

Abstract
Background and Aim:
A substantial proportion of pediatric liver transplant recipients develop subclinical chronic allograft injury. We studied whether there are distinct patterns of injury based on histopathology features and identified associated immunological profiles.
Methods:
We conducted a cross-sectional study of 157 stable, long-term pediatric recipients of transplanted livers (70 boys; less than 6 years old; mean 8.9±3.46 years after liver transplant) who underwent liver biopsy analysis from August 13, 2012 through May 1, 2014. Subjects received livers from a living or deceased donor and had normal results from liver tests for more than 4 years after receiving transplant. Liver biopsies were scored by a central pathologist; an unsupervised hierarchical cluster analysis of histologic features was used to sort biopsies into 3 clusters. We conducted transcriptional and cytometric analyses of liver tissue samples and performed a systems biology analysis that incorporated clinical, serologic, histologic, and transcriptional data.
Results:
The mean level of alanine aminotransferase in subjects was 27.6±14.57 U/L and the mean level of gamma-glutamyl transferase was 17.4±7.93 U/L. Cluster 1 was characterized by interface activity (n=34), cluster 2 was characterized by periportal or perivenular fibrosis without interface activity (n=45), and cluster 3 had neither feature (n=78). We identified a module of genes whose expression correlated with levels of alanine aminotransferase, class II donor-specific antibody, portal inflammation, interface activity, perivenular inflammation, portal and perivenular fibrosis, and cluster assignment. The module was enriched in genes that regulate T-cell mediated rejection (TCMR) of liver and other transplanted organs. Functional pathway analysis revealed over-representation of TCMR gene sets for cluster 1 but not clusters 2 or 3.
Conclusion:
In an analysis of biopsies from an apparently homogeneous group of stable, long-term pediatric liver transplant recipients with consistently normal results from liver tests, we found evidence of chronic graft injury (inflammation and/or fibrosis). Biopsies with interface activity had a gene expression pattern associated with TCMR.
Keywords: ALT, DSA, immune response, prognostic factor
Graphical Abstract
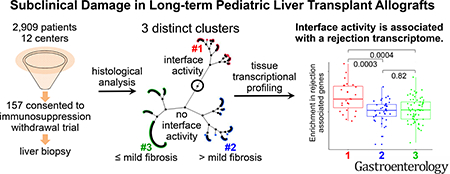
There are now thousands of long-term pediatric liver transplant recipients who apparently enjoy excellent health with no biochemical evidence of allograft injury. While this success is worthy of celebration, it has also generated new questions as to how best to care for these young patients. The goal of securing excellent health over many decades requires constant consideration of optimal immunosuppression to ensure efficacy while minimizing toxicity. The risks associated with low dose but chronic immunosuppression have been described1–7 leading to substantial efforts directed at drug minimization. Indeed, several prospective, multicenter clinical trials have reported complete discontinuation of immunosuppression without progressive allograft damage in select adult and pediatric recipients.8–11 In contrast, multiple centers around the world have reported that liver allografts of patients maintained on standard of care immunosuppression frequently harbor subclinical inflammation and/or fibrosis.12–18 Moreover, the prevalence and severity of allograft histopathology has been reported to increase over time such that, 10 years after transplant, normal histology may be present in only 30% of patients while bridging fibrosis or cirrhosis may approach 60%.13, 15, 16 Together, these reports have suggested that the observed abnormalities reflect an active and ongoing immune response, implicating chronic but imprecisely-defined immune mechanisms.13, 16, 19–24 Consequently, clinicians have been left with a challenging quandary when managing stable patients with consistently normal results of liver tests on modest immunosuppression doses: reduce immunosuppression at the risk of exacerbating silent, immune-mediated allograft injury, stay the course with uncertainty as to whether dosing is appropriate, or escalate immunosuppression unnecessarily, increasing the risk of known toxicities.
Based on the contradictory literature regarding optimal immunosuppression for pediatric liver transplant recipients who appear stable by clinical and biochemical criteria, we hypothesized that these patients are not homogeneous but would sort into distinct histopathological phenotypes reflecting specific mechanisms of chronic graft injury. The aim of this study was to identify these phenotypes and elucidate their associated immunologic profiles. We utilized prospectively collected data and biospecimens at the time of screening for participation in iWITH (NCT01638559), a prospective, multicenter, North American trial of immunosuppression withdrawal for stable pediatric liver transplant recipients. We believe that a clear description and plausible explanation of histopathological phenotypes can have an immediate impact on clinical decision-making and inform the future design of rational interventions to maximize allograft longevity.
METHODS
Study Design
We carried out a cross sectional study using data (donor, recipient and transplant) and biospecimens collected at the time of liver biopsy, the final eligibility assessment for participation in iWITH (NCT01638559), an immunosuppression withdrawal trial conducted at 12 pediatric liver transplant centers in North America (Table 1). The analysis population included all 157 patients who provided ageappropriate informed assent and consent (parent/legal guardian) to iWITH and underwent a liver biopsy between August 13, 2012 and May 1, 2014.
Table 1:
Characteristics of 157 subjects undergoing iWITH eligibility biopsy
| Characteristic* | ||||
|---|---|---|---|---|
| Donor | Age (years) | 15.7 (15.44) | ||
| Male gender | 81 (51.6) | |||
| Race (n=132) | White | 105 (79.5) | ||
| Black | 19 (14.4) | |||
| Other | 8 (6.1) | |||
| Deceased | 110 (70.1) | |||
| Recipient | Age at transplant (years) | 1.8 (1.70) | ||
| Male gender | 79 (50.3) | |||
| Race (n=152) | White | 128 (84.2) | ||
| Black | 11 (7.2) | |||
| Other | 13 (8.6) | |||
| Transplant indication | Acute Liver Failure | 11 (7.0) | ||
| Biliary Atresia | 86 (54.8) | |||
| Tumor | 8 (5.1) | |||
| Metabolic Liver Disease | 18 (11.5) | |||
| Other | 34 (21.7) | |||
| Transplant | Whole graft | 73 (46.5) | ||
| Previous rejection episodes | 0 | 96 (61.1) | ||
| 1 | 40 (25.5) | |||
| 2 or more | 21 (13.4) | |||
| Time since last rejection (years) | 7.3 (3.22) | |||
| At Study Entry | Age (years) | 10.7 (3.50) | ||
| Time since transplant (years) | 8.9 (3.46) | |||
| ALT (U/L) | 27.6 (14.57) | |||
| GGT (U/L) | 17.4 (7.93) | |||
| Quantitative IgG (n=125; mg/dL) | 701.0 (194.95) | |||
| ANA ≥1:40 (n=133) | 34 (25.6) | |||
| ASMA = 1:80 (n=133) | 5 (3.8) | |||
| α-AT1R antibody (n=119; U/mL) | 35.9 (21.49) | |||
| α-ETAR antibody (n=119; U/mL) | 35.2 (21.39) | |||
| Class II DSA positive (n=144; MFI ≥2,000) | 80 (55.6) | |||
| Class II DSA (n=80) | Number of DSAs | 1 | 42 (52.5) | |
| 2 | 28 (35.0) | |||
| 3 or more | 10 (12.5) | |||
| Maximum MFI >20,000 | 37 (46.2) | |||
| MFI sum >20,000 | 45 (56.2) | |||
Continuous variables are summarized using mean and standard deviation (SD). Categorical variables are summarized by counts and percentages.
