

Abstract
T cells play a crucial role in the adaptive immune system, and their maturation process is tightly regulated. Adenosine deaminase acting on RNA 1 (ADAR1) is the enzyme responsible for adenosine‐to‐inosine RNA editing in dsRNAs, and loss of ADAR1 activates the innate immune sensing response via melanoma differentiation‐associated protein 5 (MDA5), which interprets unedited dsRNA as non‐self. Although ADAR1 is highly expressed in the thymus, its role in the adaptive immune system, especially in T cells, remains elusive. Here, we demonstrate that T cell‐specific deletion of Adar1 in mice causes abnormal thymic T cell maturation including impaired negative selection and autoimmunity such as spontaneous colitis. This is caused by excessive expression of interferon‐stimulated genes, which reduces T cell receptor (TCR) signal transduction, due to a failure of RNA editing in ADAR1‐deficient thymocytes. Intriguingly, concurrent deletion of MDA5 restores thymocyte maturation and prevents colitis. These findings suggest that prevention of MDA5 sensing of endogenous dsRNA by ADAR1‐mediated RNA editing is required for preventing both innate immune responses and T cell‐mediated autoimmunity.
Keywords: MDA5, negative selection, RNA editing, spontaneous colitis, T cell maturation
Subject Categories: Immunology, RNA Biology
Introduction
Adenosine (A)‐to‐inosine (I) RNA editing is the most common RNA modification in mammals 1. Adenosine deaminase acting on RNA 1 (ADAR1) is one of the enzymes responsible for this type of RNA editing and is composed of two isoforms, the long p150 and short p110, which arise through alternative splicing or the use of different promoters 2, 3. The ADAR1 p150 isoform, which is induced as part of the type I interferon (IFN) response, is localized predominantly in the cytoplasm, whereas the constitutively expressed ADAR1 p110 isoform is present in the nucleus. Given that ADARs recognize double‐stranded RNAs (dsRNAs) as targets, most endogenous editing sites have been identified in repetitive elements such as short‐interspersed nuclear elements (SINEs) 4. Mutations in the ADAR1 gene cause Aicardi–Goutières syndrome (AGS), a severe early‐onset autoimmune disease that mimics infection with the aberrant production of type I IFN, and which shares features with systemic lupus erythematosus (SLE) 5. Furthermore, Adar1 knockout (KO) mice (A1 −/− mice), Adar1 p150‐specific KO mice, and Adar1 knock‐in (KI) mice that harbor the editing‐inactive E861A point mutation (A1 KI/KI mice) are all embryonic lethal and show aberrant activation of the type I IFN signaling pathway 6, 7, 8. Of note, recent studies have demonstrated that the concurrent deletion of either melanoma differentiation‐associated protein 5 (MDA5; encoded by Ifih1) or the downstream mitochondrial antiviral signaling protein (MAVS) rescued this embryonic lethality and normalized the type I IFN signaling pathway 8, 9, 10. MDA5 is an essential initiator of the innate immune response through sensing viral dsRNA in the cytoplasm upon infection, and the subsequent aggregation of MAVS leads to the transcription of numerous type I IFN‐stimulated genes (ISGs) 11, 12. Consequently, it has been suggested that RNA editing mediated by ADAR1, especially the p150 isoform, inhibits activation of the innate immune system by preventing MDA5 sensing endogenous dsRNAs as non‐self and subsequently stimulating the expression of type I ISGs 8, 9, 10.
Given that ADAR1 is essential for development, the complete or cell‐specific KO of Adar1 resulted in many defects including the loss of embryonic fetal liver hematopoietic cells, a reduced number of B cells in the bone marrow and spleen, and widespread apoptosis 10, 13, 14, 15. However, the number of T cells in the spleen of Adar1/Mavs double‐KO mice was preserved 10. Therefore, the role of ADAR1‐mediated RNA editing in T cells remains unclear.
The adaptive immune system is essential for host defense against pathogens. It is mediated by T and B cells that develop sequentially from progenitor cells to express a diverse repertoire of antigen‐specific receptors for the recognition and elimination of pathogens 16. The B cell lineage matures within the bone marrow. In contrast, T cell progenitors, which originate from fetal liver and adult bone marrow, migrate to the thymus and undergo three stages of maturation that are defined by the expression of CD4 and CD8. The initial double‐negative (DN; CD4−CD8−) stage progresses through the double‐positive (DP; CD4+CD8+) stage to either the CD4+CD8− single‐positive (4SP) or CD4−CD8+ single‐positive (8SP) stage 17. During these maturation stages, thymocytes bearing T cell receptors (TCRs) that recognize self‐peptides displayed by major histocompatibility complex (MHC) molecules with moderate affinity receive a survival signal (positive selection), whereas those with high affinity are eliminated to induce self‐tolerance (negative selection) 18. Then, naïve T cells traffick to secondary lymphoid organs such as the lymph nodes and spleen, where antigen presentation activates naïve T cells via engagement of the TCR and co‐stimulatory receptor CD28, leading to proliferation and differentiation into effector T (Teff) cells such as T helper 1 (Th1) and Th17 cells 19. Finally, these effector cells migrate into extra‐lymphoid tissues such as the skin, lungs, and intestines for host defense against pathogens 20. In contrast, regulatory T (Treg) cells, which are generated in the thymus as functionally mature T cells or differentiated from naïve T cells in peripheral tissues, are indispensable for the suppression of excessive immune responses to pathogens and maintenance of unresponsiveness to self‐antigens 21. Given that these processes of T cell maturation are tightly regulated, impairment of a certain process, such as negative selection, can trigger for autoimmune diseases, which can be accompanied by the uncontrolled differentiation and activation of Th1 and Th17 cells 22, 23, 24, 25. However, the mechanisms that underlie T cell maturation remain largely unknown.
In this study, we report that ADAR1‐mediated RNA editing regulates thymic T cell maturation, which includes negative selection. We found that ADAR1 is highly expressed in the mouse thymus and its expression is upregulated during T cell maturation, especially at the 4SP stage. The CD4+ T cell‐specific deletion of Adar1 in mice reduced the populations of 4SP and 8SP thymocytes accompanied with the impaired selection of T cells, which led to the induction of autoimmunity, such as spontaneous colitis with the accumulation of Th1 and Th17 cells in the lamina propria. These abnormalities were caused by excessive expression of ISGs, leading to reduced TCR signal transduction, via aberrant activation of the MDA5 pathway caused by a failure to upregulate RNA editing. Therefore, abnormal thymic T cell maturation and spontaneous colitis were ameliorated by the concurrent deletion of MDA5. These findings indicate that prevention of the sensing of endogenous dsRNAs by MDA5 by ADAR1‐mediated RNA editing is required for appropriate thymic T cell maturation including negative selection to avoid the induction of T cell‐mediated autoimmunity.
Results
ADAR1 is abundant in the mouse thymus, and its expression is upregulated during thymic T cell maturation
We evaluated the protein levels of ADAR1 and ADAR2, the other enzymatically active member of the ADAR family in mammals, in various organs of an adult wild‐type (WT) mouse. In the brain, ADAR1 p110 and ADAR2 were highly expressed, but ADAR1 p150 was barely detectable (Fig 1A). In contrast, we found that ADAR1 p150 was especially abundant in the thymus and spleen, although the expression level of ADAR2 was quite low, which suggests a potential role of ADAR1 in T cells. Therefore, to examine whether ADAR1 transcript and isoform expression is dynamic during thymic T cell maturation, we quantified the expression level of ADAR1 mRNA at the three major stages of maturation. The level of ADAR1 p150 mRNA, as well as that of total ADAR1 mRNA, increased approximately twofold during the transition from DN to DP thymocytes (Fig 1B and C). Subsequently, ADAR1 expression was upregulated further from the DP stage to the 4SP stage (Fig 1B and C), whereas no increase was observed in 8SP thymocytes. In accordance, the amount of ADAR1 p150 protein, but not ADAR1 p110 protein, was significantly increased at the 4SP stage (Fig EV1A). These findings suggest that the expression of ADAR1 is regulated in a maturation stage‐dependent manner. We observed a similar upregulation of ADAR2 mRNA from the DP stage to the 4SP stage, although its expression level was low (Fig EV1B). Next, we examined the mRNA expression level of various type I ISGs including Ifih1 mRNA, which encodes MDA5, at each stage. Although the developmental pattern of expression differed slightly among ISGs, in general the mRNA expression was highly upregulated during the transition from the DP stage to the 4SP stage (Fig 1D). Ifna1 and Ifnb1 mRNA, which respectively encode IFNα and IFNβ, were not detected at any stage by quantitative RT–PCR (qRT–PCR). Taken together, these results suggest that the expression of ADAR1 and ISGs, including MDA5, is upregulated in a stage‐dependent manner during thymic T cell maturation, especially at the 4SP stage.
Figure 1. Stage‐dependent upregulation of the expression of ADAR1 and interferon‐stimulated genes during thymic T cell maturation.
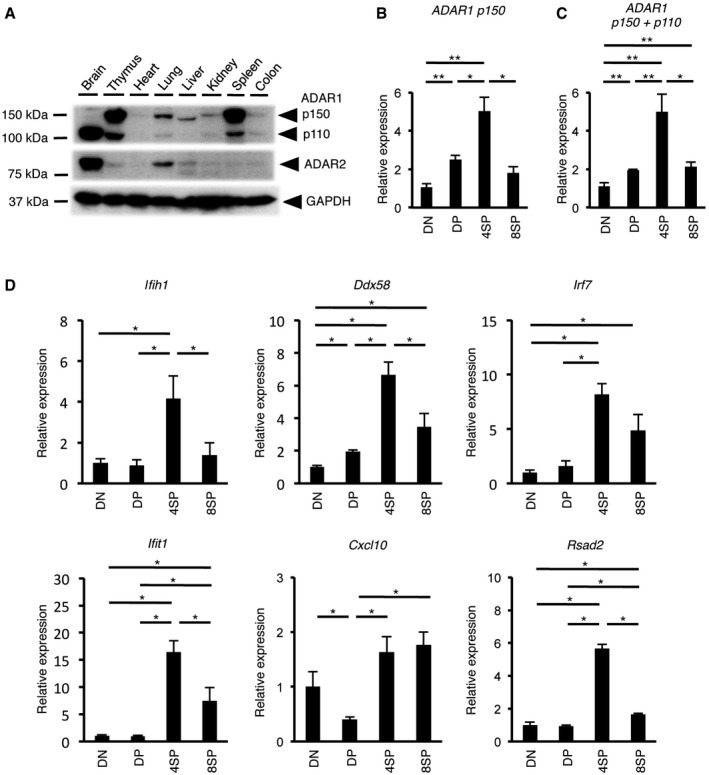
-
AImmunoblot analysis of ADAR1 p110, ADAR1 p150, and ADAR2 expression in various organs isolated from wild‐type (WT) mice. The expression of GAPDH is shown as a reference.
-
B, CThe relative expression of ADAR1 p150 mRNA (B) and total ADAR1 mRNA (C) in sorted DN, DP, 4SP, and 8SP thymocytes isolated from WT mice. The relative expression of ADAR1 p150 mRNA (B) and total ADAR1 mRNA (C) was normalized to the level of expression in DN thymocytes (DN values set to 1). Values represent the relative gene expression normalized to GAPDH mRNA and are displayed as the mean ± SEM (n = 5; Mann–Whitney U‐test, *P < 0.05, **P < 0.01).
-
DThe relative expression of type I interferon‐stimulated genes (ISGs) in sorted DN, DP, 4SP, and 8SP thymocytes isolated from WT mice. Values represent the relative gene expression normalized to GAPDH mRNA and are displayed as the mean ± SEM (n = 3; Mann–Whitney U‐test, *P < 0.05). The relative expression of ISGs was normalized against the level of expression in DN thymocytes (DN values set to 1).
Figure EV1. The expression of ADARs during thymic T cell maturation. Data related to Fig 1 .
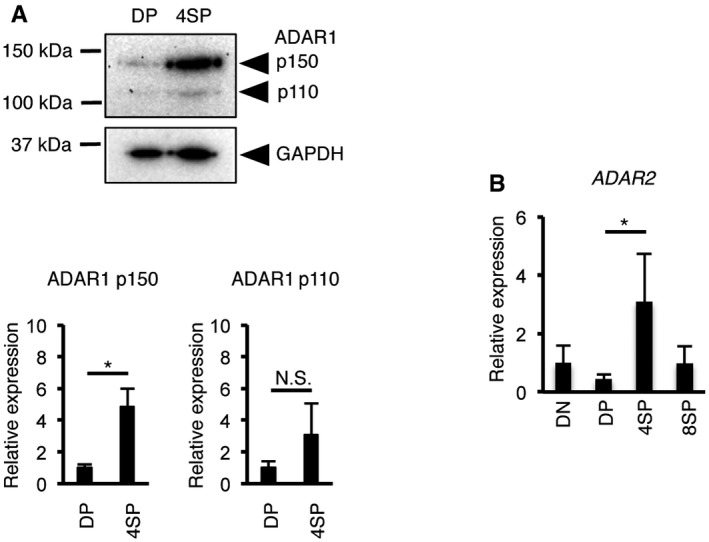
- Immunoblot analysis of ADAR1 expression in DP and 4SP thymocytes isolated from wild‐type (WT) mice. Representative images are shown (upper panel). The band intensity of each ADAR1 isoform was normalized to that of GAPDH and is displayed as the mean ± SEM (n = 3; Mann–Whitney U‐test, *P < 0.05) (lower panel). The mean value for the relative expression of ADAR1 in DP thymocytes was set as 1.
