of digestive organ involvement encouraged the spread of novel treatment strategies, such as vemurafenib, or other molecular targeted therapies.17 Additionally, the prognostic value of the increased C-reactive protein (CRP) level at onset suggested the significance of the inflammatory nature of ECD pathophysiology.
Methods
Study design and participants
We conducted a postal questionnaire-based, multicenter retrospective study on ECD. A questionnaire was simultaneously sent to the hematology, dermatology, respiratory medicine, orthopedics, and pathology departments of the involved hospitals in Japan in July 2014, which were certified by each Japanese society. All cases were diagnosed according to the histopathological findings consistent with ECD, which typically contain infiltration of foamy or lipid-laden histiocytes and which were positive for CD68, CD163 and negative for CD1a and Langerin on immunohistochemical staining. The questionnaire was also sent to hospital departments containing a member who published a paper or presented at an academic conference on ECD cases. The samples of the ECD lesions from the peripheral blood and/or bone marrow were also collected, if available. This study was performed in accordance with the Declaration of Helsinki and the Ethical Guidelines for Biomedical Research Involving Human Subjects enforced on March 29, 2001. This study was approved by the ethics committees of the University of Tokyo and each participating institution. Written informed consent was obtained from all patients whose ECD samples were collected. No definitive diagnostic criteria for ECD have been published; therefore, the diagnoses were self-reported by each institute based on the pathological, radiological, and clinical findings. The digestive organ means gastrointestinal tract plus the accessory organs of digestion (the pancreas, liver, and gallbladder).
Mutation analysis
Genomic DNA was extracted from each formalin-fixed, paraffin-embedded tissue and peripheral blood specimen using the QIAamp DNA FFPE Tissue Kit and DNA Mini Kit (Qiagen, Hilden, Germany), respectively. Polymerase chain reaction (PCR) for the detection of BRAF V600E mutation was performed using primers (TACCTAAACTCTTCATAATGCTTGC, GTAACTCAGCAGCATCTCAGGG) as previously reported.18 The products were purified with Illustra ExoStar (GE Healthcare, Tokyo, Japan), and Sanger sequencing was conducted using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and the ABI Prism 3100 Genetic Analyzer (Life Technologies, Carlsbad, CA, USA).
Statistical analysis
The numerical and categorical variables were compared using the t-test and Fisher’s exact test, respectively. Three patients diagnosed at autopsy were excluded from the analyses of the interval from onset to diagnosis. The survival time was calculated through the Kaplan–Meier method, and the log-rank test was utilized for the evaluation of the significant differences. The effect of various parameters on survival was evaluated through univariate and multivariate analyses with the use of the Cox proportional hazards regression model. ECD-related death was defined as death associated with ECD (such as heart failure due to cardiac involvement and increased intracranial pressure caused by brain mass etc.). The cumulative incidence of ECD-related death was calculated in a competing risks model. The factors with P<0.05 in the univariate analyses were included in the multivariate analysis of the survival. Hazard ratio (HR) was estimated with 95% confidence intervals (CI), and the respective P values were reported from these analyses. Differences were considered statistically significant at P values <0.05. Statistical analyses were performed using R version 3.3.2 (The R Foundation for Statistical Computing, Vienna, Austria).
Results
Patient characteristics and affected organs
The questionnaire was sent to 3850 departments, of which 52% (2007 departments) responded. We confirmed that in Japan 75 patients have ECD, and detailed data were collected from 45 patients. One patient was excluded from the analyses because of insufficient pathological validity. Table 1 shows the clinical characteristics of the remaining 44 patients. The first signs of the disease are described in Online Supplementary Table S1. The median age at initial onset was 51 (range: 23–76) years. Among the 44 patients, 28 were males (63.6%) and 16 were females (36.4%). Five, eight, nine, and 22 patients were diagnosed before 1999, between 2000 and 2004, between 2005 and 2009, and after 2010, respectively. No association between time of diagnosis and clinical features, other than treatment choice, was observed. Three patients were diagnosed at autopsy or after death, and the median and mean time from onset to diagnosis was 12 (range: 1–89) and 20.7 months (Online Supplementary Figure S1). No significant difference between interval from onset to diagnosis and presence of organ involvement, age, year of diagnosis, and any other clinical features were detected.