Subjects: Key Inclusion/Exclusion Criteria
Subjects (<18 years) were ≥4 years after primary living or deceased donor liver transplantation for nonviral and non-autoimmune liver disease at ≤6 years of age who underwent screening liver biopsy for iWITH. Participants were required to have alanine aminotransferase (ALT) and gamma-glutamyl transferase (GGT) consistently less than 50 IU/L based on medical record review by the site principal investigator and to be stably maintained on calcineurin inhibitor monotherapy without rejection during the preceding two years (Figure 1).
Figure 1:

Selection of 157 study participants
Routine Histology, C4d Scoring, Multiplex Quantum Dot (Qdot) Immunolabelling, and Automated Image Analysis
High resolution 40Xwhole slide images (WSI) of formalin-fixed, paraffin-embedded, H&E and Masson’s trichrome-stained 4μm tissue sections of eligibility liver biopsies were prospectively scored for 42 histopathologic criteria by a central pathologist without knowledge of any clinical or serological data other than the date of transplantation and original disease. Fibrosis was assessed in several ways: Ishak stage (0–6), individual compartmental scores (periportal, sinusoidal, and perivenular; 0–3 each), 3-compartment sum (0–9) known as the liver allograft fibrosis score (LAFSc) 16 and quantitatively using a combination of pixel area morphometry and/or tissue tethered cytometry25 after Qdot multiplex panel immunostaining [trichrome and smooth muscle actin (SMA) staining] (Supplementary Figure 1). C4d deposition was evaluated on frozen biopsies using indirect immunofluorescence staining for C4d (mouse monoclonal Quidel, San Diego, CA, 1:50) in distinct vascular endothelial compartments and surrounding stroma (portal vein and capillary, portal stroma, hepatic artery, sinusoid, central vein and stroma); each was separately scored (0=none; 1=minimal; 2=focal; 3=diffuse) and summed for a total C4d score (0–18). Batched slide sets were multiplex-stained and evaluated as described in the Supplementary Methods.
Derivation of Histopathological clusters
Subjects’ histologic scores were classified by an unsupervised hierarchical cluster analysis using Ward’s minimum-variance method with standardized data points. Ten features were initially considered, but only 5 exhibited sufficient variability to be considered further: interface activity, perivenular fibrosis, fibrosis stage, lobular inflammation, and portal inflammation. Subjects were categorized into 3 clusters using 3, 4 and 5 variable models and results were compared to determine the best classification model using 3 criteria, R square, cubic clustering, and pseudo-F statistic, to determine goodness of fit.
Analysis of liver tissue gene expression data
Affymetrix U219 microarray data was available from 133 of the 157 liver biopsies and analyses were batched to minimize bias. Differential expression was computed employing Significant Analysis of Microarray (SAM)26 and expressed as False Discovery Rate (FDR). We used Weighted Gene Correlation Network Analysis (WGCNA; software package available from R28)27 to identify the key biological networks associated with the demographic, clinical serological and histological features of the study subjects. This is a widely used, unsupervised, and exploratory data mining technique that reduces the multi-dimensionality of the gene expression dataset by defining modules of co-expressed genes and integrates external variables (e.g. clinical or histological traits) by establishing weighted correlations with the gene modules. Validation gene expression experiments for a set of 800 pre-defined genes were conducted on 148 RNA samples utilizing a Nanostring nCounter platform. A detailed description of the gene expression experiments is provided in the Supplementary Methods.
Autoantibody assessment
Samples isolated from plasma or serum collection tubes were assayed for quantitative serum immunoglobulin G (IgG), α-nuclear antibodies (ANA), α-smooth muscle antibodies (ASMA), α-angiotensin II type 1 receptor (AT1R) antibody and α-endothelin type A receptor (ETAR) antibody. (EIA-AT1RX/EIA-ETAR, One Lambda, Canoga Park, CA).
HLA typing and alloantibody characterization
HLA typing data was retrieved from United Network for Organ Sharing for 106 deceased and 24 living donors. HLA typing was performed by SSP or SSO (One Lambda, Canoga Park, CA) for 20 living donors and 154 recipients. HLA typing data was unavailable for 7 donors (4 deceased; 3 living) and 3 recipients. HLA mismatch data is presented in Supplementary Table 1.
Screening and specificity analysis for donor-specific HLA antibody (DSA) against HLA antigens was determined using FlowPRA®Screening and LabScreen® Single Antigen™ (One Lambda, Canoga Park, CA). FlowPRA® was acquired on a FACSCanto II (Becton-Dickinson, San Jose CA.) and Single Antigen™ bead assessments were performed on the LABScan 200 instrument (Luminex Corp. Austin, TX). Samples with sufficient volume were also tested using C1qScreen™ (One Lambda Inc., Canoga Park, CA, USA) acquired on a LabScan3D instrument (Luminex Corp., Austin, TX). HLA pattern analysis and bead mean fluorescence intensity (MFI) >2000 for LabScreen® and >5000 for C1qScreen™ determined positivity. Only class II DSA data are presented as the preponderance of literature indicates their primacy in chronic allograft damage.22, 28, 29 HLA-DRB1-DQB1-DQA1 linkage-disequilibrium data was used to assign donor specificity for HLA-DQ antibodies for subjects with limited donor HLA-DQ typing.
Statistical Analysis
Descriptive statistics including mean, standard deviation (SD), and quartiles were determined for continuous variables and frequencies and percentages were determined for categorical variables. Comparisons of categorical variables between two groups used the Fisher exact test while comparisons of continuous variables with two groups used the two-sample t-test. Comparisons of continuous variables with more than two groups used ANOVA.
Multivariable logistic regression analyses were used to study associations between histological features, C4d scores and serological profiles. Univariable and multivariable logistic regression analyses were used to identify selected predictors of subjects’ histologic cluster assignment among clinical and serologic factors along with their interactions. Small amounts of missing data in risk factors were accommodated in the multivariable analyses by case-wise deletion. Significant predictors at the 0.10 level in the univariable analyses were included in the multivariable models. Backwards variable elimination using 0.10 as the threshold for retention resulted in the final multivariable model. The model was internally validated using 1000 bootstrapped resamples to produce an optimism-adjusted area under the curve. Statistical significance was set at an alpha level of 0.05. All statistical analyses were performed using SAS software (version 9.4, SAS Institute, Inc., Cary, NC, USA). All authors had access to the study data and reviewed and approved the final manuscript.
RESULTS
Characteristics of enrolled subjects
Subjects (79 boys; 84% white) were a mean (SD) 1.8 (1.70) years old at transplant and 10.7 (3.50) years old at enrollment (Table 1). They underwent living (n=47; 30%) or deceased [whole n=73 (47%); partial n=37 (24%)] donor liver transplant predominantly for biliary atresia (55%). A modest proportion received induction immunosuppression (n=21; 13%). At enrollment, all were on calcineurin inhibitor monotherapy with mean (SD) ALT of 27.6 (14.57) U/L and GGT of 17.4 (7.93) U/L.