- The relative expression of ADAR2 mRNA in sorted double‐negative (DN), double‐positive (DP), CD4+ single‐positive (4SP), and CD8+ single‐positive (8SP) thymocytes isolated from wild‐type (WT) mice. The relative expression of ADAR2 mRNA was normalized to the level of expression in DN thymocytes (DN values set to 1). Values represent the relative gene expression normalized to GAPDH mRNA and are displayed as the mean ± SEM (n = 3; Mann–Whitney U‐test, *P < 0.05).
T cell‐specific deletion of ADAR1 leads to aberrant thymic T cell maturation
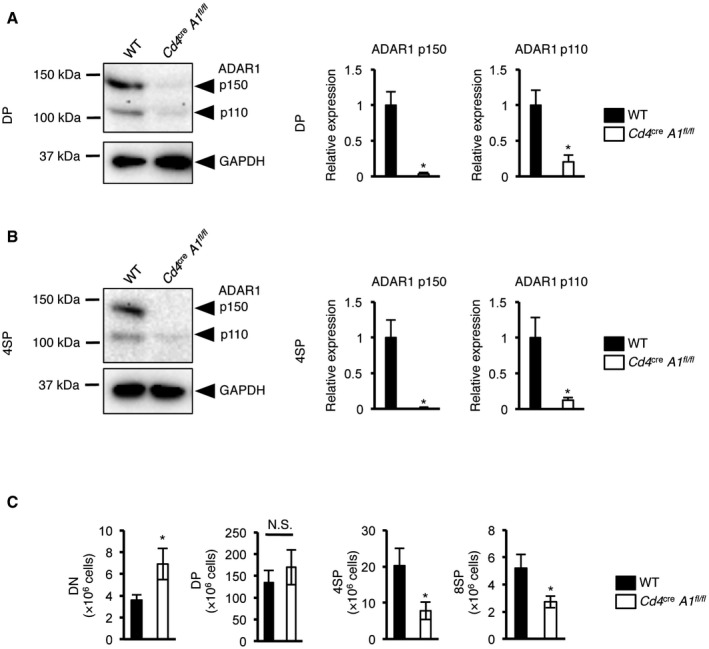
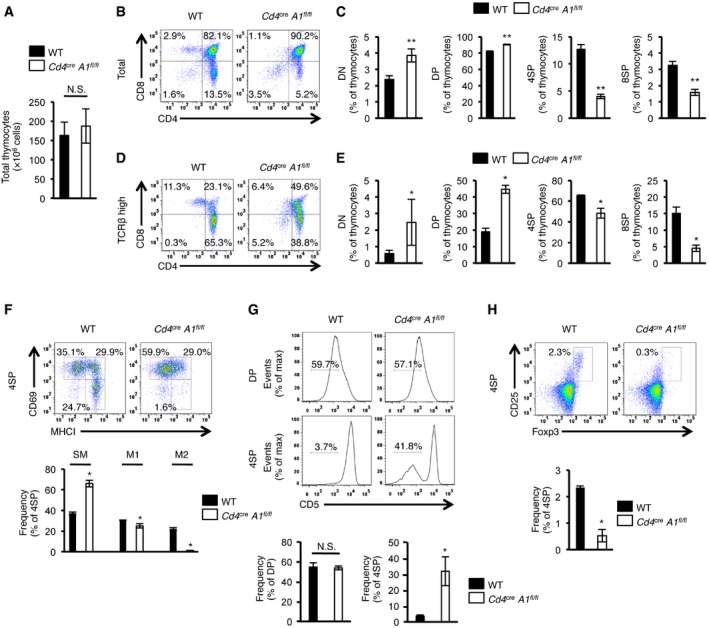
To investigate the biological significance of ADAR1‐mediated RNA editing in T cell maturation, we crossed Adar1 flox/flox (A1 fl/fl) mice with Cd4 cre mice, in which Cre recombinase is expressed from the DP stage onward under the control of the T cell‐specific Cd4 promoter 26 to create mice in which ADAR1 is specifically knocked out in CD4+ T cells (Cd4 cre A1 fl/fl mice; Fig EV2A and B). In Cd4 cre A1 fl/fl mice, although there was no significant difference in the total number of thymocytes, the proportions of 4SP and 8SP thymocytes were significantly decreased, whereas DN and DP thymocytes were slightly increased (Figs 2A–C and EV2C). These trends were also observed even when the analyzed targets were restricted to thymocytes with a high expression of TCRβ (Fig 2D and E). Then, to define the late stage of T cell maturation in Cd4 cre A1 fl/fl mice, we divided 4SP thymocytes further into three populations based on CD69 and MHC‐I expression: semi‐mature (SM; CD69+MHC‐I−), mature1 (M1; CD69+MHC‐I+), and mature2 (M2; CD69−MHC‐I+), as previously described 27. This analysis demonstrated that ADAR1‐deficient SM thymocytes were significantly accumulated and consequently the proportions of M1 and M2 thymocytes were reduced (Fig 2F), suggesting that proliferation during the late stage of T cell maturation is incompetent in Cd4 cre A1 fl/fl mice. We also observed that ADAR1‐deficient 4SP thymocytes, but not DP thymocytes, exhibited lower expression of CD5 (Fig 2G), suggesting that the TCR signal strength is impaired in ADAR1‐deficient 4SP thymocytes 28. To support this finding, we analyzed the proportion of thymic CD4+Foxp3+ Treg cells, which are critical for the maintenance of central immune tolerance and which require potent TCR signaling intensity for their generation 29. As expected, thymic Treg cells were barely detectable in Cd4 cre A1 fl/fl mice (Fig 2H). Collectively, these results suggest that ADAR1 deficiency in T cells leads to severe development defects, especially at the late stage of thymic T cell maturation, with unresponsive signatures to TCR activation.
Figure EV2. The protein level of ADAR1 in ADAR1‐deficient thymocytes. Data related to Fig 2 .
-
A, BImmunoblot analysis of ADAR1 expression in DP (A) and 4SP (B) thymocytes isolated from wild‐type (WT) and CD4+ cell‐specific Adar1 knockout (Cd4 cre A1 fl/fl) mice. Representative images are shown (left panel). The band intensity of each ADAR1 isoform was normalized to that of GAPDH and is displayed as the mean ± SEM (n = 3; Mann–Whitney U‐test, *P < 0.05; right panel). The mean value for the relative expression of ADAR1 in WT thymocytes was set as 1.
-
CThe absolute number of double‐negative (DN), double‐positive (DP), CD4+ single‐positive (4SP), and CD8+ single‐positive (8SP) thymocytes isolated from wild‐type (WT) and Cd4 cre A1 fl/fl mice. Values are displayed as the mean ± SEM (n = 7 for each group; Mann–Whitney U‐test, *P < 0.05, N.S., not significant).
Figure 2. Inhibition of T cell maturation by deletion of ADAR1 in the thymus.
-
ATotal number of thymocytes in wild‐type (WT) and CD4+ cell‐specific Adar1 knockout (Cd4 cre A1 fl/fl) mice. Values are displayed as the mean ± SEM (n = 7 for each group; Mann–Whitney U‐test, N.S., not significant).
-
B, CThymocytes isolated from WT and Cd4 cre A1 fl/fl mice were stained with anti‐CD4 and anti‐CD8 antibodies and analyzed by flow cytometry. Representative images are shown (B). The percentages of DN, DP, 4SP, and 8SP thymocytes were compared between WT and Cd4 cre A1 fl/fl mice (C). Values are displayed as the mean ± SEM (n = 7 for each group; Mann–Whitney U‐test, **P < 0.01).
-
D, EThymocytes isolated from WT and Cd4 cre A1 fl/fl mice were stained with anti‐CD4, anti‐CD8, and anti‐TCRβ antibodies and were gated by the high expression of TCRβ (TCRβhigh). Representative images are shown (D). The percentages of DN, DP, 4SP, and 8SP thymocytes were compared between WT and Cd4 cre A1 fl/fl mice (E). Values are displayed as the mean ± SEM (n = 3 for each group; Mann–Whitney U‐test, *P < 0.05).
-
FThymocytes isolated from WT and Cd4 cre A1 fl/fl mice were stained with anti‐CD4, anti‐CD8, anti‐CD69, and anti‐MHC‐I antibodies and were gated on 4SP thymocytes. Representative images are shown (upper panel). The percentages of CD69+MHC‐I− (SM), CD69+MHC‐I+ (M1), and CD69−MHC‐I− (M2) thymocytes were compared between WT and Cd4 cre A1 fl/fl mice (lower panel). Values are displayed as the mean ± SEM (n = 3 for each group; Mann–Whitney U‐test, *P < 0.05).
-
GThymocytes isolated from WT and Cd4 cre A1 fl/fl mice were stained with antibodies against CD4, CD8 and CD5 and analyzed by flow cytometry. Representative flow cytometric images for DP and 4SP thymocytes are shown (upper panel). The percentages of CD5− DP and 4SP thymocytes were compared between WT and Cd4 cre A1 fl/fl mice (lower panel). Values are displayed as the mean ± SEM (n = 3 for each group; Mann–Whitney U‐test, *P < 0.05, N.S., not significant).
-
HThymocytes isolated from WT and Cd4 cre A1 fl/fl mice were stained with anti‐CD4, anti‐CD8, anti‐CD25, and anti‐Foxp3 antibodies and were gated on 4SP thymocytes. Representative images are shown (upper panel). The percentages of CD25+Foxp3+ 4SP thymocytes (thymic Treg cells) were compared between WT and Cd4 cre A1 fl/fl mice (lower panel). Values are displayed as the mean ± SEM (n = 3 for each group; Mann–Whitney U‐test, *P < 0.05).
ADAR1 deficiency changes the thresholds for thymic T cell selection by inhibiting TCR signal transduction
During thymic T cell maturation, the appropriate T cell selection requires the achievement of a certain threshold of TCR signal strength. Therefore, we assessed the efficiency of both positive and negative selection in Cd4 cre A1 fl/fl mice by crossing them with HY‐TCR + mice, which express a transgenic TCR that recognizes the male‐specific HY antigen 30. The number of DP and 8SP thymocytes in male Cd4 cre A1 fl/fl HY‐TCR + mice was significantly higher than that in HY‐TCR + mice (Fig 3A), suggesting that negative selection is impaired in ADAR1‐deficient thymocytes. In contrast, the number of 8SP thymocytes in female Cd4 cre A1 fl/fl HY‐TCR + mice was significantly lower than that in HY‐TCR + mice (Fig 3B), suggesting that positive selection is also impaired in ADAR1‐deficient thymocytes because of aberrant TCR signal transduction. Indeed, when the TCR was stimulated with anti‐CD3 and anti‐CD28 antibodies, apoptosis was barely induced and proliferative activity was significantly reduced in ADAR1‐deficient thymocytes (Fig 3C, and Appendix Fig S1A and B). The mRNA expressions of anti‐apoptotic genes, including Bcl2 and Mcl1, were significantly increased in ADAR1‐deficient 4SP thymocytes (Fig 3D). In addition, although the activation of ZAP70 and PLCγ1 through phosphorylation is required in the early steps of TCR signaling 31, ADAR1 deficiency strongly inhibits their phosphorylation upon TCR stimulation (Fig 3E). Collectively, these results suggest that thymic T cell selection during maturation is impaired because of the reduced intensity of TCR signaling in ADAR1‐deficient T cells.
Figure 3. Impaired thymic selection in CD4+ cell‐specific Adar1 knockout mice.
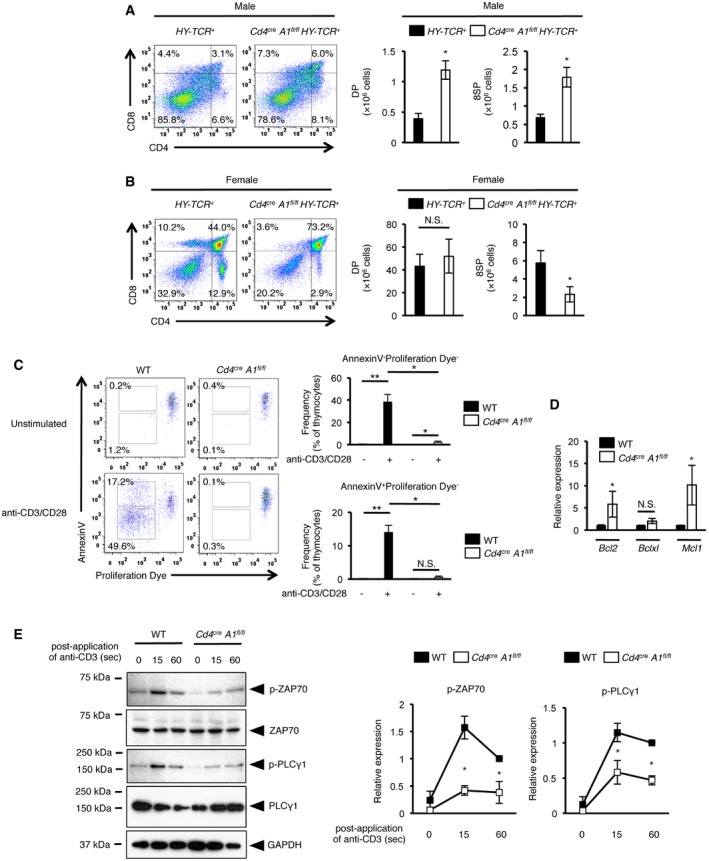
-
A, BCD4+ cell‐specific Adar1 knockout (Cd4 cre A1 fl/fl) mice were crossed with HY‐TCR + mice to generate Cd4 cre A1 fl/fl HY‐TCR + mice. Thymocytes isolated from male (A) and female (B) HY‐TCR + or Cd4 cre A1 fl/fl HY‐TCR + mice were stained with anti‐CD4 and anti‐CD8 antibodies and analyzed by flow cytometry. Representative images are shown (left panels). The absolute number of DP and 8SP thymocytes was compared between HY‐TCR + and Cd4 cre A1 fl/fl HY‐TCR + mice (right panels). Values are displayed as the mean ± SEM (n = 4 for each group; Mann–Whitney U‐test, *P < 0.05, N.S., not significant).