Table 1.
Clinical characteristics of ECD patients
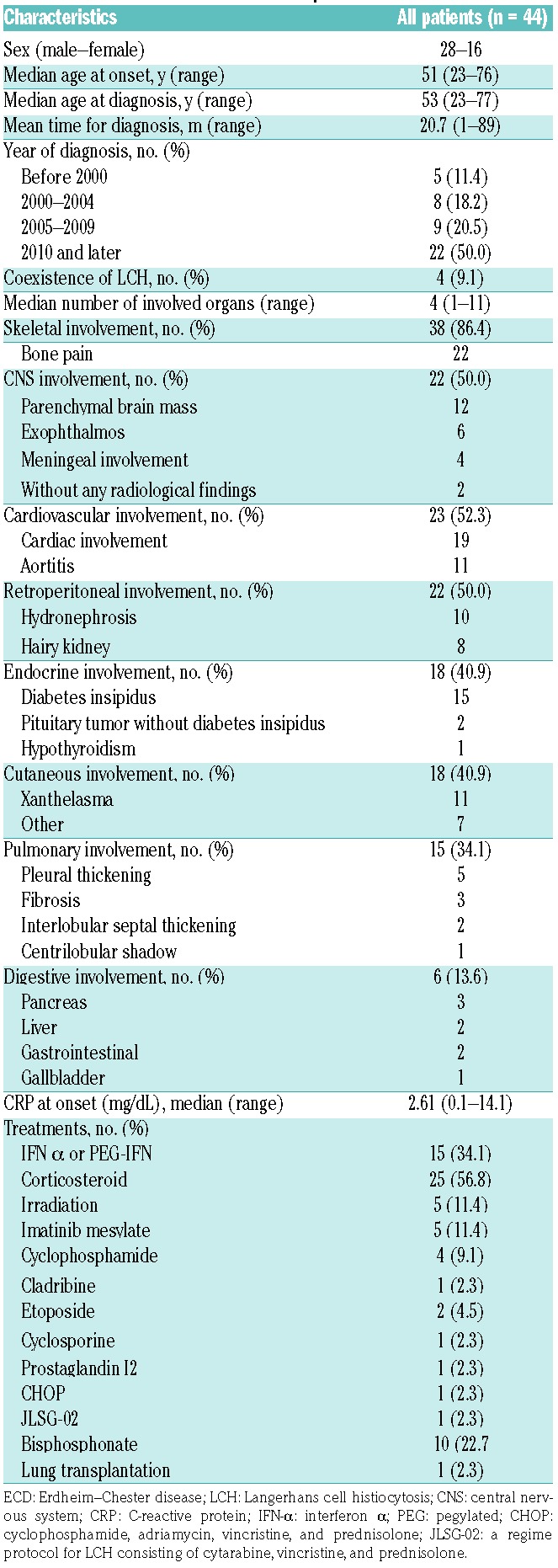
Fifteen patients (33.3%) were treated with interferon α (IFN-α) or pegylated IFN. Twenty-five patients (57.8%) were administered with corticosteroid. Patients who were diagnosed after 2005 were more frequently administered IFN than those diagnosed before 2005 (43.8% vs. 7.7%, P=0.034). No patient was administered with vemurafenib or other BRAF inhibitors.