Inflammation, fibrosis, and C4d scores for 157 eligibility biopsies
The 157 eligibility biopsies were assessed for necroinflammatory activity and fibrosis (Figures 2A and 2B). Lymphocytic inflammation was common in the portal/periportal area (59% mild; 5% moderate) but less so in the perivenular (17% mild) area. A minority showed interface activity (21% mild; 1% moderate), lobular (23% mild; 1% moderate) or perivenular inflammation (17% mild). As expected, inflammation and fibrosis typically occurred together and were spatially associated. Biopsies with portal inflammation and interface activity had higher Ishak fibrosis stages while biopsies with perivenular inflammation had higher perivenular fibrosis scores. However, fibrosis and inflammation were occasionally disconnected: some biopsies showed mild or moderate portal or perivenular fibrosis but no inflammation while others showed mild portal inflammation without interface activity and low Ishak fibrosis stage and LAFSc.
Figure 2: Key histological features of 157 biopsies.

(A) Heat map and (B) frequencies of histological features.
C4d scores, total and by compartment, are shown in Supplementary Figures 2A and 2B. The two stromal compartments, portal and perivenular, were most frequently positive. Portal stromal staining was associated with portal inflammation (p=0.03); perivenular stromal staining was associated with perivenular inflammation (p=0.009) (Supplementary Table 2). The distribution and intensity of microvascular endothelial cell C4d staining followed both HLA class II target antigen expression30–32 and blood flow: portal capillaries > sinusoids > central vein. No other significant associations with routine histopathology findings were detected.
Auto- and alloantibody profiles of subjects
ANA and ASMA were positive in 26% (34/133) and 4% (5/133), respectively; mean (SD) IgG was 701.0 (194.95) mg/dL (Table 1; Supplementary Figure 3). Most subjects were positive for α-AT1R and α-ETAR antibodies (68.1% and 66.4%, respectively) with mean (SD) concentrations of 35.9 (21.49) and 35.2 (21.39) U/mL, respectively. Subjects positive for ANA were older at study entry with a longer interval since transplant. In contrast, those positive for α-AT1R and α-ETAR antibodies were younger at time of study entry with a shorter interval since transplant. Associations between clinical characteristics and autoantibody profile are shown in Supplementary Table 3A.
For class II DSA, 80 of 144 (55.6%) tested subjects were positive with mean (SD) MFI sum of 26,699 (16,674). Forty-two subjects had a single class II DSA, 28 had 2 class II DSAs, and 10 had 3 or more class II DSAs (Table 1). Among the 130 class II DSAs identified, 38.5% had MFI >20,000 and 68.5% had specificity against DQ antigens. Thirty-seven of 80 (46.2%) subjects had at least one class II DSA with MFI >20,000 (data not shown); eight additional subjects with >1 DSA, had a DSA sum >20,000 MFI (data not shown). Finally, among the 80 subjects with class II DSA, 61 subjects had sufficient serum to test complement binding capacity. The majority (78.7%) tested positive, with mean C1q MFI >20,000; data not shown). Notably, no associations were identified between class II DSA parameters and clinical characteristics including age at transplant or study entry, interval since transplant, living or deceased donor recipient, or history of previous rejection (Supplementary Table 3B).
Associations between serological profiles, histological features, and C4d scores
Autoantibody parameters (quantitative IgG, ANA, ASMA, α-AT1R, and α-ETAR) in isolation did not show any association with either histological features or C4d scores. For analyses of class II DSA, we selected the sum of class II DSA MFI as the representative variable after testing positive/negative, maximum, and sum. A model including all serological variables confirmed the strong and dominant association between class II DSA MFI sum >20,000 and histological features as well as C4d scores (Supplementary Table 4). Compared to those with no class II DSA, those with class II DSA MFI sum >20,000 were at increased risk of higher Ishak fibrosis stage (OR 4.53; 95% CI 1.78–11.53; p=0.001), portal inflammation grade (OR 3.59; 95% CI 1.30–9.93; p=0.01), and C4d scores [portal capillary (OR 5.11; 95% CI 1.9813.20; p<0.001), sinusoidal (OR 4.40; 95% CI 1.49–12.98; p=0.007; total (OR 4.73; 95% CI 1.95–11.48; p<0.001)].
157 biopsies sort into three distinct histopathological clusters
An unsupervised hierarchical cluster analysis identified three clusters based on three histological features (Figure 3). Cluster 1 (n=34) was defined by portal inflammation with interface activity, often associated with variable degrees of fibrosis; cluster 2 (n=45) was characterized by significant Ishak and/or perivenular fibrosis but without interface activity; cluster 3 (n=78) were near normal, exhibiting neither interface activity nor significant fibrosis (Figure 3A). As evident in the constellation plot, cluster 1 clearly diverges from clusters 2 and 3 (Figure 3B). Given the reported association of DSA with chronic graft injury, we compared the prevalence of class II DSA among the clusters. When compared to cluster 2 and 3, a greater proportion of subjects in cluster 1 had class II DSA. The mean class II DSA MFI was higher in cluster 1 versus cluster 2 or 3. Moreover, a higher percentage of subjects in cluster 1, compared to clusters 2 and 3, had class II DSA MFI maximum >20,000 and class II DSA MFI sum >20,000 (Supplementary Figures 4A and 4B).
Figure 3: 157 biopsies divide into three histological clusters.

The three variables defining the clusters are Ishak fibrosis stage, interface activity and perivenular fibrosis.
(A) Three-dimensional bubble plot: Bubble size is proportional to the number of observations at that coordinate.
(B) Constellation plot: Each subject is represented by a color-outlined point. Line lengths represent distances between clusters and points. Axis scales are relative distance measures.
Quantitative determination of fibrosis, APCs, leukocytes, and APC: leukocyte pairings
Differences among the three histopathological clusters were next explored utilizing immunohistochemical and multiplex staining. Fibrosis severity, as quantified by both trichrome and SMA staining area, showed the expected trend among clusters (clusters 1 and 2 > cluster 3), but the differences were not statistically significant (data not shown). Cell counts within virtual antigenpresenting foci, which largely correspond to portal tracts (Methods; Figure 4A-C), readily distinguished the clusters. APCs (CD34-/CD45-/class II+), leukocytes (CD34-/CD45+(high)/class II+/−), and APCleukocyte pairings, defined as an APC within 5 microns of a leukocyte, were quantified and compared. The number of APCs was significantly higher in clusters 1 and 2 compared to cluster 3 (Figure 4D). Total leukocyte counts were distinctly higher in cluster 1 but similar in clusters 2 and 3 (Figure 4E). The number of APC-leukocyte pairings differed among clusters, being highest in cluster 1 and lowest in cluster 3 (Figure 4F). Analyses over the total biopsy area, as opposed to the virtual antigen-presenting foci, showed less significant differences among the clusters (data not shown).
Figure 4: Counts of antigen-presenting cells, leukocytes, and APC-leukocyte pairings in virtual antigenpresenting foci.

A) H&E section (left) and multiplex Qdot immunostaining of CD34/CD45/class II panel (right) illustrating a “virtual antigen-presenting foci” localized primarily to portal tracts (yellow circles)
B) High magnification (white box, Panel A) of an antigen-presenting focus within a portal tract illustrating CD34+ portal capillaries in the left panel (green), CD45+(high) leukocytes in the middle panel (teal), and a pairing of a CD34-/CD45-/class II+ APC (red) and a CD34-CD45+(high)/class II-(teal)/CD34-/CD45+(high)/ class II+ leukocyte (white) pairing in the right panel. The small white circle in the right panel identifies the APC-leukocyte pairing.
