

Abstract
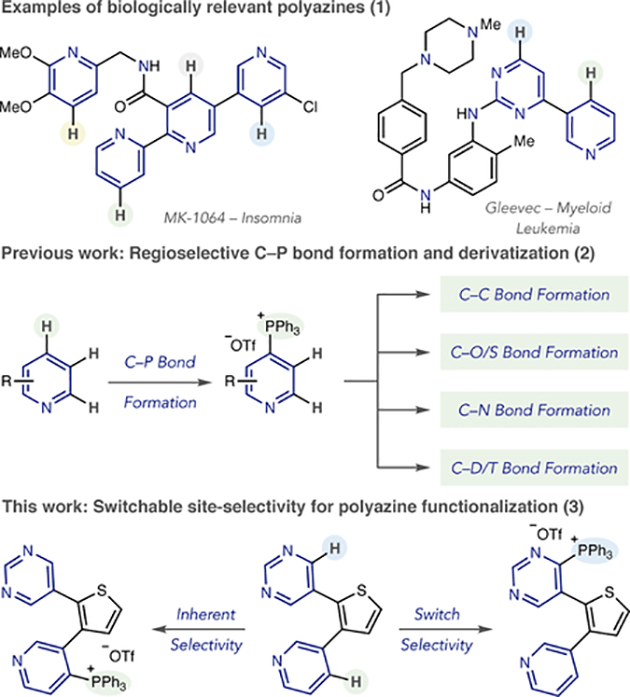
Many drug fragments and therapeutic compounds contain multiple pyridines and diazines. Developing site-selective reactions where specific C–H bonds can be transformed in polyazine structures would enable rapid access to valuable derivatives. We present a study that addresses this challenge by selectively installing a phosphonium ion as a versatile functional handle. Inherent factors that control site-selectivity are described along with mechanistically driven approaches for site-selective switching, where the C–+PPh3 group can be predictably installed at other positions in the polyazine system. Simple protocols, readily available reagents and application to complex drug-like molecules make this approach appealing to medicinal chemists.
Graphical Abstract
INTRODUCTION
When molecules have multiple occurrences of the same functional group or structural motif, site-selective reactions give immediate access to valuable derivatives without requiring prior synthesis from a simpler precursor.1 This superior efficiency is particularly relevant for biologically active molecules where structures are often complex and rapid access to analogues for structure activity relationship (SAR) studies expedites drug discovery.2 However, reaction development is a significant challenge in this regime; reactivity differences between sites is often dependent on subtle steric and electronic factors that must be recognized by the chemical entities involved in the process.
Significant progress has been made in selective alcohol functionalization of polyhydroxylated molecules and examples of polyene and polyarene derivatization have emerged.3–5 While metal-catalyzed C–H activation reactions have shown potential for site-selectivity between distinct classes of heteroaromatics, there are no general approaches to distinguish between molecules containing multiple pyridines and diazines.6–8 Polyazines are often found in therapeutic compounds (eq 1), and they are also abundant in medicinal chemistry libraries as lead compounds and drug fragments.9 Given the importance of polyazines in pharmaceuticals, site-selective functionalization strategies would be highly desirable.10 We herein report a study that addresses this challenge by selectively installing a phosphonium ion as a generic functional handle in polyazine structures. Furthermore, analyzing the reaction mechanism for C–P bond-formation has revealed several distinct strategies for switching site-selective selectivity; simple changes in reaction conditions allow the phosphonium ion to be installed at different positions in the scaffold.11