-
CThymocytes isolated from WT and Cd4 cre A1 fl/fl mice were labeled with Cell Proliferation Dye and then stimulated with plate‐bound anti‐CD3 and anti‐CD28 antibodies for 4 days. Apoptotic (Annexin V+) and proliferating thymocytes (reduced intensity of Proliferation Dye) were determined by flow cytometry. Representative flow cytometric images are shown (left panel). The percentages of either Annexin V+ or Annexin V− proliferating thymocytes were compared between WT (n = 5) and Cd4 cre A1 fl/fl (n = 4) mice (right panel). Values are displayed as the mean ± SEM (Mann–Whitney U‐test, *P < 0.05, **P < 0.01, N.S., not significant).
-
DThe relative expression of anti‐apoptotic genes in sorted 4SP thymocytes isolated from WT and Cd4 cre A1 fl/fl mice. Values represent the relative gene expression normalized to Actinβ mRNA and are displayed as the mean ± SEM (n = 3; Mann–Whitney U‐test, *P < 0.05, N.S., not significant). The mean value for the relative expression of each anti‐apoptotic gene in WT 4SP thymocytes was set as 1.
-
EImmunoblot analysis of total and phosphorylated ZAP70 and PLCγ1 expression in anti‐CD3‐stimulated thymocytes isolated from WT and Cd4 cre A1 fl/fl mice. The expression of GAPDH is shown as a reference. Representative images are shown (left panel). The band intensity of p‐ZAP70 and p‐PLCγ1 was normalized to that of total ZAP70 and PLCγ1, respectively, and is displayed as the mean ± SEM (n = 3; Mann–Whitney U‐test, *P < 0.05) (right panel) by setting the mean value of stimulated WT samples at 60 s as 1.
CD4+ T cell‐specific knockout of ADAR1 induces autoimmunity in mice
The establishment of central tolerance by negative selection is essential for the elimination of self‐reactive T cells to prevent autoimmunity 22, 23, 24, 25. Intriguingly, by 4 months of age, 40% of Cd4 cre A1 fl/fl mice in which T cell selection was impaired exhibited the symptoms of spontaneous colitis such as diarrhea, bloody stools, and rectal prolapse (Fig 4A). Histological analysis indicated a pathology of severe colitis, including extensive epithelial hyperplasia and the infiltration of inflammatory cells (Fig 4B and C). In addition, inflammatory symptoms were sometimes observed in other tissues, including the small intestine and lung in aged Cd4 cre A1 fl/fl mice (Fig EV3). To determine the mechanism underlying autoimmunity, we focused on colitis and assessed the infiltration of each CD4+ cell subset including Th1 (CD4+IFNγ+), Th17 (CD4+IL17+), and Treg cells in the colonic lamina propria. Cd4 cre A1 fl/fl mice exhibited an accumulation of pathogenic Th1 and Th17 cells in the lamina propria (Fig 4D). The number of CD4+ cells was significantly reduced in the spleen, similar to the pattern observed in the thymus of Cd4 cre A1 fl/fl mice (Figs 2B–E and EV4A). However, the proportions of Th1 and Th17 cells among either CD4+ cells or total splenocytes were significantly upregulated in Cd4 cre A1 fl/fl mice (Figs 4E and EV4B). We also noted the upregulated expression of Tbet and Rorγt mRNAs, which encode the master regulators of Th1 and Th17 cells, respectively (Fig EV4C). In addition, IL17 production was significantly increased in ADAR1‐deficient splenic CD4+ cells with the similar tendency of upregulation of IFNγ production (Fig EV4D). Treg cells were also accumulated in the colonic lamina propria of Cd4 cre A1 fl/fl mice (Fig 4D). Of note, the proportion of Treg cells among CD4+ cells was slightly but significantly higher in the spleen of Cd4 cre A1 fl/fl mice (Fig 4F), although the proportion was not increased among total splenocytes (Fig EV4E). These data suggest that T cells were expanded in response to local inflammation, although the reactivity of Th1 and Th17 cells was more enhanced than that of Treg cells.
Figure 4. Spontaneous colitis observed in CD4+ cell‐specific Adar1 knockout mice.
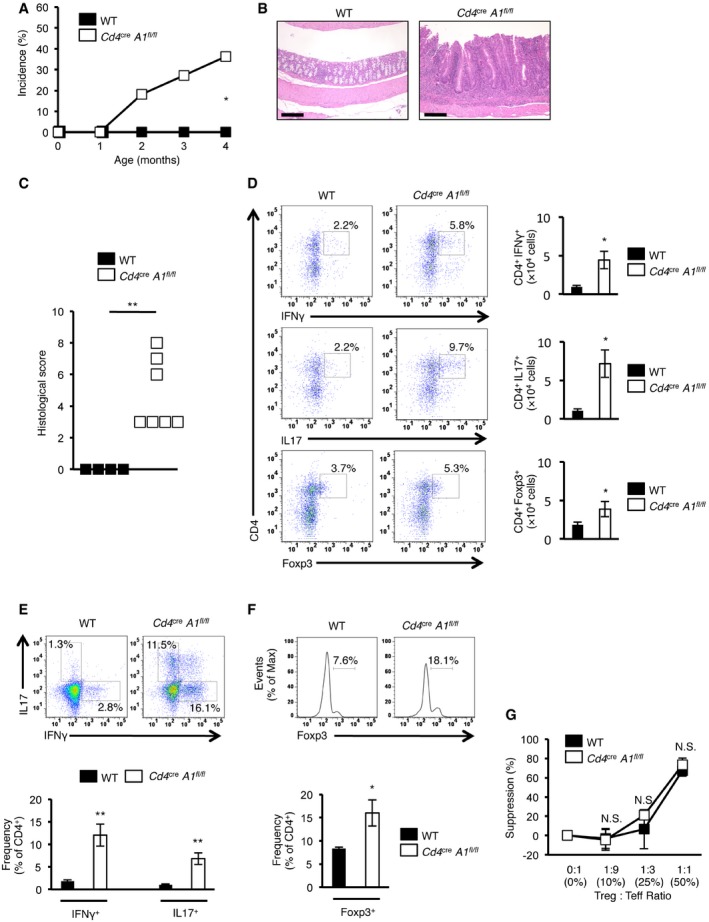
- Incidence of spontaneous colitis observed in wild‐type (WT) and CD4+ cell‐specific Adar1 knockout (Cd4 cre A1 fl/fl) mice (n = 11 for each group; Fisher's exact test, *P < 0.05).
- Representative images of HE staining of the colon dissected from a WT and Cd4 cre A1 fl/fl mouse. Scale bar, 200 μm.
- Histological scores for colitis were compared between WT (n = 4) and Cd4 cre A1 fl/fl (n = 7) mice (Mann–Whitney U‐test, **P < 0.01).
- Lymphocytes isolated from the colonic lamina propria of WT and Cd4 cre A1 fl/fl mice were stained with anti‐IFNγ, anti‐IL17, or anti‐Foxp3 antibodies together with anti‐CD4 antibodies and analyzed by flow cytometry. Representative images are shown (left panel). The absolute number of IFNγ‐, IL17‐, or Foxp3‐expressing CD4+ T cells was compared between WT and Cd4 cre A1 fl/fl mice (right panel). Values are displayed as the mean ± SEM (n = 3 for each group; Mann–Whitney U‐test, *P < 0.05).
- Splenocytes isolated from WT and Cd4 cre A1 fl/fl mice were stained with anti‐IFNγ and anti‐IL17 together with anti‐CD4 antibodies and were gated on CD4+ cells. Representative flow cytometric images are shown (upper panel). The percentages of IFNγ‐ and IL17‐secreting CD4+ T cells were compared between WT and Cd4 cre A1 fl/fl mice (lower panel). Values are displayed as the mean ± SEM (n = 6 for each group; Mann–Whitney U‐test, **P < 0.01).
- Splenocytes isolated from WT and Cd4 cre A1 fl/fl mice were stained with anti‐Foxp3 together with anti‐CD4 antibodies and were gated on CD4+ cells. Representative flow cytometric images are shown (upper panel). The percentages of Foxp3‐expressing CD4+ T cells were compared between WT and Cd4 cre A1 fl/fl mice (lower panel). Values are displayed as the mean ± SEM (n = 4 for each group; Mann–Whitney U‐test, *P < 0.05).
- Effector T (Teff) cells isolated from the spleen of a WT mouse were stimulated with soluble anti‐CD3 (5 μg/ml) in the presence of mitomycin C‐treated splenocytes and were cultured with regulatory T (Treg) cells isolated from WT and Cd4 cre A1 fl/fl mice at the indicated ratios. After 72 h, the proliferative activity was measured. Values are displayed as the mean ± SEM (n = 3 for each group; Mann–Whitney U‐test, N.S., not significant). The mean value for the suppression of proliferative activity of Teff cells in a condition without Treg cells was set as 0%.
Figure EV3. Inflammatory symptoms observed in the small intestine and lung but not in the skin in aged Cd4cre A1 fl/fl mice. Data related to Fig 4 .
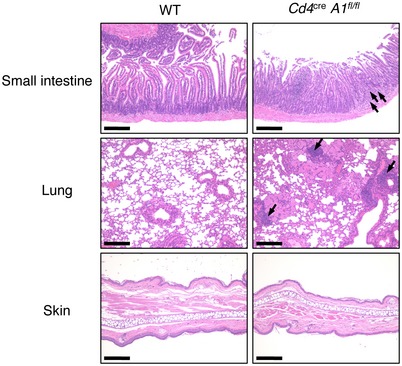
Representative images of HE staining of the small intestines, lungs, and skin dissected from wild‐type (WT) and CD4+ cell‐specific Adar1 knockout (Cd4 cre A1 fl/fl) mice. Infiltration of lymphocytes is indicated by arrows. Scale bar, 200 μm.
Figure EV4. Lymphopenia and abnormal activation of T cells in spleen of CD4+ cell‐specific Adar1 knockout mice. Data related to Fig 4 .
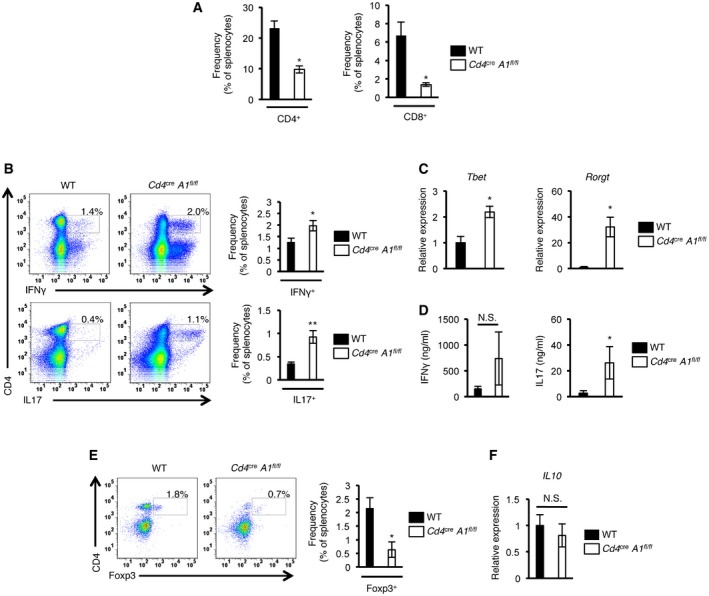
- The proportions of CD4+ and CD8+ splenocytes isolated from wild‐type (WT) and CD4+ cell‐specific Adar1 knockout (Cd4 cre A1 fl/fl) mice. Values are displayed as the mean ± SEM (n = 4 for each group; Mann–Whitney U‐test, *P < 0.05).
- Splenocytes isolated from WT and Cd4 cre A1 fl/fl mice were stained with either anti‐IFNγ or anti‐IL17 antibodies together with anti‐CD4 antibodies. Representative flow cytometric images are shown (left panel). The percentages of IFNγ‐ and IL17‐secreting CD4+ T cells were compared between WT and Cd4 cre A1 fl/fl mice (right panel). Values are displayed as the mean ± SEM (n = 6 for each group; Mann–Whitney U‐test, *P < 0.05, **P < 0.01).
- The relative expressions of Tbet and Rorγt mRNAs in splenic CD4+ T cells isolated from WT and Cd4 cre A1 fl/fl mice. Values represent the relative gene expressions normalized to Actinβ mRNA and are displayed as the mean ± SEM (n = 3; Mann–Whitney U‐test, *P < 0.05). The mean value for the relative expression of Tbet and Rorγt mRNAs in WT splenic CD4+ T cells was set as 1.