C) Statistical pairwise comparison of clusters for total APCs (CD34-/CD45-/class II+; red cells; upper panel), total leukocytes (CD34-/CD45+/class II +/−; white or teal cells; middle panel), and APCleukocyte pairings (lower panel).
The histopathological features defining the three clusters are associated with a distinct module of coexpressed genes
To further identify the biological underpinnings of the subclinical histological and immunohistochemical abnormalities, we conducted whole genome transcriptional analysis of the liver tissue samples and applied WGCNA to identify modules of co-expressed genes that were correlated with demographic, clinical, serological and histological traits of interest. Out of the thirty-three distinct modules identified, one module comprising 194 genes, which we will refer to as the “salmon” module (Figure 5A; black arrow) showed significant correlation with cluster assignment, class II DSA, interface activity, Ishak fibrosis stage, portal inflammation, perivenular fibrosis, perivenular inflammation, serum ALT and, importantly, was not influenced by clinical confounders such as recipient age or time after transplant. This module of 194 genes (Supplementary Table 5) was enriched in pathways related to cytokinecytokine receptors, chemokines, and allograft rejection, among others (Figure 5B). Moreover, 50 of the 194 genes had been previously described as being associated with T cell mediated rejection (TCMR) in microarray studies involving liver, kidney, lung, or heart transplantation (e.g. CXCL9, CXCL10, HLA-DOB, CD3E, GZMB, PRF1, CD74).33–35 The overall transcript levels of this selected gene module across the whole liver tissue microarray data set significantly correlated with the LAFSc, suggesting a potential pathogenic role of these genes in liver allograft fibrosis (Figure 5C).
Figure 5: Microarray transcriptional analysis of liver biopsy samples shows that cluster 1 is enriched in rejection-associated molecular pathways.

(A) Weighted gene co-expression network analysis of the liver transcriptome: X-axis: external traits of interest; Y-axis: 33 identified gene modules. The color intensity is proportional to the magnitude of the Pearson correlation coefficient. Asterisks denote statistical significance at p value <0.01. The black arrow signals the selected “salmon” 194-gene module (Supplementary Table 5).
(B) Gene-pathway association network visualizing the relationships between the top 10 KEGG pathways and core genes significantly enriched in the 194-gene “salmon” module. The size of the circled pathways reflects the associated p values of the terms: more significant pathways are larger.
(C) Scatter plot and best-fit line showing the correlation between the scaled sum of the normalized expression levels of the 194 genes of the “salmon” module and the LAFSc. Dots represent individual samples and their color the cluster assignment. r corresponds to the Pearson correlation coefficient.
(D) Quantile-quantile plots of expected versus observed scores comparing the distribution of microarray gene expression values for clusters 1, 2, and 3. Differentially expressed genes as computed by the SAM package deviate from the diagonal. Up-regulated versus down-regulated genes at FDR<5% are red open circles in the upper right versus green open circles in the lower left.
(E) CI plot displaying the mean and 95% CI of the activity (log2ratio) of each transplantation-related gene set of interest plotted and color-coded according to their FDR-corrected p values.
Transcriptional pathways involved in TCMR differentiate cluster 1 from clusters 2 and 3
To better understand the differences among the three histological clusters we conducted pairwise comparisons of their transcriptomes. Cluster 1 significantly differed from cluster 3 and, to a lesser degree, from cluster 2. In contrast, clusters 2 and 3 only showed minimal transcriptional differences (Figure 5D). These results and, in particular, the lack of significant differences in pro-inflammatory gene expression between clusters 2 and 3 were confirmed on a Nanostring platform (Supplementary Figure 5 and Supplementary Tables 6A–C).
To identify the pathways associated with the differential gene expression between cluster 1 and clusters 2 and 3, we conducted functional pairwise analysis employing the QuSAGE method on a set of transcriptional pathways known to be involved in allograft immunopathogenesis across a variety of clinical and animal transplant settings. As a relevant control, we also included a 13-gene signature previously described as highly specific for TCMR in stable liver recipients undergoing immunosuppression withdrawal (Supplementary Table 7).33 The liver rejection gene set was significantly associated with cluster 1 but not clusters 2 or 3. In addition, the following gene sets known to be involved in TCMR across a variety of clinical and experimental settings were also over-represented in cluster 1: GRIT1 (IFN-γ dependent rejection-associated transcripts), QCAT (cytotoxic T lymphocyte associated transcripts), TCB (T cell specific transcripts), and BAT (B cell specific transcripts). Cluster 1 was also significantly enriched in the IGT gene set (immunoglobulin associated transcripts) as compared to clusters 2 and 3, and in the MCAT gene set (mast cell associated transcripts) as compared to cluster 3, both of which are known be associated with allograft fibrosis but not with rejection.35 IGT was the only gene set over-represented in cluster 2 as compared to cluster 3 (Figure 5E).
We conducted a more extensive pathway analysis to better delineate the functional differences between cluster 2 and 3. The two clusters significantly differed in the over-representation of canonical pathways involved in fibrogenesis. In addition, cluster 2 was significantly enriched in a gene set specific for stellate cells whose expression has been associated with survival in non-transplanted patients with chronic liver disease36. In contrast, only minimal differences in inflammatory canonical pathways between the two clusters were noted (Supplementary Table 8).
To explore the impact of circulating class II DSA on the molecular profile of the liver allograft, we employed the Nanostring dataset to compute the liver gene expression differences between class II DSA positive versus negative individuals across the study cohort (Supplementary Table 9). The results greatly overlapped with the expression differences between cluster 1 and the other two clusters, likely reflecting the different prevalence of class II DSA among the three clusters. However, after fitting a linear model incorporating CD45+ cells to account for the imbalance in the degree of inflammatory infiltrate, we identified CCL18 and CXCL9 as transcriptional markers independently associated with class II DSA (FDR 0.0007 and 0.009, respectively).
ALT and MFI sum of class II DSA MFI >20,000 independently predict assignment into cluster 1
Finally, the mechanistic insight suggesting that subjects in cluster 1 are experiencing alloimmune graft injury motivated us to utilize logistic regression analyses to identify clinical and/or serological risk factors (Table 2). Univariable models identified deceased donor (OR 4.03; 95% CI 1.33–12.20; p=0.01), nonbiliary atresia transplant indication (OR 2.38; 95% CI 1.08–5.26; p=0.03), decreased time between transplant and eligibility biopsy (OR 0.88 per year increment; 95% CI 0.78–0.99; p=0.03) and increased ALT (OR 1.06 per 1 IU/L increment; 95% CI 1.02–1.11; p=0.009) as associated with assignment into cluster 1 versus clusters 2 and 3. ANA and ASMA positivity were not associated with cluster 1 assignment but both α-AT1R (OR 1.03; 95% CI 1.01–1.05; p=0.01) and α-ETAR antibodies (OR 1.03; 95% CI 1.00–1.05; p=0.02) were associated with cluster 1 assignment. Class II DSA presence (OR 2.90; 95% CI 1.14–7.34; p=0.02) was also associated with assignment into cluster 1. The association was stronger when class II DSA MFI sum was >20,000 (OR 4.49; 95% CI 1.67–12.14; p=0.003) and strongest when class II DSA maximum was >20,000 (OR 5.55; 95% CI 2.00–15.44; p=0.001). We selected to use the sum of class II DSA MFI as the class II DSA variable. The final multivariable model shows that two factors, ALT (OR 1.07 per U/L; 95% CI 1.02–1.13; p=0.01) and the sum of class II DSA MFI >20,000 (OR 5.11; 95% CI 1.82–14.41; p=0.002), were independently associated with assignment into cluster 1 versus 2 and 3 (Table 2).
