

Abstract
Purpose
Low plasma ACTH in critically ill patients may be explained by shock/inflammation-induced hypothalamus-pituitary damage or by feedback inhibition exerted by elevated plasma free cortisol. One can expect augmented/prolonged ACTH-responses to CRH injection with hypothalamic damage, immediately suppressed responses with pituitary damage, and delayed decreased responses in prolonged critical illness with feedback inhibition.
Methods
This randomized, double-blind, placebo-controlled crossover cohort study, compared ACTH responses to 100 µg IV CRH and placebo in 3 cohorts of 40 matched patients in the acute (ICU-day 3–6), subacute (ICU-day 7–16) or prolonged phase (ICU-day 17–28) of critical illness, with 20 demographically matched healthy subjects. CRH or placebo was injected in random order on two consecutive days. Blood was sampled repeatedly over 135 min and AUC responses to placebo were subtracted from those to CRH.
Results
Patients had normal mean ± SEM plasma ACTH concentrations (25.5 ± 1.6 versus 24.8 ± 3.6 pg/ml in healthy subjects, P = 0.54) but elevated free cortisol concentrations (3.11 ± 0.27 versus 0.58 ± 0.05 µg/dl in healthy subjects, P < 0.0001). The order of the CRH/placebo injections did not affect the ACTH responses, hence results were pooled. Patients in the acute phase of illness had normal mean ± SEM ACTH responses (5149 ± 848 pg/mL min versus 4120 ± 688 pg/mL min in healthy subjects; P = 0.77), whereas those in the subacute (2333 ± 387 pg/mL min, P = 0.01) and prolonged phases (2441 ± 685 pg/mL min, P = 0.001) were low, irrespective of sepsis/septic shock or risk of death.
Conclusions
Suppressed ACTH responses to CRH in the more prolonged phases, but not acute phase, of critical illness are compatible with feedback inhibition exerted by elevated free cortisol, rather than by cellular damage to hypothalamus and/or pituitary.
Electronic supplementary material
The online version of this article (10.1007/s00134-018-5427-y) contains supplementary material, which is available to authorized users.
Keywords: Septic shock, CRH, Hypothalamus, ACTH, Cortisol, Pituitary
Take-home message
| Prolonged feedback inhibition exerted by sustained elevated free cortisol, and not inflammation/shock induced hypothalamic or pituitary cell damage, explained suppressed ACTH responses to CRH exclusively in the more prolonged phases of critical illness. |
Introduction
Patients suffering from critical illnesses typically reveal high plasma (free)cortisol concentrations and low-normal plasma adrenocorticotropic hormone (ACTH). The absence of elevated plasma ACTH, particularly in patients with severe infections, has been interpreted as caused by inflammation or hypoperfusion-induced damage to cells of the hypothalamus whereby synthesis of corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) is hampered [1–4]. Shock or inflammation could also directly damage the anterior pituitary gland [4]. Also, direct inhibition at the hypothalamus and/or pituitary level by various drugs have been suggested [2, 4, 5]. However, an alternative explanation could be that high circulating free cortisol levels, brought about by suppressed cortisol binding proteins and by reduced cortisol breakdown [6], exert negative feedback inhibition at the pituitary and/or the hypothalamic level, as such lowering ACTH, CRH, and AVP expression/secretion [7]. Nevertheless, during critical illness, ACTH secretion is not completely suppressed unlike what is observed with high doses of exogenous glucocorticoids or in patients with adrenal Cushing’s syndrome [8, 9]. This could be explained by other concomitant central activation, such as via stress induced AVP increase which could potentiate CRH effects [9–12]. Also, during the first weeks of critical illness, the frequency of the ACTH and cortisol pulses was found to be normal, whereas pulse amplitudes were lower than normal [13]. However, a recent study has shown that a central suppression of ACTH is present during critical illness [14]. Suppressed ACTH sustained over an extended period of time could predispose to adrenocortical atrophy and dysfunction [15].
Differentiation between hypothalamic lesions, damage to the pituitary corticotropes and adrenal/ectopic causes of Cushing’s syndrome can be done by analyzing plasma ACTH (and cortisol) responses to an intravenous CRH bolus injection [16]. If during critical illness, the hypothalamus would be acutely damaged by shock or inflammation, and the anterior pituitary gland would be intact, one would expect augmented/prolonged ACTH responses [17]. If the pituitary would be acutely damaged by shock or inflammation, suppressed ACTH responses would be expected from the early phase onward [17]. Alternatively, if ACTH is suppressed by feedback inhibition at the level of the pituitary and hypothalamus, as in patients with adrenal/ectopic Cushing’s syndrome or on high doses of glucocorticoids, the ACTH responses to a CRH injection expectedly depend on the duration of hypercortisolism, with initially normal ACTH responses to CRH injection followed by lowered ACTH responses in the prolonged phase of illness [18]. Although the test is commonly used in the setting of Cushing’s syndrome, only few studies have been performed in critically ill patients, none of which investigated the impact of duration of illness [19–22].
We hypothesized that a longer duration of elevated circulating free cortisol, brought about by suppressed cortisol binding proteins and by reduced cortisol breakdown, reduces ACTH responses to a CRH injection specifically in the prolonged phase of critical illness, irrespective of the presence of sepsis/septic shock and irrespective of risk of death. To test this hypothesis, we performed a randomized, double-blind, placebo-controlled crossover cohort study to compare the ACTH (and cortisol) responses to a synthetic human CRH-analogue, in the acute, subacute and prolonged phases of critical illness with those of healthy subjects, in relation to presence of sepsis/septic shock and risk of death.