- Splenic CD4+ T cells isolated from WT and Cd4 cre A1 fl/fl mice were stimulated with plate‐bound anti‐CD3 and anti‐CD28 antibodies for 72 h, and the levels of IFNγ and IL17 in supernatants were measured by ELISA. Values are displayed as the mean ± SEM (n = 5 for each group; Mann–Whitney U‐test, *P < 0.05, N.S., not significant).
- Splenocytes isolated from WT and Cd4 cre A1 fl/fl mice were stained with anti‐Foxp3 antibodies together with anti‐CD4 antibodies. Representative flow cytometric images are shown (left panel). The percentages of Foxp3‐expressing CD4+ T cells were compared between WT and Cd4 cre A1 fl/fl mice (right panel). Values are displayed as the mean ± SEM (n = 4 for each group; Mann–Whitney U‐test, *P < 0.05).
- The relative expression of IL10 mRNA in mesenteric lymph node (MLN)‐derived Treg cells isolated from WT and Cd4 cre A1 fl/fl mice. Values represent the relative gene expression normalized to GAPDH mRNA and are displayed as the mean ± SEM (n = 3; Mann–Whitney U‐test, N.S., not significant). The mean value for the relative expression of IL10 mRNA in WT MLN‐derived Treg cells was set as 1.
Next, we investigated whether ADAR1 was dispensable for the optimal suppressive function of Treg cells by performing an in vitro Treg cell suppression assay. This assay demonstrated that the ability of ADAR1‐deficient Treg cells to inhibit the proliferation of Teff cells was normal (Fig 4G). In addition, production of the anti‐inflammatory cytokine IL10 in Treg cells isolated from the mesenteric lymph nodes of Cd4 cre A1 fl/fl mice was not altered, at least, at the mRNA level (Fig EV4F). Collectively, these findings suggest that the severe colitis observed in Cd4 cre A1 fl/fl mice was not caused by the aberrant differentiation or suppressive function of peripheral Treg cells but rather because of the accumulation of self‐reactive T cells that were generated by impaired negative selection.
Upregulation of ADAR1‐mediated RNA editing during thymic T cell maturation suppresses the excessive induction of ISG expression
To assess changes in the frequency of RNA editing during thymic T cell maturation, we performed RNA‐seq analysis using total RNA extracted from WT 4SP thymocytes in triplicate. This resulted in the identification of 870 potential RNA editing sites (Dataset EV1). RNA editing was rare in the 5′‐untranslated region (UTR) of mRNAs, and the majority of edited sites were annotated to the intronic regions of mRNAs and intergenic regions (Fig 5A). This distribution is similar to that of human RNA editing sites as identified in the DARNED database 32. We performed gene ontology (GO) analysis of those genes that contained potential RNA editing sites, and identified a significant enrichment in the categories related to transcription and the immune system, including the TCR signaling pathway (Fig 5B). To validate the data, we randomly selected several potential RNA editing sites and quantified the editing frequency in DP and 4SP thymocytes by Sanger sequencing. Although all the sites examined were already edited to a certain extent at the DP stage, the editing frequency at the majority of these sites increased significantly during the transition from DP to 4SP thymocytes, correlating with the increased expression of ADAR1 at the 4SP stage (Figs 1B and C, and 5C and D, and Appendix Fig S2A–C). As expected, the editing frequency of all the sites was reduced markedly in ADAR1‐deficient 4SP thymocytes (Fig 5C and D). Taken together, the data suggest that ADAR1‐mediated RNA editing is upregulated from the DP to 4SP stage, which correlates with the increased expression of ADAR1 during this transition.
Figure 5. Upregulation of ADAR1‐mediated RNA editing during thymic T cell maturation.
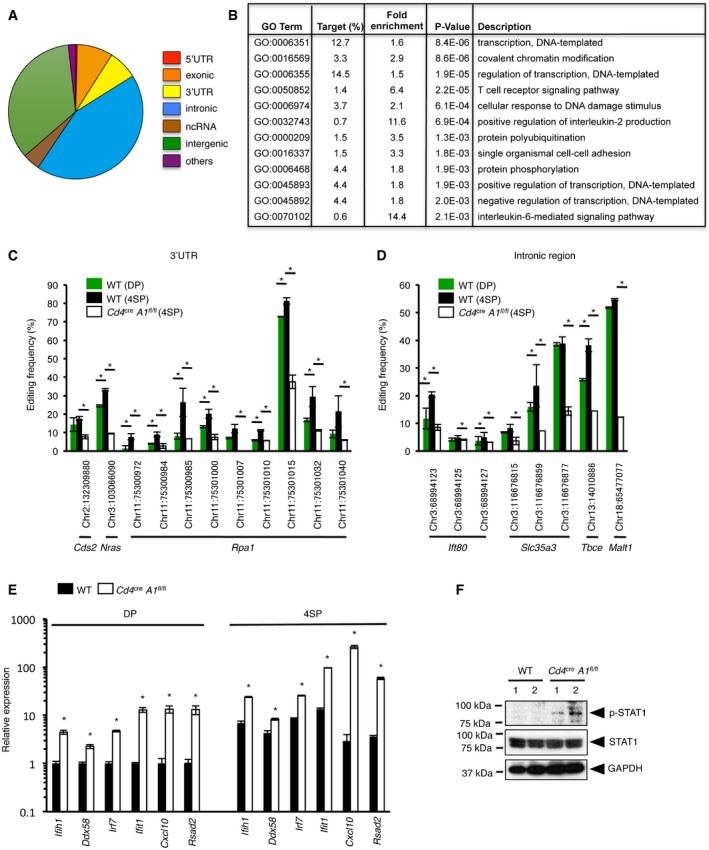
-
ADistribution of the potential RNA editing sites found in 4SP thymocytes.
-
BThe top 12 categories of biological processes, which are ranked according to the P‐value, for the genes that contained potential RNA editing sites at the 4SP stage. Fisher's exact test was used for the statistical analysis. GO, gene ontology.
-
C, DRNA editing in the 3′UTR (C) and intronic region (D) of each gene indicated in the figures with their chromosomal location was validated by direct Sanger sequencing, and the frequency of editing was compared among wild‐type (WT) DP thymocytes, WT 4SP thymocytes, and 4SP thymocytes isolated from CD4+ cell‐specific Adar1 knockout (Cd4 cre A1 fl/fl) mice. Values are displayed as the mean ± SEM (n = 3 for each group; Mann–Whitney U‐test, *P < 0.05).
-
EThe relative expression of each interferon‐stimulated gene (ISG) indicated in the figure in sorted DP and 4SP thymocytes isolated from WT and Cd4 cre A1 fl/fl mice. Values represent the relative gene expression normalized to GAPDH mRNA and are displayed as the mean ± SEM (n = 3; Mann–Whitney U‐test, *P < 0.05). The mean value for the relative expression of each ISG in WT DP thymocytes was set as 1.
-
FImmunoblot analysis of total and phosphorylated STAT1 expression in thymocytes isolated from WT and Cd4 cre A1 fl/fl mice (n = 2 for each group). The expression of GAPDH is shown as a reference.
Next, we examined changes in type I ISG expression caused by ADAR1 depletion in T cells. The expression of all the type I ISGs examined was upregulated significantly, and for some of these genes, expression was increased more than 10‐fold at the DP stage in ADAR1‐deficient T cells (Fig 5E). This increase in ISG expression was enhanced further at the 4SP stage and, as an example, the expression of Cxcl10 mRNA was increased more than 100‐fold in ADAR1‐deficient T cells at this stage. It was reported that treatment with type I IFN abrogated T cell proliferation upon TCR activation 33. However, among type I IFNs, IFNα is specifically secreted from plasmacytoid dendritic cells 34, 35, 36, and we confirmed no substantial detection of both IfnaI and IfnbI genes in thymocytes at all four stages by qRT–PCR and no substantial detection of IFNβ production in ADAR1‐deficient T cells by ELISA. Furthermore, treatment with anti‐type I IFN receptor (IFNR) antibody did not rescue the diminished proliferative activity in ADAR1‐deficient T cells (Fig EV5). Therefore, we hypothesized that impaired TCR signal transduction in ADAR1‐deficient T cells is caused by enhanced ISG expression, which mimics treatment with type I IFN. Given that the anti‐proliferative effect of type I IFN was reported to require the activation of STAT1 37, 38, we examined the phosphorylation status of STAT1 in thymocytes. Although no phosphorylation was detected in WT thymocytes, we found that ADAR1‐deficient thymocytes constitutively expressed phosphorylated STAT1 (Fig 5F). Taken together, these results suggest that defects in the upregulation of ADAR1‐mediated RNA editing result in excessive ISG expression, especially at the 4SP stage, leading to impaired TCR signal transduction.
Figure EV5. Blockade of type I IFN signaling failed to rescue the diminished proliferative activiy in ADAR1‐deficient thymocytes. Data related to Fig 5 .
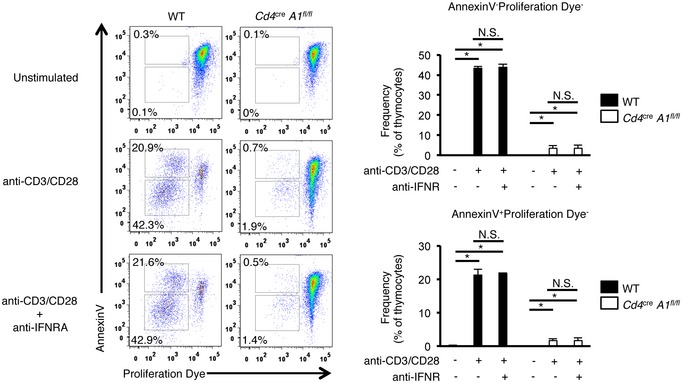
Thymocytes isolated from wild‐type (WT) and CD4+ cell‐specific Adar1 knockout (Cd4 cre A1 fl/fl) mice were labeled with Cell Proliferation Dye and then stimulated with plate‐bound anti‐CD3 and anti‐CD28 antibodies in the presence or absence of anti‐IFNAR1 antibody for 4 days. Apoptotic (Annexin V+) and proliferative thymocytes were determined by flow cytometry. Representative flow cytometric images are shown (left panel). The percentages of Annexin V+ or proliferative thymocytes were compared between WT and Cd4 cre A1 fl/fl mice (right panel). Values are displayed as the mean ± SEM (n = 3 for each group; Mann–Whitney U‐test, *P < 0.05, N.S., not significant).
Deletion of MDA5 rescues aberrant thymic T cell maturation and prevents autoimmunity induced by ADAR1 deficiency
Although MDA5 plays a critical role in the innate immune system, it is also highly expressed in the thymus from the embryonic stages as well as in mature T cells 39, likely sensing non‐self dsRNAs, for instance, to detect viral infection. Given that ADAR1‐mediated RNA editing and the expression of MDA5 were upregulated during thymic T cell maturation and that ADAR1 deficiency induced elevated ISG expression in thymocytes, especially at the 4SP stage, we investigated the involvement of the MDA5 signaling pathway in the aberrant maturation of T cells and the development of spontaneous colitis in Cd4 cre A1 fl/fl mice by crossing Cd4 cre A1 fl/fl mice with Ifih1 −/− mice. We confirmed no expression of MDA5 in thymocytes collected from Ifih1 −/− mice (Appendix Fig S3). As expected, we also confirmed that there were no differences in T cell development or ISG expression between WT and Ifih1 −/− mice in the absence of infections, and therefore, we used Ifih1 −/− mice as the control in the following analyses. Concurrent deletion of MDA5 restored the populations of DN, DP, 4SP, and 8SP thymocytes, which were reduced in Cd4 cre A1 fl/fl mice, to the level found in Ifih1 −/− mice (Fig 6A). In addition, the overexpression of type I ISGs in ADAR1‐deficient 4SP thymocytes was abrogated by the deletion of MDA5 (Fig 6B). Therefore, we investigated the contribution of the MDA5 signaling pathway on aberrant TCR signal transduction found in ADAR1‐deficient thymocytes. As expected, the level of ZAP70 and PLCγ1 phosphorylation upon TCR stimulation was restored by the deletion of MDA5 (Fig 6C). Consequently, we observed the partial but significant proliferative activity of ADAR1/MDA5‐deficient T cells upon TCR stimulation (Fig 6D). In addition, the composition of SM, M1, and M2 thymocyte populations was largely restored (Fig 6E). Finally, we observed the complete recovery of thymic Treg cells, which were diminished in Cd4 cre A1 fl/fl mice, following the deletion of MDA5 (Fig 6F).
Figure 6. Concurrent deletion of MDA5 rescues impaired thymic T cell maturation in CD4+ cell‐specific Adar1 knockout mice.