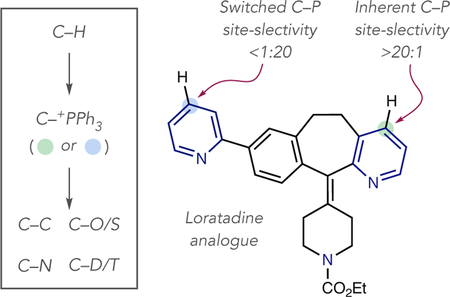
Our laboratory has shown that pyridines and diazines can be selectively converted into heterocyclic phosphonium salts using simple reagents and reaction protocols.12 The process has significant scope, including structures representative of drug fragments, and can be used as a tool for late-stage functionalization. C–P bond formation is exclusively selective for the 4-position of pyridines in most cases, and the phosphonium ion can be subsequently used as a functional handle to form C–C, C–O, C–S, C–N and C–D/T bonds (eq 2).13 During our investigations, we observed that site-selective phosphonium salt formation is possible, although the precise reasons for the selective outcomes were not always clear. Our purpose in this study is to systematically evaluate how substitution patterns, steric and electronic effects determine the inherent site of C–P bond formation and devise methods to predictably switch site-selectivity to other positions in polyazines (eq 3). Our hypothesis was that all three stages of the mechanism for C–P bond-formation could influence site-selectivity (Scheme 1): first, nucleophilic attack by the heterocyclic nitrogen on Tf2O; second, phosphine addition to the activated pyridinium triflyl salt; third, base-mediated elimination to reform the aromatic system. Furthermore, reaction parameters, such as order of reagent addition, were also envisioned to affect site-selectivity. Based on this mecha nistic approach, we herein present distinct strategies for switchable control of C–P bond formation in polyazines.
Scheme 1.

Mechanistic stages of phosphonium ion formation.
RESULTS AND DISCUSSION
An Acylation-Blocking Strategy.
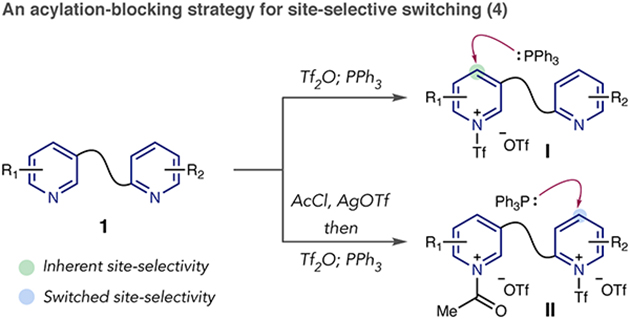
We reasoned that stage 1 of C–P bond formation would be influenced by steric and electronic properties of the nucleophilic azines, and a site-selective bias would occur when one azine is inherently more reactive towards Tf2O. As shown in equation 4, PPh3 would selectively add to one heterocycle in a polyazine (1) if a single triflyl salt isomer forms (I). Furthermore, we envisioned a blocking strategy for site-selective switching, where the more reactive heterocycle is acylated prior to adding Tf2O.11 In bis-salt (II), PPh3 should add to the more electrophilic triflyl pyridinium over the acyl pyridinium salt, resulting in a selectivity switch between different sites in a polyazine system.