Methods
Study participants and sample size calculation
This randomized, double-blind, placebo-controlled crossover cohort study was performed in 5 medical/surgical ICUs at the University Hospitals of Leuven, Belgium. The study aimed at comparing 3 cohorts of unique adult (age ≥ 18 years) critically ill patients, matched for demographics, comorbidities and type/severity of critical illness upon ICU admission (Table 1), assessed in the acute (ICU day 3–6), subacute (ICU day 7–16) or prolonged phase (ICU day 17–28) of critical illness, with demographically matched healthy control subjects. Time cohorts were a priori chosen based on (a) the increasing risk of developing hypercortisolemia-induced central suppression with increasing duration of exogenous glucocorticoid treatment which necessitates treatment tapering rather than stopping [23] and (b) on the observation that adrenal glands harvested from prolonged ICU patients who died after a median (IQR) ICU stay of 16 (13–21) days showed signs of adrenal atrophy, that was absent when patients died after an ICU stay of 2 (1–5) days [15]. All patients with a stabilized condition, after resuscitation, for at least 48 h as judged by the treating physician, and with an expected stay in ICU for at least another 48 h, were screened for eligibility. Exclusion criteria were (details provided in the Supplementary Material) treatment with systemic glucocorticoids, etomidate, azoles or other drugs predisposing to adrenal insufficiency, no vital organ support, no arterial or central venous catheter in place, referral from another ICU, cerebral/pituitary/adrenal disorders with impact on the neuroendocrine system, being pregnant or nursing, enrollment in another trial, or expected death within 12 h (Fig. 1a).
Table 1.
Participant characteristics
| Healthy subjects (n = 20) |
P value* | Acute phase of cie (n = 40) |
Subacute phase of cie (n = 40) |
Prolonged phase of cie (n = 40) |
P value** | |
|---|---|---|---|---|---|---|
| Demography and anthropometry | ||||||
| Male gender—no. (%) | 13 (65) | 0.94 | 28 (70) | 24 (60) | 25 (63) | 0.62 |
| Age—year (mean ± SEM) | 63 ± 3 | 0.58 | 67 ± 2 | 65 ± 2 | 62 ± 2 | 0.30 |
| BMIa—kg/m2 (mean ± SEM) | 26.2 ± 0.7 | 0.94 | 25.2 ± 0.8 | 25.6 ± 0.8 | 27.6 ± 1.3 | 0.20 |
| Admission characteristics | ||||||
| Diabetes mellitus—no. (%) | 6 (15) | 6 (15) | 8 (20) | 0.79 | ||
| Malignancy—no. (%) | 6 (15) | 12 (30) | 13 (33) | 0.14 | ||
| APACHE II scoreb—(mean ± SEM) | 30 ± 1 | 31 ± 1 | 31 ± 1 | 0.78 | ||
| Emergency admission—no. (%) | 8 (20) | 8 (20) | 9 (23) | 0.95 | ||
| Diagnostic admission categories | 1.00 | |||||
| Cardiac surgery—no. (%) | 9 (23) | 9 (23) | 9 (23) | |||
| Complicated other surgery—no. (%) | 10 (25) | 10 (25) | 10 (25) | |||
| Multiple trauma and burns—no. (%) | 16 (40) | 16 (40) | 16 (40) | |||
| Medical—no. (%) | 5 (12) | 5 (12) | 5 (12) | |||
| ICUc day on testday 1—median and IQR | 4 (3–5) | 9 (7–12) | 19 (17–22) | < 0.0001 | ||
| Patient characteristics on testday 1 | ||||||
| On mechanical ventilatory support—no. (%) | 35 (88) | 30 (75) | 25 (63) | 0.03 | ||
| On renal replacement therapy—no. (%) | 6 (15) | 5 (13) | 6 (15) | 0.93 | ||
| Infection—no. (%) | 30 (75) | 32 (80) | 33 (83) | 0.70 | ||
| Sepsisd—no. (%) | 27 (68) | 30 (75) | 30 (75) | 0.69 | ||
| Septic shockd—no. (%) | 21 (53) | 15 (38) | 19 (48) | 0.38 | ||
| Plasma ACTH—pg/mL [median (IQR)] | 21 (15–33) | 0.54 | 14 (11–32) | 19 (14–27) | 27 (17–41) | 0.009 |
| Plasma total cortisol—µg/dL [median (IQR)] | 13 (11–16) | < 0.0001 | 24 (16–33) | 24 (20–31) | 23 (17–29) | 0.47 |
| Plasma free cortisol—µg/dL [median (IQR)] | 0.5 (0.4–0.7) | < 0.0001 | 2.6 (1.2–4.6) | 2.1 (1.4–4.5) | 2.0 (1.1–3.0) | 0.26 |
| Plasma CBG—µg/mL (mean ± SEM) | 52 ± 2 | < 0.0001 | 37 ± 1 | 41 ± 1 | 43 ± 1 | 0.002 |
| Plasma albumin—g/dL (mean ± SEM) | 6.4 ± 0.1 | < 0.0001 | 3.9 ± 0.2 | 3.9 ± 0.1 | 3.8 ± 0.1 | 0.86 |
| Clinical outcomes | ||||||
| Days in ICU—median (IQR) | 11 (8–18) | 18 (13–26) | 30 (28–44) | < 0.0001 | ||
| ICU non-survivor—no. (%) | 8 (20) | 9 (23) | 9 (23) | 0.95 | ||
*The comparison between healthy subjects and all patients
**The comparison between patient cohorts. For all patients combined, total median (IQR) plasma ACTH was 20 (13–35) pg/ml, plasma cortisol 24 (18–30) µg/dL and free cortisol 2.3 (1.3–3.4) µg/dL
aThe body-mass index (BMI) is the weight in kilograms divided by the square of the height in meters
bThe Acute Physiology and Chronic Health Evaluation II (APACHE II) score reflects severity of illness, with higher values indicating more severe illness, and can range from 0 to 71 [50]
cICU denotes intensive care unit
dIncidence of sepsis and septic shock was defined according to [51, 52]
eci denotes critical illness
Fig. 1.