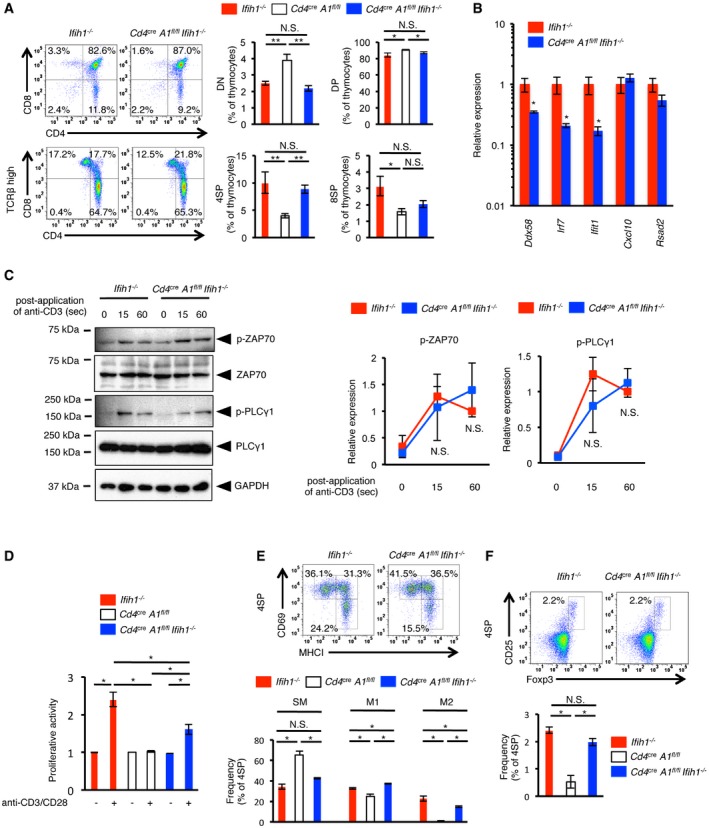
- CD4+ cell‐specific Adar1 knockout (Cd4 cre A1 fl/fl) mice were crossed with MDA5 knockout (Ifih1 −/−) mice to generate Cd4 cre A1 fl/fl Ifih1 −/− mice. Thymocytes isolated from Ifih1 −/− and Cd4 cre A1 fl/fl Ifih1 −/− mice were stained with anti‐CD4, anti‐CD8, and anti‐TCRβ antibodies and analyzed by flow cytometry. Representative images are shown (left panel). The percentages of DN, DP, 4SP, and 8SP thymocytes were compared among Ifih1 −/− (n = 4), Cd4 cre A1 fl/fl Ifih1 −/− (n = 5), and Cd4 cre A1 fl/fl (n = 7) mice (right panel). The same data shown in Fig 2C were used for Cd4 cre A1 fl/fl mice. Values are displayed as the mean ± SEM (Mann–Whitney U‐test, *P < 0.05, **P < 0.01, N.S., not significant).
- The relative expression of each interferon‐stimulated gene (ISG) indicated in the figure in sorted 4SP thymocytes isolated from Ifih1 −/− and Cd4 cre A1 fl/fl Ifih1 −/− mice. Values represent the relative gene expression normalized to GAPDH mRNA and are displayed as the mean ± SEM (n = 3; Mann–Whitney U‐test, *P < 0.05). The mean value for the relative expression of each ISG in Ifih1 −/− 4SP thymocytes was set as 1.
- Immunoblot analysis of total and phosphorylated ZAP70 and PLCγ1 expression in anti‐CD3‐stimulated thymocytes isolated from Ifih1 −/− and Cd4 cre A1 fl/fl Ifih1 −/− mice. The expression of GAPDH is shown as a reference. Representative images are shown (left panel). The band intensity of p‐ZAP70 and p‐PLCγ1 was normalized to that of total ZAP70 and PLCγ1, respectively, and is displayed as the mean ± SEM (n = 3; Mann–Whitney U‐test, N.S., not significant) (right panel) by setting the mean value of stimulated Ifih1 −/− samples at 60 s as 1.
- Thymocytes isolated from Ifih1 −/− and Cd4 cre A1 fl/fl Ifih1 −/− mice were stimulated with plate‐bound anti‐CD3 (5 μg/ml) and anti‐CD28 (1 μg/ml) antibodies for 72 h, and the proliferative activity was measured. Values are displayed as the mean ± SEM (n = 3 for each group; Mann–Whitney U‐test, *P < 0.05). The mean value for the activity of Ifih1 −/− thymocytes in an unstimulated condition was set as 1. For comparative analysis, the same data shown in Appendix Fig S1B were used for Cd4 cre A1 fl/fl mice.
- Thymocytes isolated from Ifih1 −/− and Cd4 cre A1 fl/fl Ifih1 −/− mice were stained with anti‐CD4, anti‐CD8, anti‐CD69, and anti‐MHC‐I antibodies and were gated on 4SP thymocytes. Representative images are shown (upper panel). The percentages of CD69+MHC‐I− (SM), CD69+MHC‐I+ (M1), and CD69−MHC‐I− (M2) thymocytes were compared among Ifih1 −/− (n = 5), Cd4 cre A1 fl/fl Ifih1 −/− (n = 3), and Cd4 cre A1 fl/fl (n = 3) mice (lower panel). The same data shown in Fig 2F were used for Cd4 cre A1 fl/fl mice. Values are displayed as the mean ± SEM (Mann–Whitney U‐test, *P < 0.05, N.S., not significant).
- Thymocytes isolated from Ifih1 −/− and Cd4 cre A1 fl/fl Ifih1 −/− mice were stained with anti‐CD4, anti‐CD8, anti‐CD25, and anti‐Foxp3 antibodies and were gated on 4SP thymocytes. Representative images are shown (upper panel). The percentages of CD25+Foxp3+ 4SP thymocytes (thymic Treg cells) were compared among Ifih1 −/− , Cd4 cre A1 fl/fl Ifih1 −/−, and Cd4 cre A1 fl/fl mice (lower panel). The same data shown in Fig 2H were used for Cd4 cre A1 fl/fl mice. Values are displayed as the mean ± SEM (n = 3 for each group; Mann–Whitney U‐test, *P < 0.05, N.S., not significant).
Next, we analyzed the effect of MDA5 deletion on the peripheral T cell population. We did not detect any significant difference in the proportion of Th1 cells among splenic CD4+ T cells between Cd4 cre A1 fl/fl Ifih1 −/− and Ifih1 −/− control mice (Fig 7A). Whereas the proportion of Th17 cells was slightly, but significantly, elevated in Cd4 cre A1 fl/fl Ifih1 −/− mice, this increase was significantly ameliorated when compared with Cd4 cre A1 fl/fl mice (Fig 7A). Of note, spontaneous colitis was not observed in any Cd4 cre A1 fl/fl Ifih1 −/− mouse (Fig 7B–D). These findings suggest that the aberrant activation of MDA5 signaling pathway is involved in the induction of excessive ISG expression and reduced TCR signaling transduction found in ADAR1‐deficient T cells, leading to abnormal thymic T cell maturation as well as the induction of autoimmune colitis.
Figure 7. Disruption of the MDA5 signaling pathway ameliorates colitis in CD4+ cell‐specific Adar1 knockout mice.
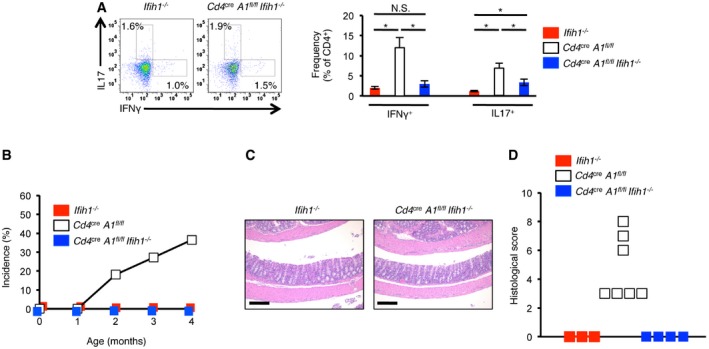
- Splenocytes isolated from Ifih1 −/− and Cd4 cre A1 fl/fl Ifih1 −/− mice were stained with anti‐IFNγ and anti‐IL17 together with anti‐CD4 antibodies and were gated on CD4+ cells. Representative flow cytometric images are shown (left panel). The percentages of IFNγ‐ and IL17‐secreting CD4+ T cells were compared among Ifih1 −/− (n = 4), Cd4 cre A1 fl/fl Ifih1 −/− (n = 5), and Cd4 cre A1 fl/fl mice (n = 6) (right panel). The same data shown in Fig 4E were used for Cd4 cre A1 fl/fl mice. Values are displayed as the mean ± SEM (Mann–Whitney U‐test, *P < 0.05, N.S., not significant).
- No incidence of spontaneous colitis observed in Ifih1 −/− (n = 5) and Cd4 cre A1 fl/fl Ifih1 −/− (n = 11) mice. For side‐by‐side comparison with Cd4 cre A1 fl/fl mice, the data shown in Fig 4A were included.
- Representative images of HE staining of colon dissected from an Ifih1 −/− and a Cd4 cre A1 fl/fl Ifih1 −/− mouse. Scale bar, 200 μm.
- Histological scores for the colitis in Ifih1 −/− (n = 3) and Cd4 cre A1 fl/fl Ifih1 −/− (n = 4) mice. For side‐by‐side comparison with Cd4 cre A1 fl/fl mice, the data shown in Fig 4C were included.
Finally, given that some abnormalities found in ADAR1‐deficient T cells were not completely recovered by the concurrent deletion of MDA5, we investigated T cell development in A1 KI/KI Ifih1 −/− mice. T cells developed normally in the thymus of A1 KI/KI Ifih1 −/− mice (Appendix Fig S4A). In addition, the proportion of Th1 and Th17 cells in splenic CD4+ T cells was not elevated in A1 KI/KI Ifih1 −/− mice (Appendix Fig S4B), which suggested that the functions of ADAR1‐mediated RNA editing that are independent of the MDA5 signaling pathway, for instance, changing amino acid residues by RNA editing in coding regions, are dispensable for T cell maturation.
Discussion
Given that A1 −/− mice, Adar1 p150‐specific KO mice, and A1 KI/KI mice are all embryonic lethal, the role of ADAR1‐mediated RNA editing in T cells had been undetermined 6, 7, 8. In this study, we created CD4+ cell‐specific Adar1 knockout mice and demonstrated that ADAR1‐mediated RNA editing was essential for optimal thymic T cell maturation, including negative selection, to prevent autoimmunity, providing a novel insight into the role of the ADAR1/MDA5 axis in adaptive immunity. We found that ADAR1 is highly expressed in the thymus and the expression levels of ADAR1 and type I ISGs, including MDA5, were increased during thymic T cell maturation, especially from the DP stage to the 4SP stage. In accordance, we observed the upregulation of ADAR1‐mediated RNA editing at this stage. During their maturation, thymocytes move from the cortex of the thymus into the medulla, which expresses type I IFN abundantly in the absence of infection 40, 41, 42. Intriguingly, a recent study demonstrated that 4SP and 8SP cells from Ifnar1 −/− mice, which lack the IFNα receptor 1, exhibit normal proliferative ability but have deficient responses to cytokines such as IL6 and type II IFN 27. This evidence suggests that the increased expression of type I ISGs during the late stages of T cell maturation is required to establish the functional properties of T cells. Therefore, increased RNA editing might be a compensatory mechanism to prevent the recognition of endogenous dsRNAs as non‐self by MDA5, whose expression is developmentally upregulated, and this mechanism is essential for correct T cell maturation. In agreement, we showed that ADAR1 deficiency induces the aberrant activation of MDA5 in thymocytes resulting in the overexpression of type I ISGs, which is similar to the previous findings that embryonic lethality and elevated ISG expression found in A1 −/− mice, Adar1 p150‐specific KO mice, and A1 KI/KI mice are ameliorated by concurrent deletion of MDA5 8, 9, 10.
The recognition of viral dsRNA by MDA5 is usually mediated by innate immune cells, such as dendritic cells and macrophages 11, 12. However, MDA5 is also highly expressed in the thymus from the embryonic stages and in mature T cells 39, likely sensing non‐self dsRNAs, given that the T cell can be the target of certain viruses. Therefore, in the absence of viral infections, we observed no abnormality in T cell development in Ifih1 −/− mice. Nevertheless, we reveal for the first time that the presence of unedited endogenous dsRNAs triggered the aberrant activation of MDA5 in ADAR1‐deficient T cells, which was deleterious for their maturation. This is unique because ADAR1‐deficient cells usually exhibit widespread apoptosis with the enhanced expression of type I ISGs, which results in a severe reduction in the number of corresponding cells, such as hematopoietic stem cells, erythroid cells, B cell lineage cells, and hepatocytes 6, 10, 13, 14, 15, 43, 44, 45. It is well known that treatment with poly(I:C), which mimics viral dsRNA and is commonly used as an MDA5 ligand, triggers pro‐apoptotic signaling in various cells 46. In contrast, although it has been reported that treatment with poly(I:C) affects thymic involution or T cell differentiation in mice, in vitro treatment with poly(I:C) has no effect on Th1 or Th17 differentiation 47, 48, 49. Therefore, the in vivo effect of MDA5 activation on T cells is considered as a secondary effect derived from the activated primary target cells. We demonstrated that the T cell‐specific deletion of ADAR1 activated the MDA5 signaling pathway in T cells, which arrested their maturation rather than induced apoptosis, although type I ISGs were increased in expression as expected. The in vitro activation of TCR further strengthened our interpretation that the induction of apoptosis was inhibited in ADAR1‐deficient thymocytes because of reduced TCR signaling intensity, which altered the thresholds for negative selection. Consequently, self‐reactive thymocytes were not eliminated, leading to autoimmune colitis. Of note, we found that STAT1 was highly phosphorylated and active in ADAR1‐deficient thymocytes, which is essential for the type I IFN‐mediated abrogation of T cell proliferation upon TCR activation 33, 37, 38. However, type I IFN was not detected in ADAR1‐deficient thymocytes and blocking type I IFN signaling failed to rescue the diminished proliferative activity in ADAR1‐deficient thymocytes. These observations suggest that STAT1 phosphorylation and enhanced ISG expression are induced likely in a type I IFN‐independent manner. Indeed, several studies demonstrated the presence of type I IFN‐independent non‐canonical pathways for STAT1 phosphorylation and ISG expression 50, 51. Nevertheless, there is a possibility that a very subtle amount of type I IFN, which was produced in ADAR1‐deficient T cells at the undetectable level, might contribute to these abnormalities. In addition, type I IFN secreted from cells other than thymocytes might play an additional role in the enhancement of ISG expression. Taken together, the enhanced ISG expression induced by the loss of ADAR1‐mediated RNA editing, mimicking treatment with type I IFN, may account for the molecular mechanisms that underlie the reduced TCR signal transduction in ADAR1‐deficient thymocytes.