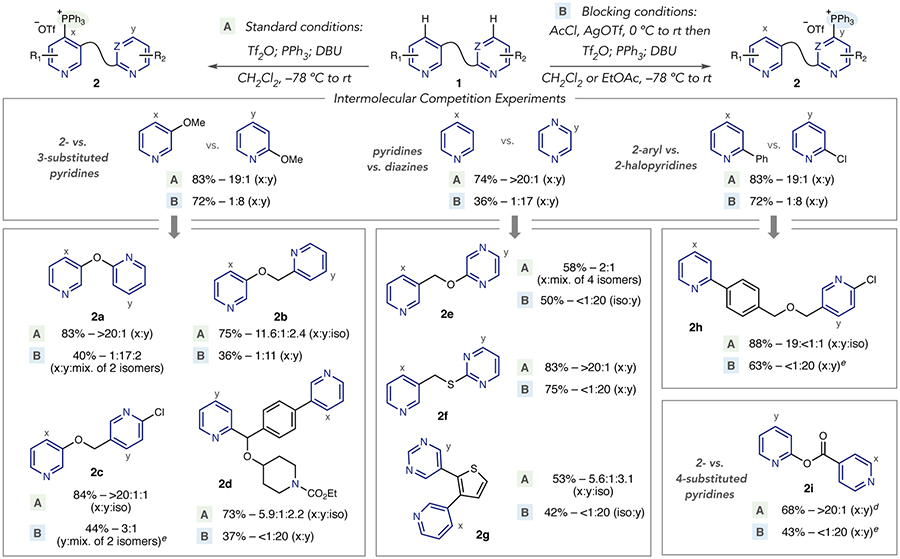
To rapidly identify inherent substrate biases and test the acylation-blocking strategy, we performed intermolecular competition experiments between pairs of azines (Table 1). Distinguishing between pyridines with 2- vs. 3-substitution patterns would apply to a large range of biologically relevant poylazine structures. Under standard conditions, a competition between 2- vs. 3-methoxy pyridine shows a clear bias towards the 3-substituted isomer, presumably due to the absence of steric crowding at the pyridine nitrogen. To test the acylation-blocking strategy, the pair of pyridines was first subjected to acetyl chloride and AgOTf at 0 °C in CH2Cl2, and stirred at room temperature for 1 hour.14 The mixture is then cooled to – 78 °C and the phosphonium salt forming protocol was conducted as normal.15 The combined phosphonium salts were isolated in good yield, with a significant reversal in selectivity towards the 2-substituted isomer.
Table 1.
 |
Isolated yields are reported of salt mixtures are reported.
Ratios were calculated from crude1H or31P NMR spectra. Ratios of isolated products can differ from the crude reaction mixture.
iso refers to an unidentified phosphonium salt isomer.
Yields calculated by1H NMR using 1,3,5-trimethoxybenzene as an internal standard.
P(p-OMePh)3 used.
After validating the acylation-blocking strategy in an intermolecular system, we anticipated that this switchable selectivity would translate into a set of bipyridines embedded with 2- and 3-substituents (Table 1). Phosphonium ion derivatives of a bis-pyridyl ether can be obtained on either heterocycle with good control of selectivity (2a). Bipyridine salt 2b shows an expected site-selectivity bias under standard conditions; surprisingly, a 2-position isomer is tentatively assigned as the next significant phosphonium species (vide infra). 2-Chloro substituents deter reactivity in the presence of 3-oxypyridines as shown in 2c; selectivity can be switched in this case to a moderate level on the chloro-substituted ring. A large number of derivatives could subsequently be obtained using the C–P and C–Cl bonds as functional handles. A derivative of an anti-histamine, bepotastine,16 bearing a piperidine ring and benzhydril stereocenter, is representative of a small molecule drug, or drug fragment, where site-selectivity is challenging. The inherent site of C–P bond formation shows a modest bias towards the 3-substituted pyridine (2d). The acylation-blocking strategy can be used to switch selectivity and provide usable quantities of the phosphonium ion on the 2-substituted ring.
An intermolecular competition reaction confirmed that phosphonium ion formation selectively occurs on pyridine over pyrazine, which reflects the significant difference in nucleophilicity of these heterocycles (Table 1). Gratifyingly, the selectivity trend can be reversed via the acylation-blocking protocol. Two pyridine-diazine systems, linked by oxymethylene and thiomethylene groups, respectively, were tested for the potential for site-selective switching. We were disappointed to observe that C–P bond formation in 2e did not match the intermolecular experiment (vide infra), and an unbiased mixture of salts was formed. However, acylation-blocking resulted in C–P bond formation on the pyrazine ring as the only detectable isomer. The pyridine-pyrimidine system showed greater reliability using both standard and switching conditions (2f); single phosphonium salt isomers were detected in the crude31P NMRs of the reaction mixtures and the salts were isolated in good yields. A thiophene-linked system, containing pyri-dine and pyrimidine rings, was moderately selective under standard conditions (2g). Like 2b and bepotastine derivative 2d, a 2-position salt is a significant component of the reaction mixture, and it is important to note that we have only seen significant levels of 2-position selectivity in polyazines. Acylation-blocking was highly selective for the pyrimidine ring in moderate yield with the remainder of the mass balance being unreacted starting material.