The median number of involved organs per patient was four (range: 1–11). The lesions were mainly found in the bone (86.4%), the CNS (50.0%), the cardiovascular system (52.3%), the kidney and retroperitoneal organs (50.0%), the endocrine organs (40.9%), the skin (40.9%), the lungs (34.1%), and the digestive organs (13.6%). Oropharyngeal lesions in three patients (6.8%), lymphadenopathy in three patients (6.8%), splenomegaly in two patients (4.5%), and breast lesions in two patients (4.5%) were also detected. In one patient, the testes, prostate, bladder, spinal cord, glottis, and bone marrow were also affected. These unusually affected organs were exclusively observed in patients with multiple (three or more) lesions and, in many cases, who were diagnosed at autopsy. Thirty-eight patients had skeletal lesions, of which 22 had bone pain. Four patients (9.1%) had simultaneous or heterochronic LCH lesions. No patient was diagnosed as having immunoglobulin (Ig)G4-related disease as the clinical and pathological findings did not correspond to the disease, although specific examinations such as serum IgG-4 levels and immunohistochemistry for IgG4 were not carried out on the majority of patients. Among the 18 patients with endocrine involvement, 15 had diabetes insipidus (DI), two had pituitary tumors without obvious DI and one had hypothyroidism. Twenty-three patients had cardiovascular involvement including 19 with cardiac involvement and 11 with aortitis (seven had both). Among the 22 patients with CNS involvement, six had exophthalmos, 12 had parenchymal brain mass and four had meningeal involvement. Two patients had no radiological finding of CNS but their neurological symptoms (delirium and cerebellar ataxia) were considered to be symptoms of ECD.
In addition, one patient had low-risk myelodysplastic syndrome (MDS) prior to ECD. He did not receive any treatment for MDS and one year later, after the detection of an abnormal pulmonary shadow, ECD was diagnosed through partial pneumonectomy. The shadow shrank after cyclosporine and prednisolone therapy, but he died of pneumonia 4.5 years after the ECD diagnosis.
Survival
The median follow-up period of survivors was 5.17 (range: 1.0–21.1) years. Eighteen out of 44 patients died during the clinical course of their illness, and the cause of death was associated with ECD in 13 patients (72.2%, Online Supplementary Table S2). Moreover, two other patients died from infection, possibly due to the use of corticosteroid therapy as a treatment for ECD.
The median survival from the initial onset, which was determined using the Kaplan–Meier method, was 10.42 years (Figure 1A). The log-rank test results revealed that the prognostic factors for this disease were age >60 years old at the onset (P<0.0001), lack of bone lesions (P<0.0001), and the involvement of the CNS (P=0.0010) and digestive organs (P=0.017) (Figure 1B-E). In contrast, the presence of cardiovascular disease, endocrinosis, and pulmonary, kidney/retroperitoneal and cutaneous lesions was not considered as a prognostic factor (Online Supplementary Figure S2). Patients with only one ECD lesion, all of whom had a bone lesion, tended to have better survival after the onset of the disease than those with multiple lesions, although this difference was not statistically significant, perhaps because the number of patients who had a single lesion was only five (P=0.064).
Figure 1.

Survival curves of patients with Erdheim–Chester disease. Kaplan– Meier estimation for survival from onset of (A) all 44 patients based on (B) age and presence of (C) bone lesions, (D) central nervous system involvement, and (E) digestive involvement. CNS: central nervous system; DO: digestive organ.
The univariate analyses, which were conducted using Cox proportional hazards regression model, revealed that age >60 years (P=0.00035), the presence of CNS involvement (P=0.0021) and digestive organ disease (P=0.0022), and the absence of bone lesions (P=0.0042) were significant poor prognostic factors for survival from the onset (Table 2). Sex, year of diagnosis, and existence of cardiovascular, endocrine, cutaneous, or pulmonary involvement and kidney/retroperitoneal lesions did not have an effect on survival.
Table 2.
Univariate and multivariate analyses for survival.

The multivariate analysis showed that age >60 years (HR, 25.9; 95% CI, 2.82–237; P=0.0040) and the presence of digestive organ involvement (HR, 4.74; P=0.043) were correlated with worse survival.