Table 2:
Clinical and serological factors associated with assignment into cluster 1
| Characteristic | Reference | OR | 95% CI | P Value | ||
|---|---|---|---|---|---|---|
| Univariable | ||||||
| Donor | Age (per year) | 0.98 | 0.95–1.01 | 0.14 | ||
| Deceased | Living | 4.03 | 1.33–12.20 | 0.01 | ||
| Recipient | Female | Male | 0.47 | 0.21–1.04 | 0.06 | |
| Biliary Atresia | Other | 0.42 | 0.19–0.93 | 0.03 | ||
| Transplant | Whole graft | Partial graft | 1.89 | 0.87–4.08 | 0.11 | |
| Whole graft | Living graft | 4.06 | 1.29–12.77 | 0.02 | ||
| Deceased partial graft | 3.98 | 1.14–13.97 | 0.03 | |||
| Induction immunosuppression | None | 0.55 | 0.15–1.99 | 0.36 | ||
| Study Entry | Recipient age (per year) | 0.90 | 0.81–1.01 | 0.07 | ||
| Time since transplant (per year) | 0.88 | 0.78–0.99 | 0.03 | |||
| ALT (per U/L) | 1.06 | 1.02–1.11 | 0.009 | |||
| GGT (per U/L) | 1.02 | 0.97–1.06 | 0.51 | |||
| ANA ≥ 1:40 | Negative | 1.62 | 0.65–4.05 | 0.30 | ||
| ASMA = 1:80 | Negative | 0.98 | 0.10–9.15 | 0.99 | ||
| α-AT1R antibody (per U/mL) | 1.03 | 1.01–1.05 | 0.01 | |||
| α-ETAR antibody (per U/mL) | 1.03 | 1.00–1.05 | 0.02 | |||
| Class II DSA | Positive | Negative | 2.90 | 1.14–7.34 | 0.02 | |
| Maximum MFI ≤ 20,000 | No class II DSA | 1.32 | 0.41–4.24 | 0.64 | ||
| Maximum MFI > 20,000 | 5.55 | 2.00–15.44 | 0.001 | |||
| MFI sum ≤ 20,000 | No class II DSA | 1.36 | 0.40–4.64 | 0.63 | ||
| MFI sum > 20,000 | 4.49 | 1.67–12.14 | 0.003 | |||
| Multivariable | ||||||
| ALT (per U/L) | 1.07 | 1.02–1.13 | 0.01 | |||
| Class II DSA | MFI sum ≤ 20,000 | No class II DSA | 1.50 | 0.43–5.26 | 0.53 | |
| 5.11 | 1.82–14.41 | 0.002 | ||||
| MFI sum > 20,000 | ||||||
DISCUSSION
Long-term sustainability of allograft and patient health is the primary challenge facing the liver transplant community. This challenge is undoubtedly the lengthiest and steepest for pediatric recipients. To address this challenge, we conducted a cross-sectional study of well-characterized, stable liver transplant recipients who had consented to enter a trial of complete immunosuppression withdrawal. As hypothesized, we were able to define, among this clinically homogeneous cohort, three distinct histopathological clusters, differentiated by the presence and severity of interface activity and/or fibrosis. To explore potential mechanism(s) underlying the distinctions among the clusters, we employed an unsupervised systems biology approach to analyze the liver tissue transcriptional patterns associated with clinical, serological, and histological features. This strategy allowed an unbiased assessment of the important parameters influencing the expression profiles as well as the identification of potential confounders. A module of co-expressed genes dominated by transcripts strongly associated with rejection34, 35 was significantly correlated with class II DSA, interface activity, and fibrosis. Direct comparisons between clusters revealed that rejection-associated transcripts were predominantly increased in cluster 1, the cluster characterized by interface activity. This finding was confirmed at the functional pathway level: we showed that IFN-γ-regulated gene signatures known to be associated with TCMR in liver, kidney, and heart transplantation were significantly enriched.35, 37 The suggestion that interface activity may reflect subclinical rejection has implications for the optimal management of immunosuppression for this subset of patients that merits future testing. The approach is clearly different from efforts to withdraw immunosuppression for those without interface activity who ultimately participated in the iWITH trial. Our description of distinct histopathological phenotypes and provision of their associated molecular patterns is a necessary step towards the personalization of immunosuppression management necessary to simultaneously optimize patient and graft health and longevity.
After liver transplantation, it is understood that the inflammatory damage of TCMR is orchestrated by effector T cells engaging alloantigen-bearing APCs and parenchymal cells, preferentially in the portal areas.31 We found that the transcript levels of the set of 194 co-expressed genes directly and significantly correlated with the magnitude of leukocyte infiltration. Furthermore, liver biopsies from cluster 1 exhibited the highest number of APC-leukocyte pairings – leukocytes in close proximity to, and potentially interacting with, APCs. Although cluster 1 subjects also had the highest prevalence of class II DSA, we did not observe a significant enrichment in intra-graft endothelial or natural killer cell related gene signatures, which have been described as selective for antibody-mediated rejection (ABMR) in kidney and heart transplantation.38, 39 The lack of up-regulation of classic ABMR-specific signatures strongly suggests that endothelial damage is not the primary pathogenic feature of the inflammatory changes observed in cluster 1 biopsies but does not exclude the involvement of DSA in immunopathogenesis. First, liver, compared to kidney and heart, allografts respond differently to DSA. Moreover, the histological features of chronic ABMR in liver transplantation remain incompletely defined.31 As a result, liver-specific transcriptional signatures discriminating chronic ABMR from TCMR cannot be derived. Second, data from experimental animal models indicate that, in addition to their capacity to induce immunopathology by binding HLA molecules in the endothelium and activating complement and natural killer cells, alloantibodies can promote allograft rejection by an alternative mechanism that enhances the expansion and effector function of donor-specific T cells.40 In support of a link between humoral sensitization and anti-donor T cell mediated responses, recent data from kidney transplant patients with chronic ABMR41 demonstrate that the magnitude of indirectly-primed CD4+ T cell responses (i.e. T cell responses elicited by recipient APCs presenting peptide fragments of donor graft antigens) correlates with the progression of kidney allograft dysfunction. Third, our observation that class II DSA was associated with expression of T cell chemokines such as CCL8 and CXCL9, after adjusting for the magnitude of inflammation, suggests a contributory role of class II DSA to the development of liver T cell infiltration. Taken together, these results indicate that patients with subclinical histological abnormalities that include interface activity (cluster 1) constitute a distinct phenotype that recapitulates the molecular mechanisms described in allograft rejection. Late subclinical histopathological abnormalities are prevalent in many different solid organ allografts and contribute to long-term allograft structural decline.13, 19–23, 28, 29, 42–48 Whether, in liver transplantation, this is exclusively a T cell mediated process, or, more likely, a mixed process influenced by both humoral sensitization and T cell alloreactivity cannot as yet be determined.