Selecting between different 2-substituted pyridines is a challenge due to comparable steric effects. We examined an electronically biased system by competing 2-phenylpyridine with 2-chloropyridine; the preference for phenyl substitution indicates that a carbon-bearing group is more favorable to an inductively withdrawing substituent. A representative fragment, containing a 2-aryl and 2-chloro motif was synthesized, and under standard conditions selectivity for the 2-aryl pyridine was observed (2h). In a1H NMR study, only the 2-arylpyridine ring protons were downshifted after adding Tf2O, in line with the observed selectivity.17 Both pyridine rings showed downfield shifts under the acylation-blocking conditions, and this protocol results in a complete reversal of selectivity towards the chloro-substituted pyridine in 2h. Notably, P(p-OMePh)3 provided higher yields than PPh3 for this system.18 Bipyridine 2i shows that a 2-acyloxypyridine is selectively functionalized in a 2,4-substituted bipyridine and that selectively can again be reversed using the blocking strategy. P(p-OMePh)3 was again superior to PPh3 in this case.18
Base-Mediated Switching.
The nature of the organic base can be tuned to control site-selectivity by exploiting structural differences during stage 3 of the reaction mechanism (Scheme 1). DBU and NEt3 are typically used, with DBU generally providing higher yields for most substrates in the reaction scope. We made an unexpected observation for certain 3-substituted pyridines that suggested site-selectivity may be controlled by base selection (Scheme 2A). Both DBU and NEt3 are effective for 3-methoxypyridine, however no product is obtained for 3-methyl and 3-phenylpyridine when NEt3 is used as a base. We hypothesized that a difference in acidity, or steric effects, preclude elimination of the triflyl anion in dearomatized intermediate III when an alkyl or aryl group is present. Scheme 2B illustrates the challenge for 3,3-bipyridine systems such as 1j; an unselective outcome is observed under standard conditions due to a similar proclivity of each pyridine nitrogen to react with Tf2O (2j). A lack of steric and electronic bias also precludes using the acylation-blocking strategy, so we conceived of an approach using base dependent selectivity. Using two equivalents each of Tf2O and PPh3, double-dearomatized bis-salt IV is a putative intermediate where only the oxy-substituted form is susceptible to elimination by NEt3. Low temperature31P NMR shows two major resonances at 21.93 ppm and 19.53 ppm in a 1:1 ratio indicating that a bisphosphonium salt is present. Adding NEt3 results in complete C–P bond site-selectivity for the 3-oxypyridine, while the 3-alkylpyridine is reformed under the reaction conditions.
Scheme 2. Effect of base on reactivity and site-selectivitya, b.

aYields and ratios reported as in Table 1.bYields calculated by1H NMR spectroscopy of the crude reaction mixture.