The autoimmune disease AGS is caused by mutations in the genes involved in cellular nucleotide metabolism including TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and IFIH1 in addition to ADAR1 52. Among these genes, the disruption of TREX1 in mice leads to postnatal mortality caused by severe inflammatory myocarditis with elevated type I IFN 53. Intriguingly, this mortality was rescued by the concurrent deletion of RAG2 (recombination‐activating gene 2), which is essential for lymphocyte development, although the interferonopathy was not ameliorated 54. This evidence suggests that lymphocytes may participate in the pathogenesis of AGS. However, the role of T cells in the disease mechanism remains unknown. Our findings demonstrated that the T cell‐specific deletion of ADAR1 in mice impaired the process of negative selection, which resulted in defects in the induction of self‐tolerance. Consequently, spontaneous colitis was induced with the accumulation of Th1 and Th17 cells, which suggests that ADAR1‐mediated RNA editing contributes to the prevention of innate immune responses as well as T cell‐mediated autoimmunity by establishing thymic self‐tolerance. Indeed, inflammatory gastrointestinal problems including colitis are sometimes observed in patients with AGS 55. Therefore, it is necessary to elucidate whether T cell development is impaired in patients with AGS. This information should provide a novel insight into the disease mechanism and strategies for the treatment of AGS.
Materials and Methods
Mouse care/maintenance
Mice were maintained on a 12/12‐h light/dark cycle at a temperature of 23 ± 1.5°C with a humidity of 45 ± 15%. All experimental procedures that involved mice were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Osaka University.
Mutant mice
A1 fl/fl mice and Cd4 cre mice were kindly provided by Prof. Kazuko Nishikura (The Wistar Institute) and Prof. Osamu Takeuchi (Kyoto University), respectively. To generate Cd4 cre A1 fl/fl mice, A1 fl/fl mice were crossed with Cd4 cre mice. Although the Cd4 cre transgene and the ADAR1 gene are located on the same chromosome, repeated crossing resulted in successful homologous recombination. Ifih1 −/− mice and HY‐TCR + mice were obtained from the Oriental Bio Service, Japan, and the European Mouse Mutant Archive (EMMA) repository, respectively. Cre‐negative floxed mice (A1 fl/fl and A1 fl/+) or Ifih1 −/− mice were used as controls. A1 KI/KI mice were reported previously 8. All the mice used in this study were on a C57BL/6 background.
Histological evaluation
Hematoxylin and eosin (HE) staining of mouse colons, small intestines, lungs, and skin was performed at New Histo. Science Laboratory Co. Ltd. (Tokyo, Japan).
Diagnosis and evaluation of colitis
Mice with signs of colitis, including diarrhea, bloody stools, and rectal prolapse, were diagnosed with colitis. HE staining of mouse colons was assessed as described previously 56. In brief, each colon was scored by three parameters, namely the severity of mucosal epithelial changes (0–3), degree of inflammation (0–4), and extent of pathology (0–4). The histological score for each colonic section was calculated as the total score from the three parameters.
Isolation of colonic lamina propria lymphocytes
Mouse lymphocytes from the colonic lamina propria were isolated as described previously 57 with some modifications. In brief, the entire colon was dissected and incubated at 37°C for 20 min in Hanks’ balanced salt solution (Thermo Fisher Scientific) with 5 mM EDTA and 5% fetal bovine serum (FBS) to remove mucus. After washing five times with Dulbecco's phosphate‐buffered saline (DPBS), the colon was minced and digested with RPMI 1640 medium (Sigma) that contained 1 mg/ml collagenase D (Sigma), 1 mg/ml Dispase (Thermo Fisher Scientific), DNase I (Sigma), and 5% FBS at 37°C for 90 min. Then, Percoll (GE Healthcare) was added to the sample and centrifuged at 780 × g for 20 min at 25°C. Lymphocytes were recovered from the interface between the 40 and 75% Percoll layers.
Flow cytometry
Fluorescence‐conjugated antibodies against CD4 (RM4‐5; BD), CD8 (53‐6.7; BD), TCRβ (H57‐597; BD), CD5 (53‐7.3; BioLegend), CD25 (PC61; BioLegend), CD45RB (C363‐16A; BioLegend), CD69 (H1.2F3; BioLegend), IL17 (TC11‐18H10; BD), MHC‐I (AF6‐88.5; BioLegend), IFNγ (XMG1.2; BD), and Foxp3 (MF23; BD) were purchased. For surface staining, cells were incubated with each antibody for 30 min at 4°C and washed with DPBS. Intracellular cytokine staining was performed as described previously 58 with some modifications. Briefly, splenocytes or lamina propria lymphocytes were stimulated with 50 ng/ml phorbol‐12‐myristate‐13‐acetate (PMA; Calbiochem) and 800 ng/ml ionomycin (Calbiochem) for 4 h in the presence of GolgiStop (BD) before staining with anti‐CD4 antibody. The cells were fixed and permeabilized using Fix Buffer I (BD) and Perm/Wash Buffer I (BD) in accordance with the manufacturer's protocol, and then incubated with antibodies against IFNγ and IL17. Intracellular Foxp3 staining was performed using anti‐Foxp3 antibody and a Mouse Foxp3 Buffer Set (BD) in accordance with the manufacturer's protocol. Stained cells were analyzed by using a FACSCanto II flow cytometer (BD) and FlowJo software (Tree Star). TCRβhigh was defined as the brightest staining of TCRβ‐expressing cells.
Enzyme‐linked immunosorbent assay (ELISA)
Mouse IFNβ, IFNγ, and IL17 levels from culture supernatants were determined by ELISA kits according to the manufacturer's instructions (BioLegend).
Detection of apoptosis and measurement of proliferative activity
Apoptosis was detected by Annexin V staining. In brief, total thymocytes were resuspended with Annexin V Binding Buffer (BD) and incubated with APC‐conjugated Annexin V (BD) for 15 min at 25°C in the dark. Then, stained cells were analyzed using a FACSCanto II flow cytometer. In the stimulation assay, thymocytes were stimulated with anti‐CD3 antibody (R&D Systems) at the indicated concentrations together with 1/5 dilution of soluble anti‐CD28 antibodies (BioLegend) for 24 h at 37°C prior to apoptosis detection. For the measurement of proliferative activity, total thymocytes were stimulated with 5 μg/ml plate‐bound anti‐CD3 antibody (R&D Systems) and 1 μg/ml soluble anti‐CD28 antibody (BioLegend) for 72 h at 37°C. Subsequently, proliferative activity was determined by using a CellTiter 96 AQueous One Solution Cell Proliferation Assay Kit (Promega) in accordance with the manufacturer's protocol. To monitor apoptosis and proliferative activity simultaneously, total thymocytes were labeled with Cell Proliferation Dye eFluor™ 450 according to the manufacturer's protocol (eBioscience) and then stimulated with 5 μg/ml plate‐bound anti‐CD3 antibody (BioLegend) and 1 μg/ml soluble anti‐CD28 antibody (BioLegend) in the presence or absence of 10 μg/ml anti‐IFNAR1 antibody (BioLegend) for 4 days at 37°C. Then, cells were incubated with APC‐conjugated Annexin V for 15 min at 25°C in the dark and analyzed using a FACSCanto II flow cytometer. The intensity of Cell Proliferation Dye is reduced during proliferation.
Detection of TCR signal transduction
Thymocytes at a concentration of 1 × 107 cells/ml were incubated with 5 μg/ml biotinylated anti‐CD3 antibody (BioLegend) for 30 min at 4°C as previously described 59. Then, the thymocytes were washed and stimulated with 20 μg/ml streptavidin (BioLegend) for 15–60 s at 37°C. Lysis buffer 1 (1% Nonidet P‐40, 50 mM Tris–HCl, pH 7.5, 250 mM NaCl, 5 mM EDTA, 0.5 mM PMSF, 0.5 mM DTT) was quickly added to the stimulated cells. After centrifugation, the supernatants were mixed with SDS sample buffer and boiled for 5 min. The samples were stored at −20°C until use.
In vitro Treg cell suppression assay
To prepare CD4+CD45RBhighCD25− (Teff) cells and CD4+CD45RBlowCD25+ (Treg) cells, total splenocytes were stained with fluorescence‐conjugated antibodies against CD4 (RM4‐5; BD), CD45RB (C363‐16A; BioLegend), and CD25 (PC61; BioLegend) and subjected to cell sorting using an SH800 Cell Sorter (Sony). CD45RBhigh and CD45RBlow were defined as approximately 30–40% of the highest and 30–40% of the lowest populations, respectively. Isolated Teff cells were cultured for 3 days with Treg cells at the indicated ratios in the presence of 1 μg/ml soluble anti‐CD3 (BioLegend) and with mitomycin C (Nacalai Tesque)‐treated splenocytes. Subsequently, proliferative activity was determined by using a CellTiter 96 AQueous One Solution Cell Proliferation Assay Kit (Promega) in accordance with the manufacturer's protocol. Suppression was calculated using the following formula: [(Teff proliferation without Treg − Teff proliferation with Treg)/Teff proliferation without Treg] × 100.
Preparation of whole cell lysates
To prepare a suspension of single cells, the thymus was mashed through a 70‐μm cell strainer (Greiner). Then, thymocytes were lysed with Lysis buffer 2 (20 mM Tris–HCl, pH 7.9, 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM PMSF, 0.5 mM DTT), frozen in liquid nitrogen, and then thawed on ice five times. After centrifugation, the supernatants were boiled for 5 min, transferred to a new 1.5‐ml tube, and stored at −80°C until use. The tissue lysates from various organs were prepared as described previously 60.
Western blot analysis
Lysates were separated using sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis (PAGE), transferred to a polyvinylidene difluoride (PVDF) membrane (Bio‐Rad), and immunoblotted with primary antibodies using the SNAP i.d.® 2.0 Protein Detection System (Merck Millipore). The primary antibodies used for Western blotting were as follows: mouse monoclonal anti‐ADAR1 antibody (15.8.6; Santa Cruz Biotechnology), mouse monoclonal anti‐ADAR2 antibody (1.3.1; Santa Cruz Biotechnology), rabbit monoclonal anti‐MDA5 antibody (D74E4; Cell Signaling Technology), rabbit polyclonal anti‐PLCγ1 antibody (Cell Signaling Technology), rabbit monoclonal anti‐phospho‐PLCγ1 antibody (D6M9S; Cell Signaling Technology), rabbit monoclonal anti‐STAT1 antibody (D1K9Y; Cell Signaling Technology), rabbit monoclonal anti‐phospho‐STAT1 antibody (D4A7; Cell Signaling Technology), mouse monoclonal anti‐ZAP70 antibody (1E7.2; BioLegend), rabbit monoclonal anti‐phospho‐ZAP70 antibody (65E4; Cell Signaling Technology), mouse monoclonal anti‐α‐tubulin (B‐5‐1‐2; Sigma), and mouse monoclonal anti‐GAPDH (M171‐3; MBL). The intensity of each band was quantified by using ImageJ software.