ECD-related death
Next, we analyzed the association between the cumulative incidence of ECD-related death and clinical factors (Figure 2A-D). ECD-related death was significantly associated with age >60 years (P<0.0001), digestive organ (P<0.0001), CNS (P=0.0062), and skeletal involvement (P=0.0072). Cardiovascular (P=0.071), cutaneous (P=0.26), retroperitoneal (P=0.27), respiratory (P=0.28), endocrine involvement (P=0.83) and sex (P=0.89) were not significantly associated with ECD-related death.
Figure 2.

Cumulative incidence of ECD-related death. Competing risks models revealed the cumulative incidence of ECD-related death based on (A) age and presence of (B) digestive organs, (C) CNS, (D) skeletal involvement. DO: digestive organ; CNS: central nervous system.
ECD without skeletal lesion
Considering that the prevalence of skeletal lesions in our study was relatively low compared with that of previous studies in Western countries (>95%),5,19 we compared the clinical data of ECD patients with and without skeletal lesions. No obvious clinical difference based on skeletal involvement was observed, except that patients with skeletal lesions had a better outcome than those without (Figure 1C and Table 3).
Table 3.
Skeletal involvement and clinical factors.

ECD with digestive involvement
Given that ECD with digestive involvement was associated with worse survival, we examined the characteristics of ECD patients with digestive disease (Table 4). The digestive organs concerned were the liver (50.0%), pancreas (50.0%), gastrointestinal tract (33.3%), and gallbladder (16.7%). Causes of death were arrhythmia in one patient with cardiac tamponade, heart and renal failure in one patient with pericardial effusion and hydronephrosis, sepsis due to rectal perforation in one patient with gastrointestinal involvement of ECD, and ECD (details unknown) in one patient. An autopsy was carried out in four patients who died during the clinical course of digestive disease, and the involvement of ECD cells was histologically proven. Histological samples were available in two patients and both were BRAFV600E positive.
Table 4.
Digestive involvement and clinical factors.

No correlation was found between the existence of digestive organ disease and the CRP level at onset (P=0.99), age at onset (P=0.11), sex (P=0.39), year of diagnosis (P=1), and the presence of specific organ involvement.
CNS involvement and associated clinical factors
Considering that the CNS was revealed to be a “risk organ”, based on the result of our analysis and that of a previous study,16 we compared the characteristics of patients with and without CNS disease to determine those were are at risk of CNS involvement (Table 5). Patients with CNS disease displayed significantly higher age at onset than those without (median, 62 [range, 23–76] years vs. 45 [range, 25–70] years; P=0.033). Additionally, the existence of CNS involvement was correlated with the presence of cardiovascular lesions (P=0.015). No statistically significant association between the presence of CNS involvement and sex (P=0.75), year of diagnosis (P=1.0), and existence of bone (P=0.19) and skin lesions (P=0.36), endocrinosis (P=0.12), pulmonary (P=0.20) and digestive organ involvement (P=0.66), or kidney/retroperitoneal disease (P=0.13) was detected.
Table 5.
CNS involvement and clinical factors.

CRP level and outcome
The CRP level at onset was higher than the upper normal limit in 29 (85.3%) out of 34 patients with sufficient laboratory data, with a median CRP level at onset of 2.61 mg/dL (range: 0.10–14.1). Interestingly, a >3.0 mg/dL CRP level at onset was associated with a worse outcome (median survival, 14.6 vs. 7.4 years; P=0.016; Figure 3A). The cumulative incidence of ECD-related death was also associated with a higher CRP level at onset (P=0.043; Figure 3B). We also compared the CRP level at onset with that after the administration of first-line therapy in 25 patients whose clinical data were available. The results of the Wilcoxon signed-rank test revealed that CRP levels tended to decline following the initial treatment (median at onset and after initial therapy: 2.38 [range, 0.10–14.1] mg/dL and 1.16 [range, 0.01–34.6] mg/dL, respectively; P=0.051; Figure 3C). In addition, patients whose CRP levels reduced by more than 2.0 mg/dL after first-line therapy had a better outcome compared with patients whose CRP levels dropped by less than 2.0 mg/dL or increased after first-line treatment (median survival, 11.3 vs. 7.4 years; P=0.045) (Figure 3D).