In contrast to the interface activity characteristics of cluster 1, the pathogenesis of cluster 2 lesions, periportal and/or perivenular fibrosis without inflammation, remains more difficult to elucidate. There were only subtle transcriptional differences between clusters 2 and 3 in pro-inflammatory and rejection gene sets. Although cluster 2 compared to cluster 3 biopsies exhibited increased numbers of APCs and APC-leukocyte pairings, the differences were smaller than those between clusters 1 and 3. These small differences could reflect fibrotic changes or, alternatively, spatial or temporal sampling issues. Nevertheless, in sum, our data suggest that an active alloimmune response may not be the basis for the separation of cluster 2 from 3. Further research is necessary to determine the mechanism(s) underlying the “bland” fibrosis characteristic of cluster 2.
While this study is the product of a comprehensive mechanistic effort, there remain limitations. First and foremost, the liver biopsies were from a highly selected subset of pediatric liver transplant recipients. The prospective application of extensive inclusion/exclusion criteria specified by iWITH resulted in the enrollment of a homogeneous cohort characterized by clinical stability and consistently normal liver tests. Moreover, all subjects necessarily came from participating clinical sites, 12 large volume and mature pediatric liver transplant centers specifically selected for experience and infrastructure to conduct a complex clinical trial. Therefore, our study cohort can arguably be described as “clinically ideal” and not representative of the general population of pediatric liver transplant recipients. This context, however, may ironically increase the importance of our findings. It is highly likely that our study, underestimates the prevalence of cluster 1 and overestimates the prevalence of cluster 3 in the general population. Second, our study’s cross-sectional design does not shed any light on the evolution of the observed histopathological changes either prior to or, perhaps more importantly, since the time point studied. It is now critically important to determine if the necroinflammatory changes characteristic of cluster 1 translate into progressive fibrosis that can shorten allograft longevity. Our well-characterized cohort provides the foundation for a future longitudinal study based on sequential biospecimens. Third, the inclusion of deceased donor recipients lacking high resolution donor HLA typing data limited the fidelity of the DSA dataset. The deficits are mitigated, at least in part, by the overall harmony of our results with the literature regarding associations between class II DSA and chronic inflammatory allograft damage.13, 19–23, 28, 42–46
In summary, our data offers a plausible rationale for the chronic, inflammatory changes that have been repeatedly described but not explained in apparently stable, long-term pediatric liver allografts.12–18 We show, at the molecular level, interface activity connotes subclinical rejection. Our findings highlight that consistently normal results of liver tests may hide a spectrum of histopathology that can only be accurately exposed by tissue examination and support the necessity of liver biopsy to guide personalized immunosuppression decision-making.49 For patients whose biopsies harbor neither inflammation nor fibrosis, immunosuppression dose reduction may be reasonable, based on the consistently reported safety of attempted immunosuppression withdrawal.8, 9, 50 For patients whose biopsies show fibrosis in the absence of inflammation, our data does not support any recommendations. Lastly, for patients whose biopsies show interface hepatitis, our data indicates that dose reduction may be unwise. Although the intuitive response may be to escalate immunosuppression, data evidencing the benefit of this approach is lacking. Clearly, the next steps are to delineate the natural history of the histopathological phenotypes that we have described which will then inform the design and justify the testing of targeted interventions to optimize allograft health and longevity.
Supplementary Material
Supplementary Methods Routine Histology, C4d Scoring, Multiplex Quantum Dot (Qdot) Immunolabelling, and Automated Image Analysis
Supplementary Figure 1: Multiplex staining and image analysis (A) Example of CD34/Ki-67 (MIB-1)/SMA multiplex stain illustrating a small portal tract in the center with a proliferating SMA+/Ki-67 (MIB-1)+ putative periportal stellate cell shown in the lower right inset.
(B) Color deconvolution using pixel area morphometry was used to quantify liver fibrosis, shown over-laid on the original image.
(C) Correlation between LAFSc and Trichrome Area/Total biopsy area.
Supplementary Figures 2A and B: C4d scores of 157 eligibility biopsies. (A) Heat map and (B) frequencies of C4d scores.
Supplementary Figure 3: Autoantibody profiles of enrolled subjects (A) ANA and ASMA (n=133). Numbers and percentages are shown. (B) α-AT1R and α-ETAR antibodies (n=119). The threshold defining positive is 17 U/ml.
Supplementary Figure 4: Comparison of class II DSA MFI by cluster The bar graph shows the proportion of each cluster for each class II DSA MFI value. The numbers shown are absolute numbers.
(A) Maximum detected class II DSA MFI
(B) Sum of class II DSA MFI
Supplementary Figure 5: Nanostring liver biopsy differential gene expression between clusters 1, 2, and 3 Expression profiles of the 109 genes differentially expressed in at least one comparison at p-value <0.05 and fold change >1.5 are expressed as a matrix view of gene expression data where rows represent genes and columns represent hybridized samples. The intensity of each color denotes the standardized ratio between each value and the average expression of each gene across all samples. Red or green colored pixels correspond to an increased or decreased abundance of the mRNA in the indicated sample
ACKNOWLEDGEMENTS
The authors would like to extend their appreciation towards all of the patients and their families that participated in this study. We wish to thank the following: Sharon Blaschka, Crystal Lala, Kalpana Harish, Allan Kirk, Allison Priore, iWITH co-investigators and research coordinators, and the staff at all iWITH core laboratories for their contributions. We acknowledge the support received by the National Institutes of Health’s (NIH) National Center for Advancing Translational Sciences from the following institutions: University of California, San Francisco, San Francisco, CA; Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL; Columbia University, New York, NY; Cincinnati Children’s Hospital, Cincinnati, OH; Children’s Hospital Colorado, Denver, CO; The Children’s Hospital of Philadelphia, Philadelphia, PA; St. Louis Children’s Hospital, St. Louis, MO and Emory University School of Medicine, Atlanta, GA.