Flowchart of the study participants and study design. a Flowchart of the study participants. b Randomization into crossover subgroups. ICU denotes intensive care unit. *Blood sample
The sample size of the study was determined based on an estimated effect size of a long duration of critical illness on the ACTH responses to corticorelin, a synthetic human CRH analogue that is further referred to as CRH. Twenty unique patients per cohort would allow to detect, with an alpha error of 1% or less and a power of 80% or more, a suppression of the ACTH response to CRH in long-stay critically ill patients of the same size (± 60% decrease) as previously reported for Cushing’s patients on replacement hydrocortisone treatment 7–9 days after surgical removal of the tumor, in comparison with the response of 20 healthy volunteers [18]. To further account for confounding by various illness-related aspects, the required number of patients was doubled to 40 unique patients per cohort (total of 120) (Fig. 1a). Recruitment of patients was performed in permuted blocks of 10. If a patient, who had been included in a certain time cohort was still in ICU and eligible for including in a later time cohort, this patient was tested again. That later test was used as the unique test if the respective block of 10 was not yet completed. This was done for pragmatic reasons, as recruitment for the first cohorts goes faster due to the larger available patient population. The results from the repeated tests within the same patient were not included in the primary analysis but were analyzed separately as a secondary, additional, longitudinal analysis of the impact of duration of illness. Screening for eligible patients started on July 1, 2016, and continued until the preset number of 40 patients in all 3 cohorts was reached (May 10, 2018), with comparable proportions of 4 diagnostic categories (Table 1; Fig. 1a). The study protocol was in accordance with the 1964 Declaration of Helsinki and its later amendments, was approved by the Institutional Ethical Review Board (S58941) and made available prior to study start (ISRCTN14587520).
Clinical data, study design, and sample collection
Demographic, ICU admission and patient characteristics at study inclusion in a time cohort, and patient outcomes were documented (Table 1). After obtaining written informed consent from the healthy volunteers and from the patients or the patients’ next of kin, intravenous injections of either 100 µg of the synthetic human CRH analogue (CRH Ferring®) in 1 ml 0.9%NaCl or of placebo (1 ml 0.9%NaCl) were given on two consecutive days at 11:00 AM, in a random order (Fig. 1b). Concealment of order assignment was ensured by the use of a central computerized randomization system. The randomization was stratified in permuted blocks of 2 according to cohort number and the 4 diagnostic admission categories. The block size was unknown to the medical and research teams. Members from the clinical staff who were not involved in the study or patient care, were responsible for preparation and blinding of study medication. Patients, healthy subjects, and the research team were blinded for CRH or placebo injection. For sample collection see Supplementary Material. Plasma ACTH concentrations were measured with a double-monoclonal immunoradiometric assay (Brahms Diagnostics, Berlin). Total plasma cortisol concentrations (Immunotech, Prague, Czech Republic) and plasma cortisol-binding-globulin (CBG) concentrations (Riazen, Louvain-La-Neuve) were quantified by competitive radio-immunoassay. Plasma albumin was quantified by the bromocresol green colorimetric method (Sigma-Aldrich, St. Louis, MO). Plasma free cortisol was calculated using the Coolens’ formula adapted for albumin and CBG concentrations, which has been previously validated as representative of measured free cortisol concentrations in the ICU context [13, 24].
Data and statistical analyses
Within the crossover design, each patient or healthy subject served as his/her own control. First, it was investigated whether the order of administration of placebo and CRH affected the hormonal responses and if this was not the case, the results for placebo and CRH could be pooled for further analysis. To determine the change in the area under the curve (AUC) of plasma ACTH and (free)cortisol in response to placebo or CRH, the plasma concentrations of sample 1 and 2 (before injection) were averaged and served as baseline, after which the AUC was calculated by the trapezoidal rule, on the placebo and the CRH test day. The AUC of the hormone responses to placebo were than subtracted from the AUC of the hormone responses to CRH, to determine the “delta AUC”, which is further referred to as the “incremental hormone response”. In addition, plasma half-life of ACTH and of cortisol were estimated by dividing ln2 by the estimated elimination rate constant, calculated from the slope of the regression line of the log-transformed linear decline of the concentration over time [25].
All data are presented as mean ± standard error of the mean (SEM), median and interquartile range (IQR), or numbers and percentages. Comparisons of normally distributed data were performed with use of unpaired Student’s t tests, and Wilcoxon rank-sum test was used to compare non-normally distributed data. Proportions were compared with the use of Chi-square tests. To compare time-series, repeated measures ANOVA was used, where necessary after transformation to obtain a near-normal distribution. Statistical analyses were performed with use of JMP® Pro 13.0.0 (SAS Institute, Cary, NC, USA). Two-sided P values at or below 0.05 were considered to indicate statistical significance.
Results
Patient characteristics and baseline plasma concentrations of ACTH and (free)cortisol
One hundred and twenty critically ill patients and 20 healthy subjects were studied (Table 1). The 3 time cohorts (median 4 days, 9 days or 19 days in ICU) had equal proportions of patients within the 4 admission diagnostic categories and of emergency admissions and had similar admission APACHE II scores. For each time cohort, as compared with healthy subjects, patients had similar morning plasma ACTH concentrations, higher plasma total and free cortisol concentrations, lower plasma cortisol binding proteins (CBG and albumin) concentrations (Table 1). With increasing time in ICU, plasma ACTH and CBG concentrations increased slightly, whereas plasma total and free cortisol remained high and albumin concentrations remained low. Of the 120 patients, 87 (73%) suffered from sepsis and 55 (46%) suffered from septic shock at study inclusion, 26 (22%) patients died in the ICU, and 42 (35%) died while in hospital.
Plasma incremental ACTH responses to CRH over time in ICU
For patients as well as healthy subjects, the order of the CRH/placebo injections did not affect the ACTH responses (P = 0.15 for the acute phase, P = 0.08 for the subacute phase, P = 1.00 for the prolonged phase, and P = 0.16 for the healthy subjects) (Fig. 2). Accordingly, results could be pooled for further analysis.
Fig. 2.