Figure 3.

C-reactive protein (CRP) at onset and clinical outcome. (A) Kaplan–Meier estimation for survival from onset and (B) the cumulative incidence of ECD-related death of 34 patients with sufficient clinical data. (C) Comparison of the CRP level before and after administration of first-line therapy. (D) Kaplan–Meier estimation for survival from onset according to the decline of CRP levels after first-line therapy.
BRAFV600E mutation
Thirteen patients had available histological samples on ECD lesions (Online Supplementary Table S3). The genomic DNA was extracted from the samples, which were then subjected to direct sequencing. A BRAFV600E mutation was detected in 11 out of 14 patients (78%). None of the BRAF mutations were detected through Sanger sequencing of the genomic DNA extracted from the peripheral blood or bone marrow samples. Moreover, no correlation between BRAF mutation status and age, CRP level at onset, and other clinical factors were observed.
Discussion
Our nationwide study broadly investigated ECD patients and analyzed the clinical data of 44 patients. ECD is so rare that few reports on multiple ECD patient studies have been published and little evidence about the clinical characteristics or prognostic factors of this disease is available. The study herein is one of the largest in terms of the number of patients with ECD involved in our research.2,6,7,16 In this study, the duration between onset and diagnosis seems to be shorter compared with previous studies, in which many patients were diagnosed several years after initial onset. It might reflect an increased familiarity with ECD in recent years, although the exact reason is unclear.20 IFN has recently been recommended as first-line therapy, and BRAF inhibitors are also strong candidates for the treatment of ECD.5,16,21 In our study, IFN was administered to a very small proportion of patients during the clinical course of the disease, and no patients received BRAF inhibitors, which is partially attributed to the Japanese insurance system. Instead, many patients were prescribed corticosteroid, which is believed to temporarily alleviate the symptoms, although it is not recommended by the consensus guidelines.5 Two patients died from infection (one pneumonia and one invasive pulmonary aspergillosis), possibly due to immunosuppression induced by corticosteroid administration for the treatment of ECD. In our study, the mortality rate was relatively high compared with a recent report which showed a five year survival rate of 82.7 %.4 However, the outcome in our cohort was slightly better than that in patients who were not administered IFN in a previous report,16 perhaps due to the improvement of supportive care. To improve the prognosis of patients with ECD, more detailed analyses and prospective studies of the pathophysiology of ECD are required. Given that future studies on ECD may not include patients who were not administered with IFN and/or BRAF inhibitors, this study could serve as an important physician’s compass that reveals the baseline clinical behaviors of ECD.
CNS involvement was a significant poor prognostic fac tor in our series, which seems to be consistent with the previous study reporting that CNS involvement was associated with resistance to IFN.16 Our study revealed that ECD patients with CNS lesions were significantly older and had cardiovascular lesions more frequently. These factors might affect the efficacy of IFN, as a cardiovascular lesion has also been suggested to contribute to the ineffectiveness of IFN therapy.22 Furthermore, the side effects of IFN, such as delirium, is known to be more frequently observed in older patients than in younger ones, despite intense psychiatric support.23 Future study is necessary to identify patients who are expected to benefit the most from treatment with IFN.
The prevalence of affected organs, median age at onset and male predominance were roughly comparable with that of previous reports in Western countries,5,7 excepting the article by Cavalli et al. which found that CNS manifestations in younger patients are relatively common,7 and the fact that the percentage of patients with skeletal disease in our cohort was relatively low.5 The coexistence of LCH was also relatively rare in our series. Considering the close relationship between ECD and LCH, perhaps some cases were overlooked.24 This dissociation might be attributed to coincidence, ethnicity, or other genetic/epigenetic diversity. No statistically significant clinical difference was found between ECD patients with and without bone involvement, which suggests that ECD cases have common features regardless of this factor. We were unable to obtain evidence that demonstrates whether ECD without bone lesions should be classified as a distinctive entity.