FUNDING
This research was primarily supported by U01-AI-100807 awarded by the National Institute of Allergy and Infectious Diseases (NIAID) and the National Institute for Diabetes and Digestive and Kidney Diseases. In addition, the research was also performed as a project of the Clinical Trials in Organ Transplantation in Children (U01-AI-104347), a collaborative clinical research project headquartered at NIAID and as a project of the Immune Tolerance Network (UM1AI109565), an international clinical research consortium headquartered at the Benaroya Research Institute and supported by NIAID. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Abbreviations used in this manuscript:
- ABMR
antibody mediated rejection
- ALT
alanine aminotransferase
- ANA
anti-nuclear antibody
- ASMA
anti-smooth muscle antibody
- APC
antigen presenting cell
- AT1R
angiotensin 2 type 1 receptor
- BAT
B cell specific transcripts
- DSA
donor specific HLA antibody
- ETAR
endothelin
- FDR
false discovery rate
- GGT
gamma-glutamyl transferase
- GRIT1 IFN-γ
dependent rejection-associated transcripts
- IgG
immunoglobulin
- IGT
immunoglobulin associated transcripts
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LAFSc
liver allograft fibrosis score
- LIMMA
linear models for microarray analyses
- MCAT
mast cell associated transcripts
- MFI
mean fluorescence intensity
- QCAT
cytotoxic T lymphocyte associated transcripts
- Qdot
quantum dot
- SAM
significant analysis of microarray
- SD
standard deviation
- SMA
smooth muscle actin
- TCB
T cell specific transcripts
- TCMR
T cell mediated rejection
- WGCNA
whole genome correlated network analysis
- WSI
whole slide images
Footnotes
CONFLICTS OF INTEREST
A.M.J. is on the speakers’ bureau and receives honoraria from One Lambda. The authors disclose no other conflicts
| Concept Design | Data Acquisition | Analysis/Data Interpretation | Manuscript Drafting | Critical Revision | Statistical Analysis | Obtained Funding | Technical Support | Material Support | Study Supervision | |
|---|---|---|---|---|---|---|---|---|---|---|
| Feng | X | X | X | X | X | X | X | X | ||
| Bucuvalas | X | X | X | X | X | X | X | X | ||
| Demetris | X | X | X | X | X | X | ||||
| Burrell | X | X | X | X | ||||||
| Spain | X | X | X | X | X | |||||
| Kanaparthi | X | X | X | X | X | |||||
| Magee | X | X | X | X | X | |||||
| Ikle | X | X | X | X | X | |||||
| Lesniak | X | X | X | X | X | |||||
| Lozano | X | X | X | X | X | |||||
| Alonso | X | X | X | |||||||
| Bray | X | X | X | |||||||
| Bridges | X | X | X | |||||||
| Doo | X | X | X | |||||||
| Gebel | X | X | X | |||||||
| Gupta | X | X | X | |||||||
| Himes | X | X | X | |||||||
| Jackson | X | X | X | X | X | |||||
| Lobritto | X | X | X | |||||||
| Mazariegos | X | X | X | X | ||||||
| Ng | X | X | X | |||||||
| Rand | X | X | X | |||||||
| Sherker | X | X | X | |||||||
| Sundaram | X | X | X | |||||||
| Turmelle | X | X | X | |||||||
| Sanchez-Fueyo | X | X | X | X | X | X |
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Campbell KM, Yazigi N, Ryckman FC, et al. High prevalence of renal dysfunction in long-term survivors after pediatric liver transplantation. J Pediatr 2006;148:475–80. [DOI] [PubMed] [Google Scholar]
- 2.Hathout E, Alonso E, Anand R, et al. Post-transplant diabetes mellitus in pediatric liver transplantation. Pediatr Transplant 2009;13:599–605. [DOI] [PubMed] [Google Scholar]
- 3.Shepherd RW, Turmelle Y, Nadler M, et al. Risk factors for rejection and infection in pediatric liver transplantation. Am J Transplant 2008;8:396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soltys KA, Mazariegos GV, Squires RH, et al. Late graft loss or death in pediatric liver transplantation: an analysis of the SPLIT database. Am J Transplant 2007;7:2165–71. [DOI] [PubMed] [Google Scholar]
- 5.Yanik EL SJ, Shiels MS, et al. Cancer Risk After Pediatric Solid Organ Transplantation. Pediatrics 2017;5:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ng VL, Alonso EM, Bucuvalas JC, et al. Health status of children alive 10 years after pediatric liver transplantation performed in the US and Canada: report of the studies of pediatric liver transplantation experience. J Pediatr 2012;160:820–6 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laryea M, Watt KD, Molinari M, et al. Metabolic syndrome in liver transplant recipients: prevalence and association with major vascular events. Liver Transpl 2007;13:1109–14. [DOI] [PubMed] [Google Scholar]
- 8.Benitez C, Londono MC, Miquel R, et al. Prospective multicenter clinical trial of immunosuppressive drug withdrawal in stable adult liver transplant recipients. Hepatology 2013;58:1824–35. [DOI] [PubMed] [Google Scholar]
- 9.Feng S, Ekong UD, Lobritto SJ, et al. Complete immunosuppression withdrawal and subsequent allograft function among pediatric recipients of parental living donor liver transplants. JAMA 2012;307:283–93. [DOI] [PubMed] [Google Scholar]
- 10.Kelly D, Verkade HJ, Rajanayagam J, et al. Late graft hepatitis and fibrosis in pediatric liver allograft recipients: Current concepts and future developments. Liver Transpl 2016;22:1593–1602. [DOI] [PubMed] [Google Scholar]
- 11.Feng S, Bucuvalas J. Tolerance after liver transplantation: Where are we? Liver Transpl 2017;23:1601–1614. [DOI] [PubMed] [Google Scholar]
- 12.Ekong UD, Melin-Aldana H, Seshadri R, et al. Graft histology characteristics in long-term survivors of pediatric liver transplantation. Liver Transpl 2008;14:1582–7. [DOI] [PubMed] [Google Scholar]
- 13.Evans HM, Kelly DA, McKiernan PJ, et al. Progressive histological damage in liver allografts following pediatric liver transplantation. Hepatology 2006;43:1109–17. [DOI] [PubMed] [Google Scholar]
- 14.Herzog D, Soglio DB, Fournet JC, et al. Interface hepatitis is associated with a high incidence of late graft fibrosis in a group of tightly monitored pediatric orthotopic liver transplantation patients. Liver Transpl 2008;14:946–55. [DOI] [PubMed] [Google Scholar]
- 15.Scheenstra R, Peeters PM, Verkade HJ, et al. Graft fibrosis after pediatric liver transplantation: ten years of follow-up. Hepatology 2009;49:880–6. [DOI] [PubMed] [Google Scholar]
- 16.Venturi C, Sempoux C, Quinones JA, et al. Dynamics of allograft fibrosis in pediatric liver transplantation. Am J Transplant 2014;14:1648–56. [DOI] [PubMed] [Google Scholar]
- 17.Syn WK, Nightingale P, Gunson B, et al. Natural history of unexplained chronic hepatitis after liver transplantation. Liver Transpl 2007;13:984–9. [DOI] [PubMed] [Google Scholar]
- 18.Briem-Richter A, Ganschow R, Sornsakrin M, et al. Liver allograft pathology in healthy pediatric liver transplant recipients. Pediatr Transplant 2013;17:543–9. [DOI] [PubMed] [Google Scholar]
- 19.Pongpaibul A, Venick RS, McDiarmid SV, et al. Histopathology of de novo autoimmune hepatitis. Liver Transpl 2012;18:811–8. [DOI] [PubMed] [Google Scholar]
- 20.Ohe H, Uchida Y, Yoshizawa A, et al. Association of anti-human leukocyte antigen and antiangiotensin II type 1 receptor antibodies with liver allograft fibrosis after immunosuppression withdrawal. Transplantation 2014;98:1105–11. [DOI] [PubMed] [Google Scholar]
- 21.Venturi C, Sempoux C, Bueno J, et al. Novel histologic scoring system for long-term allograft fibrosis after liver transplantation in children. Am J Transplant 2012;12:2986–96. [DOI] [PubMed] [Google Scholar]
- 22.Miyagawa-Hayashino A, Yoshizawa A, Uchida Y, et al. Progressive graft fibrosis and donorspecific human leukocyte antigen antibodies in pediatric late liver allografts. Liver Transpl 2012;18:1333–42. [DOI] [PubMed] [Google Scholar]