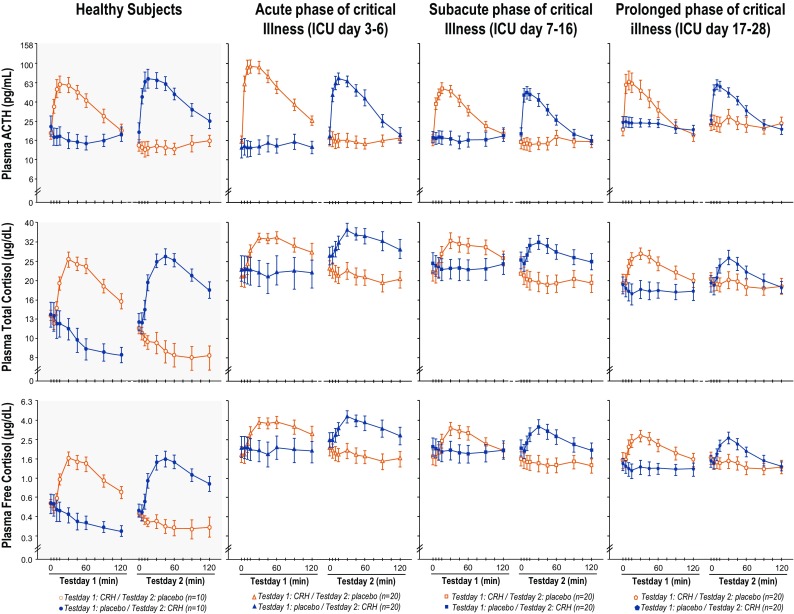
Plasma ACTH, total and free cortisol concentrations after CRH or placebo injection over time in ICU. Data are shown as mean ± SEM on a logarithmic scale. ICU denotes intensive care unit
As compared with ACTH responses of healthy subjects, the ACTH responses of patients in the acute phase of critical illness were similar, whereas those in the subacute and the prolonged phases were lower (P ≤ 0.05; Fig. 3a). The mean ACTH responses to CRH decreased by 55% from the acute to the subacute phase (P = 0.007), and remained constant from the subacute to the prolonged phase (P = 0.44; Fig. 3a).
Fig. 3.

Incremental a ACTH, b total cortisol and c free cortisol responses to CRH and placebo in 3 patient cohorts. The AUC hormone responses to placebo were subtracted from the AUC hormone responses to CRH and indicate the incremental hormone responses. Data are shown as mean ± SEM on a logarithmic scale. ICU denotes intensive care unit. The horizontal blue-shaded areas represent the mean ± SEM incremental hormone responses from the 20 healthy subjects. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.0001 for the comparisons with healthy subjects. The numerical P values are those for the comparisons between patient cohorts
Of the 120 unique patients, 30 patients were tested more than once. Of these 30 patients, 19 were tested in the acute and the subacute phase, and 14 were tested in the subacute and prolonged phase. Longitudinal analyses of these repetitive tests within patients confirmed the results of the unique patient cohorts, with a decrease of the mean ACTH responses to CRH by 60% from the acute to the subacute phase (P = 0.01) and no further change from the subacute to the prolonged phase (P = 0.74).
Plasma incremental (free)cortisol responses to CRH over time in ICU
As compared with total cortisol responses to CRH of healthy subjects, total cortisol responses of patients in the acute (P ≤ 0.05), subacute (P ≤ 0.05), and prolonged (P ≤ 0.001) phases of critical illness were always lower, whereas the free cortisol responses were always normal (Fig. 3b–c). As compared with the acute phase of critical illness, total cortisol responses to CRH tended to further lower (P = 0.08) and free cortisol responses further lowered (P = 0.05) in the prolonged phase (Fig. 3b–c).
In healthy subjects, the ACTH responses to CRH correlated positively with the total cortisol responses (P = 0.001, R2 = 0.43) and with free cortisol responses (P = 0.004, R2 = 0.37). Patients also showed positive correlations between ACTH and total cortisol responses (P = 0.001, R2 = 0.09) and between ACTH- and free cortisol responses (P = 0.0003, R2 = 0.10), but these correlations were much weaker than in healthy subjects.
Estimated half-life of plasma ACTH and (free)cortisol over time in ICU
The estimated plasma half-life of ACTH in patients was always similar to that in healthy subjects (P = 0.57, Fig. S1). The estimated plasma half-life of total cortisol was a mean 3.25-fold longer in patients than in controls (P = 0.0002), and the estimated plasma half-life of free cortisol was a mean 3.10-fold longer in patients than in controls (P = 0.006).
Comparison of survivors with non-survivors, and patients with and without sepsis/septic shock
The ACTH responses were always similar for hospital survivors and non-survivors, for patients with and without sepsis, and for patients with and without septic shock (Fig. 4). This also applied to the (free)cortisol responses (data not shown). The ACTH responses were also similar for patients who did or did not require renal replacement therapy or mechanical ventilatory support (data not shown).
Fig. 4.

Incremental ACTH responses to CRH and placebo in 3 patient cohorts, in a survivors and non-survivors, b patients with and without sepsis, and c patients with and without septic shock. The AUC ACTH responses to placebo were subtracted from the AUC ACTH responses to CRH and indicate the incremental ACTH responses. Data are shown as mean ± SEM on a logarithmic scale. ICU denotes intensive care unit. The horizontal blue-shaded areas represent the mean ± SEM incremental hormone responses from the 20 healthy subjects. The numerical P values are those for the comparisons between patient groups
Side effects of CRH injection
None of the patients revealed hemodynamic instability in response to any of the test injections, whereas a sense of flushing was reported by all healthy subjects on one of the 2 study days.
Discussion
In the presence of low/normal baseline plasma ACTH and increased plasma (free)cortisol concentrations, incremental ACTH responses to CRH in patients in the acute phase of critical illness were normal, whereas ACTH responses became ± 55% lower than normal in the later phases, irrespective of the presence of sepsis/septic shock or risk of death. Interestingly, the total cortisol responses to CRH were always lower than in healthy subjects whereas the free cortisol responses were always normal, in line with increased cortisol distribution volume during critical illness [6, 14]. The time courses of the ACTH responses to CRH were thus compatible with prolonged feed-back inhibition exerted by elevated free cortisol, rather than with hypothalamic and/or pituitary cell damage, similarly as seen with prolonged exposure to exogenous glucocorticoids [23]. These findings generate the hypothesis that CRH could offer potential for prevention of central hypoadrenalism in ICU patients who require intensive care for several weeks, for whom it has been shown that free cortisol levels are no longer elevated [14]. The absence of hemodynamic instability in response to the CRH injections in the patients of this study is an important safety aspect for future studies.