Quantitative RT–PCR (qRT–PCR) analysis
To separate total thymocytes into DN, DP, 4SP, and 8SP fractions, thymocytes were stained with fluorescence‐conjugated antibodies against CD4 (RM4‐5; BD) and CD8 (53‐6.7; BD) and subjected to cell sorting using an SH800 Cell Sorter (Sony). Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific) in accordance with the manufacturer's protocol. For the qRT–PCR, an aliquot of total RNA (50–500 ng) was subjected to reverse transcription using a ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo). The qRT–PCR was performed as described previously 61 using an ABI Prism 7900HT Real‐Time PCR System (Applied Biosystems) and THUNDERBIRD Probe qPCR Mix (Toyobo) with the following primers and probes obtained from Integrated DNA Technologies: ADAR1 p150‐primer1 (5′‐AGTACGACTGTGTCTGGTGA‐3′), ADAR1 p150‐primer2 (5′‐AGGAAACGAAAGTGAACTCTGG‐3′), ADAR1 p150‐probe (5′‐/56‐FAM/CCTGAACCC/ZEN/TTGAGACATAGTGCCG/3IABkFQ/‐3′); ADAR1 p150 + p110‐primer1 (5′‐ACTACAGGACATGGTACGGA‐3′), ADAR1 p150 + p110‐primer2 (5′‐CCTGTCTTTGAAAATCCCAAGC‐3′), ADAR1 p150 + p110‐probe (5′‐/56‐FAM/TGCCTTCCC/ZEN/CATTCTCCACCTTG/3IABkFQ/‐3′); ADAR2‐primer1 (5′‐TCACCGTTGATACACTTCGTC‐3′), ADAR2‐primer2 (5′‐AGTGCTCTCTGGAGTAGTGAT‐3′), ADAR2‐probe (5′‐/56‐FAM/CCTTGGCAT/ZEN/CTTTGACATCTGTACCTGT/3IABkFQ/‐3′); Ifih1‐primer1 (5′‐CCTTCTGCACAATCCTTCTCA‐3′), Ifih1‐primer2 (5′‐TCTTGGACACTTGCTTCGAG‐3′), Ifih1‐probe (5′‐/56‐FAM/TCTCTTACA/ZEN/CCTGACTCATTCCCGCT/3IABkFQ/‐3′); Ddx58‐primer1 (5′‐CCTTGTTGTTCTTCTCAGCCT‐3′), Ddx58‐primer2 (5′‐CCACCTACATCCTCAGCTACA‐3′), Ddx58‐probe (5′‐/56‐FAM/TGTACTGCA/ZEN/CCTCCTCATCCTCGA/3IABkFQ/‐3′); Irf7‐primer1 (5′‐CCAATAGCCAGTCTCCAAACAG‐3′), Irf7‐primer2 (5′‐GCATCACAGAGTAGTAGCATCT‐3′), Irf7‐probe (5′‐/56‐FAM/CTCACTTCA/ZEN/GCCATTGCTCCGTCT/3IABkFQ/‐3′); Ifit1‐primer1 (5′‐TGAAGCAGATTCTCCATGACC‐3′), Ifit1‐primer2 (5′‐GCAAGAGAGCAGAGAGTCAAG‐3′), Ifit1‐probe (5′‐/56‐FAM/ACAGCTACC/ZEN/ACCTTTACAGCAACCAT/3IABkFQ/‐3′); Cxcl10‐primer1 (5′‐TGATTTCAAGCTTCCCTATGGC‐3′), Cxcl10‐primer2 (5′‐ATTTTCTGCCTCATCCTGCT‐3′), Cxcl10‐probe (5′‐/56‐FAM/ATCCCTCTC/ZEN/GCAAGGACGGTC/3IABkFQ/‐3′); Rsad2‐primer1 (5′‐ACGCCAACATCCAGAATAGAC‐3′), Rsad2‐primer2 (5′‐CCAGAAGATGAAAGACTCCTACC‐3′), Rsad2‐probe (5′‐/56‐FAM/CCGGTACAG/ZEN/TTCAGAAAGCGCATATATTCA/3IABkFQ/‐3′); IL10‐primer1 (5′‐ATGGCCTTGTAGACACCTTG‐3′), IL10‐primer2 (5′‐GTCATCGATTTCTCCCCTGTG‐3′), IL10‐probe (5′‐/56‐FAM/ATCACTCTT/ZEN/CACCTGCTCCACTGC/3IABkFQ/‐3′); IFNA1‐primer1 (5′‐AGGAGTGTCAAGGCTCTCTT‐3′), IFNA1‐primer2 (5′‐GCGGTGCTGAGCTACTG‐3′), IFNA1‐probe (5′‐/56‐FAM/CTGAGGTTA/ZEN/TGAGTCTGAGGAAGGTCACA/3IABkFQ/‐3′); IFNB1‐primer1 (5′‐GGCATCAACTGACAGGTCTT‐3′), IFNB1‐primer2 (5′‐ACTCATGAAGTACAACAGCTACG‐3′), IFNB1‐probe (5′‐/56‐FAM/ATCTCTGCT/ZEN/CGGACCACCATCC/3IABkFQ/‐3′); and GAPDH‐primer1 (5′‐GTGGAGTCATACTGGAACATGTAG‐3′), GAPDH‐primer2 (5′‐AATGGTGAAGGTCGGTGTG‐3′), GAPDH‐probe (5′‐/56‐FAM/TGCAAATGG/ZEN/CAGCCCTGGTG/3IABkFQ/‐3′). The expression level of each mRNA relative to that of GAPDH mRNA was calculated by the ∆∆C t method as described previously 62. In some experiments, qRT–PCR was performed using THUNDERBIRD SYBR qPCR Mix (Toyobo) with the following primers: Bcl2‐primer1 (5′‐GTGGTGGAGGAACTCTTCAGGGATG‐3′), Bcl2‐primer2 (5′‐GGTCTTCAGAGACAGCCAGGAGAAATC‐3′); Bclxl‐primer1 (5′‐GTAGTGAATGAACTCTTTCGGGATGG‐3′), Bclxl‐primer2 (5′‐ACCAGCCACAGTCATGCCCGTCAGG‐3′); Mcl‐primer1 (5′‐GTAATGGTCCATGTTTTCAAAGATG‐3′), Mcl‐primer2 (5′‐AAGCCAGCAGCACATTTCTGATGCC‐3′); Tbet‐primer1 (5′‐CAACAACCCCTTTGCCAAAG‐3′), Tbet‐primer2 (5′‐TCCCCCAAGCAGTTGACAGT‐3′); Rorγt‐primer1 (5′‐CCGCTGAGAGGGCTTCAC‐3′), Rorγt‐primer2 (5′‐TGCAGGAGTAGGCCACATTACA‐3′); Actinβ‐primer1 (5′‐GCCAACCGTGAAAAGATGAC‐3′), and Actinβ‐primer2 (5′‐CATCACAATGCCTGTGGTAC‐3′). The expression level of each mRNA relative to that of Actinβ mRNA was calculated as described above.
Total RNA‐sequencing analysis
Total RNA was extracted using TRIzol reagent in accordance with the manufacturer's instructions. After ribosomal RNAs was removed using a Ribo‐Zero rRNA Removal Kit (Illumina), a strand‐specific RNA library was prepared using SureSelect Strand‐Specific RNA (Agilent) in accordance with the manufacturer's instructions. The library samples were then subjected to deep sequencing using a HiSeq 3000 Sequencing System (Illumina).
Mapping and analyzing RNA‐seq data to identify editing sites
We analyzed the RNA‐seq data as described previously 8, 45 with some modifications. First, a fast and sensitive spliced alignment program HISAT2 63 was used to map RNA‐seq reads onto the mm9/NCBIM37 mouse genomic sequence obtained from Ensembl 64. Potential PCR duplicates were then removed from the resulting alignments by using Samtools rmdup 65. Before calling variants from the results, GATK IndelRealigner and BaseRecalibrator 66 were applied to perform insertion/deletion (indel) local realignment and base recalibration, respectively, to filter out as many false‐positive variants as possible. These processes were done using information of known indels and single nucleotide polymorphisms (SNPs) 67. Then, we ran GATK HaplotypeCaller to obtain variants compared with the reference genome, which were subsequently filtered by using our own criteria. More specifically, we removed (i) variants that overlapped with known indels and SNPs, and (ii) variants with quality depth (QD) <2, Fisher strand (FS) >30, root mean square of the mapping quality (MQ) <40, and variant call quality (QUAL) <20 for SNPs, and QD <2 and FS >200 for indels. The remaining set of variants was annotated by running ANNOVAR 68 with the RefSeq gene model. Samtools mpileup along with our own Python script was used to add the depth of coverage and editing ratio to each editing site in the annotated file. It should be noted that the set of variants reported by GATK is on the forward strand only, and thus, the results need to contain both A‐to‐G variants and T‐to‐C variants, which might be located on the reverse strand as A‐to‐G. The results were compared with the RepeatMasker output for mice available from the UCSC Genome Browser 69 to determine whether the detected editing sites were in repeat regions.
Gene ontology analysis
The genes that contained potential RNA editing sites were subjected to GO enrichment using DAVID 70.
Validation of RNA editing by Sanger sequencing
The editing frequency of each specific site was determined as described previously 60. In brief, 250 ng of each total RNA was treated with 0.1 U/μl DNase I (Thermo Fisher Scientific) at 37°C for 15 min and denatured at 65°C for 15 min. Reverse transcription was performed using the SuperScript III First‐Strand Synthesis System (Thermo Fisher Scientific) with random hexamers (Thermo Fisher Scientific). Then, synthesized cDNA was amplified with Phusion Hot Start High‐Fidelity DNA Polymerase (Thermo Fisher Scientific) and the following primers: Cds2, 5′‐CCAAGGGCTTGCATGCATTA‐3′ and 5′‐CAGTCTCAACCACGGGGAAA‐3′; Ift80, 5′‐ACCTGTGCTCATACCGTCAA‐3′ and 5′‐TGCTGCCTTCCCTTGTGAAA‐3′; Nras, 5′‐TTGGAGGCTTGATGTTTGCC‐3′ and 5′‐GGTCTCAGCAGCTTCAGGAA‐3′; Slc35a3, 5′‐GCTCAAGGACCTGGCAAGTA‐3′ and 5′‐CCAAACCAAGGGTAGCCACT‐3′; Rpa1, 5′‐ACCTTGTTGCCATTCTAAGCA‐3′ and 5′‐GGGAGCAAGTACCACTGGTG‐3′; Tbce, 5′‐CCCTGCAGGAGTTTGTGACA‐3′ and 5′‐ACCACTTCATCCCAAGTTGACA‐3′; and Malt1, 5′‐AAGAATGATGTGTGGGGCCT‐3′ and 5′‐TGAGACTTCAGCTGTGCTCC‐3′. After cleanup with illustra ExoProStar (GE Healthcare), the PCR products were directly sequenced using the following primers: Cds2, 5′‐TGCATGCATTATCTCCTTTAATCCAC‐3′; Ift80, 5′‐CCGTCAAGTTCTCTTCTGCTCTT‐3′; Nras, 5′‐TGCCCTGATAATTCATTAGTGGGT‐3′; Slc35a3, 5′‐GGCAAGTAGAGAAGTGTCAAAGG‐3′; Rpa1, 5′‐TGGTGGCCAGAACGGAAGA‐3′; Tbce, 5′‐TGACACTTGGTTGTCCTTTGC‐3′; and Malt1, 5′‐TGTGGGGCCTGTAAATATTGCT‐3′. The editing frequency was calculated as the % ratio of the “G” peak over the sum of the “G” and “A” peaks in the sequencing chromatogram.
Statistical analyses
The Mann–Whitney U‐test or Fisher's exact test was used as described in each figure legend. All values are displayed as the mean ± standard error of the mean (SEM). Statistical significance is displayed as P < 0.05 (*) or P < 0.01 (**).
Data availability
The RNA‐seq data used in this study are available through the DNA Data Bank of Japan (DDBJ) under accession number http://info:x-wiley/ena/DRA005762? (http://trace.ddbj.nig.ac.jp/DRASearch/submission?acc=DRA005762).
Author contributions
TN and YKaw designed the study. CRW provided the A1KI/KI mice and discussed the results with TN and YKaw. YS performed the RNA‐seq analysis, and YKat performed all the bioinformatics analysis. TN, JIK, TV, and YKaw performed all the other experiments, and YKaw conceived and discussed the projects. TN and YKaw wrote the manuscript. All authors agree to the content of the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Review Process File
Acknowledgements
We thank Drs. K. Nishikura (The Wistar Institute) and O. Takeuchi (Kyoto University) for providing A1 fl/fl mice and Cd4 cre mice, respectively. We thank Drs K. Takeda, H. Kayama, and I. Chinen for technical advice and all the staff in the Center for Medical Research and Education, Graduate School of Medicine, Osaka University, for technical support. Computations were partially performed on the NIG Supercomputer at ROIS National Institute of Genetics, Japan. This work was supported by Grants‐in‐Aid KAKENHI (25293201, 25110719, and 16H06279 to Y. Kawahara; 18K15186, 16H06279, 15K19126, and 255768 to T.N.; and 18K11526 and 15K00401 to Y. Kato) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan and by grants from the Takeda Science Foundation, the SENSHIN Medical Research Foundation, and the Mochida Memorial Foundation for Medical and Pharmaceutical Research (to Y. Kawahara), Nagao Memorial Fund (to T.N.), and Victorian Cancer Agency Mid‐Career Fellowship (to C.R.W.).
EMBO Reports (2018) 19: e46303
See also: https://doi.org/10.15252/embr.201847237 (December 2018)
Contributor Information
Taisuke Nakahama, Email: nakahama@rna.med.osaka-u.ac.jp.
Yukio Kawahara, Email: ykawahara@rna.med.osaka-u.ac.jp.