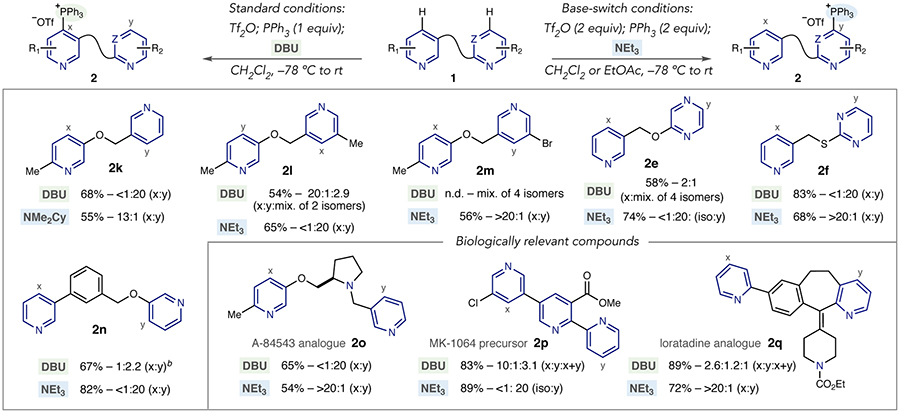
A series of oxymethyl-linked bipyridine systems was first selected to explore base effects on site-selectivity (Table 2). Under standard conditions, the 3-substituted pyridine is preferred over the 2,5-system in 2k. Using NMe2Cy under base-switch conditions results in phosphonium ion formation on the opposite pyridine ring; NEt3 was also effective in this system but in lower yield.19 Under standard conditions, the 3,5-disubstituted pyridine ring is preferred in 2l and the crude ratio improves to >20:1 after purification; base-switching conditions give access to the 3-oxy pyridine case with excellent selectivity. A bromo-containing bipyridine resulted in a complex mixture of four phosphonium isomers under the standard protocol (2m) but a site-selective outcome is observed using base-switch conditions. The same base effect is also observed between pyridines and diazines, as shown in examples 2e and 2f. As previously described in Table 1, standard conditions are less effective for pyridine-pyrazine 2e, with several phosphonium isomers observed (vide infra). However, base-switching conditions result in excellent selectivity for a single site on the pyrazine ring. Pyridine-pyrimidine salt 2f is also amenable to this base-switching strategy. A 3-aryl linker can be used as a controlling element in site-selective phosphonium ion formation (2n).
Table 2.
 |
Yields and ratios reported as in Table 1.
Yield calculated by1H NMR using 1,3,5-trimethoxybenzene as an internal standard.
x+y represents a bis-phosphonium salt.
We next selected a set of biologically active complex polyazines to test the base-switching strategy. An analogue of A-84543,20 a nicotinic acetylcholine receptor agonist, can be selectively converted into phosphonium ion derivatives on each pyridine ring with complete control of regio- and site-selectivity (2o). The tripyridine system in an MK-1064 precursor is a challenging target for site selective functionalization. Under standard conditions, moderate selectivity for the 3,5-disubstituted ring is observed, along with a mixture of a bisphosphonium salt and substitution on the 2-substituted pyri-dine (2p, vide infra). The 3,5-system can, however, be recognized by NEt3 under the base-switching conditions resulting in exclusive C–P bond formation on the 2-substituted ring. A loratadine analogue shows poor selectivity under standard conditions (2q), but the presence of a 2,3-fused carbocycle enables NEt3 to distinguish between the two pyridines with complete site-selectivity.
Order of Reagent Addition.
Several examples in this study are moderately site-selective under standard conditions and we have found that the order that the reagents are added can significantly affect these outcomes. Under the standard protocol, the polyazine is stirred with Tf2O for 30 minutes at –78 °C before PPh3 is added. This order is chosen to minimize an unwanted reaction between Tf2O and PPh3 that we have observed results in Ph3P=O. We revisited phosphonium salt formation on loratadine analogue (1q), where modest site-selectivity was observed under standard conditions (Table 2 and Scheme 3A). When the order of addition is reversed, and PPh3 is added before Tf2O, site-selectivity is significantly increased and the product phosphonium salt is isolated in good yield with minor amounts of Ph3P=O formed. This dichotomy can be rationalized by an equilibrium mixture of pyridinium-Tf salt isomers V, VI and VII formed under standard conditions (Scheme 3B). When 1q is stirred with Tf2O, the1H NMR spectrum displays a downfield shift of all pyridine protons indicating that a rapidly interconverting mixture of Tf salt isomers is present (see Supporting Information for more details). Site-selectivity is then related to the relative populations of V, VI and VII. When PPh3 is present before Tf2O is added, we propose that site-selectivity reflects a kinetic regime where pyridinium-Tf salt V forms fastest, and rapidly reacts with PPh3.
Scheme 3. Impact of reagent order on site-selectivitya,b,c,d,e.