- 23.Wozniak LJ, Hickey MJ, Venick RS, et al. Donor-specific HLA Antibodies Are Associated With Late Allograft Dysfunction After Pediatric Liver Transplantation. Transplantation 2015;99:1416–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Leary JG, Demetris AJ, Philippe A, et al. Non-HLA Antibodies Impact on C4d Staining, Stellate Cell Activation and Fibrosis in Liver Allografts. Transplantation 2017;101:2399–2409. [DOI] [PubMed] [Google Scholar]
- 25.Isse K, Lesniak A, Grama K, et al. Preexisting epithelial diversity in normal human livers: a tissuetethered cytometric analysis in portal/periportal epithelial cells. Hepatology 2013;57:1632–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 2001;98:5116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Leary JG, Demetris AJ, Friedman LS, et al. The role of donor-specific HLA alloantibodies in liver transplantation. Am J Transplant 2014;14:779–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grabhorn E, Binder TM, Obrecht D, et al. Long-term Clinical Relevance of De Novo Donor-Specific Antibodies After Pediatric Liver Transplantation. Transplantation 2015;99:1876–81. [DOI] [PubMed] [Google Scholar]
- 30.Taner T, Stegall MD, Heimbach JK. Antibody-mediated rejection in liver transplantation: current controversies and future directions. Liver Transpl 2014;20:514–27. [DOI] [PubMed] [Google Scholar]
- 31.Demetris AJ, Bellamy C, Hubscher SG, et al. 2016 Comprehensive Update of the Banff Working Group on Liver Allograft Pathology: Introduction of Antibody-Mediated Rejection. Am J Transplant 2016;16:2816–2835. [DOI] [PubMed] [Google Scholar]
- 32.Troxell ML, Higgins JP, Kambham N. Evaluation of C4d staining in liver and small intestine allografts. Arch Pathol Lab Med 2006;130:1489–96. [DOI] [PubMed] [Google Scholar]
- 33.Bonaccorsi-Riani E, Pennycuick A, Londono MC, et al. Molecular Characterization of Acute Cellular Rejection Occurring During Intentional Immunosuppression Withdrawal in Liver Transplantation. Am J Transplant 2016;16:484–96. [DOI] [PubMed] [Google Scholar]
- 34.Spivey TL, Uccellini L, Ascierto ML, et al. Gene expression profiling in acute allograft rejection: challenging the immunologic constant of rejection hypothesis. J Transl Med 2011;9:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Halloran PF, Venner JM, Madill-Thomsen KS, et al. Review: The transcripts associated with organ allograft rejection. Am J Transplant 2017. [DOI] [PubMed] [Google Scholar]
- 36.Zhang DY, Goossens N, Guo J, et al. A hepatic stellate cell gene expression signature associated with outcomes in hepatitis C cirrhosis and hepatocellular carcinoma after curative resection. Gut 2016;65:1754–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pham MX, Teuteberg JJ, Kfoury AG, et al. Gene-expression profiling for rejection surveillance after cardiac transplantation. N Engl J Med 2010;362:1890–900. [DOI] [PubMed] [Google Scholar]
- 38.Haas M The Revised (2013) Banff Classification for Antibody-Mediated Rejection of Renal Allografts: Update, Difficulties, and Future Considerations. Am J Transplant 2016;16:1352–7. [DOI] [PubMed] [Google Scholar]
- 39.Haas M Simultaneous liver-kidney transplantation: shifting renal allograft gene expression from inflammation toward preservation. Kidney Int 2017;91:1010–1013. [DOI] [PubMed] [Google Scholar]
- 40.Burns AM, Chong AS. Alloantibodies prevent the induction of transplantation tolerance by enhancing alloreactive T cell priming. J Immunol 2011;186:214–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shiu KY, McLaughlin L, Rebollo-Mesa I, et al. Graft dysfunction in chronic antibody-mediated rejection correlates with B-cell-dependent indirect antidonor alloresponses and autocrine regulation of interferon-gamma production by Th1 cells. Kidney Int 2017;91:477–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaneku H, O’Leary JG, Banuelos N, et al. De novo donor-specific HLA antibodies decrease patient and graft survival in liver transplant recipients. Am J Transplant 2013;13:1541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Leary JG, Kaneku H, Banuelos N, et al. Impact of IgG3 subclass and C1q-fixing donor-specific HLA alloantibodies on rejection and survival in liver transplantation. Am J Transplant 2015;15:1003–13. [DOI] [PubMed] [Google Scholar]
- 44.Loupy A, Lefaucheur C, Vernerey D, et al. Complement-binding anti-HLA antibodies and kidneyallograft survival. N Engl J Med 2013;369:1215–26. [DOI] [PubMed] [Google Scholar]
- 45.Wiebe C, Gibson IW, Blydt-Hansen TD, et al. Evolution and clinical pathologic correlations of de novo donor-specific HLA antibody post kidney transplant. Am J Transplant 2012;12:1157–67. [DOI] [PubMed] [Google Scholar]
- 46.Chih S, Chruscinski A, Ross HJ, et al. Antibody-mediated rejection: an evolving entity in heart transplantation. J Transplant 2012;2012:210210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Leary JG, Kaneku H, Susskind BM, et al. High mean fluorescence intensity donor-specific anti-HLA antibodies associated with chronic rejection Postliver transplant. Am J Transplant 2011;11:1868–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaneku H, O’Leary JG, Taniguchi M, et al. Donor-specific human leukocyte antigen antibodies of the immunoglobulin G3 subclass are associated with chronic rejection and graft loss after liver transplantation. Liver Transpl 2012;18:984–92. [DOI] [PubMed] [Google Scholar]
- 49.Banff Working Group on Liver Allograft P. Importance of liver biopsy findings in immunosuppression management: biopsy monitoring and working criteria for patients with operational tolerance. Liver Transpl 2012;18:1154–70. [DOI] [PubMed] [Google Scholar]
- 50.Feng S, Demetris AJ, Spain KM, et al. Five-year histological and serological follow-up of operationally tolerant pediatric liver transplant recipients enrolled in WISP-R. Hepatology 2017;65:647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods Routine Histology, C4d Scoring, Multiplex Quantum Dot (Qdot) Immunolabelling, and Automated Image Analysis
Supplementary Figure 1: Multiplex staining and image analysis (A) Example of CD34/Ki-67 (MIB-1)/SMA multiplex stain illustrating a small portal tract in the center with a proliferating SMA+/Ki-67 (MIB-1)+ putative periportal stellate cell shown in the lower right inset.
(B) Color deconvolution using pixel area morphometry was used to quantify liver fibrosis, shown over-laid on the original image.
(C) Correlation between LAFSc and Trichrome Area/Total biopsy area.
Supplementary Figures 2A and B: C4d scores of 157 eligibility biopsies. (A) Heat map and (B) frequencies of C4d scores.
Supplementary Figure 3: Autoantibody profiles of enrolled subjects (A) ANA and ASMA (n=133). Numbers and percentages are shown. (B) α-AT1R and α-ETAR antibodies (n=119). The threshold defining positive is 17 U/ml.
Supplementary Figure 4: Comparison of class II DSA MFI by cluster The bar graph shows the proportion of each cluster for each class II DSA MFI value. The numbers shown are absolute numbers.
(A) Maximum detected class II DSA MFI
(B) Sum of class II DSA MFI
Supplementary Figure 5: Nanostring liver biopsy differential gene expression between clusters 1, 2, and 3 Expression profiles of the 109 genes differentially expressed in at least one comparison at p-value <0.05 and fold change >1.5 are expressed as a matrix view of gene expression data where rows represent genes and columns represent hybridized samples. The intensity of each color denotes the standardized ratio between each value and the average expression of each gene across all samples. Red or green colored pixels correspond to an increased or decreased abundance of the mRNA in the indicated sample