References
- 1. Zipeto MA, Jiang Q, Melese E, Jamieson CH (2015) RNA rewriting, recoding, and rewiring in human disease. Trends Mol Med 21: 549–559 [DOI] [PubMed] [Google Scholar]
- 2. Nishikura K (2010) Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79: 321–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. George CX, John L, Samuel CE (2014) An RNA editor, adenosine deaminase acting on double‐stranded RNA (ADAR1). J Interferon Cytokine Res 34: 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, Fligelman ZY, Shoshan A, Pollock SR, Sztybel D et al (2004) Systematic identification of abundant A‐to‐I editing sites in the human transcriptome. Nat Biotechnol 22: 1001–1005 [DOI] [PubMed] [Google Scholar]
- 5. Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA et al (2012) Mutations in ADAR1 cause Aicardi‐Goutieres syndrome associated with a type I interferon signature. Nat Genet 44: 1243–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hartner JC, Walkley CR, Lu J, Orkin SH (2009) ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol 10: 109–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Q, Khillan J, Gadue P, Nishikura K (2000) Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 290: 1765–1768 [DOI] [PubMed] [Google Scholar]
- 8. Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR (2015) RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ et al (2014) The RNA‐editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB (2015) Isoforms of RNA‐editing enzyme ADAR1 independently control nucleic acid sensor MDA5‐driven autoimmunity and multi‐organ development. Immunity 43: 933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kawai T, Akira S (2006) Innate immune recognition of viral infection. Nat Immunol 7: 131–137 [DOI] [PubMed] [Google Scholar]
- 12. Loo YM, Gale M Jr (2011) Immune signaling by RIG‐I‐like receptors. Immunity 34: 680–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hartner JC, Schmittwolf C, Kispert A, Muller AM, Higuchi M, Seeburg PH (2004) Liver disintegration in the mouse embryo caused by deficiency in the RNA‐editing enzyme ADAR1. J Biol Chem 279: 4894–4902 [DOI] [PubMed] [Google Scholar]
- 14. Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K (2004) Stress‐induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem 279: 4952–4961 [DOI] [PubMed] [Google Scholar]
- 15. Marcu‐Malina V, Goldberg S, Vax E, Amariglio N, Goldstein I, Rechavi G (2016) ADAR1 is vital for B cell lineage development in the mouse bone marrow. Oncotarget 7: 54370–54379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cooper MD, Alder MN (2006) The evolution of adaptive immune systems. Cell 124: 815–822 [DOI] [PubMed] [Google Scholar]
- 17. Germain RN (2002) T‐cell development and the CD4‐CD8 lineage decision. Nat Rev Immunol 2: 309–322 [DOI] [PubMed] [Google Scholar]
- 18. Starr TK, Jameson SC, Hogquist KA (2003) Positive and negative selection of T cells. Annu Rev Immunol 21: 139–176 [DOI] [PubMed] [Google Scholar]
- 19. Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S (2002) Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol 20: 621–667 [DOI] [PubMed] [Google Scholar]
- 20. Ley K, Kansas GS (2004) Selectins in T‐cell recruitment to non‐lymphoid tissues and sites of inflammation. Nat Rev Immunol 4: 325–335 [DOI] [PubMed] [Google Scholar]
- 21. Sakaguchi S, Yamaguchi T, Nomura T, Ono M (2008) Regulatory T cells and immune tolerance. Cell 133: 775–787 [DOI] [PubMed] [Google Scholar]
- 22. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C et al (2002) Projection of an immunological self shadow within the thymus by the aire protein. Science 298: 1395–1401 [DOI] [PubMed] [Google Scholar]
- 23. Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC (2003) Aire regulates negative selection of organ‐specific T cells. Nat Immunol 4: 350–354 [DOI] [PubMed] [Google Scholar]
- 24. Sakaguchi N, Takahashi T, Hata H, Nomura T, Tagami T, Yamazaki S, Sakihama T, Matsutani T, Negishi I, Nakatsuru S et al (2003) Altered thymic T‐cell selection due to a mutation of the ZAP‐70 gene causes autoimmune arthritis in mice. Nature 426: 454–460 [DOI] [PubMed] [Google Scholar]
- 25. Zhu J, Yamane H, Paul WE (2010) Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 28: 445–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johnson DJ, Pao LI, Dhanji S, Murakami K, Ohashi PS, Neel BG (2013) Shp1 regulates T cell homeostasis by limiting IL‐4 signals. J Exp Med 210: 1419–1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xing Y, Wang X, Jameson SC, Hogquist KA (2016) Late stages of T cell maturation in the thymus involve NF‐kappaB and tonic type I interferon signaling. Nat Immunol 17: 565–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Azzam HS, Grinberg A, Lui K, Shen H, Shores EW, Love PE (1998) CD5 expression is developmentally regulated by T cell receptor (TCR) signals and TCR avidity. J Exp Med 188: 2301–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ (2001) Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self‐peptide. Nat Immunol 2: 301–306 [DOI] [PubMed] [Google Scholar]
- 30. Kisielow P, Bluthmann H, Staerz UD, Steinmetz M, von Boehmer H (1988) Tolerance in T‐cell‐receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature 333: 742–746 [DOI] [PubMed] [Google Scholar]
- 31. Samelson LE (2002) Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu Rev Immunol 20: 371–394 [DOI] [PubMed] [Google Scholar]
- 32. Kiran A, Baranov PV (2010) DARNED: a DAtabase of RNa EDiting in humans. Bioinformatics 26: 1772–1776 [DOI] [PubMed] [Google Scholar]
- 33. Zella D, Romerio F, Curreli S, Secchiero P, Cicala C, Zagury D, Gallo RC (2000) IFN‐alpha 2b reduces IL‐2 production and IL‐2 receptor function in primary CD4+ T cells. J Immunol 164: 2296–2302 [DOI] [PubMed] [Google Scholar]
- 34. Siegal FP, Kadowaki N, Shodell M, Fitzgerald‐Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ (1999) The nature of the principal type 1 interferon‐producing cells in human blood. Science 284: 1835–1837 [DOI] [PubMed] [Google Scholar]
- 35. Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M (1999) Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med 5: 919–923 [DOI] [PubMed] [Google Scholar]
- 36. Colonna M, Trinchieri G, Liu YJ (2004) Plasmacytoid dendritic cells in immunity. Nat Immunol 5: 1219–1226 [DOI] [PubMed] [Google Scholar]
- 37. Bromberg JF, Horvath CM, Wen Z, Schreiber RD, Darnell JE Jr (1996) Transcriptionally active Stat1 is required for the antiproliferative effects of both interferon alpha and interferon gamma. Proc Natl Acad Sci USA 93: 7673–7678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tanabe Y, Nishibori T, Su L, Arduini RM, Baker DP, David M (2005) Cutting edge: role of STAT1, STAT3, and STAT5 in IFN‐alpha beta responses in T lymphocytes. J Immunol 174: 609–613 [DOI] [PubMed] [Google Scholar]
- 39. Lizio M, Harshbarger J, Abugessaisa I, Noguchi S, Kondo A, Severin J, Mungall C, Arenillas D, Mathelier A, Medvedeva YA et al (2017) Update of the FANTOM web resource: high resolution transcriptome of diverse cell types in mammals. Nucleic Acids Res 45: D737–D743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Irla M, Hollander G, Reith W (2010) Control of central self‐tolerance induction by autoreactive CD4+ thymocytes. Trends Immunol 31: 71–79 [DOI] [PubMed] [Google Scholar]
- 41. Colantonio AD, Epeldegui M, Jesiak M, Jachimowski L, Blom B, Uittenbogaart CH (2011) IFN‐alpha is constitutively expressed in the human thymus, but not in peripheral lymphoid organs. PLoS One 6: e24252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Otero DC, Baker DP, David M (2013) IRF7‐dependent IFN‐beta production in response to RANKL promotes medullary thymic epithelial cell development. J Immunol 190: 3289–3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. XuFeng R, Boyer MJ, Shen H, Li Y, Yu H, Gao Y, Yang Q, Wang Q, Cheng T (2009) ADAR1 is required for hematopoietic progenitor cell survival via RNA editing. Proc Natl Acad Sci USA 106: 17763–17768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ben‐Shoshan SO, Kagan P, Sultan M, Barabash Z, Dor C, Jacob‐Hirsch J, Harmelin A, Pappo O, Marcu‐Malina V, Ben‐Ari Z et al (2016) ADAR1 deletion induces NFkappaB and interferon signaling dependent liver inflammation and fibrosis. RNA Biol 14: 587–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liddicoat BJ, Hartner JC, Piskol R, Ramaswami G, Chalk AM, Kingsley PD, Sankaran VG, Wall M, Purton LE, Seeburg PH et al (2016) Adenosine‐to‐inosine RNA editing by ADAR1 is essential for normal murine erythropoiesis. Exp Hematol 44: 947–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Besch R, Poeck H, Hohenauer T, Senft D, Hacker G, Berking C, Hornung V, Endres S, Ruzicka T, Rothenfusser S et al (2009) Proapoptotic signaling induced by RIG‐I and MDA‐5 results in type I interferon‐independent apoptosis in human melanoma cells. J Clin Invest 119: 2399–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Anz D, Thaler R, Stephan N, Waibler Z, Trauscheid MJ, Scholz C, Kalinke U, Barchet W, Endres S, Bourquin C (2009) Activation of melanoma differentiation‐associated gene 5 causes rapid involution of the thymus. J Immunol 182: 6044–6050 [DOI] [PubMed] [Google Scholar]
- 48. Dann A, Poeck H, Croxford AL, Gaupp S, Kierdorf K, Knust M, Pfeifer D, Maihoefer C, Endres S, Kalinke U et al (2011) Cytosolic RIG‐I‐like helicases act as negative regulators of sterile inflammation in the CNS. Nat Neurosci 15: 98–106 [DOI] [PubMed] [Google Scholar]
- 49. Papadopoulou AS, Dooley J, Linterman MA, Pierson W, Ucar O, Kyewski B, Zuklys S, Hollander GA, Matthys P, Gray DH et al (2011) The thymic epithelial microRNA network elevates the threshold for infection‐associated thymic involution via miR‐29a mediated suppression of the IFN‐alpha receptor. Nat Immunol 13: 181–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dempoya J, Matsumiya T, Imaizumi T, Hayakari R, Xing F, Yoshida H, Okumura K, Satoh K (2012) Double‐stranded RNA induces biphasic STAT1 phosphorylation by both type I interferon (IFN)‐dependent and type I IFN‐independent pathways. J Virol 86: 12760–12769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wu X, Dao TVL, Huang Y, Billerbeck E, Saha D, Hoffmann HH, Wang Y, Silva LAV, Sarbanes S, Sun T et al (2018) Intrinsic immunity shapes viral resistance of stem cells. Cell 172: 423–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Crow YJ, Manel N (2015) Aicardi‐Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 15: 429–440 [DOI] [PubMed] [Google Scholar]
- 53. Morita M, Stamp G, Robins P, Dulic A, Rosewell I, Hrivnak G, Daly G, Lindahl T, Barnes DE (2004) Gene‐targeted mice lacking the Trex1 (DNase III) 3′–>5′ DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol 24: 6719–6727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stetson DB, Ko JS, Heidmann T, Medzhitov R (2008) Trex1 prevents cell‐intrinsic initiation of autoimmunity. Cell 134: 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G et al (2015) Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 167A: 296–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Burich A, Hershberg R, Waggie K, Zeng W, Brabb T, Westrich G, Viney JL, Maggio‐Price L (2001) Helicobacter‐induced inflammatory bowel disease in IL‐10‐ and T cell‐deficient mice. Am J Physiol Gastrointest Liver Physiol 281: G764–G778 [DOI] [PubMed] [Google Scholar]
- 57. Jeon SG, Kayama H, Ueda Y, Takahashi T, Asahara T, Tsuji H, Tsuji NM, Kiyono H, Ma JS, Kusu T et al (2012) Probiotic Bifidobacterium breve induces IL‐10‐producing Tr1 cells in the colon. PLoS Pathog 8: e1002714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nakahama T, Hanieh H, Nguyen NT, Chinen I, Ripley B, Millrine D, Lee S, Nyati KK, Dubey PK, Chowdhury K et al (2013) Aryl hydrocarbon receptor‐mediated induction of the microRNA‐132/212 cluster promotes interleukin‐17‐producing T‐helper cell differentiation. Proc Natl Acad Sci USA 110: 11964–11969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhou XY, Yashiro‐Ohtani Y, Toyo‐Oka K, Park CS, Tai XG, Hamaoka T, Fujiwara H (2000) CD5 costimulation up‐regulates the signaling to extracellular signal‐regulated kinase activation in CD4+CD8+ thymocytes and supports their differentiation to the CD4 lineage. J Immunol 164: 1260–1268 [DOI] [PubMed] [Google Scholar]
- 60. Miyake K, Ohta T, Nakayama H, Doe N, Terao Y, Oiki E, Nagatomo I, Yamashita Y, Abe T, Nishikura K et al (2016) CAPS1 RNA editing promotes dense core vesicle exocytosis. Cell Rep 17: 2004–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yokoshi M, Li Q, Yamamoto M, Okada H, Suzuki Y, Kawahara Y (2014) Direct binding of Ataxin‐2 to distinct elements in 3′ UTRs promotes mRNA stability and protein expression. Mol Cell 55: 186–198 [DOI] [PubMed] [Google Scholar]
- 62. Nakahama T, Kimura A, Nguyen NT, Chinen I, Hanieh H, Nohara K, Fujii‐Kuriyama Y, Kishimoto T (2011) Aryl hydrocarbon receptor deficiency in T cells suppresses the development of collagen‐induced arthritis. Proc Natl Acad Sci USA 108: 14222–14227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho‐Silva D, Clapham P, Coates G, Fitzgerald S et al (2015) Ensembl 2015. Nucleic Acids Res 43: D662–D669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M et al (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29: 308–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang K, Li M, Hakonarson H (2010) ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 38: e164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M et al (2015) The UCSC Genome Browser database: 2015 update. Nucleic Acids Res 43: D670–D681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Review Process File
Data Availability Statement
The RNA‐seq data used in this study are available through the DNA Data Bank of Japan (DDBJ) under accession number http://info:x-wiley/ena/DRA005762? (http://trace.ddbj.nig.ac.jp/DRASearch/submission?acc=DRA005762).