aYields and ratios reported as in Table 1.bYield calculated by1H NMR using 1,3,5-trimethoxybenzene as an internal standard.cx+y represents a bis-phosphonium salt.diso refers to an unidentified phosphonium salt isomer.eMixture of two undefinited isomers
Other substrates with moderate selectivity were retested, as well as additional examples, to further assess the impact of reagent order on site-selectivity (Scheme 3C). A bepotastine analogue that produced a mixture of phosphonium salt isomers can be significantly improved by reversing the order of addition (2d). Reaction efficiency and site-selectivity in pyridinepyridine and pyridine-diazine systems 2b, 2e and 2g are also improved under these conditions. An MK-1064 precursor, that resulted in a mixture of mono- and bis-phosphonium salts, forms a single C–P bond isomer on the 3,5-disubstituted pyri-dine in the crude31P NMR (2p). Bipyridine salt 2l is an inter esting case where reversing the order of Tf2O and PPh3 is detrimental to site-selectivity. Increasing stirring time with Tf2O,before adding PPh3, results in a stronger bias towards the 3,5-disubstituted pyridine and presumably indicates more time is needed to establish a biased pyridinium-Tf salt equilibrium.Adding PPh3 before Tf2O results in poor site-selectivity and suggests that kinetic Tf-salt formation is unselective. Given the impact of reagent order on site-selectivity, we recommend alternating the order that Tf2O and PPh3 are added for substrates where site-selectivity is modest.
Initial Investigations of [m,n]-Bipyridines and Pyridine-Diazines.
Bis-azine biaryls are an important class of compounds that are encountered in numerous biologically active molecules.21 Our initial investigations into site-selective C–P bond formation have shown that inherent selectivity under standard conditions is highly effective, however, applying the site-selective switching strategies described in this report have proven challenging. Scheme 4A shows three representative cases of linked pyridine-pyridine and pyridine-diazine systems. Phosphonium ion formation occurs with excellent levels of site-selectivity and is in line with predisposed reactivity with Tf2O (2r-2t). Both acylation-blocking and base-switching strategies resulted in no product formation for bipyridines 2r and 2s. Our current hypothesis is that the adjacent pyridinium salts formed in these cases have heightened electrophilicity such that PPh3 reacts with the Tf-portion of the salts, at sulfur, and is oxidized; O=PPh3 is the major phosphorus species observed in the crude31P NMR. Pyridine-diazine 2t shows that the switching strategies can be effective, albeit in low yield.
Scheme 4. Phosphine-dependent site-selectivitya,b.

aYields and ratios reported as in Table 1.bYields calculated by1H NMR spectroscopy of the crude reaction mixture.
Scheme 4B shows a 3,3’-bipyridine where site-selective switching could be triggered by an alternative approach. Under standard conditions, phosphine addition favors the 3,5-disubstituted pyridine, with P(p-OMePh)3 proving superior to PPh3 as a nucleophile (2u).22 This outcome is again expected based on Tf-salt VIII forming selectively during the reaction. As above, attempts to switch site-selectivity using the acylation-blocking strategy resulted in no formation of any phosphonium ion species. We envisioned that site-selectivity could be controlled by selective phosphine addition to a bispyridinium salt (Scheme 4B, VIII), where the less hindered 2,5-system would be the preferred site of attack. Adding 1.75 equivalents of PPh3 to a pre-stirred mixture of the bipyridine and two equivalents of Tf2O resulted in a reversal in selectivity to the 2,5-disubstituted pyridine in 50% yield. As linked bipyridines are important in the chemical sciences, we are continuing to investigate site-selective processes involving these systems.
Derivatization reactions.