The observation of a normal ACTH response to CRH in the first few days of critical illness, similarly as documented by Schroeder et al. [20], argues against a damaged hypothalamus or pituitary by hypoperfusion or inflammation [4]. The finding that presence of sepsis or septic shock did not affect ACTH responses at any time during the course of critical illness further supports this interpretation. The 55% lowering of the ACTH responses to CRH in the subacute and prolonged phase of critical illness corroborates sustained feedback inhibition by elevated circulating free cortisol and is in line with the previously documented suppressed nocturnal pulsatile ACTH secretion during critical illness [13]. Indeed, a similar degree of suppression of the ACTH response to CRH has been reported for patients after surgical treatment for Cushing’s syndrome and for patients after withdrawal of ≥ 2 weeks of therapeutic glucocorticoid treatment [18, 26]. The suppressed ACTH responses to CRH observed in the subacute/prolonged phases of critical illness is compatible with low endogenous CRH and/or low vasopressin signaling [27], that both can be suppressed by high circulating levels of glucocorticoids [7]. Of note, baseline plasma ACTH concentrations were not completely suppressed and slightly increased over time. This is in line with earlier observations [6, 14] and suggests that during critical illness, specific central stimulatory pathways are still activated [9–12]. During health, hypothalamic CRH-neurons co-express CRH and AVP, which synergistically activate distinct signaling pathways within pituitary corticotropes [28]. It is well known that AVP is only a weak direct stimulator of ACTH but a much more powerful synergizer of CRH [7], and thus AVP action may be required for a normal ACTH response to exogenous CRH [29]. Vice versa, experiments in CRH knockout mice have shown that ACTH secretion depends on CRH [23, 30]. Reactivation of hypothalamic CRH secretion is indeed crucial for the reactivation of ACTH secretion after withdrawal of chronic glucocorticoid treatment [23]. Downregulation of CRH expression, via activating the glucocorticoid receptor, can be brought about by elevated free cortisol and/or by high circulating levels of bile acids that have previously shown to characterize subacute and prolonged critical illness [31, 32]. Also, a sustained endotoxin challenge could reduce CRH expression, although the observed comparable responses in patients with sepsis and in those without sepsis does not support a primary role for endotoxin or cytokines [33]. A postmortem study of human patients who died from septic shock after an illness of approximately 1 week, reported reduced ACTH mRNA levels in the pituitary gland [1]. This suppressed ACTH gene expression occurred in the absence of a compensatory rise in the expression of CRH and vasopressin in the hypothalamus and without altered expression of the CRH-receptor 1 and the vasopressin-receptor (V1b), supporting our current findings [1]. The results of the current study however cannot rule out a direct pituitary defect due to effects of inflammation and/or hypoxia selectively in the more prolonged phases of illness.
Remarkably, in all patients, irrespective of the duration of illness, total cortisol responses to CRH were lower than normal whereas free cortisol responses were always normal. This is in line with a recent study of long-stay patients who received weekly short ACTH stimulation tests for 4 weeks in the ICU, that revealed uniformly low incremental total cortisol responses but normal incremental free cortisol responses, explained by low plasma binding and increased cortisol distribution volume [14]. In the current study, with increasing duration of critical illness, both total and free cortisol responses tended to further decrease. This could be partially explained by the suppressed ACTH release in response to CRH and/or by the onset of decline of adrenocortical function. Indeed, appropriate ACTH signaling is essential to maintain integrity and function of the adrenal cortex [34]. A post-mortem study of adrenal glands harvested from patients who had been critically ill for several weeks showed loss of zonational structure, lipid droplet depletion, and suppressed ACTH-regulated gene expression [15]. Suppressed ACTH secretion could thus negatively affect adrenal function in long-stay ICU patients [13, 35]. Such a negative effect of suppressed ACTH could also explain why critically ill patients beyond the fourth week in the ICU were recently shown to have circulating total and free cortisol levels that were not higher than those of healthy subjects, despite their severe illness and high risk of death [14]. One week after ICU discharge on the regular ward, survivors had higher than normal plasma ACTH and total and free cortisol levels, although they were recovering. This further suggested a central adrenocortical suppression during the ICU phase, which could predispose long-stay ICU patients to central adrenal insufficiency.
A first limitation of this study is that, for obvious reasons, no hypothalamic and pituitary tissues were available for quantification of expression of CRH, vasopressin, ACTH, and of the CRH-receptor 1 and vasopressin-receptor. This should be done in validated animal models of prolonged critical illness [36]. A second limitation is that one cannot exclude additional suppression at the hypothalamic level from analgo-sedative drugs that are used throughout ICU stay, of which opioids are the main component [37]. Indeed, intra-operative opioids and prolonged opioid use for chronic pain have shown to lower plasma ACTH concentrations [38–42]. Furthermore, in healthy subjects, morphine blunts the ACTH response to CRH injection at a supra-pituitary level [43]. However, given the normal ACTH responses to CRH, observed during the acute phase, when opioid doses are usually higher than in the later phases, an important role of opioids is unlikely. Third, plasma free cortisol was calculated from plasma total cortisol with the Coolens method adapted for individual albumin and CBG concentrations, and not measured with ultra-filtration and equilibrium dialysis which could have induced some bias [44]. However, as also plasma total cortisol concentrations were always higher in patients, and given the clear decrease in both albumin and CBG, increased plasma free cortisol is obvious. Finally, we observed that throughout the acute, subacute and prolonged phases of critical illness, ACTH responses were not predictive for patient outcome. However, earlier smaller studies performed either in the acute-subacute [21] or prolonged [22] phases reported higher peak ACTH, but not AUC ACTH, responses in non-survivors than in survivors, a finding that could be at least partially biased by the variation in the day of death. The strengths of the study were the randomized, double-blind, placebo-controlled crossover design, which allowed to compare matched patients in different phases of critical illness while minimizing confounders.