The digestive organ was detected as a risk organ for patients with ECD in this study. The prognostic impact of digestive involvement was not found in previous reports,5,6 perhaps due to the small number of patients with digestive ECD lesions. Although the liver and pancreas were the most commonly affected organs, none of these patients with hepatic/pancreatic involvements suffered from the relevant organ failure. Four of six patients with digestive organ involvement died of ECD, however, only one patient died of rectal perforation due to ECD involvement and the other three patients died of other affected organs. A further accumulation of cases is necessary in order to confirm the prognostic impact of digestive involvement.
The CRP level was elevated in many patients with ECD, and declined after IFN or vemurafenib treatment,8,22 similar to the finding in our cohort. In this study, we firstly revealed that the CRP level at onset as well as the drop in CRP levels after initial therapy predict the patient outcome. We have to interpret this result with caution because initial treatments in our cohort were quite heterogeneous and most of them were not standard therapy. Recently, ECD has been considered as an inflammatory myeloid neoplasm. Although the MAPK pathway and other mutations, such as PIK3CA, tend to attract attention to the neoplastic aspect of the disease, the association between CRP levels and poor prognosis evokes the necessity of vigorous research on the inflammatory characteristic of Janus-faced ECD because these mutations themselves do not necessarily induce elevated CRP. Some inflammatory markers, such as interleukin (IL)-1, IL-6 and tumor necrosis factor, have also been reported as activation markers of ECD.25–28 However, CRP levels are relatively easy to measure compared with inflammatory cytokines and might be suitable as a convenient marker to detect patients at risk.
Interestingly, low-risk MDS and ECD coexisted in one patient in our cohort. Papo et al. reported that adult histiocytic neoplasms frequently co-occur with other myeloid neoplasms (9.5%), and MAPK pathway-associated mutations (BRAF, NRAS, and other mutations) could coexist with MDS-associated mutations (IDH1/2, ASXL1, and TET2 mutations) in the hematopoietic stem/progenitor cells of ECD patients.29 Unfortunately, we were unable to acquire a sample from the patient, and the genetic information was unavailable.
Due to the retrospective nature of our study, we could not analyze some of the most important clinical information of many of the patients, such as osteosclerosis of the sinuses and BRAF or other mutations.30 It is now mandatory to systematically collect clinical findings, radiological and laboratory data, and above all histological samples for patients with ECD. A more sophisticated method of detection for the BRAF mutation is also warranted. Sanger sequencing is a classic method and still plays an important role in detecting ECD, partially owing to its low cost and convenience. However, immunohistochemistry for the BRAF V600E protein and hypersensitive methods, such as droplet digital PCR and/or targeted sequencing techniques, are becoming crucial because only a small fraction of cells in an ECD lesion actually harbor the BRAF mutation, and some studies have reported that a BRAF mutation in the peripheral blood could only be detected with ultrasensitive techniques as opposed to Sanger sequencing.15
In summary, our nationwide survey revealed the clinical characteristics of patients with ECD, and clarified the prognostic value of age and digestive involvement. The CRP level at onset also predicts the patients’ survival, perhaps reflecting the importance of the inflammatory status in the ECD condition. Additional large-scale studies are required in order to clarify the pathophysiology of ECD and improve the prognosis of the patients.
Supplementary Material
Acknowledgments
We would like to acknowledge K. Tanaka and Y. Nakada for their technical assistance.