To show that site-selective phosphonium ion formation is useful for generating a range of ana logue compounds, we performed a series of derivatization reactions (Scheme 5). Both isomers of pyridine-pyrimidine 1f are accessible in high levels of site-selectivity using standard and base-switching conditions (2f, Table 2). We first tested carbon-heteroatom bond-forming reactions using our previously reported protocols;13 heteroaryl ethers 3a and 4a are formed efficiently on both ring systems using alcohol 5. Similarly, heteroaryl thioethers can be obtained using benzyl thio-late as a nucleophile (3b and 4b). Heating with sodium azide, followed by hydrolysis of the iminophosphorane products, results in heteroaryl aniline isomers 3b and 4b. Hydrogen isotopes can be incorporated into different sites of 1m by subjecting the respective phosphonium salts to K2CO3 in the presence of a mixture of CD3OD and D2O. Finally, a sequence of reactions can be used to create drug-like scaffold 6. Selective phosphonium salt formation on the pyrimidine ring of 1u is followed by nickel-catalyzed cross-coupling to install a thiophene group. Second phosphonium ion formation selectively occurs on the pyridine ring, and C–O bond formation using azetidine-containing alcohol 5 forms compound 6.
Scheme 5. Derivatizations of phosphonium salt isomersa,b.

aIsolated yields are shown and ratios are reported as in Table 1.bThe minor isomer is a 2-position pyridine phosphonium salt.
CONCLUSIONS
This study has established factors that control site-selective phosphonium ion formation in poylazine systems. Steric and electronic factors in pyridines and diazines dictate the inherent site of C–P bond formation, which is predictable for complex polyazine systems. Examining the reaction mechanism for C–P bond formation allowed us to develop strategies for site selective switching where phosphonium ion formation can be directed to other positions in a polyazine scaffold. Acylation-blocking, base-dependent selectivity and selective phosphine addition enable control of selectivity in polyazines with distinct substitution patterns and electronic properties. The order that reagents are added also has a significant impact on site-selectivity in polyazine systems. We believe that this study will be useful in applications such as medicinal and agrochemistry as well as applications such as ligand design. Further studies into site-selectivity are ongoing in our laboratory and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by start-up funds from Colorado State University and The National Institutes of Health under award number R01 GM124094.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and spectral data (PDF)
REFERENCES
- (1).(a) Mahatthananchai J; Dumas AM; Bode JW Angew. Chem. Int. Ed 2012, 51, 10954. [DOI] [PubMed] [Google Scholar]; (b) Huang Z; Dong G Acc. Chem. Res 2017, 50, 465. [DOI] [PubMed] [Google Scholar]
- (2).(a) Silverman RB; Holladay MW Lead Discovery and Lead Modification The Organic Chemistry of Drug Design and Drug Action, 3rd Ed.; Elsevier: Waltman, MA, 2014; pp. 19–106. [Google Scholar]; (b) Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW Chem. Soc. Rev 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]
- (3).(a) Lewis CA; Miller SJ Angew. Chem. Int. Ed 2006, 45, 5616. [DOI] [PubMed] [Google Scholar]; (b) Wilcock BC; Uno BE; Bromann GL; Clark MJ; Anderson TM; Burke MD Nature Chem 2012, 4, 996. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jordan PA; Miller SJ Angew. Chem. Int. Ed 2012, 51, 2907. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Han S; Miller SJ J. Am. Chem. Soc 2013, 135, 12414. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sun X; Lee H; Tan KL Nature Chem. 2013, 5, 790. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Bastain AA; Marcozzi A; Herrmann A Nature Chem. 2012, 4, 789. [DOI] [PubMed] [Google Scholar]