Our findings open perspectives for novel strategies to protect long-stay ICU patients against the risk of developing adrenal insufficiency. If the lack of priming of the corticotropes by CRH would be responsible for reduced ACTH expression and secretion, providing CRH could potentially allow (re)activation of ACTH synthesis and release in response to any fall in cortisol and could hereby prevent adrenal atrophy in the prolonged phase of illness [45]. It has been shown that continuous infusion of CRH can reactivate ACTH secretion with preservation of circadian rhythmicity and pulsatility [46]. Studies of CRH infusion in the critically ill should probably initiate this intervention rather early, when the corticotropes are still fully responsive to CRH. If corticotropes remain sensitive to feedback inhibition, CRH infusion may not result in too high plasma cortisol and would respect any eventual tissue-specific regulation of cortisol action, which are important safety aspects. In the current study, no side effects of a CRH bolus were noted. However, caution is warranted given that CRH has also been involved in anxiety disorders, depression, memory and learning [47, 48], and is able to increase catecholamines and heart rate [49]. If a direct pituitary defect would be present in the prolonged phases of illness, which we could not exclude, CRH will not be able to prevent this.
In conclusion, the results of the CRH tests did not support the presence of shock/inflammation-induced hypothalamic and/or pituitary damage in critically ill patients. Instead, the consequences of prolonged feedback inhibition exerted by elevated (free)cortisol are compatible with suppressed ACTH responses to CRH in the prolonged phases of critical illness. These findings raise the hypothesis that CRH infusion could prevent the development of a central adrenal insufficiency in long-stay ICU patients, which should be further investigated.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank the patients and healthy volunteers for participating, the clinical staff for excellent protocol compliance and the research assistants for sample handling and data entry. We thank Ferring International Center SA, Switzerland for kindly providing study drug (CRH Ferring), to support scientific research in septic shock, and we thank dr. Johan Masure (Ferring Pharmaceuticals) for facilitating this support. This work was supported by the Research Foundation-Flanders (FWO) [Grant G091918 N to GVdB, research mandate 11W9315 N to BP]; by the Methusalem Program of the Flemish Government [METH/14/06 to GVdB and LL via KU Leuven]; by a European Research Council Advanced Grant [AdvG-2017-785809 to GVdB] from European Union’s Horizon 2020 research and innovation program.
Compliance with ethical standards
Conflicts of interest
The authors declare that they have no conflict of interest.
References
- 1.Polito A, Sonneville R, Guidoux C, Barrett L, Viltart O, Mattot V, Siami S, Lorin DLG, Chretien F, Singer M, Gray F, Annane D, Brouland JP, Sharshar T. Changes in CRH and ACTH synthesis during experimental and human septic shock. PLoSOne. 2011;6:e25905. doi: 10.1371/journal.pone.0025905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bornstein SR. Predisposing factors for adrenal insufficiency. N Engl J Med. 2009;360:2328–2339. doi: 10.1056/NEJMra0804635. [DOI] [PubMed] [Google Scholar]
- 3.Deutschman CS, Raj NR, McGuire EO, Kelz MB. Orexinergic activity modulates altered vital signs and pituitary hormone secretion in experimental sepsis. Crit Care Med. 2013;41:e368–e375. doi: 10.1097/CCM.0b013e31828e9843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Annane D. The role of ACTH and corticosteroids for sepsis and septic shock: an update. Front Endocrinol (Lausanne) 2016;7:70. doi: 10.3389/fendo.2016.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vuong C, Van Uum SH, O’Dell LE, Lutfy K, Friedman TC. The effects of opioids and opioid analogs on animal and human endocrine systems. Endocr Rev. 2010;31:98–132. doi: 10.1210/er.2009-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boonen E, Vervenne H, Meersseman P, Andrew R, Mortier L, Declercq PE, Vanwijngaerden YM, Spriet I, Wouters PJ, Vander PS, Langouche L, Vanhorebeek I, Walker BR, Van den Berghe G. Reduced cortisol metabolism during critical illness. N Engl J Med. 2013;368:1477–1488. doi: 10.1056/NEJMoa1214969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jameson J, De Groot L. Endocrinology: adult and pediatric. 7. Philadelphia: Elsevier; 2016. pp. 142–143. [Google Scholar]
- 8.Wagner-Bartak NA, Baiomy A, Habra MA, Mukhi SV, Morani AC, Korivi BR, Waguespack SG, Elsayes KM. Cushing syndrome: diagnostic workup and imaging features, with clinical and pathologic correlation. AJR Am J Roentgenol. 2017;209:19–32. doi: 10.2214/AJR.16.17290. [DOI] [PubMed] [Google Scholar]
- 9.Keller-Wood ME, Dallman MF. Corticosteroid inhibition of ACTH secretion. Endocr Rev. 1984;5:1–24. doi: 10.1210/edrv-5-1-1. [DOI] [PubMed] [Google Scholar]
- 10.Raff H, Sharma ST, Nieman LK. Physiological basis for the etiology, diagnosis, and treatment of adrenal disorders: Cushing’s syndrome, adrenal insufficiency, and congenital adrenal hyperplasia. Compr Physiol. 2014;4:739–769. doi: 10.1002/cphy.c130035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauger RL, Aguilera G. Regulation of pituitary corticotropin releasing hormone (CRH) receptors by CRH: interaction with vasopressin. Endocrinology. 1993;133:1708–1714. doi: 10.1210/endo.133.4.8404613. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe T, Oki Y, Orth DN. Kinetic actions and interactions of arginine vasopressin, angiotensin-II, and oxytocin on adrenocorticotropin secretion by rat anterior pituitary cells in the microperifusion system. Endocrinology. 1989;125:1921–1931. doi: 10.1210/endo-125-4-1921. [DOI] [PubMed] [Google Scholar]