Furthermore, we thank M. Fukuda (Asoka Hospital), H. Hiraga (Hokkaido Cancer Center), T. Yamauchi (Toki General Hospital), K. Takeuchi (Tochigi Cancer Center), Y. Shido (Hamamatsu University School of Medicine), S. Nogawa (Tokyo Dental College Ichikawa General Hospital), T. Hamada (Okayama University), N. Hayashi (Wakayama Rosai Hospital), S. Yokokura (Shin-Yamanote Hospital), T. Makiishi (Japanese Red Cross Otsu Hospital), T. Kitani (Ehime University), Y. Shibuya (Showa University Northern Yokohama Hospital), M. Yoshimitsu (Kagoshima University), J. Mitsushima (Matsue Seikyo General Hospital), S. Yano (Matsue Medical Center), M. Susa (Keio University), K. Ono (Tosei General Hospital), K. Tanaka (Okayama University), M. Yamamoto (Kochi University), K. Sato (Saitama Medical University), H. Sugano (Kochi Health Sciences Center), T. Kondo (Kyoto University), H. Takahama (National Cerebral and Cardiovascular Center), J. Nagano (Seirei Hamamatsu General Hospital), T. Baba (Kanagawa Cardiovascular and Respiratory Center), Y. Fukui (Juntendo University), K. Takenaka (Tsuchiura Kyodo General Hospital), K. Takada (Sapporo Medical University), S. Izumi (National Center for Global Health and Medicine), J. Kikuchi (Keio University), K. Takahashi (Kameda Medical Center), K. Shimizu (Higashihiroshima Medical Center), K. Takemura (Tokyo Teishin Hospital), and I. Choi (National Hospital Organization Kyushu Cancer Center) for providing the clinical data and/or patient samples. We also thank H. Kawano (Teikyo University) and the members of the Japanese Musculoskeletal Oncology Group for cooperating in the collection of clinical data and/or patient samples.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/11/1815
Funding
This study is supported by Grants-in-Aid of the Ministry of Health, Labor and Welfare of Japan (H28-Nanchi-Ippan-002 to MS, TO, IK, and MK).
References
- 1.Chester W. Uber lipoidgranulomatose. Virchows Arch Pathol Anat 1930;279:561–602. [Google Scholar]
- 2.Haroche J, Abla O. Uncommon histiocytic disorders: Rosai-Dorfman, juvenile xanthogranuloma, and Erdheim-Chester disease. Hematology Am Soc Hematol Educ Program. 2015;2015:571–578. [DOI] [PubMed] [Google Scholar]
- 3.Haroche J, Cohen-Aubart F, Charlotte F, et al. The histiocytosis Erdheim-Chester disease is an inflammatory myeloid neoplasm. Expert Rev Clin Immunol. 2015;11(9):1033–1042. [DOI] [PubMed] [Google Scholar]
- 4.Haroche J, Cohen-Aubart F, Rollins BJ, et al. Histiocytoses: emerging neoplasia behind inflammation. Lancet Oncol. 2017; 18(2):e113–e125. [DOI] [PubMed] [Google Scholar]
- 5.Diamond EL, Dagna L, Hyman DM, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood. 2014;124(4):483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Estrada-Veras JI, O’Brien KJ, Boyd LC, et al. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv. 2017;1(6):357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cavalli G, Guglielmi B, Berti A, Campochiaro C, Sabbadini MG, Dagna L. The multifaceted clinical presentations and manifestations of Erdheim-Chester disease: comprehensive review of the literature and of 10 new cases. Ann Rheum Dis. 2013; 72(10):1691–1695. [DOI] [PubMed] [Google Scholar]
- 8.Haroche J, Cohen-Aubart F, Emile JF, et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121(9):1495–1500. [DOI] [PubMed] [Google Scholar]
- 9.Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012;120(13):2700–2703. [DOI] [PubMed] [Google Scholar]