- (4).(a) Lichtor PA; Miller SJ Nature Chem. 2012, 4, 990. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pathak TP; Miller SJ J. Am. Chem. Soc 2012, 134, 6120. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Snyder SA; Gollner A; Chiriac M I. Nature 2011, 474, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).For an example of site-selective functionalization of sp3-rich compounds see: Roiban G-D.; Reetz MT Chem. Commun 2015, 51, 2208. [DOI] [PubMed] [Google Scholar]
- (6).(a) Kalyani D; Dick AR; Anani W,Q; Sanford MS Tetrahedron 2006, 62, 11483. [DOI] [PubMed] [Google Scholar]; (b) Lapointe D; Markiewicz T; Whipp CJ; Toderian A; Fagnou KJ Org. Chem 2011, 76, 749. [DOI] [PubMed] [Google Scholar]
- (7).Zoltewicz JA; Cruskie MP Jr. Tetrahedron 1995, 51, 3103. [Google Scholar]
- (8).For an example of site-selective functionalization of benzylic and heterobenzylic positions see: Cooper JC; Luo C; Kameyama R; Van Humbeck JF J. Am. Chem. Soc 2018, 140, 1243. [DOI] [PubMed] [Google Scholar]
- (9).(a) Baumann M; Baxendale IR Beilstein J. Org. Chem 2013, 9, 2265. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vitaku E; Smith DT; Njardarson JT J. Med. Chem 2014, 57, 10257. [DOI] [PubMed] [Google Scholar]
- (10).Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Nature Chem. 2018, 10, 383. [DOI] [PubMed] [Google Scholar]
- (11).Bull JA; Mousseau JJ; Pelletier G; Charette AB Chem. Rev 2012, 112, 2642. [DOI] [PubMed] [Google Scholar]
- (12).(a) Hilton MC; Dolewski RD; McNally A J. Am. Chem. Soc 2016, 138, 13806. [DOI] [PubMed] [Google Scholar]; (b) Anders E; Markus F Tetrahedron Lett. 1987, 28, 2675. [Google Scholar]; (c) Anders E; Markus F Chem. Ber 1989, 122, 113. [Google Scholar]; (d) Anders E; Markus F Chem. Ber 1989, 122, 119. [Google Scholar]; (e) Haas M; Goerls H; Anders E Synthesis 1998, 195. [Google Scholar]
- (13).(a) Zhang X; McNally A Angew. Chem. Int. Ed 2017, 56, 9833. [DOI] [PubMed] [Google Scholar]; (b) Koniarczyk JL; Hesk D; Overgard A; Davies IW; McNally AJ Am. Chem. Soc 2018, 140, 1990. [DOI] [PubMed] [Google Scholar]; (c) Anderson RG; Jett BM; McNally A Tetrahedron http://dx.doi.org/10.1016/j.tet.2017.12.040. [Google Scholar]; (d) Patel C; Mohnike ML; Hilton MC; McNally A Org. Lett 2018, 20, 2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).In the absence of AgOTf, we observe no product formation; the presence of chloride ions appears to prevent phosphonium ion formation.
- (15).The acylpyridinium salt persists at the end of the reaction so two equivalents of pyridine are added in the work up to release the product via a transamination mechanism.
- (16).Kato M; Nishida A; Aga Y; Kita J; Kudo Y; Endo T Arzneimittelforschung 1997, 47, 1116. [PubMed] [Google Scholar]
- (17).See the Supporting Information for more details.
- (18).1H NMR yields using 1,3,5-trimethoxybenzene as an internal standard: 2h, PPh3 – 28%, P(p-OMePh)3 – 72%. For 2i, PPh3 resulted in a mixture of 4 phosphonium species in the 31P NMR.
- (19).1H NMR yields for 2k using 1,3,5-trimethoxybenzene as an internal standard: NMe2Cy – 68%,13:1 (x:y), NEt3 – 28%, 2.5:1 (x:y).
- (20).Ogunjirin AE; Fortunak JM; Brown LL; Xiao Y; Dávila-García MI Neurochem. Res 2015, 40, 2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Campeau L-C; Fagnou K Chem. Soc. Rev 2007, 36, 1058–1068. [DOI] [PubMed] [Google Scholar]
- (22).1H NMR yields for 2w using 1,3,5-trimethoxybenzene as an internal standard: PPh3 – 27%, 3.5:1 (x:y), P(p-OMePh)3 – 60%, 15:1 (x:y). Tf2O was added after the phosphine in these reactions.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.