- 13.Boonen E, Meersseman P, Vervenne H, Meyfroidt G, Guiza F, Wouters PJ, Veldhuis JD, Van den Berghe G. Reduced nocturnal ACTH-driven cortisol secretion during critical illness. Am J Physiol Endocrinol Metab. 2014;306:E883–E892. doi: 10.1152/ajpendo.00009.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peeters B, Meersseman P, Vander Perre S, Wouters PJ, Vanmarcke D, Debaveye Y, Billen J, Vermeersch P, Langouche L, Van den Berghe G. Adrenocortical function during prolonged critical illness and beyond: a prospective observational study. Intensive Care Med. 2018;44:1720–1729. doi: 10.1007/s00134-018-5366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boonen E, Langouche L, Janssens T, Meersseman P, Vervenne H, De Samblanx E, Pironet Z, Van Dijck L, Vander Perre S, Derese I, Van den Berghe G. Impact of duration of critical illness on the adrenal glands of human intensive care patients. J Clin Endocrinol Metab. 2014;99:4214–4222. doi: 10.1210/jc.2014-2429. [DOI] [PubMed] [Google Scholar]
- 16.Schulte HM, Chrousos GP, Avgerinos P, Oldfield EH, Gold PW, Cutler GB, Jr, Loriaux DL. The corticotropin-releasing hormone stimulation test: a possible aid in the evaluation of patients with adrenal insufficiency. J Clin Endocrinol Metab. 1984;58:1064–1067. doi: 10.1210/jcem-58-6-1064. [DOI] [PubMed] [Google Scholar]
- 17.Jameson J, De Groot L. Endocrinology: adult and pediatric. 7. Philadelphia: Elsevier; 2016. pp. 2655–2688. [Google Scholar]
- 18.Gomez MT, Magiakou MA, Mastorakos G, Chrousos GP. The pituitary corticotroph is not the rate limiting step in the postoperative recovery of the hypothalamic-pituitary-adrenal axis in patients with Cushing syndrome. J Clin Endocrinol Metab. 1993;77:173–177. doi: 10.1210/jcem.77.1.8392083. [DOI] [PubMed] [Google Scholar]
- 19.Reincke M, Allolio B, Wurth G, Winkelmann W. The hypothalamic-pituitary-adrenal axis in critical illness: response to dexamethasone and corticotropin-releasing hormone. J Clin Endocrinol Metab. 1993;77:151–156. doi: 10.1210/jcem.77.1.8392081. [DOI] [PubMed] [Google Scholar]
- 20.Schroeder S, Wichers M, Klingmuller D, Hofer M, Lehmann LE, von Spiegel T, Hering R, Putensen C, Hoeft A, Stuber F. The hypothalamic-pituitary-adrenal axis of patients with severe sepsis: altered response to corticotropin-releasing hormone. Crit Care Med. 2001;29:310–316. doi: 10.1097/00003246-200102000-00017. [DOI] [PubMed] [Google Scholar]
- 21.Dimopoulou I, Alevizopoulou P, Dafni U, Orfanos S, Livaditi O, Tzanela M, Kotanidou A, Souvatzoglou E, Kopterides P, Mavrou I, Thalassinos N, Roussos C, Armaganidis A, Tsagarakis S. Pituitary-adrenal responses to human corticotropin-releasing hormone in critically ill patients. Intensive Care Med. 2007;33:454–459. doi: 10.1007/s00134-006-0491-0. [DOI] [PubMed] [Google Scholar]
- 22.Schuster KM, Macleod JB, Fernandez JB, Kumar M, Barquist ES. Adrenocorticotropic hormone and cortisol response to corticotropin releasing hormone in the critically ill-a novel assessment of the hypothalamic-pituitary-adrenal axis. Am J Surg. 2012;203:205–210. doi: 10.1016/j.amjsurg.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 23.Paragliola RM, Papi G, Pontecorvi A, Corsello SM. Treatment with synthetic glucocorticoids and the hypothalamus-pituitary-adrenal axis. Int J Mol Sci. 2017;18:E2201. doi: 10.3390/ijms18102201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vanhorebeek I, Peeters RP, Vander Perre S, Jans I, Wouters PJ, Skogstrand K, Hansen TK, Bouillon R, Van den Berghe G. Cortisol response to critical illness: effect of intensive insulin therapy. J Clin Endocrinol Metab. 2006;91:3803–3813. doi: 10.1210/jc.2005-2089. [DOI] [PubMed] [Google Scholar]
- 25.Charmandari E, Hindmarsh PC, Johnston A, Brook CG. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: alterations in cortisol pharmacokinetics at puberty. J Clin Endocrinol Metab. 2001;86:2701–2708. doi: 10.1210/jcem.86.6.7522. [DOI] [PubMed] [Google Scholar]
- 26.Graber AL, Ney RL, Nicholson WE, Island DP, Liddle GW. Natural history of pituitary-adrenal recovery following long-term suppression with corticosteroids. J Clin Endocrinol Metab. 1965;25:11–16. doi: 10.1210/jcem-25-1-11. [DOI] [PubMed] [Google Scholar]
- 27.Beurel E, Nemeroff CB. Interaction of stress, corticotropin-releasing factor, arginine vasopressin and behaviour. Curr Top Behav Neurosci. 2014;18:67–80. doi: 10.1007/7854_2014_306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herman JP, McKlveen JM, Ghosal S, Kopp B, Wulsin A, Makinson R, Scheimann J, Myers B. Regulation of the hypothalamic-pituitary-adrenocortical stress response. Compr Physiol. 2016;6:603–621. doi: 10.1002/cphy.c150015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levin N, Shinsako J, Dallman MF. Corticosterone acts on the brain to inhibit adrenalectomy-induced adrenocorticotropin secretion. Endocrinology. 1988;122:694–701. doi: 10.1210/endo-122-2-694. [DOI] [PubMed] [Google Scholar]
- 30.Muglia LJ, Jacobson L, Luedke C, Vogt SK, Schaefer ML, Dikkes P, Fukuda S, Sakai Y, Suda T, Majzoub JA. Corticotropin-releasing hormone links pituitary adrenocorticotropin gene expression and release during adrenal insufficiency. J Clin Investig. 2000;105:1269–1277. doi: 10.1172/JCI5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vanwijngaerden YM, Wauters J, Langouche L, Vander Perre S, Liddle C, Coulter S, Vanderborght S, Roskams T, Wilmer A, Van den Berghe G, Mesotten D. Critical illness evokes elevated circulating bile acids related to altered hepatic transporter and nuclear receptor expression. Hepatology. 2011;54:1741–1752. doi: 10.1002/hep.24582. [DOI] [PubMed] [Google Scholar]