- 10.Emile JF, Diamond EL, Helias-Rodzewicz Z, et al. Recurrent RAS and PIK3CA mutations in Erdheim-Chester disease. Blood. 2014; 124(19):3016–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Milne P, Bigley V, Bacon CM, et al. Hematopoietic origin of Langerhans cell histiocytosis and Erdheim Chester disease in adults. Blood. 2017;130(2):167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berres ML, Merad M, Allen CE. Progress in understanding the pathogenesis of Langerhans cell histiocytosis: back to histiocytosis X? Br J Haematol. 2015;169(1):3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Durham BH, Roos-Weil D, Baillou C, et al. Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood. 2017;130(2):176–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cavalli G, Biavasco R, Borgiani B, Dagna L. Oncogene-induced senescence as a new mechanism of disease: the paradigm of erdheim-chester disease. Front Immunol. 2014;5:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cangi MG, Biavasco R, Cavalli G, et al. BRAFV600E-mutation is invariably present and associated to oncogene-induced senescence in Erdheim-Chester disease. Ann Rheum Dis. 2015;74(8):1596–1602. [DOI] [PubMed] [Google Scholar]
- 16.Arnaud L, Hervier B, Neel A, et al. CNS involvement and treatment with interferon-alpha are independent prognostic factors in Erdheim-Chester disease: a multi-center survival analysis of 53 patients. Blood. 2011;117(10):2778–2782. [DOI] [PubMed] [Google Scholar]
- 17.Cohen Aubart F, Emile JF, Carrat F, et al. Targeted therapies in 54 patients with Erdheim-Chester disease, including followup after interruption (the LOVE study). Blood. 2017;130(11):1377–1380. [DOI] [PubMed] [Google Scholar]
- 18.Arcaini L, Zibellini S, Boveri E, et al. The BRAF V600E mutation in hairy cell leukemia and other mature B-cell neoplasms. Blood. 2012;119(1):188–191. [DOI] [PubMed] [Google Scholar]
- 19.Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127(22):2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haroche J, Amoura Z, Dion E, et al. Cardiovascular involvement, an overlooked feature of Erdheim-Chester disease: report of 6 new cases and a literature review. Medicine (Baltimore). 2004;83(6):371–392. [DOI] [PubMed] [Google Scholar]
- 21.Haroche J, Cohen-Aubart F, Emile JF, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAF(V600E)-mutated Erdheim-Chester disease. J Clin Oncol. 2015; 33(5):411–418. [DOI] [PubMed] [Google Scholar]
- 22.Haroche J, Amoura Z, Trad SG, et al. Variability in the efficacy of interferon-alpha in Erdheim-Chester disease by patient and site of involvement: results in eight patients. Arthritis Rheum. 2006; 54(10):3330–3336. [DOI] [PubMed] [Google Scholar]
- 23.Raison CL, Demetrashvili M, Capuron L, Miller AH. Neuropsychiatric adverse effects of interferon-alpha: recognition and management. CNS Drugs. 2005;19(2):105–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hervier B, Haroche J, Arnaud L, et al. Association of both Langerhans cell histiocytosis and Erdheim-Chester disease linked to the BRAFV600E mutation. Blood. 2014; 124(7):1119–1126. [DOI] [PubMed] [Google Scholar]
- 25.Arnaud L, Gorochov G, Charlotte F, et al. Systemic perturbation of cytokine and chemokine networks in Erdheim-Chester disease: a single-center series of 37 patients. Blood. 2011;117(10):2783–2790. [DOI] [PubMed] [Google Scholar]
- 26.Berti A, Cavalli G, Guglielmi B, et al. Tocilizumab in patients with multisystem Erdheim-Chester disease. Oncoimmunology. 2017;6(6):e1318237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dagna L, Corti A, Langheim S, et al. Tumor necrosis factor alpha as a master regulator of inflammation in Erdheim-Chester disease: rationale for the treatment of patients with infliximab. J Clin Oncol. 2012; 30(28):e286–290. [DOI] [PubMed] [Google Scholar]
- 28.Aouba A, Georgin-Lavialle S, Pagnoux C, et al. Rationale and efficacy of interleukin-1 targeting in Erdheim-Chester disease. Blood. 2010;116(20):4070–4076. [DOI] [PubMed] [Google Scholar]
- 29.Papo M, Diamond EL, Cohen-Aubart F, et al. High prevalence of myeloid neoplasms in adults with non-Langerhans cell histiocytosis. Blood. 2017;130(8):1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Drier A, Haroche J, Savatovsky J, et al. Cerebral, facial, and orbital involvement in Erdheim-Chester disease: CT and MR imaging findings. Radiology. 2010; 255(2):586–594. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.