- 32.McMillin M, Frampton G, Quinn M, Divan A, Grant S, Patel N, Newell-Rogers K, DeMorrow S. Suppression of the HPA axis during cholestasis can be attributed to hypothalamic bile acid signaling. Mol Endocrinol. 2015;29:1720–1730. doi: 10.1210/me.2015-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beishuizen A, Thijs LG. Endotoxin and the hypothalamo-pituitary-adrenal (HPA) axis. J Endotoxin Res. 2003;9:3–24. doi: 10.1179/096805103125001298. [DOI] [PubMed] [Google Scholar]
- 34.Ferreira JG, Cruz CD, Neves D, Pignatelli D. Increased extracellular signal regulated kinases phosphorylation in the adrenal gland in response to chronic ACTH treatment. J Endocrinol. 2007;192:647–658. doi: 10.1677/joe.1.06961. [DOI] [PubMed] [Google Scholar]
- 35.Stavreva DA, Wiench M, John S, Conway-Campbell BL, McKenna MA, Pooley JR, Johnson TA, Voss TC, Lightman SL, Hager GL. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat Cell Biol. 2009;11:1093–1102. doi: 10.1038/ncb1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jenniskens M, Weckx R, Dufour T, Vander Perre S, Pauwels L, Derde S, Teblick A, Guiza F, Van den Berghe G, Langouche L. The hepatic glucocorticoid receptor is crucial for cortisol homeostasis and sepsis survival in humans and male mice. Endocrinology. 2018;159:2790–2802. doi: 10.1210/en.2018-00344. [DOI] [PubMed] [Google Scholar]
- 37.Pascoe JE, Williams KL, Mukhopadhyay P, Rice KC, Woods JH, Ko MC. Effects of mu, kappa, and delta opioid receptor agonists on the function of hypothalamic-pituitary-adrenal axis in monkeys. Psychoneuroendocrinology. 2008;33:478–486. doi: 10.1016/j.psyneuen.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor T, Dluhy RG, Williams GH. Beta-endorphin suppresses adrenocorticotropin and cortisol levels in normal human subjects. J Clin Endocrinol Metab. 1983;57:592–596. doi: 10.1210/jcem-57-3-592. [DOI] [PubMed] [Google Scholar]
- 39.Aloisi AM, Aurilio C, Bachiocco V, Biasi G, Fiorenzani P, Pace MC, Paci V, Pari G, Passavanti G, Ravaioli L, Sindaco G, Vellucci R, Ceccarelli I. Endocrine consequences of opioid therapy. Psychoneuroendocrinology. 2009;34(Suppl 1):S162–S168. doi: 10.1016/j.psyneuen.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 40.Abs R, Verhelst J, Maeyaert J, Van Buyten JP, Opsomer F, Adriaensen H, Verlooy J, Van Havenbergh T, Smet M, Van Acker K. Endocrine consequences of long-term intrathecal administration of opioids. J Clin Endocrinol Metab. 2000;85:2215–2222. doi: 10.1210/jcem.85.6.6615. [DOI] [PubMed] [Google Scholar]
- 41.Shinoda T, Murakami W, Takamichi Y, Iizuka H, Tanaka M, Kuwasako Y. Effect of remifentanil infusion rate on stress response in orthopedic surgery using a tourniquet application. BMC Anesthesiol. 2013;13:14. doi: 10.1186/1471-2253-13-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watanabe K, Kashiwagi K, Kamiyama T, Yamamoto M, Fukunaga M, Inada E, Kamiyama Y. High-dose remifentanil suppresses stress response associated with pneumoperitoneum during laparoscopic colectomy. J Anesth. 2014;28:334–340. doi: 10.1007/s00540-013-1738-x. [DOI] [PubMed] [Google Scholar]
- 43.Rittmaster RS, Cutler GB, Jr, Sobel DO, Goldstein DS, Koppelman MC, Loriaux DL, Chrousos GP. Morphine inhibits the pituitary-adrenal response to ovine corticotropin-releasing hormone in normal subjects. J Clin Endocrinol Metab. 1985;60:891–895. doi: 10.1210/jcem-60-5-891. [DOI] [PubMed] [Google Scholar]
- 44.Molenaar N, Groeneveld AB, de Jong MF. Three calculations of free cortisol versus measured values in the critically ill. Clin Biochem. 2015;48:1053–1058. doi: 10.1016/j.clinbiochem.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 45.Ur E, Capstick C, McLoughlin L, Checkley S, Besser GM, Grossman A. Continuous administration of human corticotropin-releasing hormone in the absence of glucocorticoid feedback in man. Neuroendocrinology. 1995;61:191–197. doi: 10.1159/000126840. [DOI] [PubMed] [Google Scholar]
- 46.Schulte HM, Chrousos GP, Gold PW, Booth JD, Oldfield EH, Cutler GB, Jr, Loriaux DL. Continuous administration of synthetic ovine corticotropin-releasing factor in man. Physiological and pathophysiological implications. J Clin Investig. 1985;75:1781–1785. doi: 10.1172/JCI111890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Behan DP, Grigoriadis DE, Lovenberg T, Chalmers D, Heinrichs S, Liaw C, De Souza EB. Neurobiology of corticotropin releasing factor (CRF) receptors and CRF-binding protein: implications for the treatment of CNS disorders. Mol Psychiatry. 1996;1:265–277. [PubMed] [Google Scholar]
- 48.Smith SR, de Jonge L, Pellymounter M, Nguyen T, Harris R, York D, Redmann S, Rood J, Bray GA. Peripheral administration of human corticotropin-releasing hormone: a novel method to increase energy expenditure and fat oxidation in man. J Clin Endocrinol Metab. 2001;86:1991–1998. doi: 10.1210/jcem.86.5.7491. [DOI] [PubMed] [Google Scholar]
- 49.Nijsen MJ, Croiset G, Stam R, Bruijnzeel A, Diamant M, de Wied D, Wiegant VM. The role of the CRH type 1 receptor in autonomic responses to corticotropin-releasing hormone in the rat. Neuropsychopharmacology. 2000;22:388–399. doi: 10.1016/S0893-133X(99)00126-8. [DOI] [PubMed] [Google Scholar]
- 50.Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med. 1985;13:818–829. doi: 10.1097/00003246-198510000-00009. [DOI] [PubMed] [Google Scholar]
- 51.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM consensus conference committee. American college of chest physicians/society of critical care medicine. Chest. 1992;101:1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 52.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC. The third international consensus definitions for sepsis and septic shock (Sepsis-3) JAMA. 2016;315:801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.